
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron(III) and nickel(II) complexes as potential anticancer agents: synthesis, physicochemical and structural properties, cytotoxic activity and DNA interactions†
Tülay
Bal-Demirci
*a,
Gulsah
Congur
b,
Arzum
Erdem
*b,
Serap
Erdem-Kuruca
*c,
Namık
Özdemir
*d,
Kadriye
Akgün-Dar
e,
Başak
Varol
f and
Bahri
Ülküseven
a
aDepartment of Chemistry, Engineering Faculty, İstanbul University, 34320, Avcilar, İstanbul, Turkey. E-mail: tulaybal@istanbul.edu.tr
bEge University, Faculty of Pharmacy, Analytical Chemistry Department, 35100, Bornova, İzmir, Turkey. E-mail: arzum.erdem@ege.edu.tr; arzume@hotmail.com
cDepartment of Physiology, İstanbul Medical Faculty, İstanbul University, 34093, Çapa, İstanbul, Turkey. E-mail: sekuruca@istanbul.edu.tr; sererdem@yahoo.com
dDepartment of Physics, Faculty of Arts and Sciences, Ondokuz Mayıs University, 55139, Samsun, Turkey. E-mail: namiko@omu.edu.tr
eDepartment of Biology, İstanbul Science Faculty, İstanbul University, Beyazıt 34134, İstanbul, Turkey
fDepartment of Biophysic, İstanbul Medical Faculty, İstanbul University, 34093, Çapa, İstanbul, Turkey
First published on 6th May 2015
Abstract
Template reactions of 2-hydroxy-R-benzaldehyde-S-methylisothiosemicarbazones (R = 3-methoxy or 4-hydroxy) with the corresponding aldehydes in the presence of FeCl3 and NiCl2 yielded N1,N4-disalicylidene chelate complexes. The compounds were characterized by means of elemental and spectroscopic methods. The structure of complex 1 was determined by X-ray single crystal diffraction. Crystal data (Mo Kα; 296 K) are as follows: monoclinic space group P21/c, a = 12.9857(8) Å, b = 7.8019(4) Å, c = 19.1976(12) Å, β = 101.655(5)°, Z = 4. Cytotoxic effects of the compounds were evaluated by the MTT assay in K562 leukemia, ECV304 endothelial and normal mononuclear cells, and DNA fragmentation analysis using the diphenylamine reaction was performed. The DNA binding capacity of thiosemicarbazones at IC50 and different concentrations was investigated. The DNA fragmentation percentage of compound treated cells was higher than that of non-treated control cells but was higher for compound 3 (84%) compared to the others. The interaction of compounds 1–4 and DNA was investigated voltammetrically by using nucleic acid modified electrodes after the double stranded fish sperm DNA (fsDNA), or poly(dA)·poly(dT), was immobilized onto the surface of pencil graphite electrodes (PGEs). Accordingly, the oxidation signals of DNA bases, guanine and adenine, were measured by using differential pulse voltammetry (DPV). The changes in the signals of guanine and adenine were evaluated before and after the interaction process. The results indicated that compound 3 was cytotoxic at very low concentrations in K562 leukemia cells unlike other cells and that could damage the DNA double stranded form, specifically the adenine base. Therefore, it may have a selective antileukemic effect and drug potential.
1. Introduction
Thiosemicarbazone based metal complexes have remarkable biological activities such as antitumor,1–3 antiviral,4,5 antibacterial,6 antioxidant,7–9 antidiabetic10 properties and different coordination structures.11–18 The biological properties of thiosemicarbazones are often related to metal ion coordination. Most of the biological research carried out on metal complexes is related to bi- and tridentate thiosemicarbazones with palladium, platinum19,20 and copper ions.21,22 Our research in recent years demonstrated that tetradentate N2O2 type metal complexes of S-methylisothiosemicarbazones have significant biological activities such as antioxidant,8,9 insulinomimetic effects,10 cytotoxicity and proliferation ability.23,24 Interaction of (bio)molecules and nucleic acids has become the attractive topic of research, since nucleic acids are the primary pharmacological target of many anticancer compounds and conformational changes of DNA directly result in cell damage. Thiosemicarbazone molecules have been studied to understand their cytotoxic mechanisms.25–28 Determination of the interactions between thiosemicarbazones and DNA should be elucidated to help explain the mechanisms of apoptotic events and drug potential of thiosemicarbazones. Generally, the transition metals play a very important role in an organism and their complexes can interact non-covalently with nucleic acid by intercalation, groove-binding or external electrostatic binding for cations.3,7,19–22There have been numerous studies in the literature on the investigation of drug–DNA interactions by using conventional techniques such as HPLC, GC, and mass spectrometry. Considering the numerous advantages of electrochemical analysis techniques, the disadvantages of these conventional techniques arising from their complicated form of analysis have made them less preferable. Since the discovery of the electroactivity of nucleic acids in the 1960s,29 electrochemical nucleic acid biosensors, known as electrochemical genosensors, have frequently been preferred for the recognition of (bio)molecule–DNA interactions. There are several research articles on the investigation of (bio)molecule–DNA interaction by using electrochemical techniques in the literature.30–39
PGEs have some crucial properties: they are robust, single-use, practical and have a large surface area. Their combination with electrochemical detection techniques provides a fast, practical, accurate and time-saving analysis of target analytes. Moreover, the preparation and activation of PGEs require less chemicals and shorter experimental steps (i.e., 1–2 min) compared to the ones of a carbon paste electrode (CPE), a glassy carbon electrode (GCE) or a gold electrode (AuE). The usage of PGEs in combination with voltammetric techniques allows the detection of the analytes in maximum 1 hour including preparation steps. Thus, they have been widely used for electrochemical recognition including detection of drug, DNA and drug–DNA interaction,37–39 nucleic acid hybridization,40 miRNAs,41 stem cells,42 aminoacids,43 and proteins by using the aptamer–protein interaction.44,45
The cytostatic properties and cellular effects of novel diene-ruthenium(II) complexes were studied for the human cancer cell lines MCF-7 and HT-29 and Jurkat leukemia cells by Kasper et al.35 The on-line cell based biosensor was developed to monitor a time-delayed decrease in the impedance of the cell layers. Yong et al. reported a direct toxicity assessment based on chronoamperometry to detect the effect of toxic chemicals on microorganisms.36 3,5-Dichlorophenol was chosen as the reference toxicant. Then, three pesticides, ametryn, fenamiphos, endosulfan, were examined using this method. To detect toxicities of phenol and nitrophenols, an electrochemical cell based biosensor was developed, which relied on the inhibition effects of toxicants on the respiratory chain activity of microorganisms.
The synthesis, characterizations, cytotoxic potentials and DNA binding by using differential pulse voltammetry (DPV) with the pencil graphite electrode (PGE) of hydroxy- and methoxy-substituted N1,N4-disalicylidene-S-methylisothiosemicarbazone chelates (Fig. 1) were presented for the first time in this study. The cytotoxic activity of novel S-methylisothiosemicarbazones on K562 cells, ECV304 cells, normal mononuclear cells (MNC) and their IC50 values were determined and DNA fragmentation was measured in K562 cells treated at IC50 values. The interaction between thiosemicarbazones and DNA was investigated by using DPV, which is a sensitive, rapid and inexpensive electrochemical technique for the detection of drug–DNA interaction. The oxidation signals of the thiosemicarbazones and the DNA bases, guanine and adenine, were measured before/after interaction. The interaction mechanism between thiosemicarbazones and DNA was evaluated in terms of decrease ratios of the signals by DPV using PGEs.
 | ||
| Fig. 1 The compounds. R1: 3-OCH3 (L1), 4-OH (LII); M/X/R1/R2: Fe/Cl/3-OCH3/4-OH (1), Ni/—/3-OCH3/4-OH (2), Fe/Cl/4-OH/4-OCH3 (3), Ni/—/4-OH/4-OCH3 (4). | ||
2. Experimental section
2.1. Materials and methods
The oxidation signals of the DNA bases, guanine and adenine, and the thiosemicarbazone complexes (1–4) were investigated by differential pulse voltammetry (DPV) using an AUTOLAB-PSTAT 302 electrochemical system, General Purpose Electrochemical System (GPES, 4.9007 software) package (Ecochemie, The Netherlands). Raw data from Autolab were also treated using the Savitzky and Golay filter (level 2) and a moving average baseline correction (peak width 0.01) of the GPES software. The three electrode system consists of the pencil graphite electrode (PGE), an Ag/AgCl/3 M KCl reference electrode and a platinum wire as the auxiliary electrode. All measurements were performed in a Faraday cage.
All chemicals were of reagent grade and were used as commercially purchased without further purification.
2.2. Synthesis of thiosemicarbazones
3-Methoxy-(LI) and 4-hydroxy-(LII) substituted-N1-salicylidene-S-methylisothiosemicarbazones were synthesized from the corresponding salicylaldehyde (1 mmol), methyl iodide (1 mmol) and thiosemicarbazide (1 g, 1 mmol) in equimolar ratios according to the literature.23 The compounds were checked using elemental analysis and characteristic spectroscopic data. The color, yield (%), m.p. (°C), elemental analysis, IR (KBr, cm−1) and 1H-NMR (DMSO-d6, 25 °C, δ ppm) data of L1 and LII are given as follows:
LI: cream, yield (1.552 g, 93%), 164–165 °C. Anal. calc. for C10H13N3O2S (239 g mol−1): C, 50.21; H, 5.44; N, 17.57; S, 13.39, found: C, 50.25; H, 5.42; N, 17.56; S, 13.40%. IR: νa(NH) 3412, νs(NH) 3306, ν(OH) 3129, δ(NH) 1651. NMR: δ 11.58, 10.71 (cis/trans ratio: 2/1, s, 1H, OH), 8.44, 8.30 (syn/anti ratio: 2/3, s, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N1), 6.84 (s, 2H, NH2), 2.42, 2.37 (cis/trans ratio: 3/2, s, 3H, S–CH3), 3.77 (s, 3H, OCH3).
N1), 6.84 (s, 2H, NH2), 2.42, 2.37 (cis/trans ratio: 3/2, s, 3H, S–CH3), 3.77 (s, 3H, OCH3).
LII: pinkish cream, yield (1.425 g, 94%), 179–180 °C. Anal. calc. for C9H11N3O2S (225 g mol−1): C, 48.00; H, 4.89; N, 18.66; S, 14.22, found: C, 48.18; H, 4.92; N, 18.66; S, 14.27%. IR: νa(NH) 3445, νs(NH) 3337, ν(OH) 3495, δ(NH) 1624. NMR: 11.67, 11.02 (cis/trans ratio: 5/2, s, 1H, OH), 9.75 (s, 1H, OH), 8.32, 8.20 (syn/anti ratio: 2/3, s, 1H, CHN1), 6.71, 6.65 (syn/anti ratio: 1/1, s, 2H, NH2),2.42, 2.38 (cis/trans ratio: 3/2, s, 3H, S–CH3).
For the synthesis of complex 1, compound LI (1.0 g, 1 mmol) and 2,4-dihydroxybenzaldehyde (0.58 g, 1 mmol) were dissolved in 25 mL of ethanol. The mixture was added to a solution of 1.70 g (1.5 mmol) FeCl3·6H2O in ethanol (25 mL) and then 10 mL of triethylamine. After 24 h, the black precipitate was filtered off, washed with ethanol–ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 mL) and dried in vacuo over P2O5. Complex 3 was synthesized by reaction of LII (1 mmol), 2-hydroxy-4-methoxybenzaldehyde (1 mmol) and FeCl3·6H2O (1.5 mmol) using the same method.
:1, 10 mL) and dried in vacuo over P2O5. Complex 3 was synthesized by reaction of LII (1 mmol), 2-hydroxy-4-methoxybenzaldehyde (1 mmol) and FeCl3·6H2O (1.5 mmol) using the same method.
Complexes 2 and 4 were obtained by using NiCl2·6H2O instead of FeCl3·6H2O in a similar manner. The color, yield (g, %), m.p. (°C), molar conductance (Ohm−1 cm2 mol−1, in 10−3 M DMSO, 25 ± 1 °C), μeff (BM), elemental analysis, IR (KBr, cm−1), 1H-NMR (DMSO-d6, 25 °C, δ ppm) and (+) ESI-mass data of the complexes are given as follows:
1: bright black, yield (0.8776 g, 45%), >390 °C, 19.56, 5.88. Anal. Calc. for C17H17N3O5SFeCl (466.69 g mol−1): C, 43.75; H, 3.67; N, 9.00; S, 6.87; Fe, 11.97, found: C, 43.69; H, 3.65; N, 9.12; S, 6.89; Fe, 11.95%. IR: ν(CN) 1612, 1601, 1578, (C–O)arom 1158, 1131. m/z (+c ESI-MS, % relative abundance): 413 [M–Cl–H2O] (100.00), 414 [M–Cl–H2O + H] (24.47), 415 [M–Cl–H2O + 2H] (7.98).
2: red, yield (0.6080 g, 35%), 276–277 (decomp) °C, 6.45, 0.06. Anal. Calc. for C17H15N3O4SNi (415.7 g mol−1): C, 49.07; H, 3.63; N, 10.10; S, 7.71, found: C, 49.05; H, 3.68; N, 10.34; S, 7.85%. IR: ν(OH) 3445, ν(CN) 1608, 1594, ν(C–O) 1150, 1112. NMR: δ 10.85 (s, 1H, OH), 8.42 (s, 1H, CHN1), 7.97 (s, 1H, CHN4), 6.30 (dd, J = 8.69, J = 1.83, 1H, b), 6.55 (t, J = 7.78, 1H, c), 7.58 (d, J = 8.69, 1H, d), 6.26 (s, 1H, p), 6.85 (d, J = 7.77, 1H, r), 7.10 (d, J = 8.24, 1H, s), 3.74 (s, 3H, O–CH3), 2.69 (s, 3H, S–CH3)
3: bright black, yield (0.5179, 25%), >390 °C, 22.15, 5.86. Anal. Calc. for C17H17N3O5SFeCl (466.69 g mol−1): C, 43.75; H, 3.67; N, 9.00; S, 6.87; Fe, 11.97, found: C, 43.78; H, 3.69; N, 9.02; S, 6.90; Fe, 11.95%. IR: ν(OH) 3440, ν(CN) 1608, 1597, 1582 ν(C–O) 1152, 1106. m/z (+c ESI-MS, % relative abundance): 413 [M–Cl–H2O] (100.00), 414 [M–Cl–H2O + H] (23.85), 415 [M–Cl–H2O + 2H] (8.38).
4: red, yield (0.5167 g, 28%), 276–277 (decomp) °C, 8.64, 0.012. Anal. Calc. for C17H15N3O4SNi (415.7 g mol−1): C, 49.07; H, 3.63; N,10.10; S, 7.71, found: C, 49.04; H, 3.65; N, 10.02; S, 7.67%. IR: ν(OH) 3437, ν(CN) 1608, 1598, 1582 ν(C–O) 1150, 1108. NMR: δ 10.08 (s, 1H, OH), 8.22 (s, 1H, CHN1), 8.03 (s, 1H, CHN4), 6.23 (d, J = 2.29, 1H, a), 6.21 (dd, J = 8.24, J = 2.29, 1H, c), 7.61 (d, J = 9.61, 1H, d), 6.46 (d, J = 2.29, 1H, p), 6.38 (dd, J = 9.15, J = 2.28, 1H, r), 7.34 (d, J = 8.69, 1H, s), 3.80 (s, 3H, O–CH3), 2.66 (s, 3H, S–CH3).
2.3. Single-crystal structure determination
Intensity data for complex 1 were collected on a STOE IPDS II diffractometer at room temperature (296 K) using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) by applying the ω-scan method. The structure was solved by direct methods using SHELXS-201346 and refined with full-matrix least-squares calculations on F2 using SHELXL-201446 implemented in the WinGX47 program suit. The coordinates of the water H atoms were determined from a difference map and were refined isotropically, subject to a DFIX restraint of O–H = 0.82 Å. The remaining H atoms were placed geometrically and treated using a riding model, fixing the bond lengths at 0.82, 0.93 and 0.96 Å for OH, aromatic CH and CH3 atoms, respectively. The displacement parameters of the H atoms were fixed at Uiso(H) = 1.2Ueq (1.5Ueq for OH, OH2 and CH3) of their parent atoms. Data collection: X-AREA,48 cell refinement: X-AREA, data reduction: X-RED32.48 Crystal data, data collection and structure refinement details are summarized in Table 1. The general-purpose crystallographic tool PLATON49 was used for the structure analysis and presentation of the results. Molecular graphics were generated by using ORTEP-3.50| CCDC | 1001379 |
| Color/shape | Black/prism |
| Chemical formula | FeCl[(C17H15N3O4S)]·H2O |
| Formula weight | 466.69 |
| Temperature (K) | 296 |
| Wavelength (Å) | 0.71073 Mo Kα |
| Crystal system | Monoclinic |
| Space group | P21/c (No. 14) |
| Unit cell parameters | |
| a, b, c (Å) | 12.9857(8), 7.8019(4), 19.1976(12) |
| α, β, γ (°) | 90, 101.655(5), 90 |
| Volume (Å3) | 1904.9(2) |
| Z | 4 |
| D calc (g cm−3) | 1.627 |
| μ (mm−1) | 1.077 |
| Absorption correction | Integration |
| T min, Tmax | 0.8081, 0.9553 |
| F 000 | 956 |
| Crystal size (mm3) | 0.30 × 0.15 × 0.04 |
| Diffractometer/measurement method | STOE IPDS II/rotation (ω scan) |
| Index ranges | −16 ≤ h ≤ 16, −9 ≤ k ≤ 9, −23 ≤ l ≤ 23 |
| θ range for data collection (°) | 1.60 ≤ θ ≤ 25.99 |
| Reflections collected | 13160 |
| Independent/observed reflections | 3751/1643 |
| R int | 0.115 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3751/5/362 |
| Goodness-of-fit on F2 | 0.875 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0598, wR2 = 0.1075 |
| R indices (all data) | R 1 = 0.1523, wR2 = 0.1277 |
| Δρmax, Δρmin (e Å−3) | 0.63, −0.27 |
2.4. Cell cultures
The K562 chronic myeloid leukemia cell line and ECV304 human umbilical vein endothelial cell line were purchased from ATTC. In addition, mononuclear cells (MNC) were isolated from normal human peripheral blood using Histopaque 1077. The cells were cultured in DMEM (for ECV304) and IMDM (for K562 and MNC) media (Sigma) supplemented with 10% fetal calf serum (GIBCOBRL) and 1% penicillin–streptomycin. Experiments were conducted on cells seeded into 96-well culture plates at densities 105 cells per mL while maintaining the cells at 37 °C in an atmosphere of 5% CO2 in air.2.5. Cytotoxicity assay
The cytotoxic effects of the compounds were evaluated using a MTT ([3,4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay which was reduced by living cells to yield a soluble formazan product using the method of Mossman modified by our laboratory.3 Stock solution compounds were prepared at 5 mg mL−1 in DMSO. The six concentrations (50 μg mL−1, 10 μg mL−1, 5 μg mL−1, 1 μg mL−1, 0.1 μg mL−1, 0.01 μg mL−1) were prepared from each compound and 10 μL was added to wells, each one in triplicate. Then, K562, ECV304 and mononuclear cells were plated at 104 cells per well and incubated for 3 days at 37 °C, in an atmosphere of 5% CO2. Control wells were prepared without the compound. DMSO control wells were prepared in the same rates as the former compound solutions, in order to compare and eliminate their effect on absorbance. At least 3 independent experiments were conducted. After the incubation period, the acidified medium was aspirated from wells and MTT was added to 10 μL at 5 mg mL−1. Cells were incubated at 37 °C for 3 h, after which they were subsequently lysed by the addition of 100 μL−1 of acidic (0.04 M HCl) isopropanol alcohol. Overnight, plates were stored, protected from light for dissolved formazan. The next day, the optical density (OD) of the formazan was measured at a 560 nm test wavelength and a 620 nm reference wavelength using an ELISA multiwell spectrophotometer (Diagnostics Pasteur LP 400). The cytotoxicity index (CI) was calculated using the following formula compared with the control:| CI% (Cytotoxicity index) = 1 − OD treated wells/OD control wells × 100. |
Also, the inhibitory concentration of cell growth (IC50 = the concentration of the compound that inhibited 50% cells) was calculated from dose–response curves.51
2.6. Quantitative analysis of DNA fragmentation
DNA fragmentation analysis was performed by diphenylamine reaction. 2 × 106 control and compound-treated cells (IC50) were collected by centrifugation at 3000 rpm for 5 min at 4 °C. The cell pellets were lysed in 0.5 mL of lysis buffer containing 0.5% Triton X-100, 10 mM Tris-HCl and 1 mM EDTA at pH 7.4 and were centrifuged for 10 min at 13000 rpm (Sorvall-pro 80) at 4 °C. The pellets were resuspended in 0.5 mL of lysis buffer. The pellet (P) and supernatant (S) fractions were mixed with 0.5 mL of 25% CCl3COOH (TCA) and incubated for 24 h at 4 °C. The samples were centrifuged for 10 min at 13000 rpm and 4 °C and the pellets were resuspended in 160 μL of 5% TCA, followed by incubation for 15 min at 90 °C. Then, 320 μL of diphenylamine solution (150 mg diphenylamine in 10 mL of glacial acetic acid with 150 μL of sulfuric acid and 50 μL of acetaldehyde) was added to each sample, followed by incubation for 4 h at 37 °C. The proportion of fragmented DNA was calculated from absorbance at 600 nm using the formula52| Fragmented DNA% = [OD(S)/OD(S) + OD(P)] × 100. |
2.7. Caspase 3 protein determination by the immunohistochemical method
K562 cells untreated and treated at IC50 concentrations of complexes were cultured on coverslips in a 6-well plate. After 72 h, the cells growing on coverslips were fixed with cold acetone for 10 min, rinsed twice in PBS for 10 min. Endogenous peroxidases were inactivated by immersing the sections in 3% hydrogen peroxide for 10 min, washed with distilled water and PBS for 15 min, blocked with 100 mL L−1 normal goat serum for 10 min to reduce non-specific binding, and incubated with monoclonal mouse anti-human caspase 3 antibodies (1:200) at 4 °C overnight. After incubation, the cells on coverslips were incubated with biotinylated anti-mouse IgG at 37 °C for 10 min, rinsed twice in PBS for 15 min, and then incubated in streptavidin–peroxidase at 37 °C for 10 min. The chromogenic reaction was developed with diaminobenzidine (DAB). Negative controls were incubated in the absence of primary antibodies. Cells were counter stained with Mayer's hematoxylin for 45 seconds and they were covered with glycerol gelatin.53
2.8. The interaction of compounds 1–4 and DNA
DNA immobilized electrodes were immersed into the vials containing 110 μL solution of different compounds for the surface confined interaction process for 30 min then, the electrodes were rinsed with PBS (pH 7.4) for 10 s to eliminate unspecific binding.
3. Results and discussion
3.1. Chemistry
Thiosemicarbazones LI and LII are soluble in methanol, ethanol, and chlorinated hydrocarbons. The reactions of LI and LII with corresponding aldehydes in the presence of iron(III) and nickel(II) in the 1:1:1 molar ratio yielded solid complexes, 1–4. The complexes are very soluble in donor solvents such as dimethylformamide and dimethyl sulfoxide, but less soluble in ethanol and dichloromethane. The solid complexes are stable for several weeks in air. The thiosemicarbazones and complexes are in the form of very fine powder crystals (Fig. 1).
The μeff values of iron(III) complexes (1, 3) that are in the 5.86–5.88 BM range are equivalent to five unpaired electrons and so the iron(III) ion is in the high-spin state indicating the [Fe(L)Cl] structure. Magnetic measurement results of nickel(II) complexes (2, 4) showed that they are diamagnetic and have a square-planar structure.
Template reactions of the thiosemicarbazones and aldehydes can be easily monitored by means of IR and 1H NMR spectra. The ν(NH2), one of ν(OH) (for 3, 4), and also δ(NH2) bands disappeared in the infrared spectra of the complexes due to reactions of 2-hydroxy and thioamide groups. The protons of starting materials LI and LII showed the expected chemical shift values, and even the systematic signals of syn–anti and cis–trans isomers. In the NMR spectra of 2 and 4, the proton signals of 2-OH and N4H2 groups were “absent” because of chelation. Besides, the arising N4CH signal which is a singlet and equivalent to the integral value of one proton confirms the chelate formation around nickel(II). The compositions of paramagnetic iron(III) complexes, [Fe(L)Cl]·H2O, justify [M–Cl] peaks as well as other mass data. The analytical and spectral data provide evidence that the chelating N1,N4-disalicylidene-S-methylisothiosemicarbazidato ligands bonded through the ONNO donor set has been previously accomplished.10,23,24 In addition, iron(III) complexes contain one molecule of H2O in the structures [Fe(L)Cl]·H2O unlike nickel(II) complexes [NiL].
3.2. Structural description of the complex
The solid-state structure of complex 1 has been confirmed by single crystal X-ray analysis. A perspective view of the complex using the atomic numbering scheme is depicted in Fig. 2, while selected bond lengths and angles are given in Table 2. | ||
| Fig. 2 A view of complex 1 showing the atom-numbering scheme. Displacement ellipsoids are drawn at the 30% probability level and H atoms are shown as small spheres of arbitrary radii. Hydrogen bonding interactions are indicated by dashed lines. | ||
| Bond lengths (Å) | |||
| Fe1–Cl1 | 2.2093(19) | O3–C15 | 1.350(7) |
| Fe1–O1 | 1.923(4) | O4–C14 | 1.346(7) |
| Fe1–O3 | 1.874(4) | O4–C17 | 1.430(6) |
| Fe1–N1 | 2.085(4) | N1–C7 | 1.311(7) |
| Fe1–N3 | 2.080(5) | N1–C8 | 1.395(7) |
| N2–N3 | 1.399(6) | N2–C8 | 1.304(8) |
| S1–C8 | 1.730(6) | N3–C9 | 1.285(8) |
| S1–C16 | 1.769(7) | C6–C7 | 1.409(8) |
| O1–C1 | 1.314(7) | C9–C10 | 1.452(8) |
| O2–C3 | 1.338(7) | ||
| Bond angles (°) | |||
| Cl1–Fe1–O1 | 105.25(14) | C8–S1–C16 | 102.4(3) |
| Cl1–Fe1–O3 | 106.63(14) | C14–O4–C17 | 118.1(5) |
| Cl1–Fe1–N1 | 104.47(15) | C7–N1–C8 | 120.0(5) |
| Cl1–Fe1–N3 | 103.05(15) | C8–N2–N3 | 111.4(5) |
| O1–Fe1–O3 | 97.27(17) | C9–N3–N2 | 113.6(5) |
| O1–Fe1–N1 | 87.28(17) | N1–C7–C6 | 125.7(5) |
| O1–Fe1–N3 | 149.05(18) | N2–C8–N1 | 119.2(5) |
| O3–Fe1–N1 | 146.07(19) | N2–C8–S1 | 119.7(4) |
| O3–Fe1–N3 | 86.35(19) | N1–C8–S1 | 121.1(5) |
| N1–Fe1–N3 | 73.7(2) | N3–C9–C10 | 124.4(6) |
| Torsion angles (°) | |||
| C16–S1–C8–N2 | −0.2(7) | N2–N3–C9–C10 | 177.3(6) |
| C16–S1–C8–N1 | 178.2(6) | C1–C6–C7–N1 | 3.5(10) |
| N3–N2–C8–S1 | 177.8(4) | N3–C9–C10–C11 | 177.0(6) |
| C7–N1–C8–S1 | −2.5(8) | C7–N1–C8–N2 | 175.9(6) |
| C8–N1–C7–C6 | −179.0(6) | C8–N2–N3–C9 | −170.3(6) |
| N3–N2–C8–N1 | −0.6(8) | ||
Complex 1 is composed of an N1-3-methoxysalicylidene-N4-4-hydroxysalicylidene-S-methylisothiosemicarbazidato chelate with an FeIII metal centre and one Cl ligand, and crystallizes with a solvent water molecule in the asymmetric unit. The Schiff-base ligand gets doubly deprotonated to act as an O,N,N,O tetradentate ligand, coordinating via its two phenolato oxygen atoms, O1 and O3, and two azomethine nitrogen atoms, N1 and N3. A chloride ion coordinates in the fifth position.
Five-coordinate complexes have geometries ranging from square-pyramidal to trigonal-bipyramidal. For a quantitative evaluation of the extent of distortion around the five-coordinate iron center, the structural index55τ, [τ = (β − α)/60°, α and β being the two largest angles around the central atom], is employed. The τ value can be conveniently utilized to estimate the degree of distortion from square-pyramidal to trigonal-bipyramidal structures. In the case of an ideal square-pyramidal geometry, the τ value is equal to zero, while it becomes unity for a perfect trigonal-bipyramidal geometry. The value of τ for the FeIII ion is 0.05, indicating a slightly distorted square-pyramid. In the square-pyramidal geometry, the basal plane defined by the two N and two O atoms of the Schiff base ligand and the apical position occupied by a chloride ligand. Atom Fe1 is 0.511(2) Å above the best plane defined by the Schiff-base N and O donor atoms.
The Fe–N bond distances [2.080(5) and 2.085(4) Å] are relatively longer than the Fe–O bond lengths [1.874(4) and 1.923(4) Å] while the chloride ion is weakly coordinated to the iron atom at 2.2093(19) Å. All the coordination bond distances are in accordance with the literature values.56–61 The angles of O1–Fe1–N3 and O3–Fe1–N1 are 149.05(18) and 146.07(19)°, respectively. Clearly, the FeIII center deviates from the basic plane of the quadrangle pyramid.
The O- and N-donor atoms of the tetradentate ligand form three metallacycles: one five-membered FeN3C and two six-membered FeNC3O. The five-membered chelate ring adopts an envelope conformation, with atom Fe1 displaced from the N1–C8–N2–N3 mean plane by 0.394(3) Å [the puckering parameters62: Q = 0.191(5) Å and φ = 177.66(19)°], while the six membered chelate rings exhibit a half-chair conformation [the puckering parameters: Q = 0.2752(39) Å, θ = 123.84(13)° and φ = −165.13(14)° for Fe1–O1–C1–C6–C7–N1; Q = 0.2485(46) Å, θ = 62.44(15)° and φ = 13.16(15)° for Fe1–O3–C15–C10–C9–N3].
The crystal structure does not exhibit any intramolecular interactions. In the crystal structure, the molecules are packed in columns running along the b axis. The water molecule, O5W, acts as a hydrogen-bond donor to O1, O3 and O4 atoms of the complex in each column. In addition, the columns related by two fold screw axes are connected to each other by one O–H⋯Owater intermolecular interaction. The extension of these intermolecular hydrogen-bonding interactions generates infinite zigzag chains running parallel to the [010] direction (Fig. 3). The full geometry of the intermolecular interactions is given in Table 3.
 | ||
| Fig. 3 Part of the crystal structure of complex 1 showing the intermolecular interactions. For clarity, only H atoms involved in hydrogen bonding have been included. | ||
| D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) |
|---|---|---|---|---|
| a Symmetry code: i −x + 1, y − 1/2, −z + 3/2. | ||||
| O2–H2A⋯O5Wi | 0.82 | 1.85 | 2.628(7) | 158 |
| O5W–H5B⋯O1 | 0.86(2) | 2.23(2) | 3.044(6) | 158(4) |
| O5W–H5A⋯O4 | 0.86(2) | 2.13(2) | 2.936(6) | 156(5) |
| O5W–H5A⋯O3 | 0.86(2) | 2.25(5) | 2.881(6) | 130(4) |
3.3. Cytotoxic activity
Firstly, the cytotoxic potential of thiosemicarbazone derivatives (1–4) was investigated in K562 leukemia, ECV304 endothelial and normal mononuclear cells by the colorimetric MTT assay. Cytotoxicity tests imply that the chelates (3 and 4) that have 4-OCH3 and 4-OH substituents are efficiently cytotoxic for K562 cells and not cytotoxic for ECV304 and normal mononuclear cells at the same concentrations (Table 4). The iron chelate 3 caused clearly high cytotoxic effects on the leukemic cell line and these effects occurred at a very low concentration of 0.01 μg mL−1 compared with nickel chelate 4 (0.6 μg mL−1). Interestingly, the methoxy and hydroxy substituents of complexes 3 and 4 were in the same positions except the metal residue. In complexes 1 and 2, only the methoxy substituents (3-OCH3) were in the opposite position. The results obtained from this paper and our previous studies show that substituents and the position of the substituents have effects on cytotoxicity. The cytotoxic effect of the N(1)-R-salicylidene-N(4)-4-methoxysalicylidene backbone was, in the order of toxicity in the iron complexes, R: 4-OH > 4-OCH3 > 3-OCH3, and IC50: 0.01 μg mL−1, 0.5 μg mL−1, 3.5 μg mL−1, respectively. These results suggest that the 4-position and possession of an hydroxy substituent cause an increase in cytotoxicity.23,24The most severe DNA fragmentation was observed in complex 3 (84%) (Table 5). However, complexes (1 and 2) and 4 caused severe DNA fragmentation compared with control. The apoptotic effects of compounds were confirmed by immunocytochemical determination of caspase 3 protein, though not quantitative (Fig. 4). In this instance, complex 3 was more cytotoxic in very small concentrations compared with other chelates; we believed it may have a therapeutic value.
| Complexes | DNA fragmentationa (%) |
|---|---|
| a DNA fragmentation was only measured in K562 cells because mentioned concentrations of complexes (IC50) were not effective in ECV304 and normal mononuclear cells. Control cells were not treated with complexes. | |
| Control | 8 ± 0.5 |
| 1 | 78 ± 2.8 |
| 2 | 75 ± 0.1 |
| 3 | 84 ± 1.4 |
| 4 | 75 ± 1.4 |
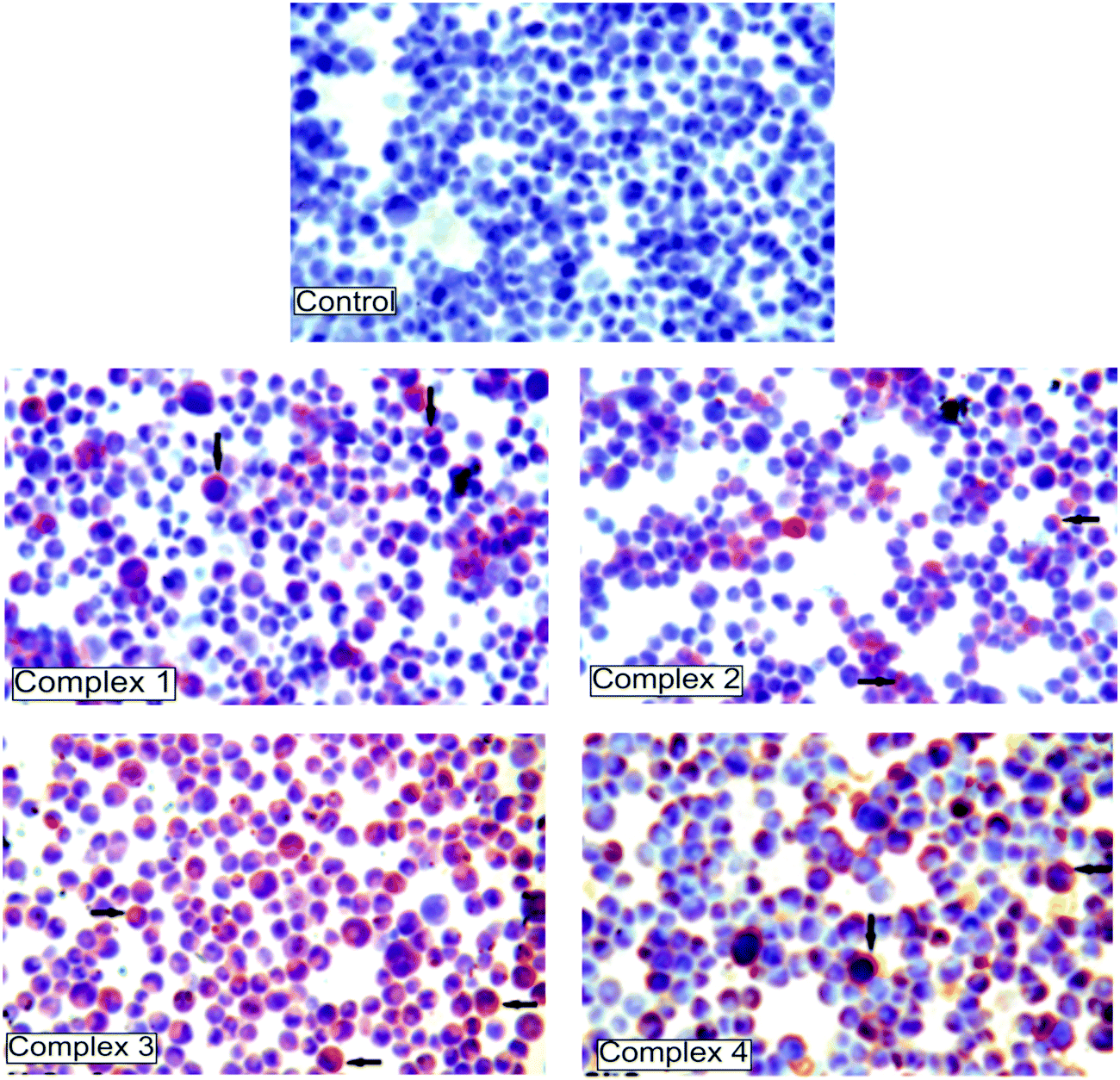 | ||
| Fig. 4 Immunohistochemistry results of the caspase-3 protein expression after 72 h treatment with complexes 1–4 in K562 cells. The brown color represents a positive staining of caspase-3. Control cells prepared without complexes and representing negative staining. | ||
3.4. Electrochemical investigation of interaction between the compounds and DNA
Firstly, the electrochemical behaviours of the thiosemicarbazone molecules, compounds 1–4, were investigated on the surface of disposable PGEs by using DPV. Compounds 1–4 were prepared at their IC50 concentrations (3.2, 4.5, 0.62 and 0.01 μg mL−1, respectively) and immobilized onto the graphite surface by passive adsorption. The oxidation signals of compounds 1, 2 and 4 were measured respectively at +0.782 V and +1.012 V (shown in Fig. 5A-a and b), +0.986 V (Fig. 5A-c) and +1.088 V (Fig. 5A-d). No signal was observed after the immobilization of compound 3 onto PGEs (Fig. 5A-e). Because the oxidation signals of compounds 1, 2 and 4 could have overlapped with the one of the guanine signals measured at +1.012 V, the interaction between compound 3 and fsDNA was performed on the surface of PGEs (Fig. 6A and B). Before the interaction process, the average oxidation signal of guanine was measured as 2.94 ± 0.4 μA (a relative standard deviation (RSD)% = 11.5%, n = 3) and then it decreased about 20.2% in the presence of the interaction of 0.01 μg mL−1 compound 3 with 20 μg mL−1 fsDNA at the PGE surface (Fig. 6A, B-b to b′). | ||
| Fig. 5 Voltammograms (A) and histograms (B) representing that the oxidation signals of compound 1 measured at +0.782 V (a) and +1.012 V (b), the oxidation signal of compound 2 measured at +0.986 V (c), compound 4 measured at +1.088 V (d) and compound 3 (e) by using DPV on the PGE surface. The control measurement was performed in ABS (f). | ||
 | ||
| Fig. 6 Voltammograms (A) and histograms (B) representing the oxidation signal of compound 3 (a, a′), guanine measured at +1.0 V (b, b′) and adenine measured at +1.2 V (c, c′) obtained before (I) and after (II) surface confined interaction between compound 3 at its IC50 value (0.01 μg mL−1) and 20 μg mL−1 fsDNA on the surface of PGE using DPV. | ||
This decrease in the guanine signal may be attributed to the ability of compound 3 since it can intercalate into the double stranded structure of DNA through its planar aromatic ring systems (Fig. 1). Moreover, a small adenine peak at +1.243 V was measured (0.34 ± 0.06 μA, RSD% = 16.5%, n = 3) in consequence of interaction of compound 3 and fsDNA whereas no adenine signal was observed before interaction (Fig. 6A-c to c′). This result indicated that the specific bindings between compound 3 and the adenine base could occur.
Due to the fact that the oxidation signals of the compounds could have overlapped with the oxidation signal of guanine, poly(dA)·poly(dT), which only has adenine and thymine bases, was used for further interaction studies. Compounds 1–3 were prepared at their IC50 levels as 4.5, 3.2, and 0.01 μg mL−1, respectively (Fig. 7); 0.1 μg mL−1 (Fig. S1, ESI†) or 1 μg mL−1 (Fig. S2, ESI†). The oxidation signals obtained after the interaction process between compounds 1–3 and 10 μg mL−1 poly(dA)·poly(dT) were recorded and the adenine signal was measured before/after the interaction process. Due to the oxidation signals of compound 1 measured at +0.782 V and +1.012 V that were not well defined and overlapped with the adenine signal, the interaction of 4.5 μg mL−1 complex 1 and 10 μg mL−1 poly(dA)·poly(dT) could not be investigated by using DPV (not shown). After interaction of 3.2 μg mL−1 compound 2 and poly(dA)·poly(dT) (Fig. 7A), the oxidation signal of compound 2 measured at +0.986 V dramatically decreased (51.4%, n = 3, Fig. 7A, I-a to II-a′) and measured as 3.4 ± 0.5 nA whereas a small decrease in the adenine signal was recorded (with RSD% as 5.6% (n = 3), Fig. 7A, I-b to II-b′). The interaction between 0.01 μg mL−1 of compound 3 and 10 μg mL−1 poly(dA)·poly(dT) was evaluated by means of the increase in the adenine signal measured at +1.2 V (Fig. 7B). The average adenine signal was measured as 4.4 ± 0.4 μA (with a RSD% = 10%, n = 3) and increased 1.7 folds (Fig. 7B, I-b to II-b′) after the interaction process. This result was consistent with the one obtained in the presence of interaction between compound 3 at its IC50 concentration level and fsDNA (Fig. 6). It was concluded that there was a specific interaction between compound 3 and the adenine base.
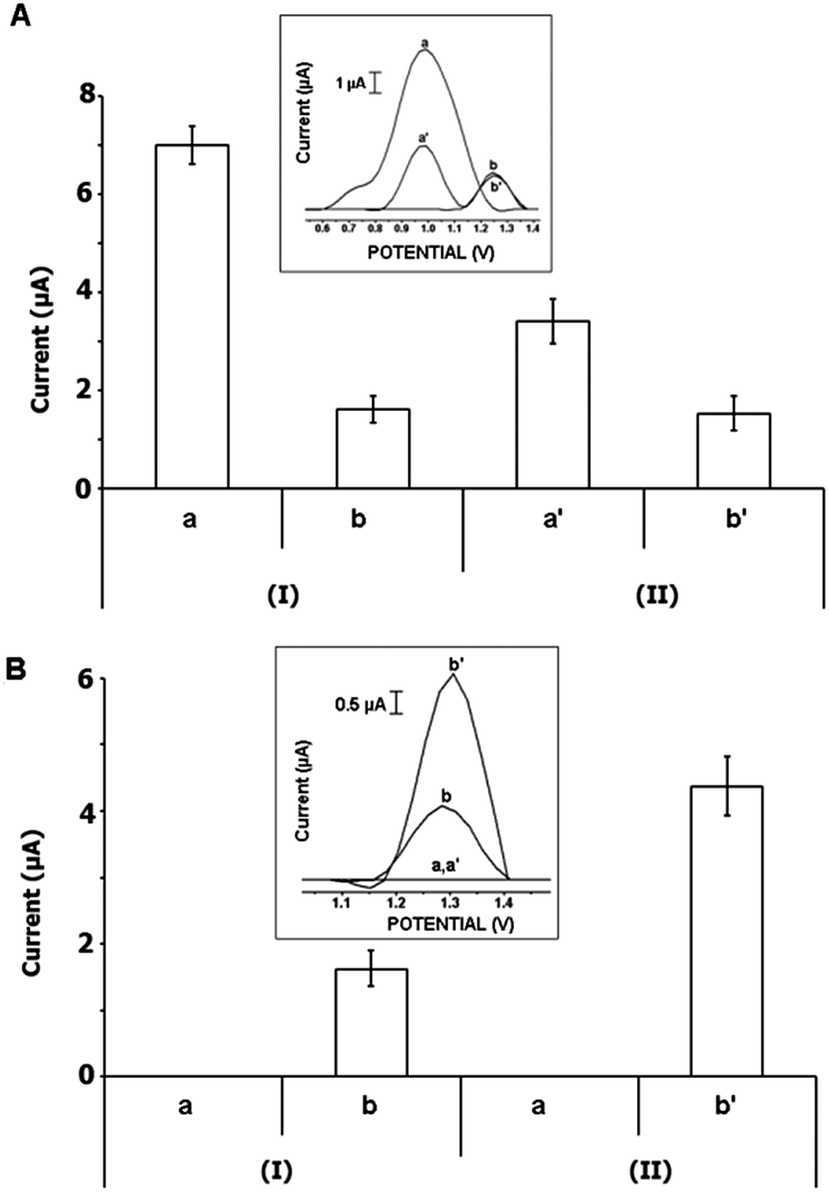 | ||
| Fig. 7 Histograms and voltammograms as insets representing the oxidation signals of compound 2 measured at +0.782 V and +1.012 V (a, a′), and adenine measured at +1.2 V (b, b′) obtained before (I) and after (II) interaction between compound 2 at the concentration level of its IC50 value (3.2 μg mL−1) and 10 μg mL−1 poly(dA)·poly(dT) (A); the oxidation signals of compound 3 (a, a′) and adenine (b, b′) obtained before (I) and after (II) interaction between compound 3 at its IC50 value (0.01 μg mL−1) and 10 μg mL−1 poly(dA)·poly(dT) (B) on the PGE surface using DPV. | ||
The interaction was then performed between 0.1 μg mL−1 of the compounds 1–3 and 10 μg mL−1 poly(dA)·poly(dT) on the PGE surface and the results are shown in Fig. S1 (ESI†). After interaction of compound 1 and poly(dA)·poly(dT) (Fig. S1-A, ESI†), a well-defined oxidation signal of compound 1 was obtained at +0.990 V (Fig. S1-A-a, ESI†). After the interaction process, the oxidation signal of compound 1 and the adenine signal decreased about 51% (Fig. S1-A, I-a to II-a′, ESI†) and 66% (Fig. S1-A, I-b to II-b′, ESI†), respectively. After interaction of compound 2 and poly(dA)·poly(dT) (Fig. S1-B, ESI†), the adenine signal was measured before/after the interaction process while the oxidation signal of compound 2 was not obtained at this concentration level; a 1.6 fold higher adenine signal was obtained after the interaction process (Fig. S1-B, I-b to II-b′, ESI†) and the average adenine signal was measured as 4.7 ± 0.6 μA (RSD% = 13.6%, n = 3). The result showed that the effect of compound 2 onto the DNA structure was concentration dependent due to the fact that no significant change was recorded at the adenine signal after the interaction of compound 2 at its IC50 value (3.2 μg mL−1) and poly(dA)·poly(dT) (Fig. 7A). Furthermore, the interaction between compound 3 and poly(dA)·poly(dT) was performed (Fig. S1-C, ESI†) and the adenine signal increased 72% (Fig. S1-C, I-b to II-b′, ESI†). It was observed that the increase in the adenine signal decreased while the concentration of compound 3 was 10 times increased in comparison to the result obtained after the interaction of compound 3 at its IC50 (0.01 μg mL−1) and poly(dA)·poly(dT) at the PGE surface (Fig. 7B).
In the last part of our study, we performed the interaction between 1 μg mL−1 of compounds 1–3 and poly(dA)·poly(dT) on the PGE surface (Fig. S2, ESI†). The oxidation signals of compound 1 and adenine were recorded (Fig. S2-A, ESI†) and the changes in compound 1 and adenine signals were evaluated. The compound 1 signal decreased about 56% (Fig. S2-A, I-a to II-a′, ESI†) while the adenine signal increased about 59% (Fig. S2-A, I-b to II-b′, ESI†). It was concluded that compound 1 affected the double stranded DNA structure and adenine in a concentration dependent manner due to the reason that the change in the oxidation signal of compound 1 was parallel to the change in the adenine signal contrary to the results obtained after interaction between 0.1 μg mL−1 compound 1 and poly(dA)·poly(dT) (Fig. S1-A, ESI†). The oxidation signal of compound 2 was observed at +0.990 V and it became zero after interaction (Fig. S2-B, I-a to II-a′, ESI†). Also, almost 2 times higher adenine signal was obtained as 4.7 ± 0.05 μA (RSD% = 0.9%, n = 3) after the interaction of compound 2 and poly(dA)·poly(dT) (Fig. S2-B, I-b to II-b′, ESI†). This result was consistent with the one obtained after interaction of 0.1 μg mL−1 compound 2 and poly(dA)·poly(dT) (Fig. S1, ESI†). The interaction between compound 3 and poly(dA)·poly(dT) was then investigated on the surface of PGE (Fig. S2-C, ESI†). The oxidation signals of compound 3 were measured at +0.557 V (Fig. S2-C, I-a, ESI†) and +1.168 V (Fig. S2-C, I-b, ESI†). The signal observed at +0.557 V sharply increased by almost 8 folds (Fig. S2-C, I-c to II-c′, ESI†), whereas the signal observed at +1.168 V overlapped with the adenine signal after the interaction process (Fig. S2-C, I-b, c to II-b′, c′, ESI†). After three repetitive measurements, the signal obtained at +0.557 V was measured as 1.7 ± 0.09 μA (RSD% = 5.7%, n = 3).
4. Conclusions
We have prepared four iron(III) and nickel(II) complexes of thiosemicarbazones. We concluded that all the complexes are cytotoxic against K562 leukemic cells while are nontoxic to ECV304 endothelial and MNC normal mononuclear cells at the same doses. The results obtained from the MTT assay showed that the cytotoxic effect of the N(1)-R-salicylidene-N(4)-4-methoxysalicylidene backbone are, in order R: 4-OH > 4-OCH3 > 3-OCH3 in iron complexes, according to IC50 values: 0.01 μg mL−1, 0.5 μg mL−1, 3.5 μg mL−1, respectively. These results suggest that the 4-position and the hydroxy substituent cause an increase in cytotoxicity.23,24 In addition, the percentage of DNA fragmentation and caspase 3 expression was corrected to IC50 values indicated cytotoxic effect caused by triggering the apoptotic pathway.The electrochemical investigation of the interactions between the compounds and double stranded DNA or poly(dA)·poly(dT) was performed at the surface of single-use PGEs which were easy-to-use, cheap, require less time and chemicals for preparation.32,39,63 Firstly, the oxidation signals of the compounds were measured by using the DPV technique. Then, the surface confined interaction process was performed in the presence of dsDNA or poly(dA)·poly(dT). The interaction mechanisms were evaluated in terms of the increase/decrease in the oxidation signals of the compounds and electroactive bases of DNA, guanine and adenine obtained after the interaction process. There was a strong evidence that compounds 1 and 2 affected the double stranded DNA structure and adenine in a concentration dependent manner. Moreover, electrochemical measurements indicated that the specific interaction between compound 3 and the adenine bases of DNA could occur at the IC50 value (0.01 μg mL−1). This interaction could occur by specific interaction between compound 3 and the adenine base of DNA and may interact with DNA through intercalation. These results indicated that compound 3 could damage the DNA double stranded form, specifically the adenine base and is not cytotoxic at same concentrations in ECV304 and mononuclear cells. Therefore, it has a selective antileukemic effect and drug potential.
We believe that the complexes may very likely be potential anticancer drugs due to their ability of binding to DNA and show a cytotoxic effect at very small concentrations in cell cultures.
Acknowledgements
This study was partly supported by TÜBİTAK (TÜBİTAK-SBAG Project number: 109S188) and was supported by the Research Fund of Istanbul University. A.E would like to express her gratitude to the Turkish Academy of Sciences (TUBA) as an Associate member for its partial support. We acknowledge the Faculty of Arts and Sciences, Ondokuz Mayıs University, Turkey, for the use of the STOE IPDS II diffractometer (purchased under grant No. F-279 of the University Research Fund).References
- D. Palanimuthu, S. Vijay Shinde, K. Somasundaram and A. G. Samuelson, J. Med. Chem., 2013, 56(3), 722–734 CrossRef CAS PubMed
.
- S. Sahni, D.-H. Bae, D. J. R. Lane, Z. Kovacevic, D. S. Kalinowski, P. J. Jansson and D. R. Richardson, J. Biol. Chem., 2014, 289, 9692–9709 CrossRef CAS PubMed
- N. T. Mossman, J. Immunol. Methods, 1983, 65, 55–63 CrossRef
- V. Mishra, S. N. Pandeya, C. Pannecouque, M. Witvrouw and E. De Clercq, Arch. Pharm., 2002, 335, 183–186 CrossRef CAS
- T. Varadinova, D. Kovala-Demertzi, M. Rupelieva, M. Demertzis and P. Genova, Acta Virol., 2002, 45, 87–94 Search PubMed
- A. S. Khan and M. Yusuf, J. Med. Chem., 2009, 44(5), 2270–2274 Search PubMed
- M. Karatepe and F. Karatas, Cell Biochem. Funct., 2006, 24, 547–554 CrossRef CAS PubMed
- T. Bal-Demirci, M. Şahin, M. Özyürek, E. Kondakçı and B. Ülküseven, Spectrochim. Acta, Part A, 2014, 126, 317–323, DOI:10.1016/j.saa.2014.02.039
- T. Bal-Demirci, M. Sahin, E. Kondakçı, M. Özyürek, B. Ülküseven and R. Apak, Spectrochim. Acta, Part A, 2015, 138, 866–872, DOI:10.1016/j.saa.2014.10.088
- R. Yanardag, T. Bal-Demirci, B. Ülküseven, S. Bolkent, S. Tunalı and Ş. Bolkent, Eur. J. Med. Chem., 2009, 44, 818–826 CrossRef CAS PubMed
- B. Ülküseven, T. Bal-Demirci, M. Akkurt, Ş. P. Yalçın and O. Büyükgüngör, Polyhedron, 2008, 27(18), 3646–3652 CrossRef PubMed
- Ş. Güveli, A. Koca, N. Özdemir, T. Bal Demirci and B. Ülküseven, New J. Chem., 2014, 38, 5582–5589 RSC
- T. Bal Demirci, Y. Köseoğlu, S. Güner and B. Ülküseven, Cent. Eur. J. Chem., 2006, 4(1), 149–159 Search PubMed
- A. Castiñeiras, N. Fernández-Hermida, R. Fernández-Rodríguez and I. García-Santos, Cryst. Growth Des., 2012, 12(3), 1432–1442 Search PubMed
- T. Bal-Demirci, Polyhedron, 2008, 27(1), 440–446 CrossRef CAS PubMed
- R. Pedrido, M. J. Romero, M. R. Bermejo, M. Martínez-Calvo, A. M. González-Noya and G. Zaragoza, Dalton Trans., 2009, 8329–8340 RSC
- B. Ülküseven, T. Bal and M. Şahin, Rev. Inorg. Chem., 2006, 26(4), 367–378 CrossRef
- T. Bal-Demirci, M. Akkurt, Ş. P. Yalçın and O. Büyükgüngör, Transition Met. Chem., 2010, 35, 95–102 CrossRef CAS PubMed
- S. Padhye, Z. Afrasiabi, E. Sinn, J. Fok, K. Mehta and N. Rath, Inorg. Chem., 2005, 44, 1154–1156 CrossRef CAS PubMed
- A. P. Rebolledo, M. Vieites, D. Gambino, O. E. Piro, E. E. Castellano, C. L. Zani, E. M. Souza-Fagundes, L. R. Teixeira, A. A. Batista and H. Beraldo, J. Inorg. Biochem., 2005, 99, 698–706 CrossRef CAS PubMed
- E. W. Ainscough, A. M. Brodie, W. A. Denny, G. J. Finlay and J. D. Ranford, J. Inorg. Biochem., 1998, 70(3–4), 175–185 CrossRef CAS
- I. H. Hall, C. B. Lackey, T. D. Kistler, R. W. Durham Jr, E. M. Jouad, M. Khan, X. D. Thanh, S. Djebbar-Sid, O. Benali-Baitich and G. M. Bouet, Pharmazie, 2000, 55, 937–941 CAS
- T. Bal Demirci, B. Atasever, Z. Solakoğlu, S. Erdem-Kuruca and B. Ülküseven, Eur. J. Med. Chem., 2007, 42, 161–167 CrossRef PubMed
- B. Atasever, B. Ülküseven, T. Bal-Demirci, S. Erdem-Kuruca and Z. Solakoğlu, Invest. New Drugs, 2010, 28, 421–432 CrossRef CAS PubMed
- J. M. Perez, V. Cerrillo, A. I. Matesanz, J. M. Millan, P. Navarro, C. Alonso and P. Souza, ChemBioChem, 2001, 2, 119–123 CrossRef CAS
- D. Kovala-Demertzi, M. A. Demertzis, E. Filiou, A. A. Pantazaki, P. N. Yadav, J. R. Miller, Y. Zheng and D. A. Kyriakidis, BioMetals, 2003, 16, 411–418 CrossRef CAS
- M. Baldini, M. Belicchi-Ferrari, F. Bisceglie, P. P. Dall'aglio, G. Pelosi, S. Pinelli and P. Tarasconi, Inorg. Chem., 2004, 43, 7170–7179 CrossRef CAS PubMed
- S. Capacchi, G. Pelosi and P. Tarasconi, J. Inorg. Biochem., 2005, 99, 1504–1513 CrossRef PubMed
- E. Palecek, Nature, 1960, 188, 656–657 CrossRef CAS PubMed
- M. Mascini, G. Bagni, M. L. Pietro, M. Ravera, S. Baracco and D. Osella, BioMetals, 2006, 19, 409–418 CrossRef CAS PubMed
- J. Wang, G. Rivas, X. Cai, H. Shiraishi, P. A. M. Farias, N. Dontha and D. Luo, Anal. Chim. Acta, 1996, 332, 139–144 CrossRef CAS
- A. Erdem and M. Ozsoz, Electroanalysis, 2002, 14, 965–974 CrossRef CAS
- S. C. B. Oliveira, O. Corduneanu and A. M. Oliveira-Brett, Bioelectrochemistry, 2008, 72, 53–58 CrossRef CAS PubMed
- F. Li, W. Chen, C. Tang and S. Zhang, Talanta, 2008, 77, 1–8 CrossRef CAS PubMed
- C. Kasper, H. Alborzinia, S. Can, I. Kitanovic, A. Meyer, Y. Geldmacher, M. Oleszak, I. Ott, S. Wolfl and W. S. Sheldrick, J. Inorg. Biochem., 2012, 106, 126–133 CrossRef CAS PubMed
- D. Yong, C. Liu, D. Yu and S. Dong, Talanta, 2011, 84, 7–12 CrossRef CAS PubMed
- G. Congur, A. Erdem and F. Mese, Bioelectrochemistry, 2015, 102, 21–28 CrossRef CAS PubMed
- A. Erdem and G. Congur, Int. J. Biol. Macromol., 2013, 61, 295–301 CrossRef CAS PubMed
- A. Erdem, M. Muti, P. Papakonstantinou, E. Canavar, H. Karadeniz, G. Congur and S. Sharma, Analyst, 2012, 137(9), 2129–2135 RSC
- F. Mese, G. Congur and A. Erdem, J. Electroanal. Chem., 2014, 719, 92–97 CrossRef CAS PubMed
- A. Erdem, G. Congur and E. Eksin, Sens. Actuators, B, 2013, 188, 1089–1095 CrossRef CAS PubMed
- A. Erdem, G. Duruksu, G. Congur and E. Karaoz, Analyst, 2013, 138, 5424–5430 RSC
- E. Eksin and A. Erdem, RSC Adv., 2015, 5, 4774–4779 RSC
- A. Erdem, E. Eksin and M. Muti, Colloids Surf., B, 2014, 115, 205–211 CrossRef CAS PubMed
- A. Erdem, G. Congur and F. Mese, Electroanalysis, 2014, 26, 1–12 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS
-
Stoe & Cie, X-AREA Version 1.18 and X-RED32 Version 1.04, Stoe & Cie, Darmstadt, Germany, 2002 Search PubMed
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed
- L. J. Farrugia, J. Appl. Chem., 1997, 30, 565 CAS
- N. T. Mossman, J. Immunol. Methods, 1983, 65, 55–63 CrossRef
- K. Burton, Biochem. J., 1956, 62, 315–323 CAS
- E. I. Taşkın, K. Akgun-Dar, A. Kapucu, A. Yagci, M. Caner and H. Dogruman, Rev. Med. Vet., 2008, 159, 123–129 Search PubMed
- M. J. R. P. Queiroz, E. M. S. Castanheira, M. S. D. Carvalho, A. S. A. Paula, M. T. Ferreira, H. Karadeniz and A. Erdem, Tetrahedron, 2008, 64, 382–391 CrossRef CAS PubMed
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC
- A. Bhunia, Y. Lan, V. Mereacre, M. T. Gamer, A. K. Powell and P. W. Roesky, Inorg. Chem., 2011, 50, 12697–12704 CrossRef CAS PubMed
- Y. Yahsi, H. Kara, L. Sorace and O. Buyukgungor, Inorg. Chim. Acta, 2011, 366, 191–197 CrossRef CAS PubMed
- C. Mukherjee, A. Stammler, H. Bögge and T. Glaser, Chem. – Eur. J., 2010, 16, 10137–10149 CrossRef CAS PubMed
- T. Kurahashi, K. Oda, M. Sugimoto, T. Ogura and H. Fujii, Inorg. Chem., 2006, 45, 7709–7721 CrossRef CAS PubMed
- K. Kiss, T. Holczbauer, M. Czugler, P. Sohár, A. Bodor and A. Csámpai, J. Organomet. Chem., 2012, 706–707, 46–51 CrossRef CAS PubMed
- H. Fujii and Y. Funahashi, Angew. Chem., Int. Ed., 2002, 114, 3790–3793 CrossRef
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS
- E. Canavar, F. Kuralay and A. Erdem, Electroanalysis, 2011, 23, 2343–2349 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1001379. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00594a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |