
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity of halfsandwich rare-earth metal methylaluminates toward potassium (2,4,6-tri-tert-butylphenyl)amide and 1-adamantylamine†
Dorothea
Schädle
a,
Markus
Enders
b,
Christoph
Schädle
a,
Cäcilia
Maichle-Mössmer
a,
Karl W.
Törnroos
c and
Reiner
Anwander
*a
aInstitut für Anorganische Chemie, Universität Tübingen, Auf der Morgenstelle 18, D-72076 Tübingen, Germany. E-mail: reiner.anwander@uni-tuebingen.de; Fax: +49 7071 29 35271; Tel: +49 7071 29 72069
bAnorganisch-Chemisches Institut, Universität Heidelberg, Im Neuenheimer Feld 270, D-69120 Heidelberg, Germany
cDepartment of Chemistry, University of Bergen, Allégaten 41, N-5007 Bergen, Norway
First published on 14th May 2015
Abstract
The equimolar reaction of potassium (2,4,6-tri-tert-butylphenyl)amide with Cp*Ln(AlMe4)2 (Cp* = 1,2,3,4,5-pentamethyl cyclopentadienyl) yielded {Cp*Ln(AlMe4)[NH(mes*)]}x (Ln = Y, La; mes* = C6H2tBu3-2,4,6). The treatment of Cp*Ln(AlMe4)[NH(mes*)] with tetrahydrofuran led to intramolecular C–H bond activation of a tBu group with the formation of Cp*YMe{NH[C6H2tBu2-2,4-(CMe2CH2)-6]}(AlMe2)(thf). A similar methyl-anilide species CpQLuMe{NH[C6H2tBu2-2,4-(CMe2CH2)-6]}(AlMe2) (CpQ = 2,3,4,5-tetramethyl-1-(8-quinolyl)cyclopentadienyl) with a C–H bond activated ligand backbone formed by the reaction of CpQLu(AlMe4)2 and K[NH(mes*)]. The reactivity of CpQY(AlMe4)2 toward H2NAd (Ad = adamantyl) ultimately led to the methyl–amide complex CpQYMe[NH(Ad)](AlMe3), corroborating the presence of competing deprotonation and donor-induced methylaluminate cleavage reactions. The halfsandwich complexes CpQLu(AlMe4)2, Cp*Y(AlMe4)[NH(mes*)], Cp*YMe{NH[C6H2tBu2-2,4-(CMe2CH2)-6]}(AlMe2)(thf), CpQLuMe{NH[C6H2tBu2-2,4-(CMe2CH2)-6]}(AlMe2), and CpQYMe[NH(Ad)](AlMe3) as well as the side-product AlMe3(H2NAd) were fully characterized by NMR/FTIR spectroscopy, elemental analysis, and X-ray crystallography.
Introduction
Halfsandwich rare-earth metal bis(hydrocarbyl) complexes1 have emerged as versatile synthesis precursors (e.g., for hydrido clusters)2 and eminent (pre)catalysts for polymerization reactions (e.g., fabrication of syndiotactic polystyrene).3 In 2003, we introduced halfsandwich rare-earth metal bis(methylaluminate) complexes as thermally quite stable variants of the aforementioned bis(alkyl) derivatives.4 Upon cationization with fluorinated phenylborane/borate reagents complexes CpRLn(AlMe4)2 promote the living 1,4-trans-selective polymerization of isoprene (synthetic gutta-percha)5 and butadiene.6 Such catalyst activation proceeds via well-established protonolysis and alkyl abstraction pathways.7 The feasibility of protonolysis reactions was also demonstrated for the syntheses of metallocenes8 as well as silica-grafted hybrid materials and respective alkoxide/siloxide model complexes.9 Furthermore, complexes of the type CpRLn(AlMe4)2 were successfully applied in C–H bond activation protocols giving access to the first structurally characterized rare-earth metal methylidyne10 and methylidene complexes.11,12 In the following, we adopted the concept of alkylaluminate-based organoaluminium-assisted deprotonation for the synthesis of rare-earth metal imide complexes.13 One major finding was that the superbulky monanionic TptBu,Me (= hydrotris(3-tert-butyl-5-methyl-pyrazolyl)borate) ligand can afford monolanthanide derivatives of the type [(TptBu,Me)Ln(NAr)(AlHMe2)] (Ar = C6H3Me2-2,6)14 and [(TptBu,Me)Ln(NR)(AlMe3)] (Ln = Y, Ho; R = tBu, adamantyl).15 The isolation of rare-earth metal complexes [(TptBu,Me)Ln=NR(dmap)] (Ln = Y, Ar = C6H3Me2-2,6; Ln = Lu, Ar = C6H3(CF3)2-3,5) with a terminally bonded imido ligand was achieved by an aluminium-free synthesis procedure.16 Prior to our studies it has been shown that the deprotonation of rare-earth metal anilide complexes with organoaluminium or alkyl lithium reagents (e.g.Scheme 1, V)17 is a viable strategy toward LnIII imide complexes.17,18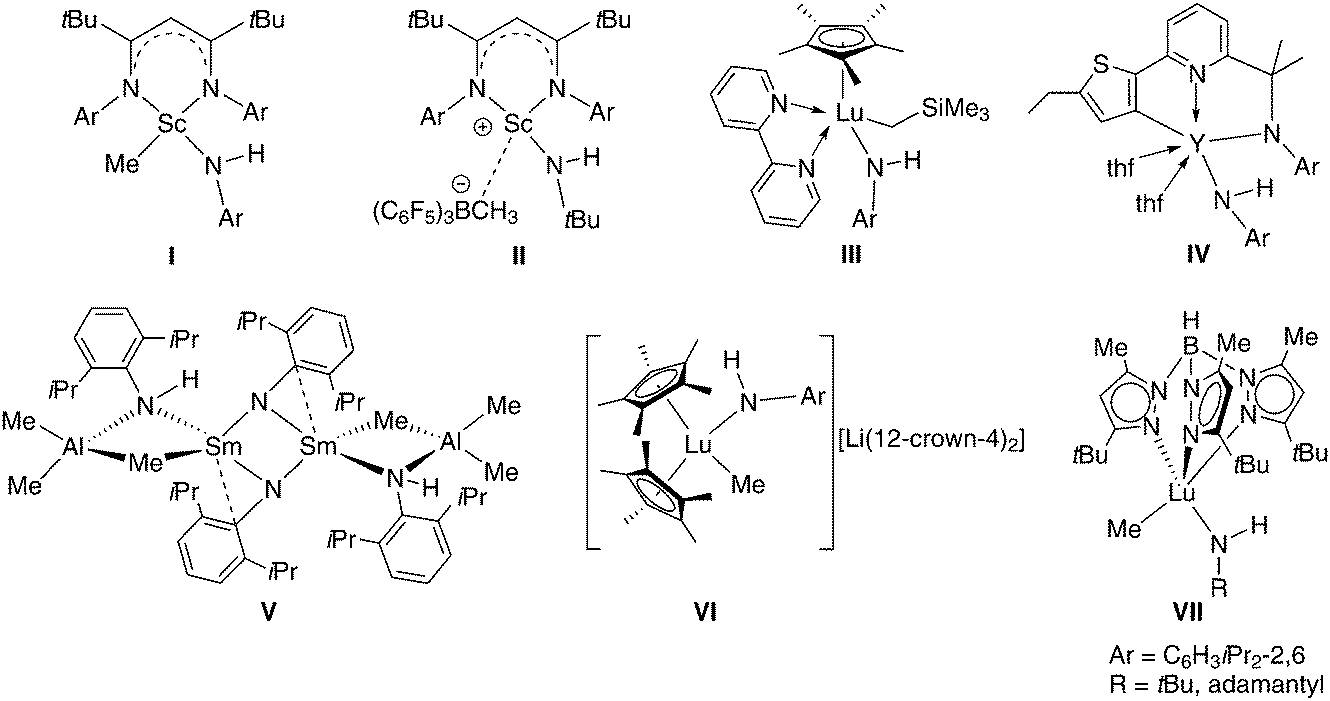 | ||
| Scheme 1 Structurally characterized rare-earth metal complexes with the amido NH functionality adjacent to Ln–C(alkyl).17,22–24 | ||
Given the plethora of group 4 imide complexes,19 rare-earth metal imide chemistry is still in its infancy.20 Mountford and coworkers examined the efficiency of deprotonation reactions utilizing, e.g. TiCl4 or Ti(NMe2)4 in amine elimination and transamination/transimination reactions, respectively.21 It is noteworthy that such synthesis protocols are not applicable for the rare-earth metals (vide infra).
Based on the successful synthesis of rare-earth metal imide complexes exploiting the synergistic effect associated with organoaluminium moieties and the superbulky monanionic TptBu,Me, we have now set out to explore cyclopentadienyl-supported rare-earth metal bis(tetramethylaluminate)s as potential precursors for imide complexes. Herein, we describe the reactivity of Cp*Ln(AlMe4)2 and CpQLn(AlMe4)2 toward potassium (2,4,6-tri-tert-butylphenyl)amide and 1-adamantylamine, respectively (Cp* = 1,2,3,4,5-pentamethylcyclopentadienyl, CpQ = 2,3,4,5-tetramethyl-1-(8-quinolyl)cyclopentadienyl). Overall, this study discloses a better understanding of why steric/electronic mismatches in isolable mixed amide–alkyl complexes hamper their conversion into the respective imide derivatives.22–24 As compiled in Scheme 1, various mixed amide–alkyl complexes (including cationic (II)22b and anionic rare-earth metal entities (VI)22c) have been accessed in the presence of a stabilizing N-donor (I,22aII,22bIV,23VII,15) and cyclopentadienyl ancillary ligands (III,24VI22c).
Results and discussion
Reactivity of Cp*Ln(AlMe4)2 (1)
The salt-metathesis reaction of half-sandwich complexes Cp*Ln(AlMe4)2 (Ln = Y (1a), La (1b))8 with 1 equivalent potassium (2,4,6-tri-tert-butylphenyl)amide, K[NH(mes*)], in n-hexane at ambient temperature yielded {Cp*Ln(AlMe4)[NH(mes*)]}x (Ln = Y (2a), x = 2; Ln = La (2b)) in 83% and 87% yield, respectively (Scheme 2). Rather surprisingly, the reaction came to a halt at the heteroleptic amide–methylaluminate complex and the subsequent deprotonation of the NH anilido functionality did not occur. The mixed amide–imide complex [Sm(NHAr)(AlMe3)(NAr)]217 is the only other example featuring an NH(Ar) ligand adjacent to an organoaluminium moiety (V, Scheme 1). The latter complex V was obtained from Sm(NHAr)3 and AlMe3. For comparison, excess addition of trimethylaluminium to Nd(NHPh)3 (Ph = phenyl) led to a completely deprotonated imide complex [Nd(AlMe4)2(NPh)(AlMe2)]2 and co-product [(NHPh)AlMe2]3 formed by anilido-methyl exchange.18a Aryloxide derivatives [Cp*Ln(AlMe4)(OArtBu,R)] (Ln = Lu, Y; R = H, Me) reminiscent of complexes 2 were obtained by the treatment of [Cp*Ln(OArtBu,R)2] with an excess of trimethylaluminium.9b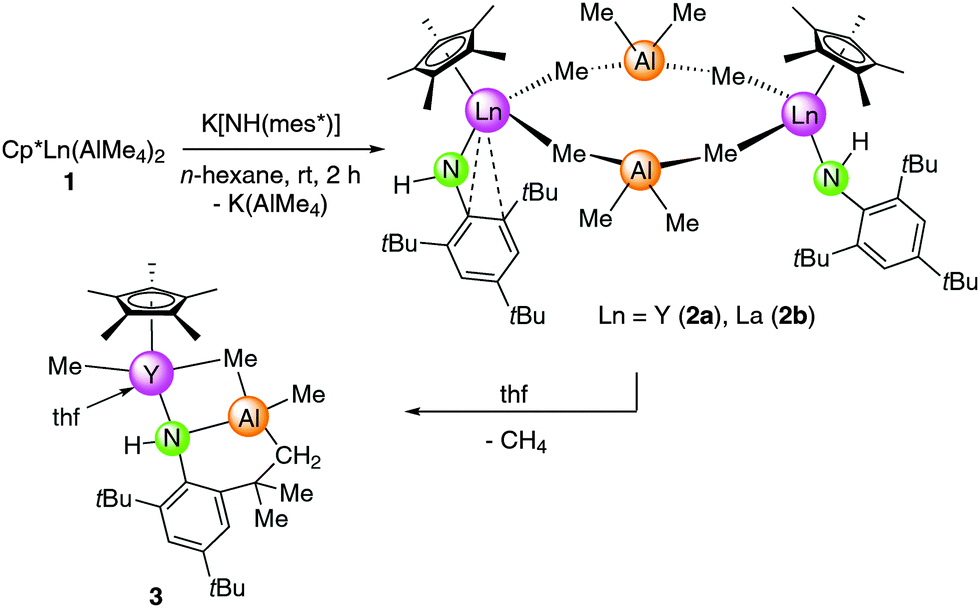 | ||
| Scheme 2 Synthesis of anilide complexes 2 and 3. | ||
Colourless crystals of 2 were sparingly soluble in aliphatic hydrocarbons but soluble in aromatic solvents. The 1H NMR spectra of complexes 2 show one set of signals for the Cp*, NH(mes*), and AlMe4 ligands. The five methyl groups of the Cp* ring appear as one singlet at 1.89 and 2.04 ppm in complexes 2a and 2b, respectively. The signals at 5.05 ppm (2a) and 5.83 ppm (2b) can be assigned to the NH functionalities. The metal-bonded methyl groups show one sharp signal at ambient temperature (2a, −0.33 ppm, 2JYH = 2.5 Hz; 2b, −0.43 ppm), which implies a rapid exchange between bridging and terminal methyl groups. For yttrium complex 2a signal splitting of the bridging and terminal methyl groups into two broad singlets at −0.37 ppm and −0.11 ppm occurred at approximately −90 °C. Variable-temperature 1H NMR studies on lanthanum complex 2b did not reveal any signal splitting at temperatures from +25 to −90 °C indicative of a higher mobility of the La–(AlMe4) moiety.
Single crystals of {Cp*Y(AlMe4)[NH(mes*)]}2 (2a) were grown from saturated n-hexane solutions at −35 °C. The X-ray crystallographic analysis revealed a structural motif as found in the solid-state structure of [Cp*2Ln(AlMe4)]8,25 (Ln = Y, La, Sm) with the yttrium metal centers being bridged by two μ2-η1:η1 coordinated AlMe4 ligands (Y1–C1–Al1 = 174.8(2)°; Y2–C3–Al1 = 169.3(2)°), while the Cp* and amido ligands are coordinated terminally (Fig. 1). The NH(mes*) ligands exhibit two distinct coordination modes with the most striking difference being the Y–N(amide)–C(aryl) bond angles of Y1–N1–C5 = 150.7(4)° and Y1–N2–C05 = 93.1(3)°. The Y–N(amido) bond lengths of 2.186(4) Å and 2.226(3) Å are in the expected range.26 The bent amido ligand features additional Ln⋯arene interactions involving two short contacts to the ipso and ortho carbon atoms of the aryl ring. Both amido ligands show secondary interactions with the Y(III) center through one of the methyl groups of the tert-butyl substituents.
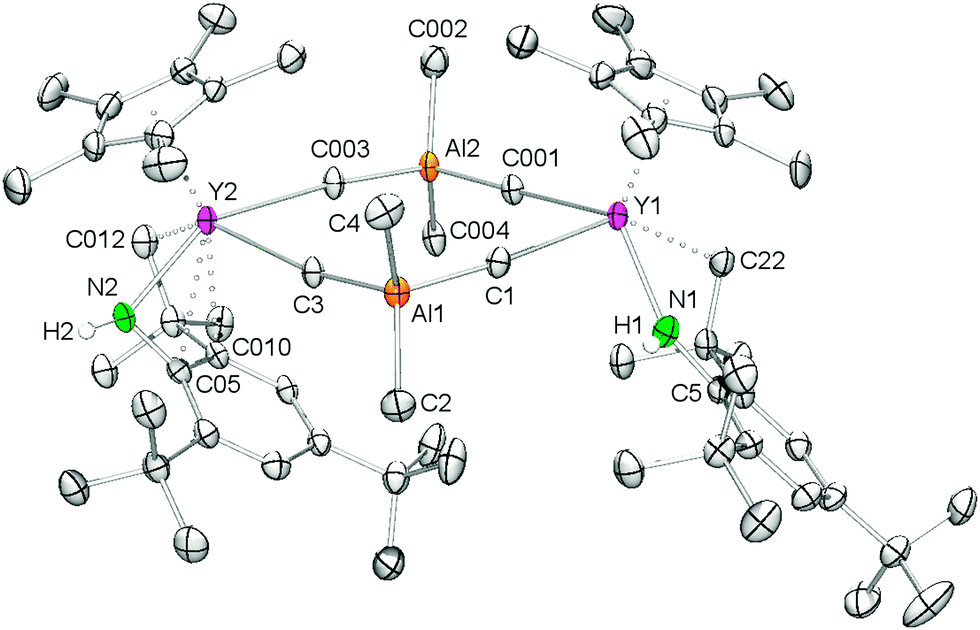 | ||
| Fig. 1 Molecular structure of 2a. Atoms are represented by atomic displacement ellipsoids set at the 50% level. Solvent molecules (n-hexane) and hydrogen atoms except for the NH protons are omitted for clarity. Selected bond distances [Å] and angles [deg]: Y1–N1 2.186(4), Y2–N2 2.226(3), Y1–C1 2.680(7), Y1–C001 2.653(4), Y2–C3 2.685(4), Y2–C003 2.769(7), Y1⋯Ct1 2.325, Y2⋯Ct2 2.328, Y1⋯C22 2.980(6), Y2⋯C012 2.819(4), Y2⋯C05 2.683(4), Y2⋯C010 2.805(4), Y1–N1–C5 150.7(4), Y2–N2–C05 93.1(3), Y1–C1–Al1 174.8(2), Y2–C3–Al1 169.3(2). | ||
Attempts to synthesize putative Cp*Ln![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Nmes* from 2avia Lewis base-induced methane elimination, as described earlier for other alkyl–amide complexes, were not successful.27 Upon addition of thf to 2a no visual changes were observed, meaning that the mixture remained as a yellow solution. However, evaporation of the solvent, drying in vacuo, and recrystallization of the residue from n-hexane produced single crystals of complex 3, revealing AlMe4 cleavage and C–H bond activation of one of the tert-butyl groups (Scheme 2, Fig. 2). The generated monomeric complex is 7-coordinate by the C–H activated primary amido ligand, (μ2-Me)AlMe{NH[C6H2tBu2-2,4-(CMe2CH2)-6]}, a η5-Cp* ligand, a thf molecule, and a terminal methyl group. The Y–N(amido) bond length of 2.489(2) Å is significantly elongated compared to 2a (Y–N(amido) = 2.186(4)/2.226(3) Å), which is due to the formation of a four-membered Y–(μ2-Me)–Al–N metallacycle. This four-membered metallacycle is fused via the Al–N bond to a six-membered metallacycle, which itself is annulated to the (anilido)phenyl ring. The Y–C bond lengths differ markedly with the terminal Y–C(CH3) distance being quite short (2.406(3) Å) similar to the diketiminate complex (L)YMe2 (L = MeC(N(C6H3iPr2-2,6))CHC(Me)NCH2CH2N(Me)CH2CH2NMe2) (av. 2.433 Å)28 and hydrotris(pyrazolyl)borate compound (TptBu,Me)YMe(AlMe4) (Y–C(CH3) = 2.382(3) Å; Y–C(μ2-CH3) = 2.715(3) Å),29 while the Y–C(μ2-CH3) (2.827(3) Å) distance is relatively long in comparison to 2a (av. 2.697 Å) and [Cp*2Y(AlMe4)]225a (2.66 Å). Unfortunately, 1H and 13C NMR spectroscopic investigations were hampered by fast decomposition of the product.
Nmes* from 2avia Lewis base-induced methane elimination, as described earlier for other alkyl–amide complexes, were not successful.27 Upon addition of thf to 2a no visual changes were observed, meaning that the mixture remained as a yellow solution. However, evaporation of the solvent, drying in vacuo, and recrystallization of the residue from n-hexane produced single crystals of complex 3, revealing AlMe4 cleavage and C–H bond activation of one of the tert-butyl groups (Scheme 2, Fig. 2). The generated monomeric complex is 7-coordinate by the C–H activated primary amido ligand, (μ2-Me)AlMe{NH[C6H2tBu2-2,4-(CMe2CH2)-6]}, a η5-Cp* ligand, a thf molecule, and a terminal methyl group. The Y–N(amido) bond length of 2.489(2) Å is significantly elongated compared to 2a (Y–N(amido) = 2.186(4)/2.226(3) Å), which is due to the formation of a four-membered Y–(μ2-Me)–Al–N metallacycle. This four-membered metallacycle is fused via the Al–N bond to a six-membered metallacycle, which itself is annulated to the (anilido)phenyl ring. The Y–C bond lengths differ markedly with the terminal Y–C(CH3) distance being quite short (2.406(3) Å) similar to the diketiminate complex (L)YMe2 (L = MeC(N(C6H3iPr2-2,6))CHC(Me)NCH2CH2N(Me)CH2CH2NMe2) (av. 2.433 Å)28 and hydrotris(pyrazolyl)borate compound (TptBu,Me)YMe(AlMe4) (Y–C(CH3) = 2.382(3) Å; Y–C(μ2-CH3) = 2.715(3) Å),29 while the Y–C(μ2-CH3) (2.827(3) Å) distance is relatively long in comparison to 2a (av. 2.697 Å) and [Cp*2Y(AlMe4)]225a (2.66 Å). Unfortunately, 1H and 13C NMR spectroscopic investigations were hampered by fast decomposition of the product.
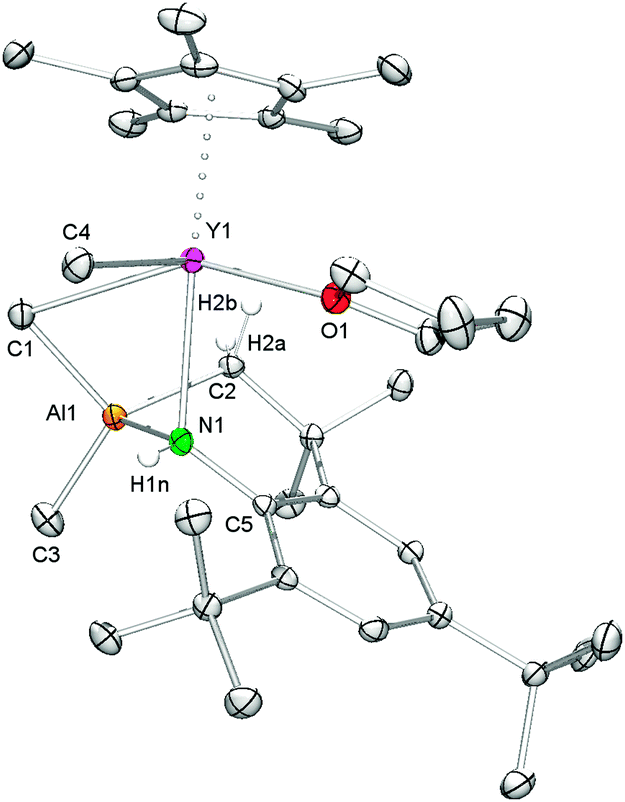 | ||
| Fig. 2 Molecular structure of 3. Atoms are represented by atomic displacement ellipsoids set at the 30% level. Hydrogen atoms except for the NH proton and the methylene protons are omitted for clarity. Selected bond distances [Å] and angles [deg]: Y1–C1 2.827(3), Y1–C4 2.406(3), Y1–N1 2.489(2), Y1–O1 2.375(2), Y1⋯Ct1 2.384, Y1⋯Al1 3.0705(9), Al1–N1 1.902(2), Al1–C1 2.014(3), Al1–C2 1.985(3), Al1–C3 1.962(3), Y1–N1–C5 123.6(2), C1–Y1–C4 80.4(1), C1–Y1–O1 151.64(8), C4–Y1–O1 92.38(9), N1–Y1–Ct1 162.03, Y1–C1–Al1–N1 39.6(1). | ||
Reactivity of CpQLn(AlMe4)2 (4 and 6)
In order to enhance the steric pressure at the rare-earth metal center during the tetramethylaluminate displacement we decided to use the N-donor substituted bis(tetramethylaluminate) precursor CpQLn(AlMe4)2 (Ln = Y, Lu). The presence of the rigid quinolyl functionality was anticipated to minimize any undesired C–H bond activation at the N-donor substitutent.32 CpQLu(AlMe4)2 was prepared by a slightly modified procedure than described earlier for the yttrium and lanthanum congeners.33Accordingly, quinolyl-substituted half-sandwich complex CpQLu(AlMe4)2 (4) was obtained via protonolysis of homoleptic Lu(AlMe4)3 with HCpQ. Brown powdery 4 is readily soluble in toluene, but only sparingly soluble in n-hexane. The 1H NMR spectrum of 4 in C6D6 at ambient temperature shows one signal for the AlMe4 ligands at −0.13 ppm, two singlets for the cyclopentadienyl methyl groups at 1.78 and 2.02 ppm and one set of signals for the quinolyl moiety.
Single crystals of 4 suitable for X-ray structure analysis were grown from a toluene–n-hexane mixture at −35 °C. As for the yttrium and lanthanum congeners33 the CpQ ligand binds to the metal center in an η5 fashion through all five carbon atoms and via the quinolyl nitrogen atom (Fig. 3). Not surprisingly, in the sterically more congested lutetium complex one of the AlMe4 ligands coordinates in an η1 fashion while the other exhibits the usually observed planar η2 coordination mode (Lu1–C24–Al2–C23 = 5.16(8)°), accounting for an overall heptacoordinate Lu(III) metal center. Such η1 coordination mode of the AlMe4 ligand with an almost linear Lu–C(μ-CH3)–Al bond angle of 166.51(9)° has been observed before in (TptBu,Me)YMe(AlMe4) (Y1–C(μ-CH3)–Al = 161.0(1)°)29 and [(ArNMe2)2NC5H3]La(AlMe4)(thf) (Ar = C6H3iPr2-2,6, La1–C(μ-CH3)–Al = 165.0(4)°),30 supported by sterically demanding tridentate (NNN)n− ligands (n = 1, 2).
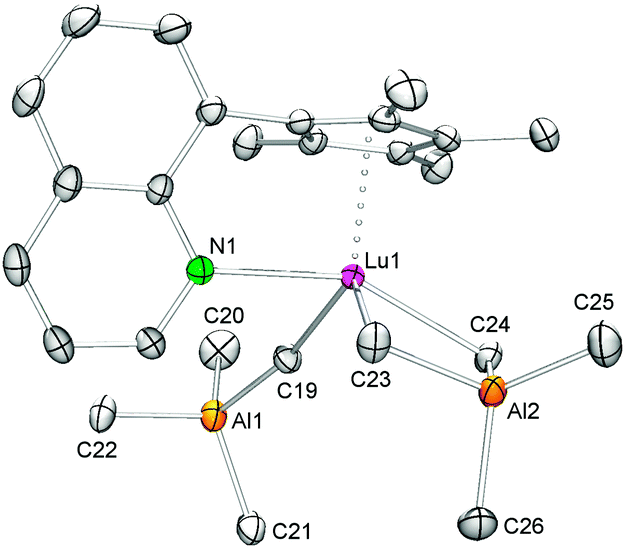 | ||
| Fig. 3 Molecular structure of 4. Atoms are represented by atomic displacement ellipsoids set at the 50% level. Hydrogen atoms are omitted for clarity. Selected bond distances [Å] and angles [deg]: Lu1–N1 2.417(1), Lu1–C19 2.539(2), Lu1–C23 2.485(2), Lu1–C24 2.524(2), Lu1⋯Ct1 2.247, Lu1⋯Al1 4.5877(6), Lu1⋯Al2 3.0296(5), Al1–C19 2.081(2), Al1–C20/21/22 1.991(2)/2.001(2)/1.997(2), Al2–C23 2.081(2), Al2–C24 2.069(2), Al2–C25/26 1.964(2)/1.968(2), N1–Lu1–Ct1 96.57, C19–Lu1–Ct1 116.25, C23–Lu1–Ct1 120.30, C24–Lu1–Ct1 110.86, Lu1–C19–Al1 166.51(9), Lu1–C23–Al2 82.63(6), Lu1–C24–Al2 81.87(6), Lu1–C24–Al2–C23 5.16(8). | ||
Interestingly, the salt metathesis reaction of CpQLu(AlMe4)2 (4) and K[NH(mes*)] led to complex CpQLuMe{NH[C6H2tBu2-2,4-(CMe2CH2)-6]}(AlMe2) (5) (Scheme 3) revealing a structural motif reminiscent of that found in 3, with the hard quinolyl donor now occupying the position of the thf coordination site. In the 1H NMR the proton of the NH functionality was clearly evidenced by a singlet at 4.76 ppm, while the presence of three singlets at 0.21, −0.25, and −0.35 ppm for the metal bonded methyl groups and two doublets at 0.85 and 0.67 ppm with a geminal coupling constant of 2JHH = 14.5 Hz for the methylene group are consistent with the solid-state structure.
 | ||
| Scheme 3 Synthesis of anilide complexes 5 and 7. | ||
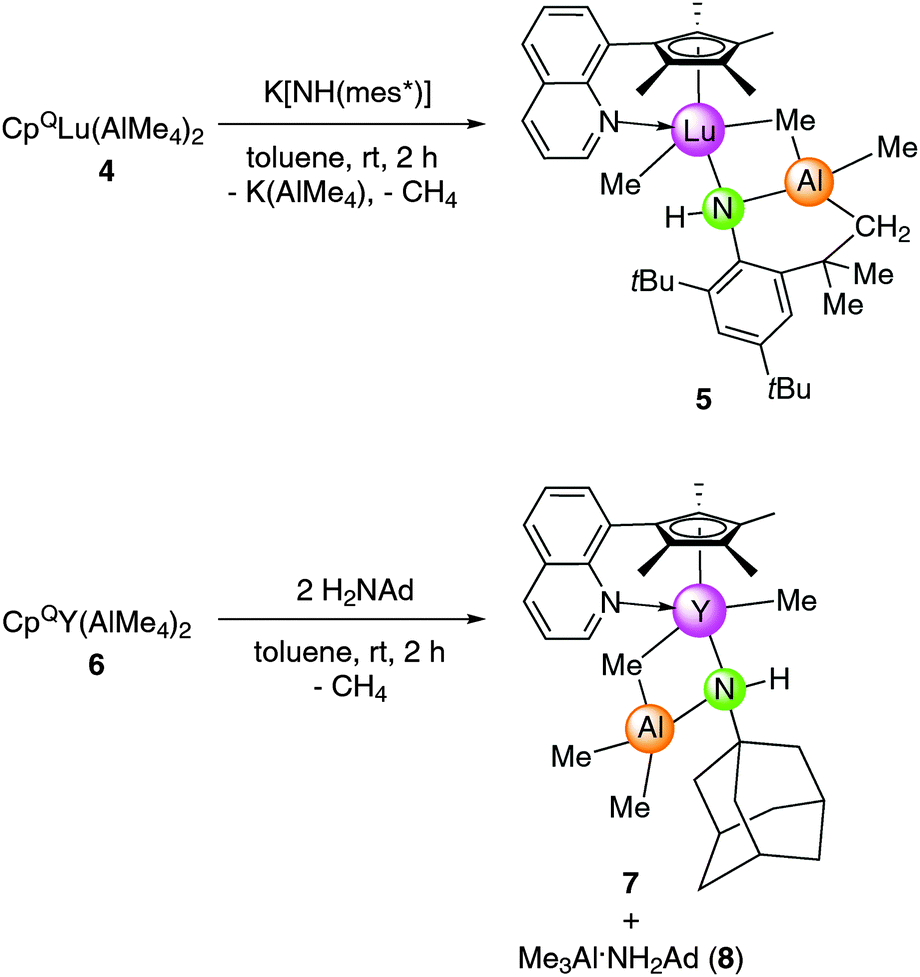
An X-ray diffraction study of 5 revealed that the lutetium metal center is coordinated by the CpQ ligand via the η5-C5Me4 moiety and the quinolyl nitrogen donor, one terminal and one bridging methyl group as well as the aluminium-linked amido ligand resulting in an overall coordination number of seven (see Fig. 4). The Lu–C(CH3) and Lu–C(μ-CH3) bond lengths of 2.334(2) Å and 2.703(2) Å, respectively, and the Lu–N(amido) distance of 2.479(2) Å compare well to those detected in complex 3 considering the difference of the Ln(III) ionic radii. For comparison, complexes Cp*2LuMe2Li(thf)322c and [Cp*2LuMe]231 show similar terminal Lu–C(CH3) bond lengths of 2.361(9) Å and 2.423(3) Å, respectively, whereas the bridging carbon bonds Lu–C(μ-CH3) can differ markedly ranging from 2.385(9)22c to 2.737(3) Å.31 The bending of the nonlinear Lu1–N1–C32(ipso) fragment of 129.0(1)° in 5 is much more pronounced than in the related complex [Cp*2LuMe(NHAr)][Li(12-crown-4)2] (Ar = C6H3iPr2-2,6) (Lu–N = 2.245(4) Å, Lu–N–C = 155(3)°).22c
 | ||
| Fig. 4 Molecular structure of 5. Atoms are represented by atomic displacement ellipsoids set at the 30% level. Hydrogen atoms except for the NH proton and the methylene protons are omitted for clarity. Selected bond distances [Å] and angles [deg]: Lu1–C20 2.703(2), Lu1–C40 2.334(2), Lu1–N1 2.479(2), Lu1–N2 2.401(2), Lu1⋯Ct1 2.339, Lu1⋯Al1 2.9694(6), Al1–N1 1.905(2), Al1–C20 2.023(2), Al1–C21 1.991(2), Al1–C39 1.964(2), Lu1–N1–C32 129.0(1), C20–Lu1–C40 89.45(8), C20–Lu1–N2 154.45(6), C20–Lu1–N1 71.84(5), C40–Lu1–N2 97.17(6), C40–Lu1–N1 86.91(6), N1–Lu1–Ct1 165.11, Lu1–C20–Al1–N1 41.27(7). | ||
Remarkably, the reaction of CpQY(AlMe4)2 with H2Nmes* or K[NH(mes*)] led to the formation of several metal-containing species. However, we have recently shown that protonolysis of complexes (TptBu,Me)YMe(MMe4) (M = Al, Ga) with primary amines/anilines, is also a viable strategy to access imide species (TptBu,Me)LnNR or (TptBu,Me)Ln(NR)(AlMe3).15,16 For assessing the feasibility of the protonolysis protocol for Cp-based systems, we reacted CpQY(AlMe4)2 with 1-adamantylamine (H2NAd). Primary amine H2NAd was selected in order to minimize any C–H bond activation at the ligand backbone. Accordingly, the reaction of 6 with H2NAd afforded amide complex CpQYMe[NH(Ad)](AlMe3) (7) together with adduct Me3Al·NH2(Ad) (8) (Scheme 3). As anticipated C–H bond activation of the amide ligand did not occur. The observed reactivity comes not as a total surprise since the reaction of Ln(AlMe4)3 (Ln = Y, Lu) with 1-adamantylamine was previously shown to produce [LnMe3]n and Me3Al·NH2(Ad). Such distinct reactivity clearly shows that the type of ancillary ligand (Cp versus Tp = tris(pyrazolyl)borate) affects organoaluminium-assisted deprotonation10,13 and Lewis base-induced aluminate cleavage34 as competing reaction pathways and the action of primary amines as Brønsted acids or Lewis bases.
Anilide species 7 is sparingly soluble in aliphatic hydrocarbons but soluble in aromatic solvents. The 1H NMR spectrum of 7 shows the expected set of signals for the coordinated CpQ ligand. The four methyl groups of the Cp ring appear as two singlets at 1.78 and 2.20 ppm and the signals of the quinolyl protons were found in the range from 6.57 to 8.88 ppm. All signals are shifted 0.1–0.3 ppm upfield compared to those of 6, indicating the coordination of the adamantylamide ligand to the rare-earth metal center. The resonances assignable to the adamantyl methylene and methine groups were detected as a doublet at 2.28 ppm (6H), a multiplet ranging from 1.79 to 1.61 ppm (6H) and a broad singlet at 2.14 ppm (3H), respectively. Only one doublet was observed for the metal-bonded methyl groups in complex 7 at ambient temperature (−0.06 ppm, 12H), which implies a high mobility of the Y/Al–CH3 moieties. Such a facile exchange of a terminal Y–CH3 and a bridging methyl group Y–CH3–M has been observed before in (TptBu,Me)YMe[(μ-Me)(GaMe3)].16 The open coordination sphere at the yttrium metal center seems to enable enhanced methyl group mobility.
Single crystals of 7 suitable for X-ray structure analysis were grown from toluene–n-hexane mixtures at −35 °C. In the solid state, complex 7 is heptacoordinate by one terminal and one bridging methyl group, the amido nitrogen atom as well as the CpQ ligand binding via the Cp ring (η5) and the nitrogen atom of the quinolyl moiety (see Fig. 5). The amido ligand and the (μ-Me)AlMe2 moiety form a heterobimetallic four-membered ring, which is considerably bent (torsion angle Y1–N2–Al3–C22 −55.70(9)°), resulting in a smaller Y⋯Al separation of 3.0417(6) Å than in precursor 6 (av. 3.1784(7) Å).33 The mean metal–ring-carbon distances Ln–C(CpQ) (av. 2.656(2) Å) and the Y–N(quinolyl) bond length (2.527(2) Å) are similar to those found in 6.33 The bond length of the terminal methyl group Y–C(CH3) (2.392(2) Å) compares well to that found in 3 while the distance of the bridging Y–C(μ-CH3) moiety (3.006(2) Å) is significantly longer than the one found in 3 (Y1–C1 = 2.827(3) Å) and markedly longer than the average distances Y–C(CH3) (2.645(2) Å) in homoaluminate precursor 6.33 The Y–N(amido) bond length of 2.428(2) Å is slightly shorter than in 3, reflecting a less sterically crowded coordination environment around the metal center.
 | ||
| Fig. 5 Molecular structure of 7. Atoms are represented by atomic displacement ellipsoids set at the 50% level. Selected bond distances [Å] and angles [deg]: Y1–C19 2.392(2), Y1⋯C22 3.006(2), Y1–N1 2.527(2), Y1–N2 2.428(2), Y1⋯Ct1 2.366, Y1⋯Al3 3.0417(6), Al3–C20 1.964(2), Al3–C21 2.007(2), Al3–C22 2.022(2), Al3–N2 1.920(2), Y1–N2–C23 126.9(1), Y1–N2–Al3 87.98(7), C19–Y1–C22 144.61(7), N1–Y1–N2 140.16(6), C19–Y1–N2 103.26(7), C19–Y1–N1 95.24(7), C22–Y1–N1 78.49(6), Y1–N2–Al3–C22 −55.70(9). | ||
In Scheme 4, we propose a plausible mechanistic scenario for the formation of half-sandwich amide complexes 3, 5, and 7: both salt metathesis involving CpRLn(AlMe4)2 and KNHR (Scheme 4a) and protonolysis involving CpRLn(AlMe4)2 and H2NR (Scheme 4b) afford intermediate I2, which in the absence of donor functionalities can be isolated as shown for 2a and 2b. The protonolysis reaction is preceded by an amine-induced tetramethylaluminate cleavage and the formation of a transient terminal Ln–CH3 moiety (I1), which reacts instantly with another amine molecule via methane elimination to generate I2. The occurrence of donor-induced tetramethylaluminate cleavage as the rate-determining step along reaction path b is supported by the formation of considerable amounts of 7 along with AlMe3(H2NR) (8) in equimolar reactions of 6 and H2NAd. Intermediate I2 is not stable in the presence of donor functionalities (quinolyl or thf), which can exert enhanced steric pressure on the amido as well as tetramethylaluminato ligands. As a consequence the amido nitrogen can approach the aluminate ligand (blue arrow), which might experience a η2–η1 coordination switch, ultimately leading to an aluminate cleavage and concomitantly to the formation of a stable Al–N bond as well as a (second) Ln–CH3 moiety (isolation of complex 7). In the case of proximal alkyl moieties, the organoaluminium fragment can trigger C–H bond activation (green arrows), as evidenced for the formation of complexes 3 and 5.
 | ||
| Scheme 4 Proposed reaction mechanism for the formation of 3, 5, and 7. | ||
Synthesis and characterization of Me3Al·NH2(Ad) (8)
Byproduct 8, which could be separated from 7 by extraction with n-hexane, was independently synthesized in quantitative yield via the addition of AlMe3 to a toluene solution of 1-adamantylamine. Recrystallization from toluene–n-hexane at −35 °C gave colourless crystals. In the solid-state, the aluminium center adopts a distorted tetrahedral coordination geometry (Fig. 6). The average Al–C(CH3) bond length of 1.983 Å and the Al–N(amine) distance of 2.032(1) Å are in accord with the corresponding metrical parameters observed for the related complex Me3Al·NH2(tBu)35 (av. Al–C = 1.96 Å, Al–N = 2.027(3) Å) and 7 (av. Al–C = 1.998 Å), but are considerably elongated compared to the Al–N(amido) bond length in 7 (Al–N = 1.920(2) Å). As expected, the 1H NMR spectrum of aluminium complex 8 in C6D6 shows one high-field signal for the methyl groups at −0.36 ppm (9H), a singlet for the NH2 functionality at 1.59 ppm (2H), a broad singlet for the methine groups (3H), a doublet at 1.20 ppm and a multiplett at 1.27 ppm for the methylene groups integrating six protons each, respectively.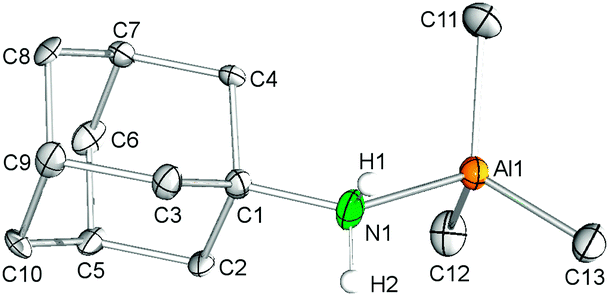 | ||
| Fig. 6 Molecular structure of 8. Atoms are represented by atomic displacement ellipsoids set at the 50% level. The disorders in the adamantyl moiety and hydrogen atoms except for the NH2 protons are omitted for clarity. Selected bond distances [Å] and angles [deg]: Al1–C11 1.988(2), Al1–C12 1.976(2), Al1–C13 1.986(2), Al1–N1 2.032(1), Al1–N1–C1 126.41(9), C12–Al1–C13 113.32(9), C12–Al1–C11 116.32(8), C13–Al1–C11 113.38(9), C12–Al1–N1 107.25(8), C13–Al1–N1 99.39(6), C11–Al1–N1 105.13(8). | ||
Conclusion
The reactions of monocyclopentadienyl rare-earth metal bis(tetramethylaluminate) complexes with 1-adamantylamine or potassium (2,4,6-tri-tert-butylphenyl)amide yield half-sandwich mixed alkyl–amide complexes. The reactivity of the metal-bonded alkyl groups toward adjacent NH functionalities appears strongly reduced and further deprotonation was not observed. This is in sharp contrast to previously examined complexes (TptBu,Me)YMe(AlMe4), which do react with primary amines/anilines to afford Lewis-acid stabilized imido derivatives (TptBu,Me)Ln(NR)(AlMe3).15 Such changed reactivity of complexes Cp*Ln(AlMe4)2 and CpQLn(AlMe4)2 might be caused by a steric misfit of the metal–alkyl and N–H groups, since the former still exhibit high reactivity as shown for the C–H activation of the NH(mes*) amido ligand backbone. Though substitution of mes* by adamantyl decreases the reactivity of the NH moiety (pKa criterion), the occurrence of undesired side-reactions is impeded. Previous studies from the Hessen and Hou groups revealed that imides could only be obtained from reactive Cp ligated rare-earth metal hydride and diene complexes via the insertion of benzonitrile.2b,36 Overall, the present study reinforces that primary alkylamines not only act as protic substrates but also as Lewis bases and that the steric and electronic properties of the ancillary ligands are crucial factors directing the formation of either heteroleptic alkyl–amide complexes or imide species.15Experimental section
General considerations
All operations were performed with rigorous exclusion of air and water, using standard Schlenk, high-vacuum, and glovebox techniques (MBraun 200B; <1 ppm O2, <1 ppm H2O). THF, toluene and n-hexane were purified by using Grubbs columns (MBraun SPS, solvent purification system) and stored inside a glovebox. C6D6 and toluene-d8 were obtained from Aldrich, degassed, dried over Na for 24 h, and filtered. 1-Adamantylamine was received from abcr and used as received. Pro-ligand HCpQ,37 heteroleptic complexes Cp*Ln(AlMe4)2 (Ln = Y, La) (1)8 and CpQY(AlMe4)2 (6),33 and potassium (2,4,6-Tri-tert-butylphenyl)amide13 were synthesized according to literature procedures. The NMR spectra of air and moisture sensitive compounds were recorded by using J. Young valve NMR tubes at variable temperature on a Bruker AVII+400 (1H: 400 MHz; 13C: 101 MHz) and a Bruker AVII+500 (1H: 500 MHz; 13C: 126 MHz). 1H and 13C shifts are referenced to internal solvent resonances and reported in parts per million relative to TMS. Coupling constants are given in Hertz. DRIFT spectra were recorded on a NICOLET 6700 FTIR spectrometer using dried KBr and KBr windows. Elemental analyses were performed on an Elementar Vario Micro Cube.General procedures for the synthesis of {Cp*Ln(AlMe4)[NH(mes*)]}x
A solution of (Cp*)Ln(AlMe4)2 in n-hexane (2 mL) was added to a stirred suspension of potassium (2,4,6-tri-tert-butylphenyl)amide K[NH(mes*)] in n-hexane (2 mL). The reaction mixture was stirred for 2 h at ambient temperature and the n-hexane solution then separated by centrifugation, decanted, and filtrated. The solid residue product and K(AlMe4) were extracted with additional n-hexane (2 × 2 ml). Compounds 2 were obtained as powders or by crystallization from n-hexane solutions at −35 °C.CpQLuMe{NH[C6H2tBu2-2,4-(CMe2CH2)-6]}(AlMe2) (5)
To a stirred suspension of K[NH(mes*)] in toluene (1 mL) a solution of 4 (87 mg, 0.14 mmol) in toluene (3 mL) was added. The brown suspension was stirred 3 h at ambient temperature and the product then separated by centrifugation, decanted, and filtrated. The solid residue product and K(AlMe4) was extracted with additional toluene (2 × 1 ml). The combined extracts were dried and washed with n-hexane (3 × 2 ml), followed by drying under reduced pressure (96 mg, 0.12 mmol, 91%, 30% crystalline yield). Crystallization from a toluene–n-hexane solution at −35 °C afforded yellow crystals of 5 suitable for X-ray diffraction analysis. 1H NMR (500 MHz, C6D6, 26 °C, TMS): δ = 8.03 (d, 1H, 3JHH = 4.0 Hz, quin-H), 7.67 (d, 1H, 3JHH = 2.3 Hz, quin-H), 7.45–7.43 (m, 3H, quin-H), 7.30 (d, 1H, 3JHH = 8.5 Hz, quin-H), 7.20 (s, 1H, Ar), 7.13–7.00 (m, 2H, quin-H and Ar), 6.41 (dd, 1H, 3JHH = 8.1 Hz and 3.2 Hz), 4.76 (s, 1H, NH), 2.28 (s, 3H, Cp CH3), 2.06 (s, 3H, Cp CH3), 2.04 (s, 3H, Cp CH3), 1.84 (s, 3H, Cp CH3), 1.53 (s, 9H, C(CH3)3), 1.49 (s, 6H, C(CH3)2), 1.36 (s, 9H, C(CH3)3), 0.85 (d, 1H, 2JHH = 14.5 Hz, Al–CH2), 0.67 (d, 1H, 2JHH = 14.5 Hz, Al–CH2), 0.21 (s, 3H, Lu–CH3), −0.25 (s, 3H, Al–CH3), −0.35 (s, 3H, Al–CH3) ppm. 13C{1H} NMR from 1H13C HSQC and HMBC (126 MHz, C6D6, 26 °C) δ = 152.3 (quin-C), 150.1 (quin-C), 145.1 (quin-C), 142.2 (Ar), 140.2 (Ar), 138.4 (Ar), 137.3 (Ar), 139.5 (quin-C), 128.6 (Ar, quin-C, overlapping with solvent signal), 126.4 (Ar, quin-C), 124.6 (Ar), 124.0 (quin-C), 122.5 (quin-C), 121.0 (quin-C), 119.2 (Cp), 117.4 (Cp), 38.8 (CH3), 37.9 (C(CH3)3), 34.9 (C(CH3)2), 34.2 (C(CH3)3), 32.4 (CH3), 31.9 (C(CH3)3), 31.5 (C(CH3)3), 29.4 (CH2), 28.8 (Al–CH3), 0.9 (Lu–CH3), −9.2 (Al–CH3) ppm. DRIFT (KBr): 3394vw (NH), 3348vw, 3045vw, 2951vs, 2902s, 2865s, 2754w, 1603vw, 1586vw, 1506m, 1475w, 1467w, 1423s, 1396w, 1362w, 1334vw, 1303vw, 1281vw, 1261w, 1237w, 1223m, 1183w, 1159w, 1119m, 841w, 829w, 821w, 806m, 788m, 776m, 757m, 738w, 699m, 687m, 656w, 648w, 626vw cm−1. Elemental analysis calcd (%) for C39H56AlLuN2 (754.82 g mol−1): C 62.06, H 7.48, N 3.71; found: C 60.53, H 5.92, N 3.72. Multiple attempts to obtain a better microanalysis failed.X-ray crystallography and crystal structure determination of 2a, 3, 4, 5, 7, and 8
Crystals of 2a and 3 were grown using standard techniques from saturated solutions using n-hexane or n-hexane–toluene (4, 5, 7 and 8) at −40 °C. Suitable crystals for X-ray structure analyses were selected in a glovebox and coated with Parabar 10312 and fixed on a nylon loop/glass fiber.X-ray data for 3 were collected on a Bruker AXS TXS rotating anode instrument using a Pt 135 CCD detector and for 4, 7 and 8 on a Bruker APEX DUO instrument equipped with an IμS microfocus sealed tube and QUAZAR optics for MoKα radiation (λ = 0.71073 Å). Data for compounds 2a and 5 were collected on a Bruker SMART APEX II instrument equipped with a fine focus sealed tube and a graphite monochromator using MoKα radiation (λ = 0.71073 Å). The Data collection strategy was determined using COSMO38 employing ω- and ϕ scans. Raw data were processed using APEX38 and SAINT,38 corrections for absorption effects were applied using SADABS.38 The structure was solved by direct methods and refined against all data by full-matrix least-squares methods on F2 using SHELXTL38 and ShelXle.39 All Graphics were produced employing ORTEP-340 and POV-Ray.41 Further details of the refinement and crystallographic data are listed in Table S1 (ESI†) and in the CIF files. CCDC 1055068–1055073.
Acknowledgements
We are grateful to the German Science Foundation for support (Grant: AN 238/15-1).Notes and references
- (a) S. Arndt and J. Okuda, Chem. Rev., 2002, 102, 1953 CrossRef CAS PubMed; (b) M. Zimmermann and R. Anwander, Chem. Rev., 2010, 110, 6194 CrossRef CAS PubMed.
- (a) O. Tardif, M. Nishiura and Z. Hou, Organometallics, 2003, 22, 1171 CrossRef CAS; (b) D. Cui, O. Tardif and Z. Hou, J. Am. Chem. Soc., 2004, 126, 1312 CrossRef CAS PubMed; (c) M. Nishiura and Z. Hou, Nat. Chem., 2010, 2, 257 CrossRef CAS PubMed; (d) M. Nishiura, J. Baldamus, T. Shima, K. Mori and Z. Hou, Chem. – Eur. J., 2011, 17, 5033 CrossRef CAS PubMed; (e) T. Shima, M. Nishiura and Z. Hou, Organometallics, 2011, 30, 2513 CrossRef CAS.
- (a) K. Beckerle and J. Okuda, Syndiotactic Polystyrene, John Wiley & Sons, Inc., 2009, pp. 125–139 Search PubMed; (b) X. Xu, Y. Chen and J. Sun, Chem. – Eur. J., 2009, 15, 846 CrossRef CAS PubMed; (c) Z. Zhang, D. Cui, B. Wang, B. Liu and Y. Yang, in Molecular Catalysis of Rare-Earth Elements, ed. P. W. Roesky, Springer, Berlin, Heidelberg, 2010, pp. 49–108 Search PubMed; (d) Z. Jian, S. Tang and D. Cui, Chem. – Eur. J., 2010, 16, 14007 CrossRef CAS PubMed; (e) Z. Jian, D. Cui and Z. Hou, Chem. – Eur. J., 2012, 18, 2674 CrossRef CAS PubMed; (f) Y. Pan, W. Rong, Z. Jian and D. Cui, Macromolecules, 2012, 45, 1248 CrossRef CAS; (g) Y. Luo, S. Chi and J. Chen, New J. Chem., 2013, 37, 2675 RSC.
- R. Anwander, M. G. Klimpel, H. M. Dietrich, D. J. Shorokhov and W. Scherer, Chem. Commun., 2003, 1008 RSC.
- (a) M. Zimmermann, K. Törnroos and R. Anwander, Angew. Chem., Int. Ed., 2008, 47, 775 CrossRef CAS PubMed; (b) M. Zimmermann, K. W. Törnroos, H. Sitzmann and R. Anwander, Chem. – Eur. J., 2008, 14, 7266 CrossRef CAS PubMed; (c) M. Zimmermann, J. Volbeda, K. W. Törnroos and R. Anwander, C. R. Chim., 2010, 13, 651 CrossRef CAS.
- D. Robert, T. P. Spaniol and J. Okuda, Eur. J. Inorg. Chem., 2008, 2801 CrossRef CAS.
- H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143 CrossRef CAS.
- H. M. Dietrich, K. W. Törnroos, E. Herdtweck and R. Anwander, Organometallics, 2009, 28, 6739 CrossRef CAS.
- (a) H. M. Dietrich, C. Zapilko, E. Herdtweck and R. Anwander, Organometallics, 2005, 24, 5767 CrossRef CAS; (b) A. Fischbach, E. Herdtweck and R. Anwander, Inorg. Chim. Acta, 2006, 359, 4855 CrossRef CAS; (c) E. L. Roux, Y. Liang, K. W. Törnroos, F. Nief and R. Anwander, Organometallics, 2012, 31, 6526 CrossRef.
- H. M. Dietrich, H. Grove, K. W. Törnroos and R. Anwander, J. Am. Chem. Soc., 2006, 128, 1458 CrossRef CAS PubMed.
- (a) H. M. Dietrich, K. W. Törnroos and R. Anwander, J. Am. Chem. Soc., 2006, 128, 9298 CrossRef CAS PubMed; (b) W.-X. Zhang, Z. Wang, M. Nishiura, Z. Xi and Z. Hou, J. Am. Chem. Soc., 2011, 133, 5712 CrossRef CAS PubMed.
- In 1979, Schumann et al. reported on the synthesis of lutetium and erbium alkylidenes, [Li][Lu(CH2SiMe3)2(CHSiMe3)] and [Er(CH2SiMe3)(CHSiMe3)]x: H. Schumann and J. Müller, J. Organomet. Chem., 1979, 169, C1 CrossRef CAS.
- D. Schädle, C. Schädle, K. W. Törnroos and R. Anwander, Organometallics, 2012, 31, 5101 CrossRef.
- C. Schädle, D. Schädle, K. Eichele and R. Anwander, Angew. Chem., Int. Ed., 2013, 52, 13238 CrossRef PubMed.
- D. Schädle, C. Maichle-Mössmer, C. Schädle and R. Anwander, Chem. – Eur. J., 2014, 21, 662 CrossRef PubMed.
- D. Schädle, M. Meermann-Zimmermann, C. Schädle, C. Maichle-Mössmer and R. Anwander, Eur. J. Inorg. Chem., 2015, 1334 CrossRef.
- J. C. Gordon, G. R. Giesbrecht, D. L. Clark, P. J. Hay, D. W. Keogh, R. Poli, B. L. Scott and J. G. Watkin, Organometallics, 2002, 21, 4726 CrossRef CAS.
- (a) W. J. Evans, M. A. Ansari, J. W. Ziller and S. I. Khan, Inorg. Chem., 1996, 35, 5435 CrossRef CAS PubMed; (b) H.-S. Chan, H.-W. Li and Z. Xie, Chem. Commun., 2002, 652 RSC.
- (a) L. H. Gade and P. Mountford, Coord. Chem. Rev., 2001, 216–217, 65 CrossRef CAS; (b) A. P. Duncan and R. G. Bergman, Chem. Rec., 2002, 2, 431 CrossRef CAS PubMed.
- (a) G. R. Giesbrecht and J. C. Gordon, Dalton Trans., 2004, 2387 RSC; (b) O. T. Summerscales and J. C. Gordon, RSC Adv., 2013, 3, 6682 RSC.
- (a) P. Mountford, Chem. Commun., 1997, 2127 RSC; (b) S. R. Dubberley, A. Friedrich, D. A. Willman, P. Mountford and U. Radius, Chem. – Eur. J., 2003, 9, 3634 CrossRef CAS PubMed; (c) S. R. Dubberley, S. Evans, C. L. Boyd and P. Mountford, Dalton Trans., 2005, 1448 RSC; (d) N. Adams, H. R. Bigmore, T. L. Blundell, C. L. Boyd, S. R. Dubberley, A. J. Sealey, A. R. Cowley, M. E. G. Skinner and P. Mountford, Inorg. Chem., 2005, 44, 2882 CrossRef CAS PubMed; (e) A. R. Fout, U. J. Kilgore and D. J. Mindiola, Chem. – Eur. J., 2007, 13, 9428 CrossRef CAS PubMed; (f) A. D. Schwarz, A. J. Nielson, N. Kaltsoyannis and P. Mountford, Chem. Sci., 2012, 3, 819 RSC.
- (a) L. K. Knight, W. E. Piers, P. Fleurat-Lessard, M. Parvez and R. McDonald, Organometallics, 2004, 23, 2087 CrossRef CAS; (b) L. K. Knight, W. E. Piers and R. McDonald, Organometallics, 2006, 25, 3289 CrossRef CAS; (c) R. K. Thomson, M. J. Monreal, J. D. Masuda, B. L. Scott and J. L. Kiplinger, J. Organomet. Chem., 2011, 696, 3966 CrossRef CAS.
- A. A. Karpov, A. V. Cherkasov, G. K. Fukin, A. S. Shavyrin, L. Luconi, G. Giambastiani and A. A. Trifonov, Organometallics, 2013, 32, 2379 CrossRef CAS.
- T. M. Cameron, J. C. Gordon and B. L. Scott, Organometallics, 2004, 23, 2995 CrossRef CAS.
- (a) M. A. Busch, R. Harlow and P. L. Watson, Inorg. Chim. Acta, 1987, 140, 15 CrossRef CAS; (b) W. J. Evans, L. R. Chamberlain, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1988, 110, 6423 CrossRef CAS.
- C. Cui, A. Shafir, J. A. R. Schmidt, A. G. Oliver and J. Arnold, Dalton Trans., 2005, 1387 RSC.
- (a) E. Lu, Y. Li and Y. Chen, Chem. Commun., 2010, 46, 4469 RSC; (b) E. Lu, J. Chu, Y. Chen, M. V. Borzov and G. Li, Chem. Commun., 2011, 47, 743 RSC; (c) W. Rong, J. Cheng, Z. Mou, H. Xie and D. Cui, Organometallics, 2013, 32, 5523 CrossRef CAS.
- J. Zhou, J. Chu, Y. Zhang, G. Yang, X. Leng and Y. Chen, Angew. Chem., Int. Ed., 2013, 52, 4243 CrossRef CAS PubMed.
- (a) R. Litlabø, M. Zimmermann, K. Saliu, J. Takats, K. W. Törnroos and R. Anwander, Angew. Chem., Int. Ed., 2008, 47, 9560 CrossRef PubMed; (b) M. Zimmermann, J. Takats, G. Kiel, K. W. Törnroos and R. Anwander, Chem. Commun., 2008, 612 RSC.
- M. Zimmermann, K. W. Törnroos and R. Anwander, Angew. Chem., Int. Ed., 2007, 46, 3126 CrossRef CAS PubMed.
- W. J. Evans, J. M. Perotti and J. W. Ziller, J. Am. Chem. Soc., 2005, 127, 3894 CrossRef CAS PubMed.
- L. N. Jende, C. Maichle-Mössmer and R. Anwander, Chem. – Eur. J., 2013, 19, 16321 CrossRef CAS PubMed.
- R. Litlabø, M. Enders, K. W. Törnroos and R. Anwander, Organometallics, 2010, 29, 2588 CrossRef.
- M. F. Lappert, J. Holton, D. G. H. Ballard, R. Pearce, J. L. Atwood and W. E. Hunter, J. C. S. Chem. Commun., 1976, 480 Search PubMed.
- D. A. Atwood and D. Rutherford, Main Group Chem., 1996, 1, 431 CrossRef CAS.
- D. J. Beetstra, A. Meetsma, B. Hessen and J. H. Teuben, Organometallics, 2003, 22, 4372 CrossRef CAS.
- M. Enders, R. Rudolph and H. Pritzkow, Chem. Ber., 1996, 129, 459 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- POV-Ray v. 3.6, Persistence of Vision Pty. Ltd., Williamstown, Victoria, Australia, 2004, http://www.povray.org/.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1055068–1055073. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00800j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |