
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rare earth 3-(4′-hydroxyphenyl)propionate complexes†
Glen B.
Deacon
a,
Peter C.
Junk
*b,
Winnie W.
Lee
ab,
Maria
Forsyth
c and
Jun
Wang
ab
aSchool of Chemistry, Monash University, Clayton, Vic. 3800, Australia
bCollege of Science, Technology & Engineering, James Cook University, Townsville, Qld 4811, Australia. E-mail: peter.junk@jcu.edu.au
cARC Centre of Excellence for Electromaterials Science, Institute for Frontier Materials, Deakin University, Burwood Campus, Australia
First published on 22nd July 2015
Abstract
The reaction of lanthanoid chlorides or nitrates with sodium 3-(4′-hydroxyphenyl)propionate (Na4hpp) in methanol or water has yielded complexes [La4(4hpp)12(H2O)6]·4H2O·MeOH (1), [Ce2(4hpp)6(H2O)3]·(H2O)·2.5(EtOH) (2a) (after crystallization from ethanol), [Ho(4hpp)3(H2O)2] (5), [Er(4hpp)3(H2O)2]·1.5(H2O) (6), and [Lu(4hpp)3]·H2O crystal composition (7), as well as heterobimetallics [NaCe2(4hpp)7(H2O)2]·3(H2O) (2b), [NaPr2(4hpp)7(H2O)2]·3(H2O) (3), and [NaNd2(4hpp)7(H2O)(MeOH)]·(H2O)·3(MeOH) (4). The structures of homometallic complexes 1, 2a, 6, and 7 reveal one-dimensional coordination polymers and vividly illustrate the effect of lanthanoid contraction with a decline in coordination numbers in the series from 9–11 (1), 9,10 (2a), 8 (6) to 7 (7) through variations in carboxylate coordination and ligation of water. Bimetallic complexes 2a and 4 each exhibit five different carboxylate binding modes as well as coordination of the 4-OH substituent of 4hpp to sodium thereby linking 1D polymer chains into a 2D network with both 9 and 10 coordinate Ln atoms and 6 coordinate sodium. Bulk products after drying lose solvent of crystallization in some cases (2a, 6), or exchange MeOH for water (4). X-ray powder diffraction indicates that bulk 2b and 3 are isotypic, as are bulk 5 and 6. In contrast to the excellent corrosion protection of lanthanum 4-hydroxycinnamate, compound 1 is ineffective in preventing the corrosion of mild steel, thereby establishing the importance of the –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– structural unit of the former in its anti-corrosion properties. However the flexible –CH2–CH2– chain of the 4hpp ligand enables the crystal engineering of its lanthanoid complexes in a wide variety of structures as well as effective crystallization for structure determination, whereas the analogous 4-hydroxycinnamates have so far evaded structural characterization except for Ln = La, Ce owing to crystallization problems.
CH– structural unit of the former in its anti-corrosion properties. However the flexible –CH2–CH2– chain of the 4hpp ligand enables the crystal engineering of its lanthanoid complexes in a wide variety of structures as well as effective crystallization for structure determination, whereas the analogous 4-hydroxycinnamates have so far evaded structural characterization except for Ln = La, Ce owing to crystallization problems.
Introduction
Although the 20% decline in ionic radii1 from La3+ to Lu3+ might be expected to lead to a reduction in coordination numbers for a series of analogous complexes, the actual situation is complicated.2 Thus in the case of cyclopentadienyl lanthanoids, the coordination numbers of [LnCp3] complexes decrease from 11 to 8 from [LaCp3] to [LuCp3] with intermediate 10 and 9 coordinate derivatives,3,4 whereas [LnCp3(thf)] (thf = tetrahydrofuran) complexes are all ten coordinate.3,4 Accordingly, it remains a challenge to harness the ionic radius reduction to engineer predetermined structural outcomes. However, use of multidentate ligands to cage or wrap the metal ions is a useful approach to block the coordination number change.5In conjunction with our studies of the anti-corrosion properties of rare earth cinnamates ([Ln(cinn)3]) and 4-substituted cinnamates [Ln(4-Xcinn)3(solv)n] (X = OH, NO2, Cl, MeO; solv = H2O/MeOH)6,7 (as potential replacements for the toxic8 commonly used9 metal chromates) where lanthanum 4-hydroxycinnamate proved to be the optimum compound,6,7 we have determined the structures of a range of rare earth cinnamates10 in addition to earlier studies.11 This revealed just two structural series, La-Dy (nine coordinate, 1-D polymer) and Dy–Lu, Y (seven coordinate, 1-D polymer), with Dy being able to crystallise in both forms.10 In the case of [Ln(4-Xcinn)3(solv)n] complexes, the better protection agents, considerable difficulty was encountered in obtaining single crystals, and only [Ce(4-OHcinn)3(MeOH)2(H2O)]·H2O was structurally characterised,12 as well as an earlier isomorphous La complex.13 Although X-ray powder diffraction suggested more than one structural class for each group with the same X substituent, crystallization difficulties frustrated establishing defined structural changes induced by lanthanoid contraction. This has led us to investigate complexation by the reduced 4-hydroxycinnamate, namely 3-(4′-hydroxyphenyl)propionate ions, in the expectation that replacing the rigid –CHCH– linker of cinnamate by a more flexible –CH2–CH2– unit would facilitate crystallization and enable engineering of a progressive structural change as the Ln3+ radius decreases. Accordingly, we now report the preparative procedures and structures of rare earth 3-(4′-hydroxyphenyl)propionates, [Ln(4hpp)3(solv)n] and bimetallics [NaLn2(4hpp)7(solv)2], where the lanthanoid contraction has been utilised to give several structural classes. In addition, [La4(4hpp)12(H2O)6] was found to be ineffective as a corrosion inhibitor thereby showing the importance of the –CHCH– unit in the effective inhibitor properties of lanthanum 4-hydroxycinnamate.
Results and discussion
Syntheses and characterisation
Metathesis reactions of rare earth metal salts with 3-(4′hydroxyphenyl)propionic acid (H(4hpp)) deprotonated in situ with aqueous NaOH, or the preformed sodium salt (mole ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) yield rare earth complexes [Ln(4hpp)3(solv)n] (Scheme 1 Ln = La (1), Ce (2a), Ho (5), Er (6), Lu (7)) (Table 1).
:3) yield rare earth complexes [Ln(4hpp)3(solv)n] (Scheme 1 Ln = La (1), Ce (2a), Ho (5), Er (6), Lu (7)) (Table 1).
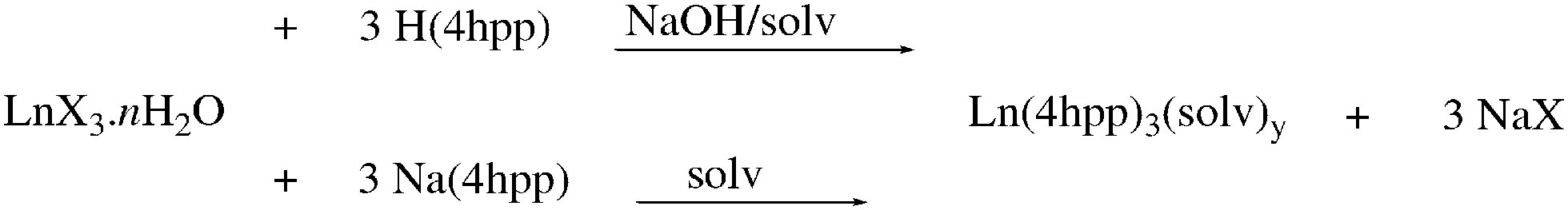 | ||
| Scheme 1 | ||
| Compounds | Main bands (cm−1) | |||
|---|---|---|---|---|
| ν(OH)water | ν as(CO2−) | ν s(CO2−) | Δν = (νas – νs)avg | |
| Na(4hpp) | 3404 m br, 3350 m br | 1556 s, 1517 s | 1417 s | 120 |
| [La4(4hpp)12(H2O)6]·4(H2O)·(MeOH) 1 | 3368 m br, 3286 m br, 3172 br m | 1529 s, 1516 s | 1419 s | 104 |
| [Ce2(4hpp)6(H2O)3]·(H2O)·2.5(EtOH) 2a | 3350 m br, 3205 m br | 1531 s, 1515 s | 1417 s | 106 |
| [NaCe2(4hpp)7(H2O)2]·3(H2O) 2b | 3606 w, 3369 m br | 1578 m, 1527 s, 1516 s | 1418 s | 122 |
| [NaPr2(4hpp)7(H2O)2]·3(H2O) 3 | 3623 w, 3271 s br | 1580 s, 1516 s | 1417 s | 131 |
| [NaNd2(4hpp)7(H2O)(MeOH)]·(H2O)·3(MeOH) 4 | 3530 m, 3256 s br | 1579 m, 1536 s, 1516 s | 1407 s | 137 |
| [Ho(4hpp)3(H2O)2] 5 | 3566 w, 3432 m br, 3220 m br | 1577 m, 1543 s, 1515 m | 1419 m | 126 |
| [Er(4hpp)3(H2O)2]·1.5(H2O) 6 | 3567 w, 3436 m br, 3221 m br | 1578 m, 1543 s, 1515 m | 1419 m | 126 |
| [Lu(4hpp)3]·(H2O) 7 | 3568 m, 3256 s br, 3197 s br | 1581 s, 1550 s, 1515 s | 1404 s | 145 |
Whilst the use of CeCl3 provided [Ce2(4hpp)6(H2O)3]·H2O·2.5EtOH (2a) in low yield, the use of Ce(NO3)3 gave the bimetallic complex [NaCe2(4hpp)7(H2O)2]·3H2O (2b). For Pr and Nd, bimetallic complexes [NaLn2(4hpp)7(solv)2] (Ln = Pr (3), Nd (4)) were obtained regardless of the use of chloride or nitrate reactants. In several cases, the bulk product was obtained as a precipitate and single crystals were obtained by slow evaporation of the filtrate (3, 6, and 7). Single crystals of 2a were derived from recrystallization of the bulk product from ethanol, and crystals of 2b were mixed with the bulk product, whilst 1 and 4 were initially isolated as crystals. Precipitated 5 was microcrystalline but single crystals could not be obtained.
The characterisation of the bulk samples of compounds 1–7 was carried out after drying over silica gel to constant weight. This resulted in the loss of solvent of crystallisation for some of the complexes from the compositions derived by X-ray crystallography of single crystals. Thus, compounds 2a and 6 lost lattice solvent from the single crystal composition whilst both lattice and coordinated methanol of single crystals of 4 were replaced by water. However, bulk 1 and 2b had the same bulk and crystal composition. On the other hand, microanalysis and TGA of bulk 7 were indicative of one more water of crystallisation than in the single crystals, suggesting that the powdered bulk material was hygroscopic.
The X-ray powder pattern of bulk 2b was in agreement with that simulated from single crystal data and was also in agreement with that of 3 indicating that 2b and 3 are isotypic (d-spacings, Experimental section, Fig. S1, ESI†). It was different from that generated from crystal data for 4, which has coordinated methanol and methanol of crystallisation. Powder data of bulk 5 and 6 are in agreement but differ from that simulated from the crystal structure of 6, which has additional water of crystallisation (Fig. S2, ESI†). Consistent with the observation that bulk 7 has more water of crystallisation than single crystals, the powder pattern of the former differs from that simulated from single crystal data (d-spacings, Experimental section).
Broad infrared bands at 3600–3200 cm−1 (Table 1) are attributable to the overlap of ν(OH) of water (both coordinated and lattice) and of the 4-hydroxy group of the 4hpp ligand. Antisymmetric carboxylate stretching gave rise to at least two intense bands at 1580–1515 cm−1, whereas the symmetric mode gave only a single band at 1420–1404 cm−1. The separation between νas(CO2)ave and νs(CO2) (145–104 cm−1) (Table 1) is close to that (120 cm−1) of the sodium salt consistent14 with the predominantly bridging nature of the carboxylate coordination (below). However, given the complexity of the carboxylate bonding (Table 2), it is fruitless to seek further structure/IR correlation. This is well illustrated by the similarity of the νas(CO2) frequencies of 4, 6, and 7, which have entirely different carboxylate coordination. The range of different bridging and chelating modes observed in the structures of this series of complexes is shown in Fig. 1.
| Compound | Ln atom | Coordination number | Ligation | Ave Ln–O (Å) |
|---|---|---|---|---|
| a Chelating. b Unidentate. c Provided for connectivity purposes only. | ||||
| 1 | La(1) | 11 | 4 × (iii)a; 1 × (iii)b, 2 × H2O | 2.67 |
| La(2) | 9 | 1 × (i); 1 × (iii)a; 4 × (iii)b; H2O | 2.54 | |
| La(3) | 9 | 1 × (i); 1 × (ii); 1 × (iii)a; 3 × (iii)b; H2O | 2.54 | |
| La(4) | 10 | 1 × (ii); 3 × (iii)a; 1 × (iii)b; 2 × H2O | 2.63 | |
| 2a | Ce(1) | 9 | 1 × (i); 1 × (ii); 1 × (iii)a; 3 × (iii)b; H2O | 2.53 |
| Ce(2) | 10 | 1 × (ii); 3 × (iii)a; 1 × (iii)b; 2 × H2O | 2.57 | |
| 2b | Ce(1) | 10 | 1 × (ii); 1 × (iii)a; 1 × (iv)a; 1 × (iv)b; 2 × (v)a | |
| Ce(2) | 9 | 1 × (i); 1 × (ii); 1 × (iii)a; 1 × (iii)b; 2 × (v)b; H2O | ||
| Na(1) | 6 | 1 × (iii)b; 1 × (iv)b; 2 × (v)b; 1 × (vi); H2O | ||
| 4 | Nd(1) | 10 | 1 × (ii); 1 × (iii)a; 1 × (iv)a; 1 × (iv)b; 2 × (v)a | 2.55 |
| Nd(2) | 9 | 1 × (i); 1 × (ii); 1 × (iii)a; 1 × (iii)b; 2 × (v)b; H2O | 2.50 | |
| Na(1) | 6 | 1 × (iii)b; 1 × (iv)b; 2 × (v)b; 1 × (vi); MeOH | ||
| 6 | Er(1) | 8 | 2 × (i); 2 × (ii); 2 × H2O | 2.36 |
| 7 | Lu(1) | 7 | 4 × (ii); 1 × (iii)a; 1 × (iii)b | 2.27 |
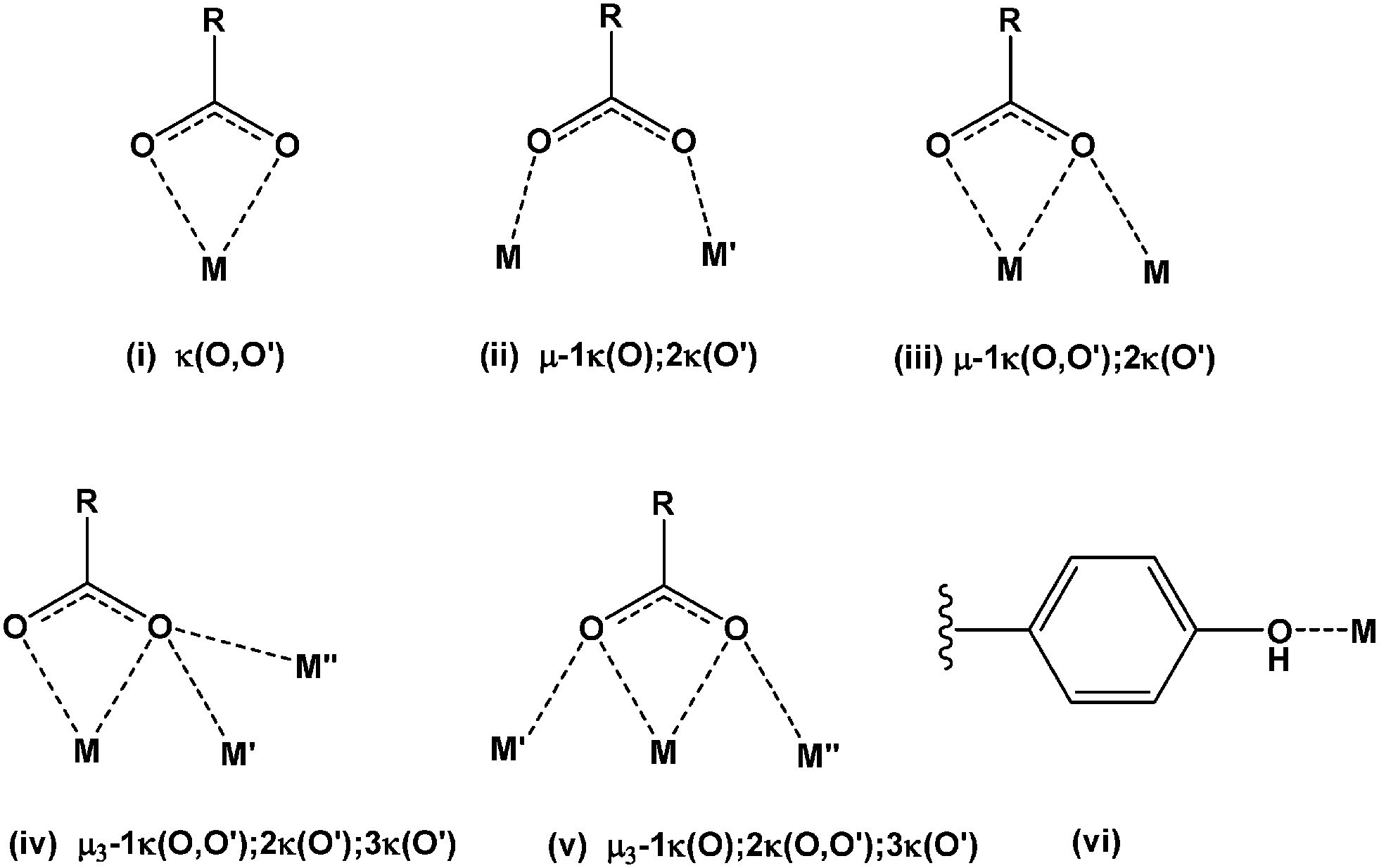 | ||
| Fig. 1 Different coordination modes observed for the 3-(4′-hydroxyphenyl)propionate ion. | ||
X-ray crystal structures
Even though coordination of the metal centres La(3) and La(4) in complex 1 corresponds to those of Ce(1) and Ce(2) in complex 2a, the eleven coordinate La(1) and nine-coordinate La(2) have no counterparts in 2a. As a result, the 1D polymeric chain in complex 2a propagates in the mirroring of the asymmetric unit in the sequence of Ce(1)#⋯Ce(1)⋯Ce(2)⋯Ce(2)# whilst complex 1 propagates in the sequence of La(4)#⋯La(1)⋯La(2)⋯La(3)⋯La(4)⋯La(1)# (Fig. 2). In the asymmetric unit of complex 1, the two nine coordinate metal atoms, La(2) and La(3), have two pairs of chelating carboxylate oxygen atoms (La(2)–O(13,14;16,17) and La(3)–O(19,20;22,23)) with O(17) and O(20) bridging the two metal centres (μ-1κ(O,O′):2κ(O′)). The other coordination sites are occupied by three other bridging carboxylate oxygen atoms (La(2)–O(5,8,11) and La(3)–O(25,29,32)) and one water molecule each. La(1) is eleven coordinate with four chelating carboxylate ligands, O(1,2;4,5;7,8;10,11), all also bridging μ-1κ(O,O′);2κ(O′), two water molecules O(1Z, 2Z), and an oxygen atom O(35)# bridging La(4)# in an adjacent asymmetric unit. La(4) differs from La(1) in that there are only three chelating carboxylate ligands O(28,29;31,32;34,35). An oxygen atom O(26) from a (μ-1κ(O);2κ(O′)) ligand (the only one in the asymmetric unit) and two water molecules make up the coordination sphere. The O(25,26) carboxylate ligand is regarded as μ-1κ(O);2κ(O′) rather than μ-1κ(O,O′);2κ(O′) as La(4)⋯O(25) is 3.191(5) Å and 0.23 Å longer than the next longest La–O distance (La(1)–O(5) 2.960(5) Å).15 A similar (μ-1κ(O):2κ(O′)) carboxylate(O(7,8)) is seen in complex 2a where the Ce(1)⋯O(8) and Ce(2)⋯O(7) values of 3.59 Å and 4.09 Å are far too long to be part of chelating interactions.1 In 1, the three longest La–O bond lengths (La(1)–O(5)); La(1)–O(8); La(4)–O(32), all >0.2 Å longer than the next longest, all involve a bridging oxygen of a chelating oxygen pair of a 1κ(O,O′):2κ(O′) ligand (Table S2 (ESI†)/Fig. 2). The range of La–O bonds falls from 0.43 Å for La(1) to 0.24 Å (La(3)) and 0.17 Å (La(2)) with a decrease in the coordination number from 11 to nine. The La–O bond length range for 1 (Table S2, ESI†) is similar to that (2.443–3.06(2) Å) of [La3(O2CPh)9(dmf)3] which has nine and 11-coordinate La atoms.16 In ten coordinate [La2(ad)3(H2O)4] (adH2 = adipic acid), the range of La–O bond lengths and the average La–O length17 are similar to those of La(4) of 1. With 2a, the average Ce–O bond length of nine-coordinate Ce(1) and ten coordinate Ce(2) relates to the corresponding values of nine coordinate Ce in cerium phenylene-1,2-dioxyacetate18 and ten coordinate Ce in cerium pimelinate, respectively.19
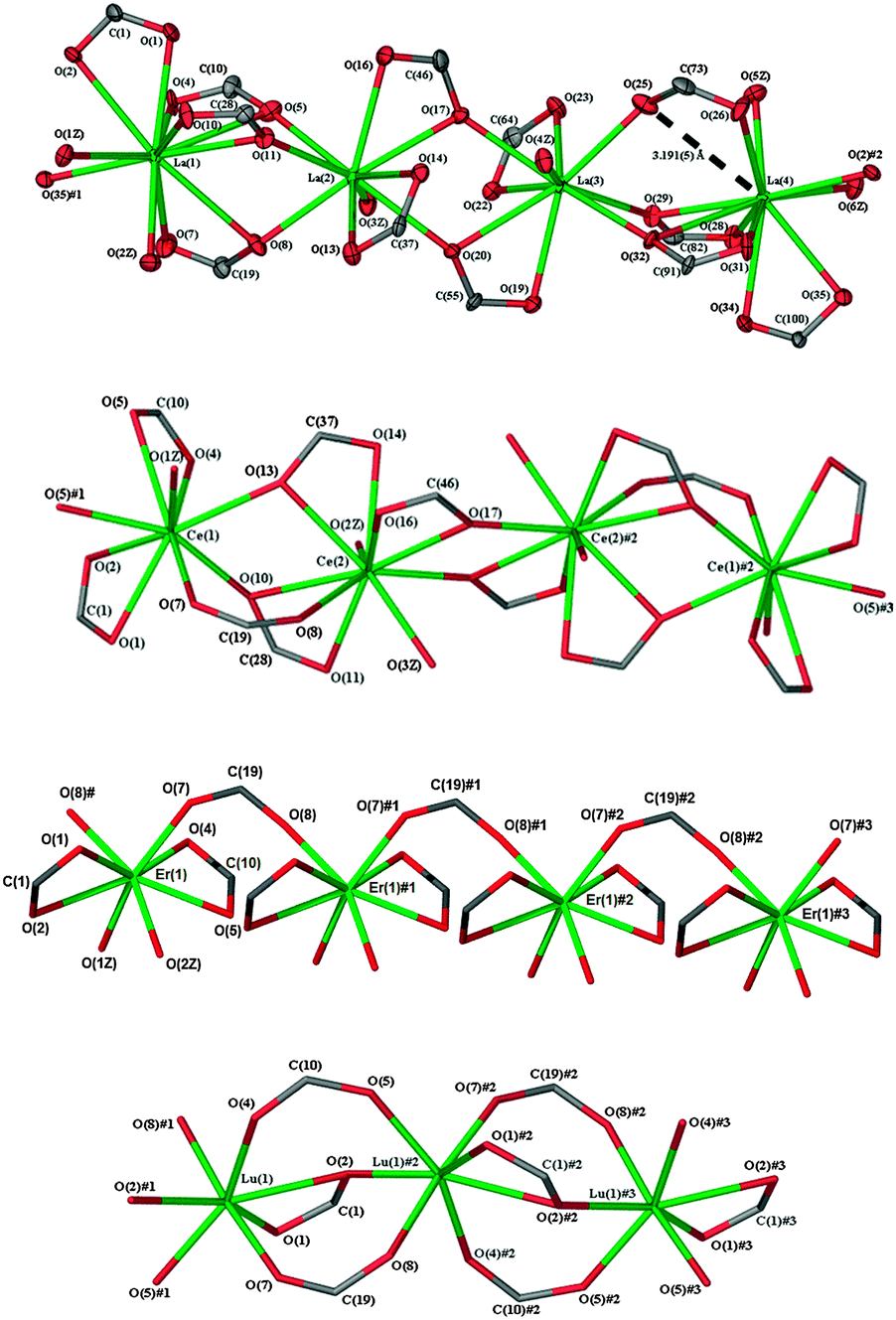 | ||
| Fig. 2 The propagation of the 1D polymeric chains of [La4(4hpp)12(H2O)6]·4(H2O)·(MeOH) (1) and [Ce2(4hpp)6(H2O)3]·2.5(EtOH)·(H2O) (2a), [Er(4hpp)3(H2O)2]·1.5(H2O) (6) and [Lu(4hpp)3].(H2O) (7). Symmetry equivalents used: complex 1 (#1 x, y − 1, z; #2 x, 1 + y, z), complex 2a (#1 1 − x, − y, 1 − z; #2 1 − x, 1 − y, 1 − z; #3 x, 1 + y, z), complex 6 (# x, 1 + x, z; #1 x − 1/2, 1/2 − y, z; #2 1/2 − x, 1/2 + y, z − 1/2), and complex 7 (#1 1 − x, 1/2 + y, 3/2 − z; #2 1 − x, y − 1/2, 3/2 − z; #3 x, y − 1, z). | ||
The asymmetric unit of complex 1 is almost linear with angles of 170.1° and 170.6° between La(1)–La(2)–La(3) and La(2)–La(3)–La(4), respectively. However, the angles formed between adjacent asymmetric units are smaller (150.5° and 150.4° between La(4)#–La(1)–La(2) and La(3)–La(4)–La(1)#, respectively). This results in a slight wave with nodes between the repeating asymmetric units in the 1D polymeric chain (Fig. 2). Most of the chelating carboxylates have varying degrees of asymmetry with the exception of ligands O(13,14) and O(16,17), which show symmetrical chelation. A similar trend is observed in complex 2a where most of the chelating carboxylate ligands are asymmetric except for ligand O(16,17). Although the asymmetric units of complexes 1 and 2a contain a (μ-1κ(O′):2κ(O′)) carboxylate bridge, the La(3)–O(25)–C(73)–O(26)–La(4) bridge is asymmetric due to a twist of the carboxylate group whilst the Ce(1)–O(7)–C(19)–O(8)–Ce(2) bridge is more symmetrical.
The influence of lanthanoid contraction is further illustrated by the decreased coordination numbers (eight and seven, respectively) of [Er(4hpp)3(H2O)2]·1.5(H2O) (6) and [Lu(4hpp)3]·(H2O) (7) (Table 2 and Fig. 2). The complexes differ markedly in the coordination modes of the carboxylate groups and only 6 has coordinated water, though both have water of crystallisation. The asymmetric unit of complex 6 has two (κ(O,O′)) carboxylate groups (O(1,2) and O(4,5)) coordinating quite symmetrically and two carboxylates bridging in the (μ-1κ(O);2κ(O′)) mode through O(7) and O(8)# to the metal centre. Compared with eight coordinate [Er2(anth)6(H2O)4]·2H2O,206 has a similar average Er–O bond length but a wider range of bond lengths by ca. 0.1 Å. The immediate coordination environment of Lu in complex 7 is one (μ-1κ(O,O′);2κ(O)) carboxylate group (chelating asymmetrically through O(1,2)) and single oxygen atoms of four bridging (μ-1κ(O);2κ(O′))carboxylate groups O(4), O(5)#1, O(7), O(8)#1 and one (μ-1κ(O);2κ(O,O′)) bridging oxygen (O(2)#1). The Lu–O bond lengths of 7 (Table S5, ESI†) cover a similar range to that (2.191–2.448 Å) of lanthanum cinnamate, also a seven coordinate unsolvated polymer,21 and 〈Lu–O〉 is 2.27 Å in each case.
The decrease in the average Ln–O bond lengths for complexes 1, 2a, 6, and 7 (Table 2) is consistent with the combination of lanthanoid contraction and the reduced coordination number at the metal centres. Coordination of water varies from 1.5/Ln (1, 2a) to 2 (6) then 0 (7). Solvent of crystallisation in the asymmetric units decreases from five (1; MeOH and 4H2O) to 3.5 (2a, 2.5EtOH and 1H2O) then 1.5 (6, 1.5H2O), and 1 (7, 1H2O), but solvation per Ln atom in the asymmetric unit (1.25, 1.75, 1.5 and 1 for 1, 2a, 6, and 7 respectively) varies little.
Both complexes 2b and 4 form a bimetallic two dimensional network with very similar ligand binding modes to the metal centres. The same five unique carboxylate binding modes are observed in both complexes. There are two μ3-1κ(O);2κ(O,O′);3κ(O′), two μ-1κ(O,O′);2κ(O′) and one each of μ3-1κ(O,O′);2κ(O′);3κ(O′), μ-1κ(O);2κ(O′) and κ(O,O′). Adjacent one dimensional polymeric chains of repeating asymmetric units (through bridged Ln(2)⋯Ln(1)⋯Ln(1)#⋯Ln(2)# chains) of these complexes are bridged by the phenolic oxygen atom of one of the (μ-1κ(O,O′):2κ(O′)) ligands (O(7,8)) to a sodium metal centre of a neighbouring chain (Fig. 3). The metal–metal distances between Nd(1)⋯Nd(2) and Nd(1)⋯Na(1) in complex 4 are 4.167 Å and 3.608 Å, respectively. There are two unique Ln metal centres with one being ten coordinate and the second being nine coordinate whilst the sodium metal centre is six coordinate in 2b and 4 (Table 2). Comparison of the Nd–O bond lengths for ten coordinate Nd(1) (Table S6, ESI†) and nine coordinate Nd(2) with those of ten coordinate Nd in the adipate complex17 and nine coordinate Nd in the anthranilate complex,20 respectively, shows that the values are similar.
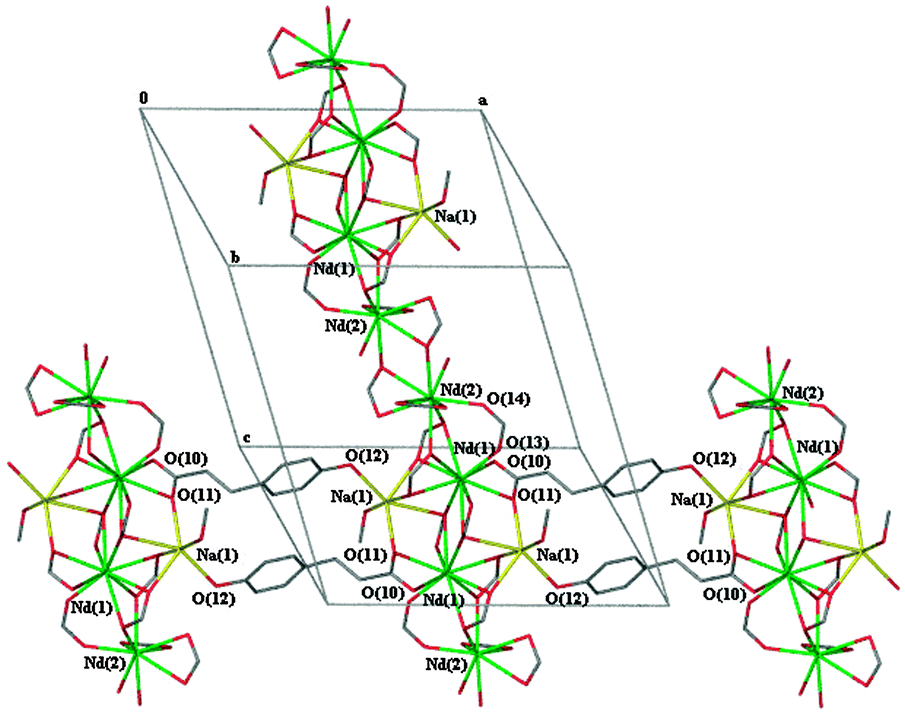 | ||
| Fig. 3 The propagation of the 2D polymeric network of [NaNd2(4hpp)7(H2O)(MeOH)]·(H2O)·3(MeOH) (4) showing the bridging of the phenolic oxygen atom to the sodium metal centre from a neighbouring chain. Non-metal atoms except those immediately surrounding the metal centres are omitted for clarity. | ||
Given the facile loss of solvent of crystallisation for 2a and 6 as indicated by microanalysis and TGA and the facile replacement of coordinated and lattice MeOH by water in 4, it is probable that bulk 2a, 4 and 6 retain the structures determined for single crystals.
Corrosion inhibition
Because of our discovery that lanthanum 4-hydroxycinnamate provides the optimum corrosion protection for mild steel from examination of a wide range of lanthanoid carboxylates including many cinnamates and substituted cinnamates,6,7 examination of the anti-corrosion properties of lanthanum 3-(4′-hydroxyphenyl)propionate 1 was of considerable interest. Both 1 and sodium 3-(4′-hydroxyphenyl)propionate were examined for their protective properties (200 ppm concentration) for a mild steel coupon in 0.01 M aqueous sodium chloride. Visual inspection of both coupons and examination under an optical microscope showed extensive corrosion (Fig. S3, ESI†), hence neither showed useful inhibitor properties at a concentration at which lanthanum 4-hydroxycinnamate is particularly effective. The sodium salt was also ineffective at 2000 ppm. Because of the poor qualitative performance, electrochemical evaluation by linear polarisation resistance or potentiodynamic polarisation was not carried out. These negative results are highly significant, as they show that the –CHCH– structural unit of the cinnamate/4-hydroxycinnamate ion is an essential structural feature for effective inhibitor performance.
Dissolution of 1 requires breakup of the coordination polymer, and the subsequent poor inhibitor performance of 1 may be related to low stability of La/4hpp complexes in solution. The mass spectrum shows no La3+ containing complexes in the +ve ESMS, and no La(4hpp) species in the −ve ESMS even though the complex was examined in the poor donor solvent methanol. Thus carboxylate coordination is completely dissociated in solution. By contrast, the effective inhibitors lanthanum 4-hydroxycinnamate,22 praseodymium 4-hydroxycinnamate23 and lanthanum 3-hydroxycinnamate23 examined in the highly coordination competitive environment of 0.01 M aqueous sodium chloride show the existence of [LnL2Cl2]−, [LnL3Cl]− and [LnL4]− complexes, which evidently account for the good protective properties.
Although Ln3+ ions have some inhibitor properties,24,25 La3+ is less effective than the optimum Ce3+,24 and 4hpp− ions have poor inhibitor properties as indicated by the sodium salt (Fig. S3, ESI†). The lack of stable La/4hpp complexes in solution obviates synergic lanthanoid carboxylate corrosion inhibition observed for lanthanum 4-hydroxycinnamates.6,7,25
Conclusions
From reactions between lanthanoid salts and sodium 3-(4′-hydroxyphenyl)propionate both homometallic [Ln(4hpp)3(H2O)n] and bimetallic [NaLn2(4hpp)7(solv)2] complexes have been obtained and representative structures determined. The former are one dimensional polymers for which there is a decrease in the coordination number progressively from Ln = La (9–11), Ce (9, 10), Er (8), and Lu (7), the last having no coordinated water. By contrast, bimetallics are 2D networks with a much wider range of carboxylate binding modes and cross-chain coordination of the 4-hydroxy substituent to sodium. The La complex is a poor inhibitor of corrosion of mild steel in contrast to the corresponding 4-hydroxycinnamates, thereby establishing that the –CHCH− functionality of the latter is vital to the excellent anti-corrosion behaviour of lanthanum 4-hydroxycinnamate.
Experimental
General details
All commercially available solvents and chemicals were obtained from Sigma-Aldrich, BDH or Ajax and used without further purification. IR spectra were collected on a Perkin-Elmer 1600 Fourier Transform Infrared Spectrometer (FTIR) as Nujol mulls on NaCl plates. CH analyses were performed in the Campbell Microanalytical Laboratory, University of Otago, New Zealand. Metal analyses were affected by complexometric titrations with EDTA using Xylenol Orange indicator. Thermogravimetric analysis (TGA) was conducted using a Perkin-Elmer Pyris TGA instrument. X-ray powder diffraction (XRD) patterns were obtained on a Philips 1140 powder diffractometer with a Cu Kα source and the X-ray diffraction screen processing software, Traces Ver. 6.6.1, was used as a graphical interface. X-ray powder simulations were undertaken using LAZY PULVERIX26 powder generation software from data obtained from low temperature single crystal X-ray measurements.X-ray crystallography
Single crystals were mounted on fine glass fibres using viscous hydrocarbon oil and collections were carried out at 123 K using an open-flow N2 Oxford Cryosystems cryostream. Single crystal X-ray crystallographic data were collected using either a Bruker X8 Apex II diffractometer or an Enraf-Nonius Kappa-CCD diffractometer, both utilising graphite monochromated Mo-Kα radiation (λ = 0.71013 Å). Collection and refinement parameters are shown in Table 3. Data were initially processed using the program SAINT27 or DENZO program28 for the respective instruments. Structures were solved using SHELXS-9729 and refined using conventional alternating least-squares methods with SHELXL-9729 or SHELXL-2014. The program X-Seed30 or Olex2 was used as a graphical interface. Anisotropic thermal parameters were refined for non-hydrogen atoms except some atoms of the disordered fragments (complex 2a) and lattice solvent molecules (complex 4). Most hydrogen atoms were calculated and refined using a riding model, but those of lattice water molecules were located from the difference Fourier maps. High mobility of lattice solvent molecules in compounds 2a and 6 results in destabilisation of the refinement, thus they were treated using the Platon/Squeeze program. Bond distances and angles are provided in the ESI.† For compound 2b, data were very poor and it is included only for connectivity purposes.| 1 | 2a | 4 | 6 | 7 | |
|---|---|---|---|---|---|
| Formula | C109H132La4O47 | C54H60Ce2O21 | C67H83NaNd2O27 | C27H31ErO11 | C27H29LuO10 |
| M | 2749.78 | 1325.26 | 1631.80 | 698.78 | 688.47 |
| Crystal system | Triclinic | Triclinic | Triclinic | Orthorhombic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
Pna21 | P21/c |
| a/Å | 15.2502(9) | 14.2020(11) | 15.328(3) | 24.425(5) | 13.345(3) |
| b/Å | 16.6406(10) | 15.9564(14) | 15.480(3) | 5.351(1) | 8.0505(16) |
| c/Å | 25.6709(14) | 16.1020(11) | 16.300(3) | 22.945(5) | 24.550(5) |
| α/° | 89.014(2) | 116.872(5) | 78.16(3) | 90 | 90 |
| β/° | 78.289(2) | 93.433(5) | 70.63(3) | 90 | 96.45(3) |
| γ/° | 64.668(3) | 92.854(5) | 86.33(3) | 90 | 90 |
| V/Å3 | 5747.7(6) | 3236.9(4) | 3571.0(14) | 2999.0(11) | 2621.0(9) |
| Z | 2 | 2 | 2 | 4 | 4 |
| μ/mm−1 | 1.547 | 1.455 | 1.524 | 2.853 | 3.824 |
| No. total reflections | 80774 |
20909 |
31950 |
26496 |
25249 |
| No. unique | 25932 |
11352 |
12470 |
6590 | 6018 |
| No. observed [I ≥ 2σI] | 20184 |
7369 | 7672 | 4452 | 4263 |
| Parameters | 1500 | 707 | 822 | 314 | 354 |
| R int | 0.0552 | 0.0627 | 0.1163 | 0.1058 | 0.1513 |
| wR2 (all data) | 0.1419 | 0.1755 | 0.1616 | 0.1225 | 0.1632 |
| R [I ≥ 2σI] | 0.0814 | 0.0676 | 0.0687 | 0.0627 | 0.0714 |
Crystallographic data (excluding structure factors) for the structures reported in this paper and associated ESI† have been deposited with the Cambridge Crystallographic Data Centre as supplementary numbers CCDC 1055961 for compound 2a, 1055962 for compound 6, 1055963 for compound 1, 1055964 for compound 7, 1055965 for compound 2b and 1055966 for compound 4.
Syntheses
Treatment of 3-(4′-hydroxyphenyl)propionic acid, H(4hpp), deprotonated in situ with aqueous sodium hydroxide or of the preformed sodium salt (1, 6), with hydrated lanthanoid chloride (1, 2a, 7) or nitrate (2b, 3–6), (mole ratio 3:1) in methanol (1–5) or water (6–7) at pH 6 gave lanthanoid 3-(4′-hydroxyphenyl)propionate complexes. The reaction mixtures were stirred at room temperature for a minimum of 3 hours. When precipitates (2b, 3, and 5–7) formed, they were filtered off and dried over silica. 1 and 4 deposited as crystals whilst crystals of 2b were mixed with the bulk precipitate. Crystals (3, 6, and 7) were obtained upon the slow evaporation of the mother liquor but the reaction with holmium nitrate (5), only gave a microcrystalline precipitate. Data reported are for the bulk precipitated samples except for complexes 1, 2a and 4 which were crystalline samples. Bulk samples were dried to a constant weight in a desiccator over silica gel.
IR (ν/cm−1): 3404 br m, 3350 br m, 1883 vw, 1658 w, 1614 w, 1601 w, 1556 s, 1517 s, 1444 s, 1417 s, 1296 w, 1256 m, 1235 m, 1177 w, 1160 w, 1105 m, 1015 w, 934 w, 826 s, 779 m, 689 m.
1H NMR (d6-DMSO): δ = 6.98 (d, 2H, 3JH–H = 8 Hz, Ar-H3′,5′), 6.66 (d, 2H, 3JH−H = 8 Hz, Ar-H2′,6′), 2.68 (t, 2H, 3JH–H = 8 Hz, H2), 2.16 (t, 2H, 3JH–H = 8 Hz, H3). The OH proton was not observed.
IR (ν/cm−1): 3368 m br, 3286 m br, 3172 m br, 1612 m, 1591 m, 1529 s, 1516 s, 1419 s, 1340 m, 1314 w, 1259 m, 1238 m, 1209 w, 1172 w, 1103 w, 1071 w, 1045 w, 1017 w, 963 w, 829 m, 695 w.
TGA weight loss: 1.33% (25–65 °C, 1 step, 1 MeOH, calculated 1.16%), 2.40% (65–163 °C, 1 step, 4H2O, calculated 2.62%), 3.66% (163–335 °C, 1 step, 6H2O, calculated 3.93%).
ESI-MS [m/z (I/I0)]: (−ve) 92.8 (10) [HOC6H4]˙−, 148.9 (100) [C6H5(CH2)2CO2]−, 165.0 (70) [(4hpp)]−, 223.0 (15), 233.0 (5) [La(OH)2CO3]−, 353.3 (4) [La(OC6H4Et)(OMe)3]−. Powder XRD [d-spacings Å (I/I0)]: 13.64 (32), 12.99 (63), 12.54 (100), 11.19 (22), 10.71 (64), 7.99 (23), 6.68 (69), 6.57 (57), 6.45 (26), 6.30 (33), 6.15 (19), 6.05 (18), 5.38 (21), 5.14 (39), 5.03 (28), 4.91 (27), 4.65 (21), 4.17 (19), 4.10 (21), 3.87 (20), 3.74 (26), 3,72 (29), 3.65 (18), 3.58 (23), 3.36 (27), 2.89 (37), 2.86 (17), 2.78 (20), 2.71 (20), 2.50 (23), 2.46 (33), 2.32 (18). d-Spacings simulated from X-ray structure: 13.34 (31), 13.10 (100), 12.54 (72), 10.92 (54), 7.91 (14), 6.73 (16), 6.55 (11), 6.50 (10), 6.27 (6), 6.01 (4), 5.46 (5), 5.07 (9), 5.01 (3), 4.87 (5), 4.63 (6), 4.43 (4), 4.41 (4), 4.03 (5), 3.75 (5), 3.70 (2), 3.65 (2), 3.64 (3), 3.52 (3), 3.42 (2), 2.52 (2), 2.18 (2).
IR (ν/cm−1): 3307 br m, 1612 w, 1595 w, 1531 s, 1515 s, 1417 s, 1312 w, 1236 m, 1171 w, 1102 w, 1043 w, 1021 w, 960 w, 830 m.
IR (ν/cm−1): 3606 w, 3369 m br, 1885 w, 1614 m, 1598 m, 1578 m, 1527 s, 1516 s, 1418 s, 1338 w, 1312 w, 1237 s, 1174 w, 1158 w, 1103 w, 1016 w, 949 w, 829 m, 788 w.
TGA weight loss: 3.99% (25–70 °C, 1 step, 3H2O, calculated 3.48%), 2.22% (70–100 °C, 1 step, 2H2O, calculated 2.32%).
Powder XRD [d-spacings Å (I/I0)]: 14.49 (84), 13.94 (100), 10.97 (44), 9.54 (33), 7.36 (29), 7.08 (39), 6.95 (35), 6.77 (26), 6.18 (22), 6.07 (26), 5.52 (20), 4.72 (20), 3.94 (20). d-Spacings simulated from the X-ray structure: 14.70 (100), 14.16 (96), 11.14 (24), 9.46 (10), 7.35 (9), 7.08 (6), 7.00 (13), 6.83 (6), 6.13 (4), 6.01 (4), 5.60 (5), 5.57 (5), 5.31 (3), 3.92 (8).
ESI-MS [m/z (I/I0)]: (+ve) 282.3 (34) [Ce(OH)2(H2O)6]+, 304.2 (100) [Ce(OC6H4(CH2)2CO2)]+, 321.9 (32) [Ce(4hpp)(OH)]+, 338.3 (25), 360.3 (80), 410.0 (15), 428.0 (10) [Ce(4hpp)(OH)3(H2O)4]+, 442.0 (14), 469.9 (20), 529.9 (12), 589.8 (12) [NaCe(4hpp)2(HCO3)(H2O)2]+; (−ve) 164.9 (81) [4hpp]−, 353.0 (100), 375.2 (15), 540.9 (15), 559.9 (11) [Ce(4hpp)2(OEt)2]−, 679.9 (18) [Ce(4hpp)3(OEt)]−, 701.8 (15) [Ce(4hpp)3(OH)(H2O)(MeOH)]−.
IR (ν/cm−1): 3623 w, 3271 s br, 1888 w, 1615 m, 1600 m, 1580 s, 1516 s, 1417 s, 1336 m, 1311 m, 1237 s, 1172 m, 1159 m, 1103 m, 1047 w, 1010 w, 949 w, 830 m, 784 w, 697 w.
TGA weight loss: 4.06% (25–70 °C, 1 step, 3H2O, calculated 3.48%), 2.25% (70–150 °C, 1 step, 2H2O, calculated 2.32%).
Powder XRD [d-spacings (I/I0)]: 14.56 (100), 14.07 (37), 10.96 (27), 9.53 (21), 7.37 (39), 6.95 (35), 6.20 (17), 5.52 (14), 4.87 (17), 4.73 (12), 4.46 (12), 3.94 (12), 3.670 (13).
IR (ν/cm−1): 3530 m, 3256 br s, 1880 w, 1617 m, 1600 m, 1579 m, 1536 s, 1516 s, 1407 s, 1332 m, 1300 w, 1237 s, 1172 m, 1155 w, 1102 w, 1073 w, 1042 w, 1015 w, 950 w, 828 m, 672 m.
TGA weight loss: 2.46% (25–115 °C, 1 step, 2H2O, calculated 2.39%).
Generated powder XRD [d-spacings Å (I/I0)]: 15.15 (93), 14.46 (100), 10.48 (28), 10.44 (24), 7.57 (17), 7.23 (11), 6.72 (12), 6.51 (5), 6.23 (4), 5.55 (5), 5.31 (3), 5.24 (6), 4.64 (4), 4.51 (5), 4.33 (4), 3.99 (3), 3.84 (6), 3.79 (3), 3.69 (3), 3.67 (5), 3.50 (5).
IR (ν/cm−1): 3562 w, 3432 m br, 3224 m br, 1612 w, 1597 w, 1577 m, 1543 s, 1515 m, 1419 m, 1340 w, 1318 w, 1302 w, 1257 m, 1230 m, 1158 w, 1104 w, 1050 w, 957 w, 828 m, 707 w, 675 m.
Powder XRD [d-spacings Å (I/I0)]: 14.50 (32), 13.44 (81), 10.50 (70), 9.69 (69), 8.17 (12), 7.59 (11), 6.56 (100), 5.81 (29), 5.62 (10), 4.84 (12), 4.68 (13), 4.50 (10), 4.22 (41), 4.16 (11), 4.01 (15), 3.89 (12), 3.40 (74), 3.75 (28), 3.63 (12), 3.20 (22), 2.97 (11), 2.86 (14), 2.80 (14), 2.66 (14), 2.63 (12), 2.53 (15), 2.45 (16), 2.39 (11), 2.35 (10), 2.26 (17), 2.08 (13).
IR (ν/cm−1): 3567 w, 3436 m br, 3221 m br, 1612 w, 1600 w, 1578 m, 1543 s, 1515 m, 1419 m, 1340 w, 1301 w, 1257 m, 1231 m, 1174 w, 1158 w, 1105 w, 1051 w, 1015 w, 956 w, 878 w, 855 w, 828 w, 707 w, 675 w.
TGA weight loss: 5.16% (20–150 °C, 1 step, 2H2O, calculated 5.15%).
Powder XRD [d-spacings Å (I/I0)]: 14.42 (35), 13.48 (83), 10.50 (88), 9.68 (64), 8.16 (16), 7.58 (16), 6.56 (100), 5.81 (29), 4.86 (14), 4.66 (19), 4.22 (43), 4.00 (19), 3.80 (62), 3.74 (34), 3.64 (16), 3.20 (26), 2.85 (20), 2.80 (19), 2.66 (17), 2.53 (21), 2.45 (21), 2.39 (16), 2.26 (18).
IR (ν/cm−1): 3568 m, 3256 s br, 3197 s br, 1613 m, 1581 s, 1550 s, 1515 s, 1404 s, 1339 m, 1318 w, 1297 w, 1255 s, 1234 s, 1174 m, 1158 m, 1105 m, 1048 w, 956 w, 824 m, 708 m.
TGA weight loss: 5.33% (20–150 °C, 1 step, 2H2O, 5.10%).
Powder XRD [d-spacings (I/I0)]: 13.71 (100), 10.47 (71), 9.34 (72), 7.88 (26), 7.57 (16), 7.00 (15), 6.42 (60), 5.62 (21), 5.18 (15), 4.99 (23), 4.61 (43), 4.50 (31), 4.24 (19), 4.09 (29), 3.95 (46), 3.89 (19), 3.73 (32), 3.55 (19), 3.25 (23), 3.21 (19), 3.13 (18), 2.92 (18).
d-Spacings simulated from X-ray structure: 13.26 (100), 12.20 (82), 9.53 (52), 8.51 (41), 7.64 (2), 6.63 (44), 6.12 (22), 6.10 (23), 5.79 (22), 5.57 (4), 5.32 (9), 5.10 (3), 4.76 (2), 4.58 (25), 4.44 (11), 4.26 (9), 3.97 (10), 3.61 (17), 3.53 (16), 3.24 (12), 3.10 (11), 2.98 (4), 2.93 (5), 2.89 (5), 2.63 (6), 2.63 (6).
Corrosion testing
Corrosion immersion tests were conducted according to ASTM G31-72.31 The appropriate weights of sample compounds were dissolved in water to give concentrations of 200 ppm (for Na(4hpp) and [La4(4hpp)12(H2O)6]·4(H2O) (1)) and 2000 ppm (for Na(4hpp)) in 0.01 M sodium chloride solution. The pH of the resultant test solutions was maintained within the range of 5–7. Mild steel alloy AS1020 coupons (cut to approx. 25 × 25 × 1 mm) used in the immersion tests were progressively polished with silica-carbide polishing papers of 240, 400, 800, 1200 and 4000 grits. The coupons were rinsed thoroughly with water and dried under flowing N2 gas and used immediately after polishing and washing to ensure optimum surface conditions.The sample and reference coupons were immersed in a standard beaker (500 mL) containing the test solutions. Two sample coupons were suspended by Teflon-string and fully immersed at mid-depth in each beaker filled with 500 mL of test solution. The immersion coupon was left in place for seven days and the level of test solution was checked intermittently during the duration of the test and was readjusted as required to replenish water loss due to evaporation. The mouth of the beaker was covered with a filter paper to prevent contaminants (dust, insects, etc.) whilst still allowing free air flow. As the corrosion of the sample coupon progresses, it is important to avoid oxygen depletion in the test solution which affects the uniformity of the environment within the solution during the test duration. The post immersion cleaning procedure involved the rinsing of the coupons first with water and then with mild sonication in clean water for approx. one minute to remove rust clinging to the test coupons' surface. They were then dried by N2 gas.
Both normal photography and an optical microscope (with up to 20× magnification) were used for the visual inspection of the tested coupons. The general uniformity of the metal surface on the coupons was compared amongst the tested coupons with the blank coupons.
References
- R. D. Shannon, Acta Crystallogr., 1976, A32, 751 CrossRef CAS.
- (a) S. A. Cotton, Scandium, Yttrium and the Lanthanides, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier Science, 2003, vol. 3, pp. 93–188 Search PubMed; (b) S. A. Cotton, Lanthanide and Actinide Chemistry, John Wiley & Sons, Chichester, 2006 Search PubMed.
- F. T. Edelmann, Scandium, Yttrium and the Lanthanide and Actinide Elements, Excluding their Zero Oxidation State Complexes, in Comprehensive Organometallic Chemistry II, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Elsevier Science, 1995, vol. 4, pp. 11–212 Search PubMed.
- G. B. Deacon, P. MacKinnon, R. S. Dickson, G. N. Pain and B. O. West, Appl. Organomet. Chem., 1990, 4, 439 CrossRef CAS PubMed.
- (a) M. Kanesato and Y. Yokoyama, Chem. Lett., 1999, 137 CrossRef CAS; (b) M. Kanesato and Y. Yokoyama, Anal. Sci., 2000, 16, 335 CrossRef CAS; (c) P. V. Bernhardt, B. M. Flanagan and M. J. Riley, Aust. J. Chem., 2000, 53, 229 CrossRef CAS; (d) P. V. Bernhardt, B. M. Flanagan and M. J. Riley, Aust. J. Chem., 2001, 54, 229 CrossRef CAS; (e) M. Seitz, A. G. Oliver and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 11153 CrossRef CAS PubMed.
- F. Blin, S. G. Leary, K. Wilson, G. B. Deacon, P. C. Junk and M. Forsyth, J. Appl. Electrochem., 2004, 34, 591 CrossRef CAS.
- F. Blin, S. G. Leary, G. B. Deacon, P. C. Junk and M. Forsyth, Corros. Sci., 2006, 48, 404 CrossRef CAS PubMed.
- (a) R. P. Farrell and P. A. Lay, Comments Inorg. Chem., 1992, 13, 133 CrossRef CAS PubMed; (b) R. Codd, C. T. Dillon, A. Levina and P. A. Lay, Coord. Chem. Rev., 2001, 216–217, 537 CrossRef CAS; (c) A. Levina, R. Codd, C. T. Dillon and P. A. Lay, Prog. Inorg. Chem., 2003, 51, 145 CAS.
- (a) E. McCafferty, J. Electrochem. Soc., 1979, 126, 385 CrossRef CAS PubMed; (b) G. P. Bierwagen and D. E. Tallman, Prog. Org. Coat., 2001, 41, 201 CrossRef CAS; (c) J. Sinko, Prog. Org. Coat., 2001, 42, 267 CrossRef CAS.
- G. B. Deacon, C. M. Forsyth, P. C. Junk, M. Hilder, S. G. Leary, C. Bromant, I. Pantenburg, G. Meyer, B. W. Skelton and A. H. White, Z. Anorg. Allg. Chem., 2008, 634, 91 CrossRef CAS PubMed.
- (a) W. Lu, L. Xiaoyang, C. Weigno, P. Wei, D. Nan, L. Keti, H. Youging and H. Shengzhi, J. Hangzhao U. (Nat. Sci.), 1997, 24, 55 CAS; (b) W. Lu, W.-G. Chen, X.-Y. Luo, N. Dong, K.-L. Liang, Y.-Q. Huang and S.-Z. Hu, Chem. J. Chin. Univ., 1997, 18, 337 CAS.
- G. B. Deacon, M. Forsyth, P. C. Junk, S. G. Leary and W. W. Lee, Z. Anorg. Allg. Chem., 2009, 635, 833 CrossRef CAS PubMed.
- H. Li and C. W. Hu, J. Solid State Chem., 2004, 177, 4501 CrossRef CAS PubMed.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
- Only one out of 14117 LaO distances in the CCDC (3.224 Å) exceeds this value with the next longest 3.133 Å significantly shorter than La(4)⋯(O5).
- H. Busskamp, G. B. Deacon, M. Hilder, P. C. Junk, U. H. Kynast, W. W. Lee and D. R. Turner, CrystEngComm, 2007, 9, 394 RSC.
- C. Bromant, H. Flemig, W. Nika, I. Pantenburg and G. Meyer, Z. Anorg. Allg. Chem., 2006, 632, 851 CrossRef CAS PubMed.
- T. Behrsing, G. B. Deacon, P. C. Junk, B. W. Skelton, A. N. Sobolev and A. H. White, Z. Anorg. Allg. Chem., 2013, 639, 41 CrossRef CAS PubMed.
- H. Flemig, I. Pantenburg and G. Meyer, Z. Anorg. Allg. Chem., 2006, 632, 2205 CrossRef CAS PubMed.
- G. B. Deacon, M. Forsyth, P. C. Junk, S. G. Leary and G. J. Moxey, Polyhedron, 2006, 25, 579 CrossRef PubMed.
- G. B. Deacon, C. M. Forsyth, P. C. Junk, M. Hilder, S. G. Leary, I. Pantenburg, G. Meyer, B. W. Skelton and A. H. White, Z. Anorg. Allg. Chem., 2008, 634, 91 CrossRef CAS PubMed.
- M. Seter, B. Hinton and M. Forsyth, J. Electrochem. Soc., 2012, 159, c181 CrossRef CAS PubMed.
- M. Seter, G. Girard, W. W. Lee, G. Deacon, P. Junk, B. Hinton and M. Forsyth, AIMS Mater. Sci., 2015, 2, 1 CrossRef.
- J. de Damborenea, A. Conde and M. A. Arenas, Corrosion Inhibition with Rare Earth Metal Compounds in Aqueous Solution, in Rare Earth-based Corrosion Inhibitors, ed. M. Forsyth and B. Hinton, Woodhead Publishing, Cambridge, 2014, ch. 3 Search PubMed.
- M. Forsyth, M. Seteer, B. Hinton, G. Deacon and P. Junk, Aust. J. Chem., 2011, 64, 812 CrossRef CAS.
- K. Yvon, W. Jeitschko and E. Parthe, J. Appl. Crystallogr., 1977, 10, 73 CrossRef.
- Apex II Version 2.1, Bruker AXS Ltd, Madison, Wisconsin, 2005 Search PubMed.
- Z. Otwinowski and W. Minor, in Macromolecular Crystallography, Pt A, ed. C. W. Carter Jr. and R. M. Sweet, 1997, vol. 276, p. 307 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
- American-Standard-Test-Methods in ASTM G31-72, Philadelphia, USA, 2004.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1055961–1055966. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00787a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |