
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of bifunctional dendrimers: preliminary use for the coating of gold surfaces and the proliferation of human osteoblasts (HOB)†
Edwin R.
de Jong
abc,
Nicole
Deloch
c,
Wolfgang
Knoll
cd,
Cédric-Olivier
Turrin
ab,
Jean-Pierre
Majoral
ab,
Anne-Marie
Caminade
*ab and
Ingo
Köper
ce
aCNRS, LCC (Laboratoire de Chimie de Coordination), 205 Route de Narbonne, BP 44099, F-31077 Toulouse Cedex 4, France. E-mail: anne-marie.caminade@lcc-toulouse.fr
bUniversité de Toulouse, UPS, INPT, F-31077 Toulouse Cedex 4, France
cMax-Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
dAustrian Institute of Technology, Donau-City-Straße 1, 1220 Vienna, Austria
eFlinders Centre for NanoScale Science and Technology, School of Chemical and Physical Sciences, Flinders University, Sturt Road, Adelaide SA 5001, Australia. E-mail: ingo.koeper@flinders.edu.au
First published on 8th July 2015
Abstract
Two different novel families of bifunctional water-soluble dendrimers are synthesized, using the specific functionalization of one function of the cyclotriphosphazene core. Dendrimers are grown from the 5 remaining functions, up to generation 2. Water-solubility is attained in the last step of the synthesis by grafting either ammonium terminal groups or carboxylate terminal groups, on generations 1 and 2 of these bifunctional dendrimers. 12 new compounds are synthesized and fully characterized, in particular by multi-nuclear NMR. The function linked to the core is thioctic acid, suitable for grafting onto gold, thus both types of water-soluble dendrimers can be used to coat gold surfaces. These macromolecular assemblies are characterized by surface plasmon resonance (SPR). In a preliminary attempt, the gold surfaces modified by either positively or negatively charged dendrimers are used for studying their interaction with cells. Exposed to human osteoblast cells (OBC), the influence of the surface coatings on the cell responses is investigated. Polycationic dendrimers provoke cell apoptosis, whereas negatively charged dendrimers support cell adhesion and proliferation.
Introduction
Dendrimers1 are macromolecules with a regularly branched and repetitive structure that are synthesized in an iterative fashion. Due to their monodispersity, well-defined shape and extremely high functionality, dendrimers are ideal nano-sized objects for functional and biocompatible surface coatings. Furthermore, dendrimers play an important role as versatile tools in nano-medicine.2 In most cases dendrimers have numerous but identical terminal groups. However, the demand on their structural complexity is increasing,3 and there is a real need for elaborating strategies toward bifunctional dendrimers. Such an approach is particularly suitable for the functionalization of the surface of materials at the nanometric scale.4Extensive studies on cell–surface interactions have indicated that surface properties such as hydrophilicity,5 surface charge, surface energy,6 protein adsorption7 and surface topography and morphology8 have a significant effect on these interactions. Typically, cationic charges enhance cell attachment, possibly through electrostatic attraction,9 as it was shown for instance for human umbilical vein endothelial cells,10 and several other types of cells.11 In addition, different cell types typically react entirely differently to an environment;12 therefore a detailed study on every surface coating is necessary. So far, dendrimers have been used only occasionally for cell-attractive surface coatings, despite the fact that their multivalency should enhance the binding avidity towards cells. Rat neurons showed a high affinity and proliferation rate on multilayered generation 4 (G4) poly(phosphorhydrazone) dendrimer surfaces that were created by a layer-by-layer deposition technique, with the neurons showing a preference for positively charged outer layers.13 Human corneal epithelial cells and mouse fibroblasts showed good adhesion on (G1–G4) dendronized surfaces with hydroxyl extremities (non-ionic) of the polyamidoamine-based (PAMAM-based) cascade structure.14 Human fibroblasts and myoblasts also adhered on surfaces coated with D-glucose terminated dendrimers,15 and embryonic stem cells were maintained in an undifferentiated state on the same surface.16 Recently, surfaces coated with PAMAM generation 7 dendrimers modified with anti-epithelial cell adhesion molecules (one of the most commonly used circulating tumour cells capturing agents) were efficiently used for the capture of circulating tumour cells in peripheral blood.17
The surface attachment of osteoblast cells is of particular interest for many biomedical applications. These cells interact with the surface of bones, as they are continuously involved in renewing and reshaping bone tissue. They are also important when a foreign material needs to be inserted into the body, for example in the case of implants. It was found that positively charged amino-coated titanium surfaces enhanced the first steps towards osteoblast adhesion, compared to negatively charged surfaces.18 This could be related to the negative charge of hyaluronan that is involved in the early adhesion process.19 TiOx surfaces modified with biotinylated fibronectin (a partial aminoacid sequence of an extracellular matrix protein) adsorbed on a streptavidin-silane self-assembled multilayer were also found effective for osteoblast adhesion.20 However, to our knowledge, no experiment to date involving osteoblasts has been carried out with dendrimer coated surfaces.
Here we report the synthesis of two new series of bifunctional dendrimers based on a phosphorus scaffold of the type poly(phosphorhydrazone), a well-established bio-compatible element.21 Gold surfaces are chosen to be coated by the dendrimers, as any modification on this surface can be easily investigated by surface plasmon resonance (SPR) spectroscopy and other surface analytical techniques.22 For this purpose, the dendrimers are functionalized with one dithiolane group linked to the core, suitable for the formation of self-assembled monolayers on gold surfaces, due to the well-known affinity of gold to thiols. Positively or negatively charged functions are used as terminal groups, to confer a hydrophilic character to the surface of the dendrimer,23 and thus to the coated surface. As it has been already shown that the nature of the charges (positive or negative) strongly influences the behaviour of the cells, the presence of charges as terminal groups of dendrimers will enable a comparison with these data.18 The formation of a self-assembled monolayer on a gold substrate is characterized by surface plasmon resonance (SPR) spectroscopy. In a preliminary attempt, the coated surfaces are exposed to human osteoblast (HOB) cells and their adhesion and proliferation are studied by optical microscopy and biochemical assays.
Results and discussion
Synthesis and characterization of bifunctional dendrimers
Hexachlorocyclotriphosphazene is an interesting core for dendrimers, as it possesses 6 functions for growing the branches, instead of 3 or 4 for most classical cores. Furthermore, we have previously shown that it is possible to differentiate one function among six, affording AB5-type compounds, suitable to elaborate non symmetrical dendrimers24 or highly dense dendrimers.25 The possibility of differentiating the reactivity of each Cl in P(X)Cl2 groups (X = S, O, or NR)26 is particularly relevant for the design of new bifunctional dendrimers. The A function is the dithiolane group for grafting onto the gold surface, and the B functions are aldehydes, which are suitable starting points for the synthesis of the branches of poly(phosphorhydrazone) dendrimers,27 and further functionalizations. The reaction of phenols with P–Cl functions is the most convenient reaction (easy and quantitative) for the functionalization of phosphorus dendrimers under basic conditions. Thus we used hydroxybenzaldehyde as the B group and we grafted tyramine to thioctic acid by peptide coupling to elaborate the A group, which contains a phenol also (compound 1, Scheme 1).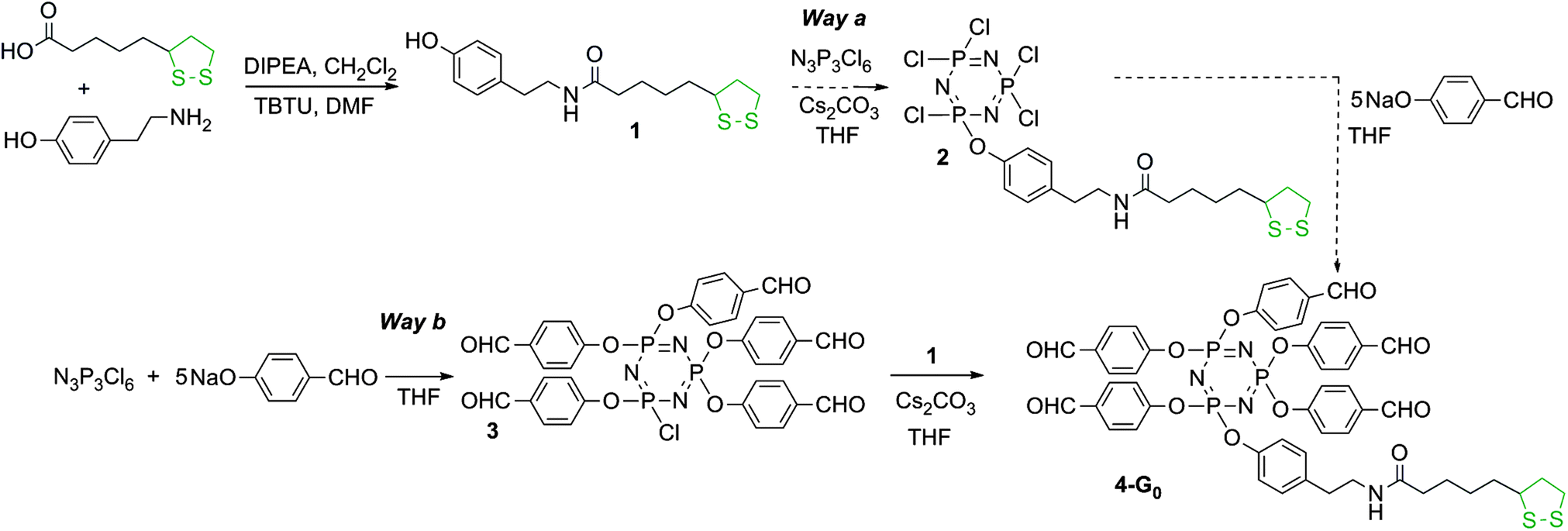 | ||
| Scheme 1 Synthesis of the dithiolane derivative 1, and two strategies for its grafting to the cyclotriphosphazene core. Only Way b affords pure compounds. | ||
In order to obtain the AB5 core, two strategies can be envisaged: either by first grafting one dithiolane derivative 1 onto the core followed by the reaction of 5 equivalents of hydroxybenzaldehyde (Way a, Scheme 1) or grafting first 5 equivalents of hydroxybenzaldehyde then the dithiolane derivative 1 (Way b, Scheme 1). We tested both the strategies. Way a yielded the expected compound 2 as the major product, characterized by the presence of one doublet at 22.4 ppm in the 31P NMR spectrum, and a pseudo triplet at 12.2 ppm (2JPP = 64 Hz), characteristic of a single substitution on N3P3Cl6. However, the 31P NMR spectrum displays also the presence of small impurities (about 5%) that could not be removed in the purification process. Way b yielded cleanly the pentaaldehyde 3,28 characterized by the presence of a pseudo triplet at 20.8 ppm and a doublet at 5.1 ppm (2JPP = 84 Hz) in the 31P NMR spectrum. The grafting of compound 1 in the second step induces the disappearance of these signals and the appearance of a pseudo singlet at 7.4 ppm for compound 4-G0. We also tried to obtain compound 4-G0 by continuing the process of Way a, i.e. by reacting 5 equivalents of hydroxybenzaldehyde with compound 2. The reaction worked, but some impurities from the previous step could not be removed. Thus, only compound 4-G0 obtained through Way b was used in the subsequent steps (Scheme 1).
The condensation of 5 equivalents of H2NNMeP(S)Cl2 resulted in the first generation bifunctional dendrimer 5-G1. The completion of the reaction is shown by the disappearance of the signals corresponding to the aldehydes both in 1H and 13C NMR. Compound 5-G1 is the starting material for pursuing the growth of the branches. Compound 5-G1 is reacted with 10 equivalents of hydroxybenzaldehyde. The completion of the reaction affording 4-G1 is detected by 31P NMR, which displays the disappearance of the intermediate singlet at 69 ppm, corresponding to the monosubstitution (P(S)Cl(OC6H4CHO)) on behalf of the appearance of a singlet at 60.4 ppm, corresponding to the full substitution. Starting from compound 4-G1, the second generation of the bifunctional dendrimer is obtained by the condensation with H2NNMeP(S)Cl2. Compound 5-G2 is isolated and characterized, as 5-G1 previously. It is in particular characterized in 31P NMR by the presence of three singlets at 8.3 ppm (N3P3), 61.9 ppm (the 5 P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S groups of the first generation), and 62.8 ppm (the 10 PS groups of the second generation). Compound 5-G2 is then reacted with 4-hydroxybenzaldehyde to afford dendrimer 4-G2 (Scheme 2). In this case also, 31P NMR displays the appearance of the intermediate singlet at 69 ppm, corresponding to the monosubstitution, which disappears when the reaction has reached completion. All these steps are compatible with the presence of the dithiolane linked to the core, as shown by 1H and 13C NMR, with unchanged signals corresponding to the CH and CH2 groups of the 5-membered ring.
S groups of the first generation), and 62.8 ppm (the 10 PS groups of the second generation). Compound 5-G2 is then reacted with 4-hydroxybenzaldehyde to afford dendrimer 4-G2 (Scheme 2). In this case also, 31P NMR displays the appearance of the intermediate singlet at 69 ppm, corresponding to the monosubstitution, which disappears when the reaction has reached completion. All these steps are compatible with the presence of the dithiolane linked to the core, as shown by 1H and 13C NMR, with unchanged signals corresponding to the CH and CH2 groups of the 5-membered ring.
 | ||
| Scheme 2 Synthesis of bifunctional dendrimers having one dithiolane function at the core, and either Cl (5-G1 and 5-G2) or CHO (4-G1 and 4-G2) terminal functions. | ||
Dendrimers 4-G1, 4-G2, 5-G1, and 5-G2 are the precursors of the hydrophilic bifunctional dendrimers suitable for grafting onto the gold surface. For this purpose, two different strategies have been applied. For obtaining positively charged dendrimers, N,N-diethylethylenediamine is reacted with the P(S)Cl2 terminal groups (1 diamine per Cl). HCl generated in the reaction is trapped by the tertiary amine moieties,29 affording directly the water-soluble bifunctional dendrimer 6-G1 from 5-G1. As the protonation of the tertiary amine is reversible, the percentage of protonation varies depending on the media, and in particular depending on the pH.13 The simultaneous presence of the two types of terminal groups is clearly detected by 1H and 13C NMR, in particular for the CH3 groups of NH+(CH2CH3)2 (major, δ 1H = 1.33 ppm (t), δ 13C = 7.8 ppm) and N(CH2CH3)2 (minor, δ 1H = 1.26 ppm (t), δ 13C = 9.0 ppm). The same type of reaction is applied to compound 5-G2, used as a precursor for the synthesis of the water-soluble bifunctional dendrimer 6-G2 (Scheme 3).
 | ||
| Scheme 3 Synthesis of water-soluble bifunctional dendrimers having one dithiolane function at the core, and 10 (6-G1) or 20 (6-G2) ammonium terminal groups. | ||
Compound 4-G1 is the starting point for affording another type of water-soluble (negatively charged) bifunctional dendrimer suitable for grafting onto gold surfaces. For this purpose, a Doebner-like reaction is carried out with malonic acid in pyridine with a catalytic amount of piperidine.30 This reaction affords compound 7-G1 ending with carboxylic acids. The characterization is performed at this step. The completion of the reaction is shown by 1H NMR, with the disappearance of the signal corresponding to the aldehydes on behalf of two doublets at 6.43 and 7.19 ppm (3JHH = 15.9 Hz), corresponding to the alkene groups. Compound 7-G1 ending with carboxylic acids is not soluble in water, contrarily to its sodium salt, obtained by the addition of NaOH. Finally, compound 4-G2 is used as a precursor for dendrimer 7-G2, obtained from the reaction with malonic acid (Scheme 4 displays the full structure of compound 7-G2, after reaction with NaOH). 31P NMR displays the presence of 3 signals corresponding to the core N3P3 (8.4 ppm) and the PS groups of the first (62.4 ppm) and second (62.0 ppm) layers. Interestingly, all these steps also are compatible with the presence of the dithiolane function linked to the core. Indeed, it is by far not trivial to have at each step of the multi-step synthesis of dendrimers two types of functions that do not interfere.
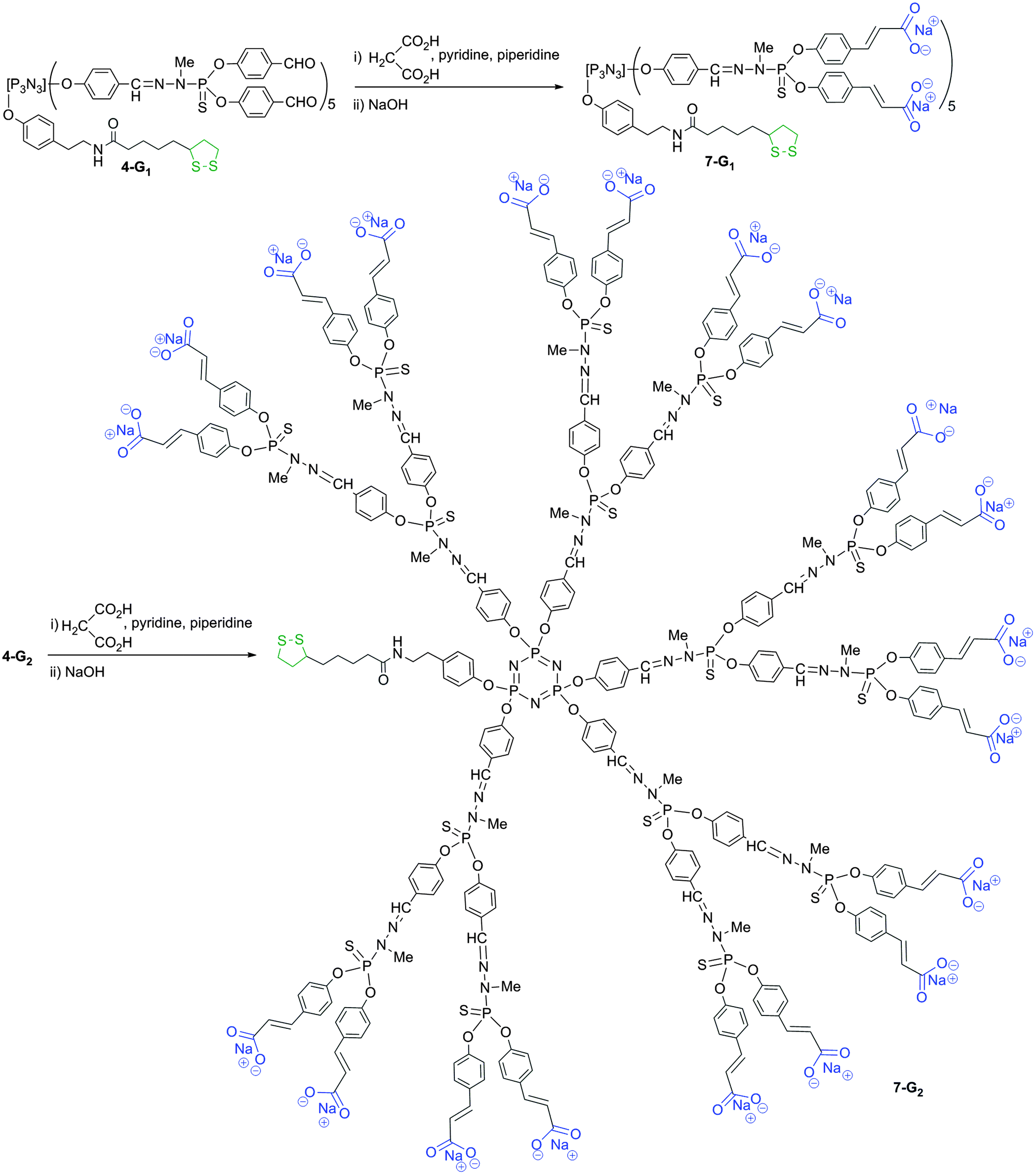 | ||
| Scheme 4 Synthesis of water-soluble bifunctional dendrimers having one dithiolane function at the core, and 10 (7-G1) or 20 (7-G2) ammonium terminal groups. | ||
Gold surface coating with the bifunctional dendrimers
Thin glass slides commonly used for optical microscopy were coated with thin layers of chromium (5 nm) and gold (48 nm). In the first attempt, dendrimers 6-G1 and 7-G1 were used in solution in water. A self-assembled layer of the bifunctional first generation dendrimer was formed on the gold surfaces. The dendrimers attach to the gold surface through the dithiolane moiety at the core, which strongly binds to gold via quasi-covalent bonds.31 The successful assembly process was characterized by contact angle and surface plasmon resonance (SPR) measurements. After about five hours, the binding process was completed. Fig. 1 displays the SPR angular scans of a gold sample coated with either dendrimer 6-G1 or dendrimer 7-G1. Assuming a refractive index of 1.532 for the dendrimer layer, film thicknesses of about 1.0 nm were obtained. This low value suggests that the hydrophobic interior of the dendrimers collapses upon contact with the aqueous environment. This is in agreement with the behaviour of phosphorus dendrimers in water measured earlier.33 Analogous experiments were attempted with the second generations of the bifunctional dendrimers 6-G2 and 7-G2 but failed, presumably due to the protection/shielding of the dithiolane induced by the branches of the dendrimers, which prevents the interaction with the gold surface.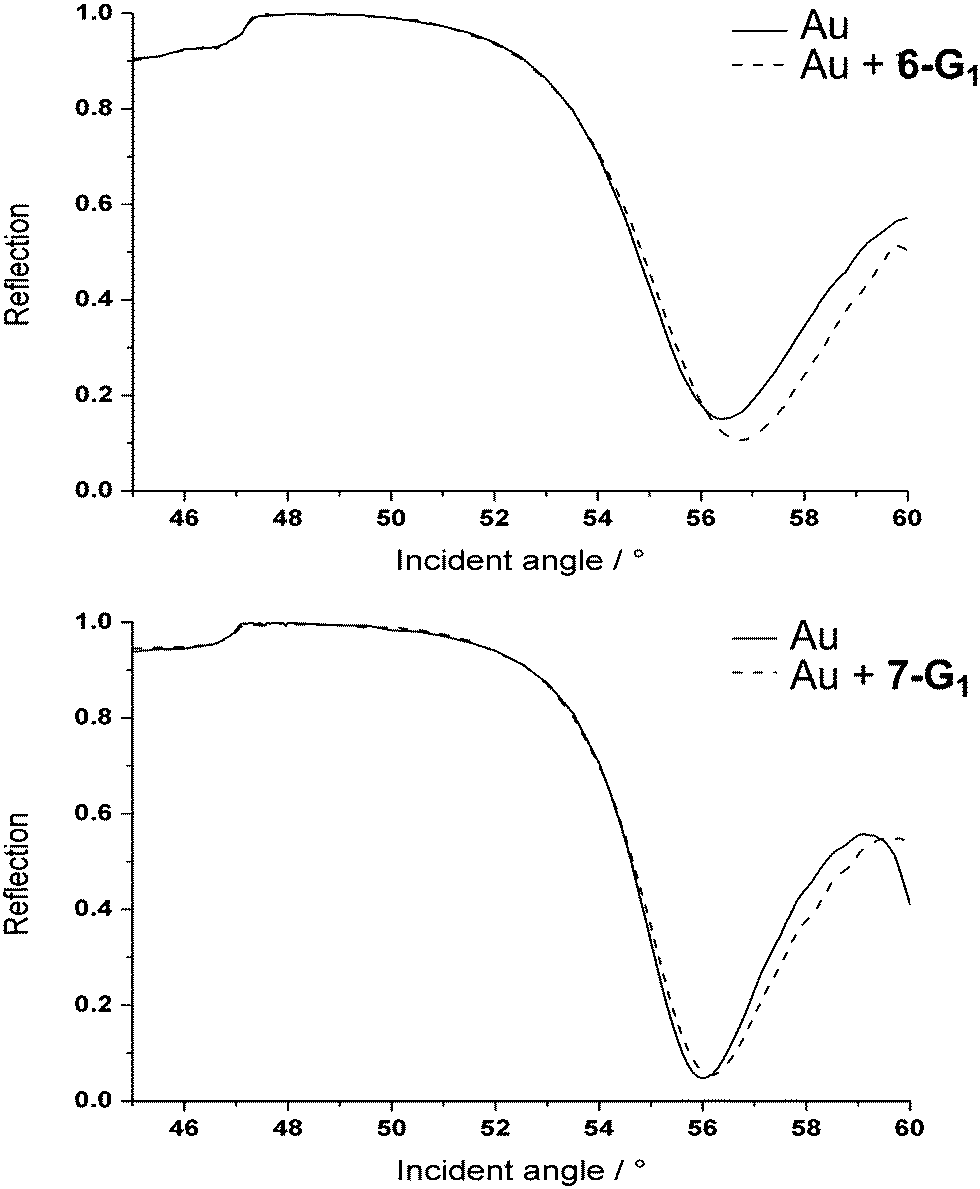 | ||
| Fig. 1 SPR angular scan of gold samples. Up: with and without a 6-G1 coating; down: with and without a 7-G1 coating. | ||
Water contact angle measurements (see ESI†) showed contact angles of approximately 50° for both the polycationic and polyanionic first generation dendrimer coatings. This value suggests that the hydrophobic interior of the dendrimers is exposed to the aqueous phase, resulting in a film without extreme polarity for both 6-G1 and 7-G1.
Human osteoblast (HOB) cell proliferation on dendrimer functionalized surfaces
In order to demonstrate the potential utility of the new water soluble bifunctional dendrimers in biology, the dendrimer-coated glass substrates were exposed to a cell culture of human osteoblast HOB cells and the proliferation behaviour of these cells was studied optically. Optical microscopy is a valuable method for assessing cell morphology during cell adhesion and proliferation. During the first 24 hours, the cells were observed several times in order to follow the cell attachment and spreading process. The HOB cells were seeded to the dendrimer coated substrates and spread similarly on the polycationic, the polyanionic and on the control substrates (Au coated glass and the petri dish plastic). After 24 hours, most of the cells had adopted their typical, stretched shape and initiated cell–cell contact. These contacts are vital for their survival and are usually made within the first few days.The cell proliferation was followed until a confluence of 80–100%. After the first few days, the cells started to show a different behaviour on the samples coated with positively charged dendrimers 6-G1 (Fig. 2). On the negatively charged dendrimers 7-G1 as well as on the control substrates, the cells attached, stretched and proliferated normally. On the positively charged dendrimers 6-G1, however, the cells proliferated less, resulting in many rounded cells and a low surface coverage. The rounded cells were probably dead cells, which concurs with the decreased proliferation rate. A few rounded cells were observed on the other samples (polyanionic 7-G1 and control), but most of them spread again. This is a common process and is related to mitosis. Moreover, on the positively charged dendrimers 6-G1 it was observed that all cells were retracting on longer time scales.
 | ||
| Fig. 2 HOB cell proliferation on control, polyanionic (7-G1) and polycationic (6-G1) samples at different stages. | ||
The microscopy images of the cells on positively charged dendrimer have been studied in more detail for the visualization of the dead cells (Fig. 3). Most cells died through apoptosis (“programmed” cell death), rather than necrosis (traumatic cell death). It was possible to observe the main apoptotic hallmarks by their appearance: chromatin condensation (3A), cell retraction (3B–3C), membrane blebbing (3C) and the final phase where apoptotic bodies are formed (3C–3D). Cell shrinkage manifests itself with an irregular cell shape and shady areas around the cells. Membrane blebbing is an advanced form of cell shrinkage, where as a result the membrane starts to deform heavily, showing large quasi-spherical protrusions. It manifests itself as small blobs attached to the cell.34 Apoptotic bodies are the leftovers of the entire process and can be identified under the microscope (phase contrast) as condensed, ring-like features. Fig. 3a and b were obtained after staining cells with DAPI (4′,6-diamidino-2-phenylindole dihydrochloride), which selectively reveals the location of the genetic material. By overlaying the obtained fluorescence images with the regular microscopy images, it was possible to distinguish between healthy cells (nucleus stained blue) and apoptotic cells (diffuse blue stain throughout the cell, Fig. 3b) and to recognize apoptotic hallmarks such as chromatin condensation and breakdown of the nuclear envelope.
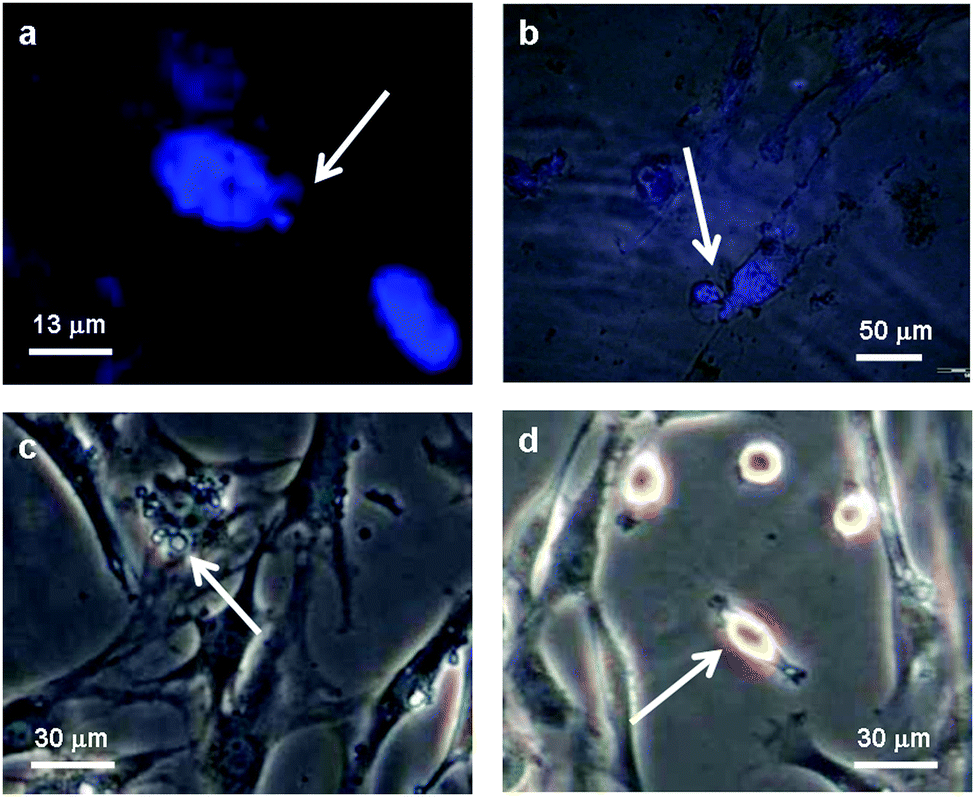 | ||
| Fig. 3 Hallmarks of apoptosis among HOB cells were only found on positively charged dendrimers (6-G1). (a) Fluorescence image of a DAPI stained cell nucleus; (b) fluorescence and optical microscopy overlay image of a DAPI stained cell; (c) and (d) optical microscopy of cells. All images were recorded after approximately 280 hours. | ||
The proliferation behaviour and health status of the cells have been quantified in terms of cell viability (Fig. 4A). The viability was determined as the ratio between the number of living and total cells. A significant difference was observed between cells that grew on polycationic 6-G1 or on polyanionic 7-G1 dendrimers. For each sample, values obtained for cells adhered on the petri dish plastic served as a positive internal reference. Cells on polyanionic dendrimers 7-G1 and positive control samples showed a very similar behaviour. This indicates that polyanionic dendrimers 7-G1 did not affect HOB cell growth as compared to any reference sample. In contrast, polycationic dendrimer 6-G1 coatings had cytotoxic effects, resulting in a decreased cell number and viability.
 | ||
| Fig. 4 Proliferation and apoptosis quantified on different dendrimer (negatively charged 7-G1; positively charged 6-G1) coated surfaces: (A) viability, (B) caspase 3/7 activity assay. These values have been obtained after approximately 280 hours of cell adhesion and proliferation. | ||
A caspase 3/7 specific assay was performed, where the luminescence of Luciferase directly reflects the presence and/or activity of the caspases 3 and 7 (Fig. 4B). These enzymes are only active when a cell undergoes apoptosis. The polyanionic dendrimers 7-G1 and the control substrates showed similar low values, clearly indicating that cells were not apoptotic. The polycationic dendrimers 6-G1, however, did strongly provoke apoptosis, confirming the microscopy observations.
Water-soluble positively charged dendrimers (polyamido amine type) and linear polymers have been described to induce apoptosis among many cell types,35,36 mostly through uptake by the cells. Polyamidoamine dendrimers and linear polycations were found to diffuse through the cell membrane and to integrate into the mitochondrial membrane. They compromised the integrity of organelles resulting in a sudden cytochrome c release, which indirectly triggers apoptosis. Furthermore, the amine content as well as the amine substitution degree (primary-quaternary) seems to affect the cytotoxicity; higher substitution degrees were found to be more lethal.35,37 In contrast to these experiments, the dendrimers in the current study were attached to a solid support. There are only few reports on apoptosis specifically induced by surface coatings.38 In general, cells are unaffected by amines or by positively charged surfaces. In fact, cells adhere preferentially to cationic surfaces due to the stronger electrostatic interaction between the surface and the cell membrane. Furthermore, similar dendrimers with diethyl ammonium groups have been electrostatically attached to a substrate and they showed no apoptotic effect on neuronal cells.13
Our results are in marked contrast with several previous reports studying the influence of the charges on the behaviour of cells, including osteoblasts. Indeed, a lower osteoblast cell proliferation on COOH-terminated titanium films was observed compared with NH2-terminated titanium films,18 and recent observations described in the literature with positively charged polymeric-dendritic poly(lysine)-coated surfaces39 also emphasize the positive role of such a surface on the adhesion of cells. However, the topology of dendrimers is different from that of small molecules and of polymers, and it is well-known that subtle changes in the periphery of dendrimers may deeply modify their properties and cytotoxicity.40 In the case of phosphorus dendrimers, positively charged dendrimers with the same ammonium terminal groups than 6-G1 (but without the thioctic arm) have been used in many cases in contact with cells, as transfection agents,29 as anti-HIV agents,41 as in vivo anti-prion agents,42 and as agents against the aggregation of Alzheimer's peptides.43 No acute toxicity was observed, except in the case of human mononuclear blood cells, the polycationic dendrimers induce changes in their morphology, and also the loss of cell attachment properties.44 Negatively charged phosphorus dendrimers have many properties for modulating in vitro the response of the human immune system,45 and have also anti-inflammatory properties in vivo.46 Phosphorus dendrimers ending by the same carboxylate groups as those of 7-G1 have been already used as non-covalent carriers of anti-HIV aminolactitol,47 and in vivo for the delivery of an anti-hypertensive drug,48 without an acute toxicity effect.
Since only surfaces covered with the polycationic dendrimers 6-G1 were cytotoxic to HOB cells, the specific interaction between the surface bound amine groups and the HOB cells caused an activation of the apoptotic pathway. Stronger electrostatic attraction between cells and substrate can be advantageous for cell initial adhesion, but most cells require mobility and cell–cell contact for proper proliferation, which is inhibited by a stronger attraction, and may account for the observed apoptosis.
Conclusions
Water-soluble generation 1 and generation 2 bifunctional dendrimers with 10 or 20 peripheral charges and one thioctic acid function linked to the core were synthesized and characterized. Only the first generations could be bound to a gold surface via the dithiolane moiety. Such a function is a convenient alternative to classical thiols for interaction with gold, and may find use in other fields, such as the coating of gold nanoparticles, which have many biological applications.49 These functionalized gold surfaces were exposed to human osteoblast (HOB) cells and probed for their ability to sustain cell proliferation. Diethylammonium terminated polycationic dendrimers induced apoptosis among HOB cells, probably triggered by the strong attractive electrostatic interaction between the cells and substrate. A caspase 3/7 activity assay confirmed the apoptotic hallmarks observed by optical microscopy. The carboxyl terminated polyanionic derivatives showed no significant effect on the proliferation of HOB cells. The two different dendrimer coatings are thus potential candidates for application in coatings of biomedical devices, if a specific cell response to the coating is required. Implants require strong cell adhesion and good cell proliferation, while other applications require a non-adhesive coating. Contrarily to polymers classically used for such purposes, the high peripheral functionality, the monodispersity and well-defined shape of dendrimers allow for fine-tuning the surface properties, with high reproducibility, as required for ideal cell–surface interactions.Experimental
Syntheses of the dendrimers
All reactions were carried out under an argon atmosphere and in freshly distilled solvents. All column chromatography experiments were carried out with silica gel 60 as the static phase. All starting compounds were purchased from Aldrich, Merck or Fluka and used as received. Solvents were dried and distilled prior to use. The references for NMR are Me4Si for 1H and 13C NMR, H3PO4 (85% in water) for 31P NMR. The numbering used for NMR assignment is shown in Fig. 5. p-Hydroxybenzaldehyde sodium salt was prepared by ion exchange with NaH. H2NNMeP(S)Cl250 and compound 328 were prepared as published previously.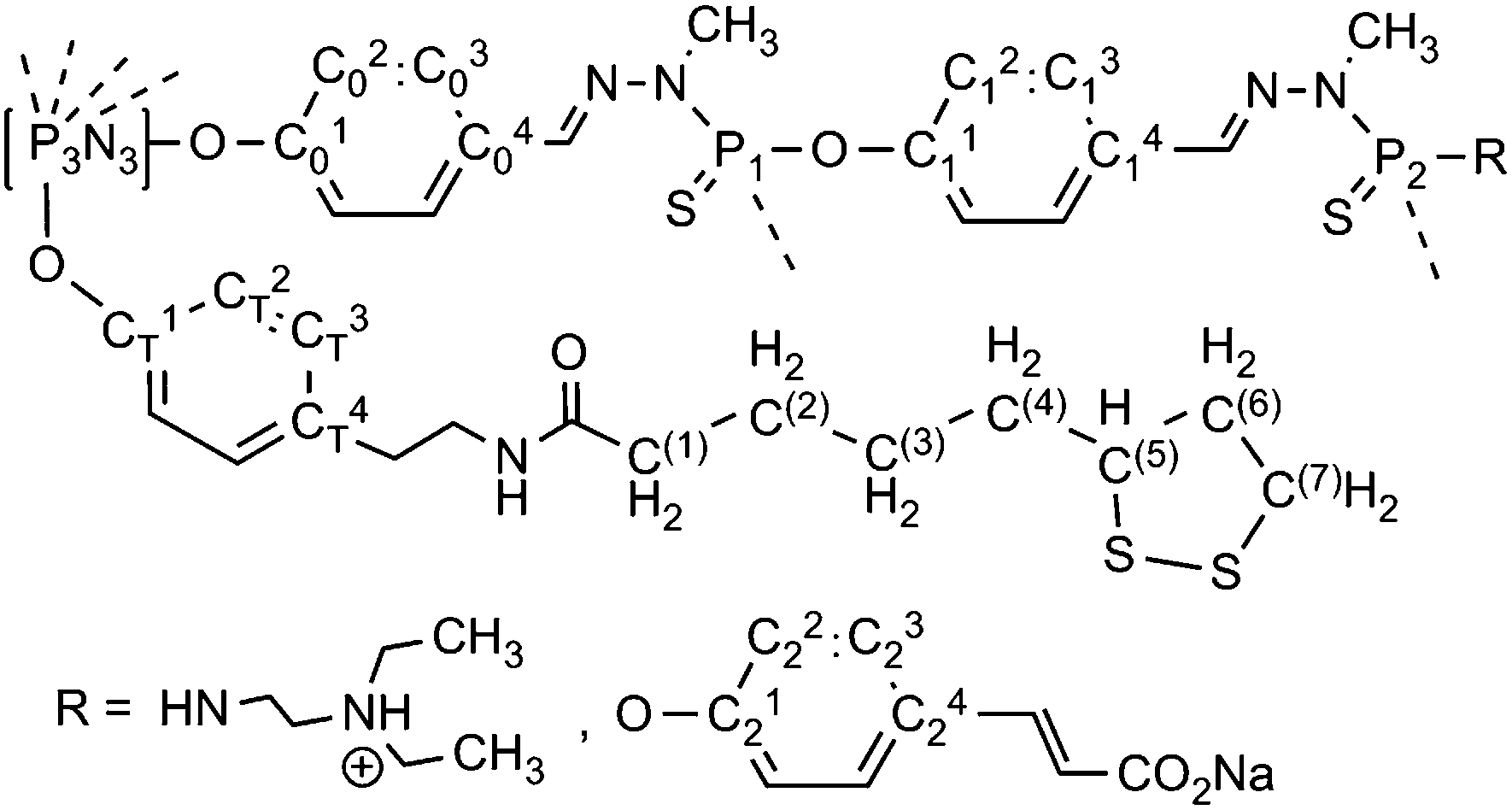 | ||
| Fig. 5 Numbering used for NMR assignment. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. This yielded the dendrimer 4-G0 (2.91 g, 2.72 mmol, 75%) as a yellow oil. 31P{1H}-NMR (101.3 MHz, CDCl3, δ): 7.4 (m, N3P3). 1H-NMR (250 MHz, CDCl3, δ): 1.41 (m, 2H, C(3)H2), 1.62 (m, 4H, C(2)H2, C(4)H2), 1.90 (m, 1H, C(6)H2), 2.12 (t, 3JHH = 7.4 Hz, 2H, C(1)H2CO), 2.40 (m, 1H, C(6)H2), 2.73 (t, 3JHH = 7.0 Hz, 2H, CH2Ar), 3.11 (m, 2H, C(7)H2S), 3.50 (m, 3H, C(5)H–S, CH2N), 5.81 (br s, 1H, NH), 6.82 (m, 2H, CT2H), 7.02 (m, 2H, CT3H), 7.28 (m, 10H, C02H), 7.80 (m, 10H, C03H), 9.95 (2s, 5H, CHO). 13C{1H}-NMR (62.9 MHz, CDCl3, δ): 25.3 (C(2/4)H2), 28.9 (C(3)H2), 34.6 (C(2/4)H2), 35.1 (CH2Ar), 36.4 (C(1)H2CO), 38.5 (C(7)H2S), 40.3 (C(6)H2), 40.5 (CH2NH), 56.4 (C(5)H), 120.7 (CT2), 121.3 (C02), 129.9 (CT3), 131.4 (C03), 133.7 (C04), 136.6 (CT4), 154.7 (C01, CT1), 172.8 (NCO), 190.5 (CHO).
N). 13C{1H}-NMR (75.5 MHz, CDCl3, δ): 25.4 (C(2/4)H2), 28.9 (C(3)H2), 32.0 (d, 2JCP = 13.2 Hz, P1–N–CH3), 34.6 (C(2/4)H2), 35.2 (CH2Ar), 36.4 (C(1)H2), 38.5 (C(7)H2S), 40.3 (C(6)H2), 40.7 (CH2NH), 56.5 (C(5)H), 121.1 (CT2), 121.4 (C02), 128.6 (C03), 129.7 (CT3), 131.3 (C04), 135.9 (CT4), 140.8 (br d, 3JCP = 20.1 Hz, CHN–N–P1), 149.0 (CT1), 151.7 (C01), 173.0 (NCO).
N–N–P1). 13C{1H}-NMR (75.5 MHz, CD3OD, δ): 7.8 (N+CH2CH3), 9.0 (NCH2CH3), 25.4 (C(2/4)H2), 28.5 (C(3)H2), 31.4 (2d, 2JCP = 10.6 Hz, P1–N–CH3), 34.4 (C(2/4)H2), 35.5 (CH2Ar), 36.2 (C(1)H2CO, CH2–N–P1), 38.0 (C(7)H2S), 40.0 (C(6)H2), 40.4 (CH2NHCO), 49.0 (N–CH2CH3), 52.2 (d, 3JCP = 6.9 Hz, P1–N–CH2CH2), 56.5 (C(5)H–S), 120.5 (CT2), 120.9 (C02), 128.0 (C03), 129.7 (CT3), 133.2 (C04), 136.4 (CT4), 137.7 (m, CHN–N–P1), 150.8 (m, C01, CT1), 174.5 (NCO).
N–N), 7.67 (s, 2H, CHN–N), 7.81 (m, 20H, C13H), 9.93 (s, 10H, CHO). 13C{1H}-NMR (62.9 MHz, CDCl3, δ): 25.4 (C(2/4)H2), 28.8 (C(3)H2), 32.8 (d, 2JCP = 12.8 Hz, P1–N–CH3), 34.6 (C(2/4)H2), 35.0 (CH2Ar), 36.2 (C(1)H2), 38.4 (C(7)H2S), 40.2 (C(6)H2), 40.5 (CH2NH), 56.5 (C(5)H–S), 121.0 (CT2), 121.4 (C02), 121.8 (d, 3JCP = 6.8 Hz, C12), 128.3 (C13), 129.7 (CT3), 131.4 (C14), 131.8 (C03), 133.7 (C04), 135.8 (CT4), 139.7 (d, 3JCP = 13.4 Hz, CHN–N–P1), 151.5 (d, 2JCP = 7.0 Hz, C01, CT1), 155.0 (d, 2JCP = 7.0 Hz, C11), 172.8 (NCO), 190.7 (CHO).
CHCO2), 6.80 (m, 2H, CT2H), 7.03 (m, 12H, CT3H, C02H), 7.16 (m, 20H, C12H), 7.19 (d, 3JHH = 15.9 Hz, 10H, CHCHCO2), 7.61 (m, 30H, C03H, C13H), 7.85 (s, 3H, CHN–N), 7.91 (s, 2H, CHN–N), 12.4 (s, 10H, COOH). 13C{1H}-NMR (75.5 MHz, DMSO-d6, δ): 25.5 (C(2/4)H2), 28.8 (C(3)H2), 33.4 (2d, 2JCP = 11.9 Hz, P1–N–CH3), 34.6 (C(2/4)H2), 34.9 (CH2Ar), 35.7 (C(1)H2), 38.5 (C(7)H2S), 56.5 (C(5)H–S), 119.9 (Ar–CHCH), 120.7 (CT2), 121.4 (C02), 121.8 (d, 3JCP = 4.1 Hz, C12), 128.7 (C03), 129.0 (CT3), 130.2 (C13), 132.2 (C14), 132.4 (C04), 137.2 (CT4), 141.1 (CHN–N), 143.1 (Ar–CHCH), 148.4 (CT1), 151.0 (C01), 151.7 (C11), 167.9 (COOH), 172.3 (NCO). (C(6)H2 and CH2NH hidden by DMSO).
N–N). 13C{1H}-NMR (75.5 MHz, CDCl3, δ): 25.3 (C(2/4)H2), 28.9 (C(3)H2), 31.8 (d, 2JCP = 13.1 Hz, P2–N–CH3), 33.1 (d, 2JCP = 12.9 Hz, P1–N–CH3), 34.6 (C(2/4)H2), 35.1 (CH2Ar), 36.4 (C(1)H2), 38.5 (C(7)H2S), 40.3 (C(6)H2), 40.5 (CH2NH), 56.5 (C(5)H–S), 121.0 (CT2), 121.4 (d, 3JCP = 6.2 Hz, C02), 121.9 (d, 3JCP = 4.3 Hz, C12), 128.3 (C03), 128.8 (C13), 129.8 (CT3), 131.6 (C14), 131.9 (C04), 135.9 (CT4), 138.9 (m, CHN–N–P1), 140.6 (d, 2JCP = 18.7 Hz, CHN–N–P2), 151.4 (m, C01), 151.8 (d, 2JCP = 7.2 Hz, C11), 172.6 (NCO).
N–N–P1,2). 13C{1H}-NMR (75.5 MHz, CDCl3, δ): 7.9 (N+CH2CH3), 9.7 (NCH2CH3), 25.1 (C(2/4)H2), 28.5 (C(3)H2), 31.3 (d, 2JCP = 9.4 Hz, P2–N–CH3), 32.5 (d, 2JCP = 12.2 Hz, P1–N–CH3), 34.4 (C(2/4)H2), 35.6 (CH2Ar), 36.2 (CH2–N–P2), 36.4 (C(1)H2), 38.1 (C(7)H2S), 40.1 (C(6)H2, CH2NHCO), 49.5 (N–CH2CH3), 52.2 (d, 3JCP = 6.9 Hz, P1–N–CH2CH2), 56.3 (C(5)H–S), 121.0 (C02, CT2), 121.4 (C12), 128.0 (C13), 128.3 (C03), 130.0 (CT3), 132.6 (C04), 133.3 (C14), 137.5 (d, 2JCP = 13.4 Hz, CHN–N–P1,2), 151.0 (m, C01, CT1, C11), 175.0 (NCO).
N–N), 7.83 (m, 40H, C23), 9.90 (s, 20H, CHO). 13C{1H}-NMR (75.5 MHz, CDCl3, δ): 25.3 (C(2/4)H2), 28.9 (C(3)H2), 33.0 (2d, 2JCP = 12.8 Hz, P1,2–N–CH3), 34.5 (C(2/4)H2), 35.0 (CH2Ar), 36.3 (C(1)H2), 38.5 (C(7)H2S), 40.3 (C(6)H2, CH2NH), 56.5 (C(5)H–S), 120.7 (CT2), 121.3 (C02), 121.3 (d, 3JCP = 4.0 Hz, C12), 122.0 (d, 3JCP = 4.8 Hz, C22), 128.4 (C0,13), 129.6 (CT3), 131.5 (C23), 132.0 (C0,14), 133.7 (C24), 135.9 (CT4), 139.6 (2d, 2JCP = 15.1 Hz, CHN–N–P1,2), 151.5 (2d, 2JCP = 7.1 Hz, C0,11), 155.1 (d, 2JCP = 7.2 Hz, C21), 172.8 (NCO), 190.7 (CHO).
CHCO2), 6.85 (m, 2H, CT2H), 6.98 (m, 12H, CT3H, C02H), 7.18 (m, 60H, C12H, C22H), 7.53 (d, 3JHH = 16.2 Hz, 20H, CHCHCO2), 7.66 (m, 70H, C03H, C13H, C23H), 7.88 (m, 15H, CHN–N), 12.4 (COOH, 20H). 13C{1H}-NMR (75.5 MHz, DMSO-d6, δ): 25.4 (C(2/4)H2), 28.7 (C(3)H2), 33.4 (m, P1,2–N–CH3), 34.0 (C(2/4)H2), 34.5 (CH2Ar), 35.6 (C(1)H2), 38.4 (C(7)H2S), 56.5 (C(5)H–S), 119.8 (Ar–CHCH), 120.6 (C02, CT2), 121.8 (d, 3JCP = 4.8 Hz, C12, C22), 128.8 (CT,0,13), 130.3 (C23), 132.2 (C24), 132.5 (C0,14), 141.1 (m, CHN–N–P1,2), 143.1 (Ar–CHCH), 151.3 (d, 5JCP = 6.9 Hz, C0,11), 151.7 (d, 5JCP = 6.9 Hz, C21), 167.9 (COOH), 172.4 (NCO). (C(6)H2 and CH2NH hidden by DMSO).
:1 ratio. This yielded the dendrimer 4-G0 (2.91 g, 2.72 mmol, 75%) as a yellow oil. 31P{1H}-NMR (101.3 MHz, CDCl3, δ): 7.4 (m, N3P3). 1H-NMR (250 MHz, CDCl3, δ): 1.41 (m, 2H, C(3)H2), 1.62 (m, 4H, C(2)H2, C(4)H2), 1.90 (m, 1H, C(6)H2), 2.12 (t, 3JHH = 7.4 Hz, 2H, C(1)H2CO), 2.40 (m, 1H, C(6)H2), 2.73 (t, 3JHH = 7.0 Hz, 2H, CH2Ar), 3.11 (m, 2H, C(7)H2S), 3.50 (m, 3H, C(5)H–S, CH2N), 5.81 (br s, 1H, NH), 6.82 (m, 2H, CT2H), 7.02 (m, 2H, CT3H), 7.28 (m, 10H, C02H), 7.80 (m, 10H, C03H), 9.95 (2s, 5H, CHO). 13C{1H}-NMR (62.9 MHz, CDCl3, δ): 25.3 (C(2/4)H2), 28.9 (C(3)H2), 34.6 (C(2/4)H2), 35.1 (CH2Ar), 36.4 (C(1)H2CO), 38.5 (C(7)H2S), 40.3 (C(6)H2), 40.5 (CH2NH), 56.4 (C(5)H), 120.7 (CT2), 121.3 (C02), 129.9 (CT3), 131.4 (C03), 133.7 (C04), 136.6 (CT4), 154.7 (C01, CT1), 172.8 (NCO), 190.5 (CHO).
N). 13C{1H}-NMR (75.5 MHz, CDCl3, δ): 25.4 (C(2/4)H2), 28.9 (C(3)H2), 32.0 (d, 2JCP = 13.2 Hz, P1–N–CH3), 34.6 (C(2/4)H2), 35.2 (CH2Ar), 36.4 (C(1)H2), 38.5 (C(7)H2S), 40.3 (C(6)H2), 40.7 (CH2NH), 56.5 (C(5)H), 121.1 (CT2), 121.4 (C02), 128.6 (C03), 129.7 (CT3), 131.3 (C04), 135.9 (CT4), 140.8 (br d, 3JCP = 20.1 Hz, CHN–N–P1), 149.0 (CT1), 151.7 (C01), 173.0 (NCO).
N–N–P1). 13C{1H}-NMR (75.5 MHz, CD3OD, δ): 7.8 (N+CH2CH3), 9.0 (NCH2CH3), 25.4 (C(2/4)H2), 28.5 (C(3)H2), 31.4 (2d, 2JCP = 10.6 Hz, P1–N–CH3), 34.4 (C(2/4)H2), 35.5 (CH2Ar), 36.2 (C(1)H2CO, CH2–N–P1), 38.0 (C(7)H2S), 40.0 (C(6)H2), 40.4 (CH2NHCO), 49.0 (N–CH2CH3), 52.2 (d, 3JCP = 6.9 Hz, P1–N–CH2CH2), 56.5 (C(5)H–S), 120.5 (CT2), 120.9 (C02), 128.0 (C03), 129.7 (CT3), 133.2 (C04), 136.4 (CT4), 137.7 (m, CHN–N–P1), 150.8 (m, C01, CT1), 174.5 (NCO).
N–N), 7.67 (s, 2H, CHN–N), 7.81 (m, 20H, C13H), 9.93 (s, 10H, CHO). 13C{1H}-NMR (62.9 MHz, CDCl3, δ): 25.4 (C(2/4)H2), 28.8 (C(3)H2), 32.8 (d, 2JCP = 12.8 Hz, P1–N–CH3), 34.6 (C(2/4)H2), 35.0 (CH2Ar), 36.2 (C(1)H2), 38.4 (C(7)H2S), 40.2 (C(6)H2), 40.5 (CH2NH), 56.5 (C(5)H–S), 121.0 (CT2), 121.4 (C02), 121.8 (d, 3JCP = 6.8 Hz, C12), 128.3 (C13), 129.7 (CT3), 131.4 (C14), 131.8 (C03), 133.7 (C04), 135.8 (CT4), 139.7 (d, 3JCP = 13.4 Hz, CHN–N–P1), 151.5 (d, 2JCP = 7.0 Hz, C01, CT1), 155.0 (d, 2JCP = 7.0 Hz, C11), 172.8 (NCO), 190.7 (CHO).
CHCO2), 6.80 (m, 2H, CT2H), 7.03 (m, 12H, CT3H, C02H), 7.16 (m, 20H, C12H), 7.19 (d, 3JHH = 15.9 Hz, 10H, CHCHCO2), 7.61 (m, 30H, C03H, C13H), 7.85 (s, 3H, CHN–N), 7.91 (s, 2H, CHN–N), 12.4 (s, 10H, COOH). 13C{1H}-NMR (75.5 MHz, DMSO-d6, δ): 25.5 (C(2/4)H2), 28.8 (C(3)H2), 33.4 (2d, 2JCP = 11.9 Hz, P1–N–CH3), 34.6 (C(2/4)H2), 34.9 (CH2Ar), 35.7 (C(1)H2), 38.5 (C(7)H2S), 56.5 (C(5)H–S), 119.9 (Ar–CHCH), 120.7 (CT2), 121.4 (C02), 121.8 (d, 3JCP = 4.1 Hz, C12), 128.7 (C03), 129.0 (CT3), 130.2 (C13), 132.2 (C14), 132.4 (C04), 137.2 (CT4), 141.1 (CHN–N), 143.1 (Ar–CHCH), 148.4 (CT1), 151.0 (C01), 151.7 (C11), 167.9 (COOH), 172.3 (NCO). (C(6)H2 and CH2NH hidden by DMSO).
N–N). 13C{1H}-NMR (75.5 MHz, CDCl3, δ): 25.3 (C(2/4)H2), 28.9 (C(3)H2), 31.8 (d, 2JCP = 13.1 Hz, P2–N–CH3), 33.1 (d, 2JCP = 12.9 Hz, P1–N–CH3), 34.6 (C(2/4)H2), 35.1 (CH2Ar), 36.4 (C(1)H2), 38.5 (C(7)H2S), 40.3 (C(6)H2), 40.5 (CH2NH), 56.5 (C(5)H–S), 121.0 (CT2), 121.4 (d, 3JCP = 6.2 Hz, C02), 121.9 (d, 3JCP = 4.3 Hz, C12), 128.3 (C03), 128.8 (C13), 129.8 (CT3), 131.6 (C14), 131.9 (C04), 135.9 (CT4), 138.9 (m, CHN–N–P1), 140.6 (d, 2JCP = 18.7 Hz, CHN–N–P2), 151.4 (m, C01), 151.8 (d, 2JCP = 7.2 Hz, C11), 172.6 (NCO).
N–N–P1,2). 13C{1H}-NMR (75.5 MHz, CDCl3, δ): 7.9 (N+CH2CH3), 9.7 (NCH2CH3), 25.1 (C(2/4)H2), 28.5 (C(3)H2), 31.3 (d, 2JCP = 9.4 Hz, P2–N–CH3), 32.5 (d, 2JCP = 12.2 Hz, P1–N–CH3), 34.4 (C(2/4)H2), 35.6 (CH2Ar), 36.2 (CH2–N–P2), 36.4 (C(1)H2), 38.1 (C(7)H2S), 40.1 (C(6)H2, CH2NHCO), 49.5 (N–CH2CH3), 52.2 (d, 3JCP = 6.9 Hz, P1–N–CH2CH2), 56.3 (C(5)H–S), 121.0 (C02, CT2), 121.4 (C12), 128.0 (C13), 128.3 (C03), 130.0 (CT3), 132.6 (C04), 133.3 (C14), 137.5 (d, 2JCP = 13.4 Hz, CHN–N–P1,2), 151.0 (m, C01, CT1, C11), 175.0 (NCO).
N–N), 7.83 (m, 40H, C23), 9.90 (s, 20H, CHO). 13C{1H}-NMR (75.5 MHz, CDCl3, δ): 25.3 (C(2/4)H2), 28.9 (C(3)H2), 33.0 (2d, 2JCP = 12.8 Hz, P1,2–N–CH3), 34.5 (C(2/4)H2), 35.0 (CH2Ar), 36.3 (C(1)H2), 38.5 (C(7)H2S), 40.3 (C(6)H2, CH2NH), 56.5 (C(5)H–S), 120.7 (CT2), 121.3 (C02), 121.3 (d, 3JCP = 4.0 Hz, C12), 122.0 (d, 3JCP = 4.8 Hz, C22), 128.4 (C0,13), 129.6 (CT3), 131.5 (C23), 132.0 (C0,14), 133.7 (C24), 135.9 (CT4), 139.6 (2d, 2JCP = 15.1 Hz, CHN–N–P1,2), 151.5 (2d, 2JCP = 7.1 Hz, C0,11), 155.1 (d, 2JCP = 7.2 Hz, C21), 172.8 (NCO), 190.7 (CHO).
CHCO2), 6.85 (m, 2H, CT2H), 6.98 (m, 12H, CT3H, C02H), 7.18 (m, 60H, C12H, C22H), 7.53 (d, 3JHH = 16.2 Hz, 20H, CHCHCO2), 7.66 (m, 70H, C03H, C13H, C23H), 7.88 (m, 15H, CHN–N), 12.4 (COOH, 20H). 13C{1H}-NMR (75.5 MHz, DMSO-d6, δ): 25.4 (C(2/4)H2), 28.7 (C(3)H2), 33.4 (m, P1,2–N–CH3), 34.0 (C(2/4)H2), 34.5 (CH2Ar), 35.6 (C(1)H2), 38.4 (C(7)H2S), 56.5 (C(5)H–S), 119.8 (Ar–CHCH), 120.6 (C02, CT2), 121.8 (d, 3JCP = 4.8 Hz, C12, C22), 128.8 (CT,0,13), 130.3 (C23), 132.2 (C24), 132.5 (C0,14), 141.1 (m, CHN–N–P1,2), 143.1 (Ar–CHCH), 151.3 (d, 5JCP = 6.9 Hz, C0,11), 151.7 (d, 5JCP = 6.9 Hz, C21), 167.9 (COOH), 172.4 (NCO). (C(6)H2 and CH2NH hidden by DMSO).
Surface plasmon resonance spectroscopy
Dendrimers were adsorbed on template stripped gold, which was prepared following the described procedure.51 The freshly prepared gold samples were mounted onto a customized surface plasmon resonance52 spectrometer and a 1 mg mL−1 aqueous solution of either 6-G1 or 7-G1 was added. The formation of the dendrimer layers was followed in real time as an increase in SPR reflectivity, which could be translated into a layer thickness. The laser used was a HeNe laser with one spectral line at 632.8 nm.Contact angle goniometry
The hydrophobicity of the different coatings was measured with an OCA 15+ (DataPhysics, Filderstadt, Germany) goniometer equipped with a CCD camera and an electronic dosing unit. For the determination of the contact angles, SCA 20 software was used. The droplet volume was 3 μL and the water was degassed before usage. Measurements were performed in 5-fold.Cell experiments
A cell bank of a commercial cell line was created (HOB, human hipbone osteoblasts, promocell, Heidelberg, Germany). HOB cells were cultured at 37 °C and 5% CO2 in commercially available HOB Growth medium, containing a promocell supplement mix. The proliferation of the cells was followed by optical inspection. The coated glass substrates were sterilized and transferred to a petri-dish containing 3 mL of growth medium and 2.0 × 104 HOB cells per mL. The growth medium was changed every 48 h.After about 300 hours, 80–100% confluence was reached and cells were trypsinated and counted. 20 μL of the solution left after trypsination were mixed with an equal amount of Trypan Blue stain (GIBCO, 0.4%). The cell suspension was transferred to a Neubauer cell counter. Subsequently, the relative number of living cells (stained brightly blue) with respect to the total number of cells (including dead cells, stained dark blue) was determined.
After trypsination, the cells were transferred from the sample surface to a 96-well Elisa plate. The cells were allowed to re-attach to the Elisa walls for approximately 24 hours in HOB growth medium. 100 μL medium was left in each Elisa well and mixed with 100 μL of the assay solution (Caspase-Glo® 3/7, Promega). Subsequently, the Elisa plate was kept in the dark for 30 minutes for the assay to reach full activity and then measured with a Promega luminometer.
50 μL of an aqueous DAPI solution (4′,6-diamidino-2-phenylindole dihydrochloride. 2 mg mL−1, Carl Roth GmbH, Karlsruhe, Germany) were added to the cell culture. Cells were imaged using an Olympus IX 70 microscope (Olympus, Center Valley, USA). Images were recorded at 400-fold magnification using a CCD camera.
Acknowledgements
The European framework program 6 (Marie-Curie EST NANOTOOL) and the Max Planck Society are gratefully acknowledged for funding (ERJ). Thanks are due also to the CNRS (Centre National de la Recherche Scientifique) for financial support, and the COST action CM1302 SIPs.Notes and references
- Dendrimers. Towards Catalytic, Material and Biomedical Uses, ed. A. M. Caminade, C. O. Turrin, R. Laurent, A. Ouali and B. Delavaux-Nicot, John Wiley & Sons, Chichester, UK, 2011 Search PubMed.
- O. Rolland, C. O. Turrin, A. M. Caminade and J. P. Majoral, New J. Chem., 2009, 33, 1809 RSC.
- L. Roglin, E. H. M. Lempens and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 102 CrossRef CAS PubMed; P. Antoni, Y. Hed, A. Nordberg, D. Nystrom, H. von Holst, A. Hult and M. Malkoch, Angew. Chem., Int. Ed., 2009, 48, 2126 CrossRef PubMed; R. Al-Hellani, J. Barner, J. P. Rabe and A. D. Schluter, Chem. – Eur. J., 2006, 12, 6542 CrossRef PubMed; H. Zong, T. P. Thomas, K. H. Lee, A. M. Desai, M. H. Li, A. Kotlyar, Y. H. Zhang, P. R. Leroueil, J. J. Gam, M. M. B. Holl and J. R. Baker, Biomacromolecules, 2012, 13, 982 CrossRef PubMed.
- K. Kanie, M. Matsubara, X. B. Zeng, F. Liu, G. Ungar, H. Nakamura and A. Muramatsu, J. Am. Chem. Soc., 2012, 134, 808 CrossRef CAS PubMed; G. Lamanna, M. Kueny-Stotz, H. Mamlouk-Chaouachi, C. Ghobril, B. Basly, A. Bertin, I. Miladi, C. Billotey, G. Pourroy, S. Begin-Colin and D. Felder-Flesch, Biomaterials, 2011, 32, 8562 CrossRef PubMed.
- T. A. Horbett, J. J. Waldburger, B. D. Ratner and A. S. Hoffman, J. Biomed. Mater. Res., 1988, 22, 383 CrossRef CAS PubMed.
- B. Hopp, N. Kresz, J. Kokavecz, T. Smausz, H. Schieferdecker, A. Doring, O. Marti and Z. Bor, Appl. Surf. Sci., 2004, 221, 437 CrossRef CAS.
- A. Dekker, T. Beugeling, H. Wind, A. Poot, A. Bantjes, J. Feijen and W. G. Van Aken, J. Mater. Sci.: Mater. Med., 1991, 2, 227 CrossRef CAS.
- V. I. Sikavitsas, J. S. Temenoff and A. G. Mikos, Biomaterials, 2001, 22, 2581 CrossRef CAS.
- K. Kato, S. Sano and Y. Ikada, Colloids Surf., B, 1995, 4, 221 CrossRef CAS; J. S. Mao, Y. L. Cui, X. H. Wang, Y. Sun, Y. J. Yin, H. M. Zhao and K. De Yao, Biomaterials, 2004, 25, 3973 CrossRef PubMed; G. Rainaldi, P. Filippini, A. Ferrante, P. L. Indovina and M. T. Santini, J. Biomed. Mater. Res., 2001, 55, 104 CrossRef; M. Wahlgren and T. Arnebrant, Trends Biotechnol., 1991, 9, 201 CrossRef.
- C. R. Wittmer, J. A. Phelps, W. M. Saltzman and P. R. Van Tassel, Biomaterials, 2007, 28, 851 CrossRef CAS PubMed.
- See for instance: R. K. Aithal, D. P. Kumaraswamy, D. K. Milis and D. Kuila, J. Biomed. Nanotechnol., 2007, 3, 254 CrossRef CAS PubMed; A. P. Zhu and N. Fang, Biomacromolecules, 2005, 6, 2607 CrossRef PubMed.
- S. F. Rose, A. L. Lewis, G. W. Hanlon and A. W. Lloyd, Biomaterials, 2004, 25, 5125 CrossRef CAS PubMed.
- J. L. Hernandez-Lopez, H. L. Khor, A. M. Caminade, J. P. Majoral, S. Mittler, W. Knoll and D. H. Kim, Thin Solid Films, 2008, 516, 1256 CrossRef CAS PubMed.
- S. R. Benhabbour, H. Sheardown and A. Adronov, Biomaterials, 2008, 29, 4177 CrossRef CAS PubMed.
- M. H. Kim, M. Kino-oka and M. Taya, Biotechnol. Adv., 2010, 28, 7 CrossRef CAS PubMed.
- S. Mashayekhan, M. H. Kim, M. Kino-oka, J. Miyazaki and M. Taya, Polymers, 2011, 3, 2078 CrossRef CAS PubMed.
- J. H. Myung, K. A. Gajjar, J. Saric, D. T. Eddington and S. Hong, Angew. Chem., Int. Ed., 2011, 50, 11769 CrossRef CAS PubMed.
- B. Finke, F. Luethen, K. Schroeder, P. D. Mueller, C. Bergemann, M. Frant, A. Ohl and B. J. Nebe, Biomaterials, 2007, 28, 4521 CrossRef CAS PubMed; Y. Cai, M. Frant, J. Bossert, G. Hildebrand, K. Liefeith and K. D. Jandt, Colloids Surf., B, 2006, 50, 1 CrossRef PubMed.
- B. Nebe, B. Finke, F. Luthen, C. Bergemann, K. Schroder, J. Rychly, K. Liefeith and A. Ohl, Biomol. Eng., 2007, 24, 447 CrossRef CAS PubMed.
- M. Lehnert, M. Gorbahn, C. Rosin, M. Klein, I. Köper, B. Al-Nawas, W. Knoll and M. Veith, Langmuir, 2011, 27, 7743 CrossRef CAS PubMed.
- A. M. Caminade, C. O. Turrin and J. P. Majoral, New J. Chem., 2010, 34, 1512 RSC.
- D. H. Kim, P. Karan, P. Goring, J. Leclaire, A. M. Caminade, J. P. Majoral, U. Gosele, M. Steinhart and W. Knoll, Small, 2005, 1, 99 CrossRef CAS PubMed; T. D. Lazzara, K. H. A. Lau, A. I. Abou-Kandil, A. M. Caminade, J. P. Majoral and W. Knoll, ACS Nano, 2010, 4, 3909 CrossRef PubMed.
- A. M. Caminade and J. P. Majoral, Prog. Polym. Sci., 2005, 30, 491 CrossRef CAS PubMed.
- O. Rolland, L. Griffe, M. Poupot, A. Maraval, A. Ouali, Y. Coppel, J. J. Fournié, G. Bacquet, C. O. Turrin, A. M. Caminade, J. P. Majoral and R. Poupot, Chem. – Eur. J., 2008, 14, 4836 CrossRef CAS PubMed; D. Riegert, A. Pla-Quintana, S. Fuchs, R. Laurent, C. O. Turrin, C. Duhayon, J. P. Majoral, A. Chaumonnot and A. M. Caminade, Eur. J. Org. Chem., 2013, 5414 CrossRef PubMed.
- V. Maraval, A. M. Caminade, J. P. Majoral and J. C. Blais, Angew. Chem., Int. Ed., 2003, 42, 1822 CrossRef CAS PubMed.
- M. Slany, A. M Caminade and J. P. Majoral, Tetrahedron Lett., 1996, 37, 9053 CrossRef CAS.
- N. Launay, A. M. Caminade, R. Lahana and J. P. Majoral, Angew. Chem., Int. Ed. Engl., 1994, 33, 1589 CrossRef PubMed.
- G. Franc, S. Mazeres, C. O. Turrin, L. Vendier, C. Duhayon, A. M. Caminade and J. P. Majoral, J. Org. Chem., 2007, 72, 8707 CrossRef CAS PubMed.
- C. Loup, M. A. Zanta, A. M. Caminade, J. P. Majoral and B. Meunier, Chem. – Eur. J., 1999, 5, 3644 CrossRef CAS.
- G. Soler-Illia, L. Rozes, M. K. Boggiano, C. Sanchez, C. O. Turrin, A. M. Caminade and J. P. Majoral, Angew. Chem., Int. Ed., 2000, 39, 4250 CrossRef.
- E. Oh, J. B. Delehanty, K. E. Sapsford, K. Susumu, R. Goswami, J. B. Blanco-Canosa, P. E. Dawson, J. Granek, M. Shoff, Q. Zhang, P. L. Goering, A. Huston and I. L. Medintz, ACS Nano, 2011, 5, 6434 CrossRef CAS PubMed.
- J. Voros, Biophys. J., 2004, 87, 553 CrossRef PubMed.
- J. Leclaire, Y. Coppel, A. M. Caminade and J. P. Majoral, J. Am. Chem. Soc., 2004, 126, 2304 CrossRef CAS PubMed.
- M. Bovellan, M. Fritzsche, C. Stevens and G. Charras, FEBS J., 2010, 277, 58 CrossRef CAS PubMed.
- D. Fischer, Y. X. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121 CrossRef CAS.
- N. A. Stasko, C. B. Johnson, M. H. Schoenfisch, T. A. Johnson and E. L. Holmuhamedov, Biomacromolecules, 2007, 8, 3853 CrossRef CAS PubMed.
- P. Ferruti, S. Knobloch, E. Ranucci, R. Duncan and E. Gianasi, Macromol. Chem. Phys., 1998, 199, 2565 CrossRef CAS.
- C. Gretzer, M. Werthen and P. Thomsen, Biomaterials, 2002, 23, 1639 CrossRef CAS; A. Terada, A. Yuasa, T. Kushimoto, S. Tsuneda, A. Katakai and M. Tamada, Microbiology, 2006, 152, 3575 CrossRef PubMed.
- C. Galli, M. Piemontese, S. T. Meikle, M. Santin, G. M. Macaluso and G. Passeri, Clin. Oral Implants Res., 2014, 25, E133 CrossRef CAS PubMed; C. Zhao, C. Pan, J. Sandstedt, Y. Fu, A. Lindahl and J. Liu, RSC Adv., 2015, 5, 42276 RSC.
- S. P. Hong, A. U. Bielinska, A. Mecke, B. Keszler, J. L. Beals, X. Y. Shi, L. Balogh, B. G. Orr, J. R. Baker and M. M. B. Holl, Bioconjugate Chem., 2004, 15, 774 CrossRef CAS PubMed; X. Y. Shi, S. H. Wang, H. P. Sun and J. R. Baker, Soft Matter, 2007, 3, 71–74 RSC; X. Y. Shi, I. Lee, X. S. Chen, M. W. Shen, S. L. Xiao, M. F. Zhu, J. R. Baker and S. H. Wang, Soft Matter, 2010, 6, 2539 RSC.
- V. Briz, M. J. Serramia, R. Madrid, A. Hameau, A. M. Caminade, J. P. Majoral and M. A. Munoz-Fernandez, Curr. Med. Chem., 2012, 19, 5044 CrossRef CAS.
- J. Solassol, C. Crozet, V. Perrier, J. Leclaire, F. Beranger, A. M. Caminade, B. Meunier, D. Dormont, J. P. Majoral and S. Lehmann, J. Gen. Virol., 2004, 85, 1791 CrossRef CAS.
- T. Wasiak, M. Ionov, K. Nieznanski, H. Nieznanska, O. Klementieva, M. Granell, J. Cladera, J. P. Majoral, A. M. Caminade and B. Klajnert, Mol. Pharm., 2012, 9, 458 CrossRef CAS PubMed; T. Wasiak, M. Marcinkowska, I. Pieszynski, M. Zablocka, A. M. Caminade, J. P. Majoral and B. Klajnert-Maculewicz, New J. Chem., 2015, 39, 4852 RSC.
- P. Gomulak, B. Klajnert, M. Bryszewska, J. P. Majoral, A. M. Caminade and J. Blasiak, Curr. Med. Chem., 2012, 19, 6233 CrossRef CAS.
- L. Griffe, M. Poupot, P. Marchand, A. Maraval, C. O. Turrin, O. Rolland, P. Metivier, G. Bacquet, J. J. Fournie, A. M. Caminade, R. Poupot and J. P. Majoral, Angew. Chem., Int. Ed., 2007, 46, 2523 CrossRef CAS PubMed; A. M. Caminade, S. Fruchon, C. O. Turrin, M. Poupot, A. Ouali, A. Maraval, M. Garzoni, M. Maly, V. Furer, V. Kovalenko, J. P. Majoral, G. M. Pavan and R. Poupot, Nat. Commun. DOI:10.1038/ncomms8722.
- M. Hayder, M. Poupot, M. Baron, D. Nigon, C. O. Turrin, A. M. Caminade, J. P. Majoral, R. A. Eisenberg, J. J. Fournie, A. Cantagrel, R. Poupot and J. L. Davignon, Sci. Transl. Med., 2011, 3, 81ra35 Search PubMed; S. Fruchon, S. Mouriot, T. Thiollier, C. Grandin, A. M. Caminade, C. O. Turrin, H. Contamin and R. Poupot, Nanotoxicology, 2015, 9, 433 CrossRef CAS PubMed.
- M. Blanzat, C. O. Turrin, A. M. Aubertin, C. Couturier-Vidal, A. M. Caminade, J. P. Majoral, I. Rico-Lattes and A. Lattes, ChemBioChem, 2005, 6, 2207 CrossRef CAS PubMed.
- G. Spataro, F. Malecaze, C. O. Turrin, V. Soler, C. Duhayon, P. P. Elena, J. P. Majoral and A. M. Caminade, Eur. J. Med. Chem., 2010, 45, 326 CrossRef CAS PubMed.
- L. A. Dykman and N. G. Khlebtsov, Chem. Rev., 2014, 114, 1258 CrossRef CAS PubMed; D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280 CrossRef PubMed; C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef PubMed; R. A. Sperling, P. Rivera Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896 RSC.
- M. L. Lartigue, N. Launay, B. Donnadieu, A. M. Caminade and J. P. Majoral, Bull. Soc. Chim. Fr., 1997, 134, 981 CAS.
- R. Naumann, S. M. Schiller, F. Giess, B. Grohe, K. B. Hartman, I. Karcher, I. Koper, J. Lubben, K. Vasilev and W. Knoll, Langmuir, 2003, 19, 5435 CrossRef CAS.
- W. Knoll, Annu. Rev. Phys. Chem., 1998, 49, 569 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of all compounds and contact angle goniometry data. See DOI: 10.1039/c5nj00620a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |