DOI:
10.1039/C5NJ00568J
(Paper)
New J. Chem., 2015,
39, 7617-7625
Investigation of the “bent sandwich-like” divalent lanthanide hydro-tris(pyrazolyl)borates Ln(TpiPr2)2 (Ln = Sm, Eu, Tm, Yb)†‡
Received
(in Montpellier, France)
6th March 2015
, Accepted 5th May 2015
First published on 6th May 2015
Abstract
The series of homoleptic lanthanide(II) “bent sandwich-like” hydro-tris(pyrazolyl)borate complexes Ln(TpiPr2)2 (Ln = Sm (1), Eu (2), Tm (3), Yb (4); TpiPr2 = hydro-tris(3,5-diisopropylpyrazolyl)borate) has been completed by the synthesis of the hitherto unknown europium and ytterbium derivatives 2 and 4. Both compounds were prepared in high yields by treatment of LnI2(THF)2 (Ln = Eu, Yb) with 2 equiv. of KTpiPr2 in a THF solution. Although the molecules are sterically highly congested, an X-ray diffraction study of bright red 4 revealed a similar bent B–Yb–B arrangement (151.1° and 153.9°, two independent molecules) as in the previously investigated Sm(II) and Tm(II) complexes 1 and 3. An initial reactivity study showed a very different behavior with acetonitrile. While 2 and 4 proved to be unreactive toward acetonitrile, the more strongly reducing Sm(II) complex 1 yielded two new products. The major product was the dark green-black acetonitrile solvate SmII(TpiPr2)2·CH3CN (5), while the second product, the colorless (TpiPr2)SmIII(3,5-iPr2pz)2(NCCH3) (6) with two 3,5-diisopropyl-pyrazolate ligands, resulted from oxidation of samarium to the trivalent state and degradation of a TpiPr2 ligand. Disappointingly, from the most reducing Tm(II) complex 3 only the ligand fragmentation product pyrazabole, [HB(3,5-iPr2pz)2]2 (7), could be isolated and the fate of the Tm containing by-product(s) remains unknown. The new compounds 4–6 were structurally authenticated through single-crystal X-ray diffraction. The europium compound 2 shows an extremely bright yellow emission in solution, which can be observed also at daylight excitation, as well as in the solid state. The high intensity is even remarkable when compared to other Eu(II) containing materials. The photoluminescence was investigated with the conclusion that the rigidity of this complex is responsible for these outstanding luminescence properties.
1. Introduction
The discovery of the divalent lanthanide sandwich complexes Ln(C5Me5)2 (Ln = Sm, Eu, Yb; C5Me5 = η5-pentamethylcyclopentadienyl) ca. 30 years ago sparked a firework of unprecedented reactivity and structures in organolanthanide chemistry.1,2 The exceptionally high reactivity of decamethylsamarocene, Sm(C5Me5)2, even allowed the isolation of the first dinitrogen complex of an f-element, (μ-N2)[Sm(C5Me5)2]2,3 and still today novel reactions of decamethylsamarocene are being uncovered.4 A fascinating structural feature of the unsolvated lanthanide sandwich complexes Ln(C5Me5)2 (Ln = Sm, Eu, Yb) is their bent metallocene structure in the solid state. This opens up the coordination sphere of the central lanthanide(II) ions and accounts for the high reactivity of these compounds. Various theoretical and spectroscopic studies have been carried out to fully understand the nature of this unexpected deviation from the normal linear sandwich structure (Scheme 1(a)). It is now generally accepted, based on computational studies, that the unusual bent sandwich structure of Ln(C5Me5)2 (Ln = Sm, Eu, Yb) is the result of attractive dispersion/van der Waals interactions.5
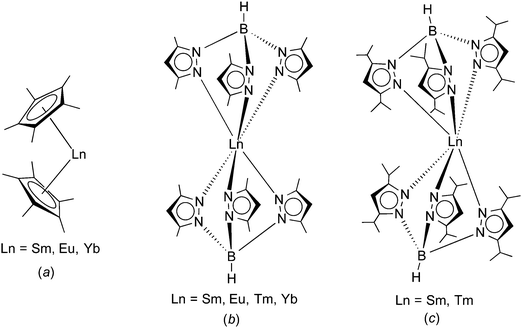 |
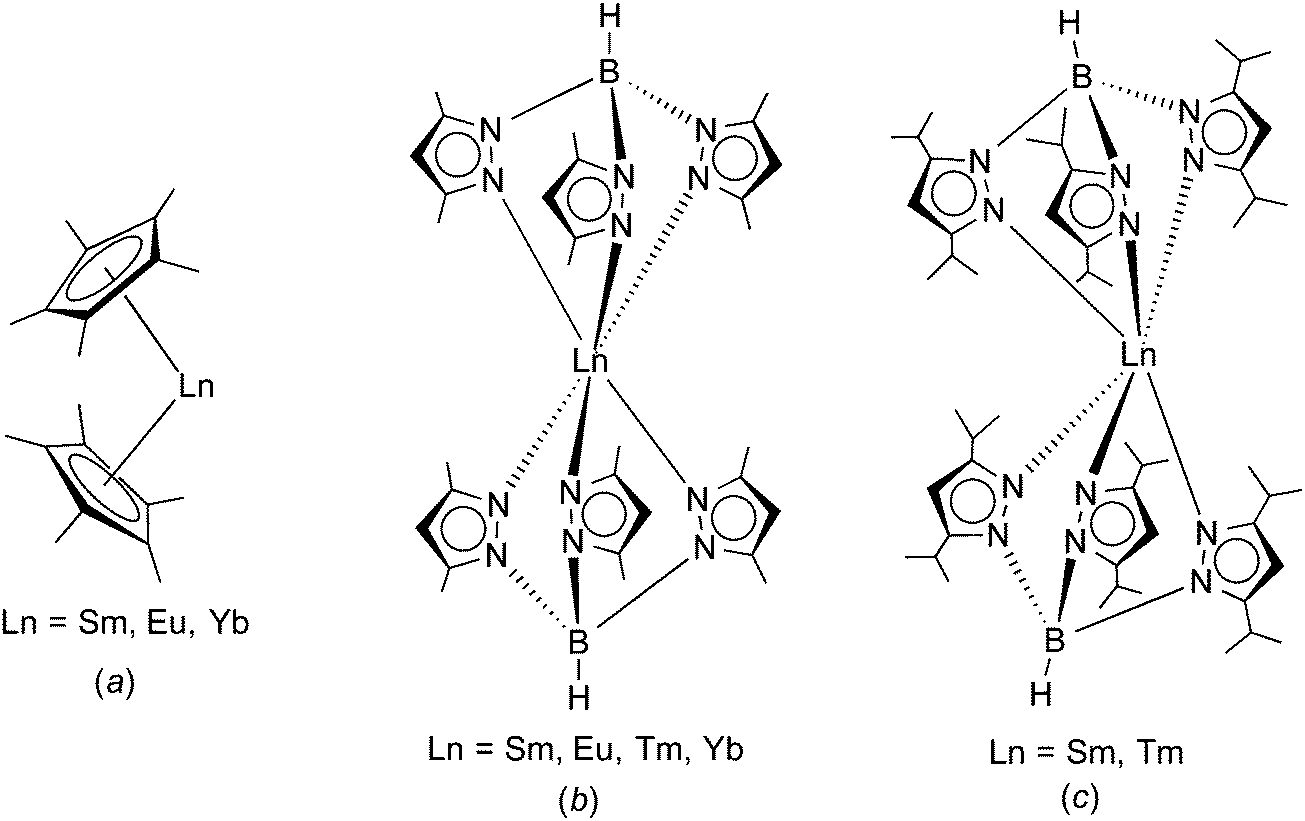
| | Scheme 1 Comparison of the molecular structures of (a) Ln(C5Me5)2 (Ln = Sm, Eu, Yb),1,2 (b) Ln(TpMe2)2 (Ln = Sm, Eu, Tm, Yb),8–11 and (c) Ln(TpiPr2)2 (Ln = Sm (1), Tm(3)).14 | |
Trofimenko's hydro-tris(pyrazolyl)borate ligands (“scorpionates”) have proven to be useful alternatives to the omnipresent cyclopentadienyl ligands.6,7 Like the cyclopentadienyls, these tridentate, monoanionic ligands can also be greatly varied in their steric bulk by changing the substituents in the 3- and 5-positions of the pyrazolyl rings. According to Trofimenko's nomenclature, the abbreviation Tp stands for the ring-unsubstituted hydro-tris(pyrazolyl)borate, whereas e.g. TpMe2 denotes the sterically more demanding hydro-tris(3,5-dimethylpyrazolyl)borate. The homoleptic divalent lanthanide complexes Ln(TpMe2)2 (Ln = Sm, Eu, Yb) have been found to adopt a highly symmetrical, trigonal antiprismatic molecular structure comprising a linear B⋯Ln⋯B arrangement (Scheme 1(b)).8–11 Apparently, this “sandwich-like” structure of Ln(TpMe2)2 is the result of the much larger cone angle of TpMe2 (239°) as compared to the C5Me5 ligand with 142°.12 Most recently, these studies have been extended to the even larger hydro-tris(3,5-diisopropylpyrazolyl)borate ligand (TpiPr2).13 It was possible to isolate homoleptic complexes of this ligand with divalent samarium and thulium.14 Rather surprisingly, crystal structure determinations revealed a “bent sandwich-like” molecular structure like Ln(C5Me5)2 as shown in Scheme 1(c). Computational studies indicated that steric repulsion between the isopropyl groups forces the TpiPr2 ligands apart and permits the formation of unusual interligand C–H⋯N hydrogen-bonding interactions that help stabilizing the structure.14
Among the “classical” divalent lanthanide ions (Sm2+, Eu2+, Yb2+) only the homoleptic samarium(II) TpiPr2 complex Sm(TpiPr2)2 (1) has previously been prepared and fully characterized.14 In this article we report the synthesis and characterization of the corresponding divalent europium and ytterbium species Eu(TpiPr2)2 (2) and Yb(TpiPr2)2 (3) as well as the behavior of the full series of Ln(TpiPr2)2 complexes toward acetonitrile, and the first results on the photoluminescence of compound 2.
2. Results and discussion
2.1 Synthesis and reactivity
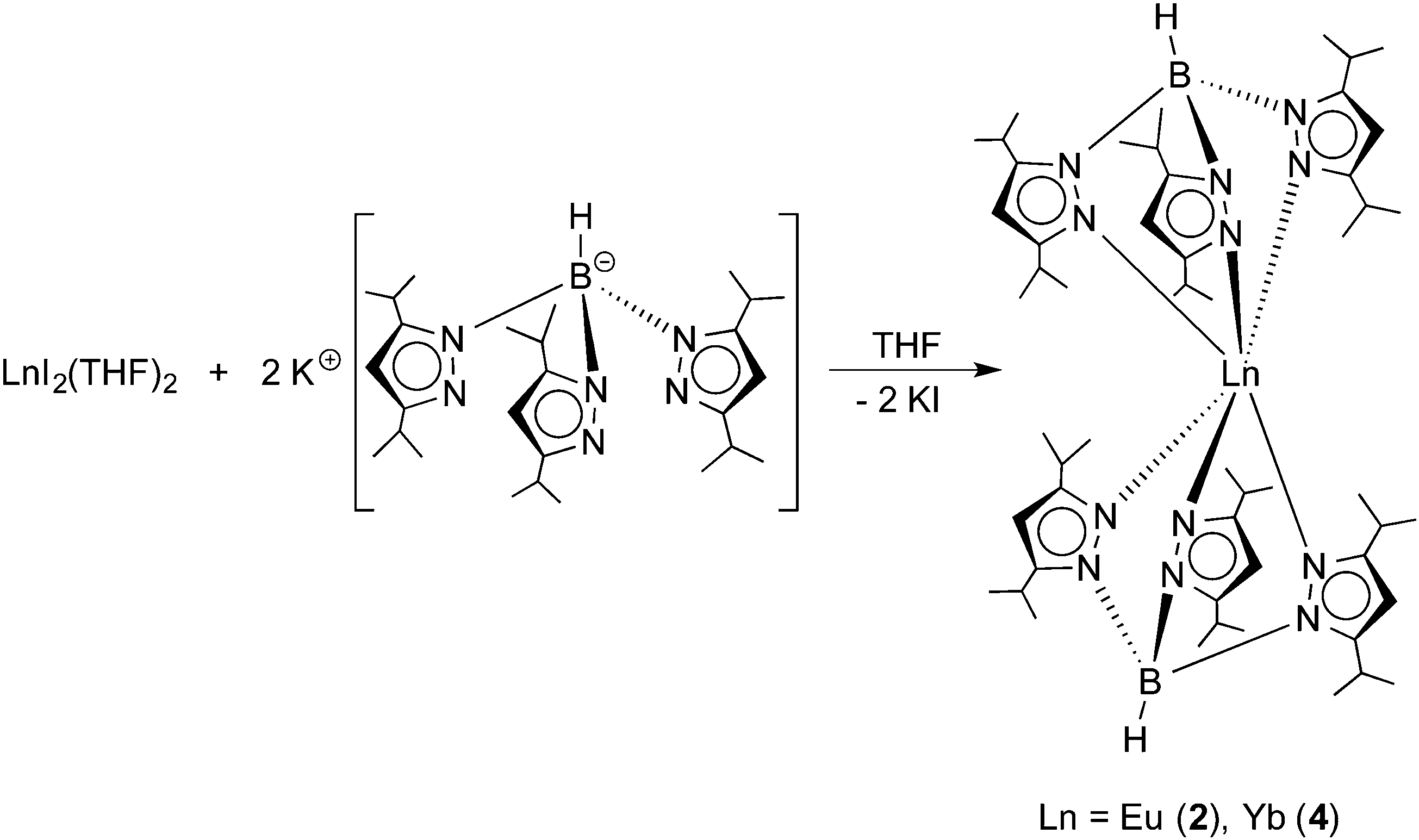
The title compounds were prepared following the synthetic route outlined in Scheme 2. Similar to the recently reported preparation of the samarium(II) and thulium(II) derivatives 1 and 3,14 reactions of EuI2(THF)2 and YbI2(THF)2 with 2 equiv. of KTpiPr2 were carried out in THF solutions at room temperature.
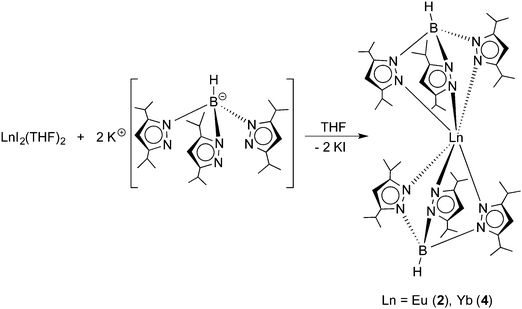 |
| | Scheme 2 Synthesis of Ln(TpiPr2)2 (Ln = Eu (2), Yb (4)). | |
Both reactions were accompanied by striking color changes to “neon-yellow” (Eu) or bright red (Yb), respectively, and formation of a white precipitate (KI). After removal of the potassium iodide by-product through filtration, the products could be readily extracted with n-pentane. Recrystallization from very concentrated solutions in n-pentane at −20 °C for 24 h afforded yellow Eu(TpiPr2)2 (2) and bright red Yb(TpiPr2)2 (4) in high yields (2: 83%, 4: 77%). Both new compounds were fully characterized by the usual combination of spectroscopic data and elemental analyses. While the 1H NMR spectrum of diamagnetic 4 showed the expected number of resonances for the TpiPr2 ligands, meaningful 1H and 13C NMR data for paramagnetic 2 could not be obtained, as noted also for Eu(C5Me5)2 and its derivatives,2e,h and Eu(TpMe2,Et)2.10 In both cases, the 11B NMR spectra showed a single broad resonance (2: δ = −7 ppm (very broad), 4: δ = −6.2 ppm). Moreover, Yb(TpiPr2)2 (4) was characterized by its 171Yb NMR spectrum. High-resolution 171Yb NMR spectroscopy is well established as a valuable tool for characterizing divalent (diamagnetic) ytterbium complexes in solution and in the solid state.15,16171Yb resonances have been reported to encompass a chemical shift dispersion of some 3000 ppm (from ca. δ +2500 to −500 ppm).15c The 171Yb spectrum of 4 comprises a singlet at δ = 619.1 ppm. An almost identical value (δ = 614 ppm) has previously been reported for the ytterbium(II) bis(trimethylsilyl)amide complex Yb[N(SiMe3)2]2(OEt2)2.15a
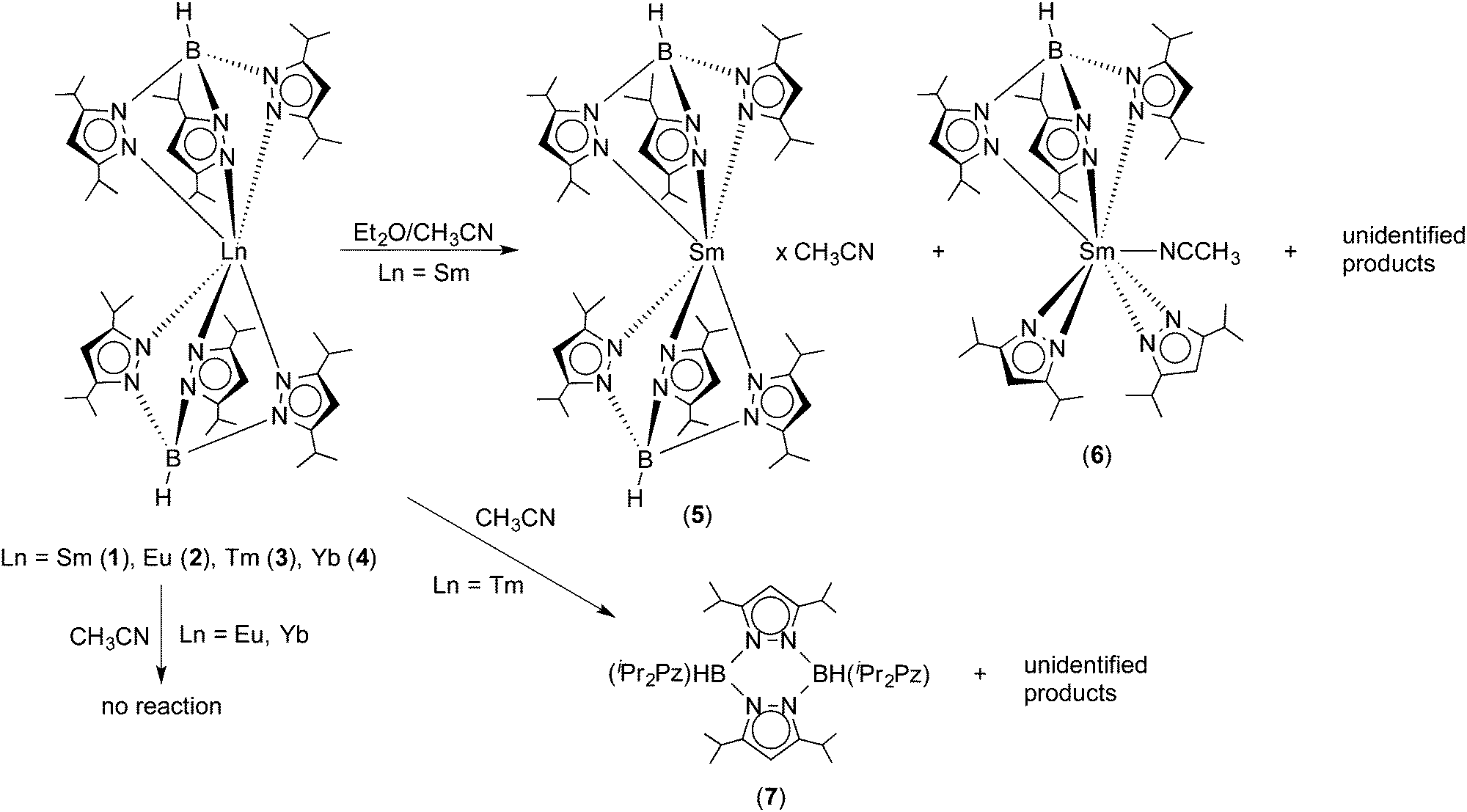
The recent structural characterization of 1 and its thulium congener 3 had already shown that these “bent sandwich-like” molecules are sterically highly congested. Thus for an initial reactivity study, the reagent acetonitrile was chosen in order to find out if a small, rod-like molecule such as CH3CN could enter the coordination sphere and bind to the central Ln2+ ions in 1–4. Surprisingly, no reaction with acetonitrile was observed for the europium and ytterbium complexes 2 and 4 even upon slight warming. Both complexes produced clear solutions in dry acetonitrile, from which they could be recovered unchanged by evaporation or cooling. In fact, acetonitrile appears to be a suitable solvent for recrystallizing bulk samples of 2 and 4. This is not the case for the Sm(II) and Tm(II) complexes 1 and 3. Unexpectedly, and curiously the Sm(II) complex is virtually insoluble in acetonitrile. Addition of acetonitrile to solid 1 produces an almost colorless supernatant and a very dark green, almost black solid. The latter was shown to be unchanged Sm(TpiPr2)2 by 1H NMR spectroscopy. To study the behavior of Sm(TpiPr2)2 toward acetonitrile, acetonitrile was added to a dark green solution of 1 in diethyl ether. Concentration of the solution by slow evaporation at RT in the dry-box resulted in the formation of two types of crystals, dark green and colorless; the former was shown to be SmII(TpiPr2)2·CH3CN (5), with a solvate molecule of CH3CN in the lattice, while the latter proved to be the partially ligand fragmented Sm(III) complex, (TpiPr2)SmIII(3,5-iPr2pz)2(NCCH3) (6), with a coordinated NCCH3 ligand. The most reducing Tm(II) complex 3 dissolved in acetonitrile and gave a dark, plum-red solution which slowly bleached with time, indicating oxidation of Tm(II) to Tm(III). Multiple attempts to grow crystals from various solvent mixtures only resulted in the formation of colorless blocks which were shown, by X-ray diffraction, to be the pyrazabole derivative [HB(3,5-iPr2pz)]2 (7). No thulium-containing product could be isolated. Scheme 3 summarizes the results of this initial reactivity study of 1–4 toward acetonitrile.
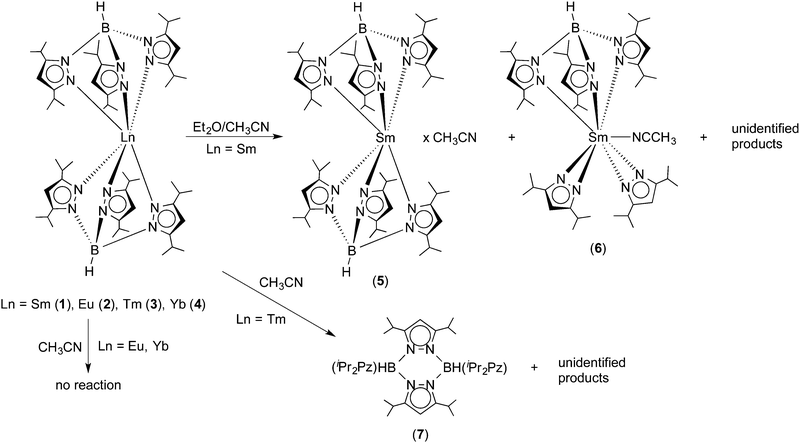 |
| | Scheme 3 Reactivity of Ln(TpiPr2)2 (Ln = Sm (1), Eu (2), Tm (3), Yb (4)) toward acetonitrile. | |
2.2 X-ray crystallography
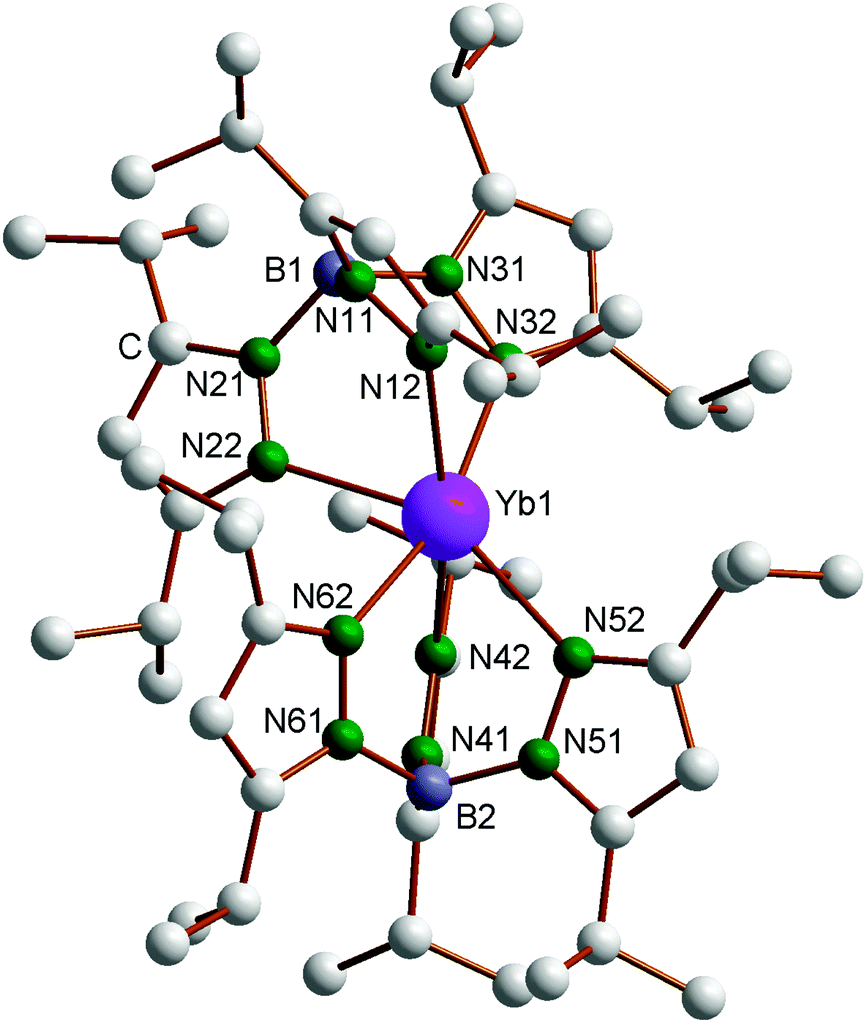
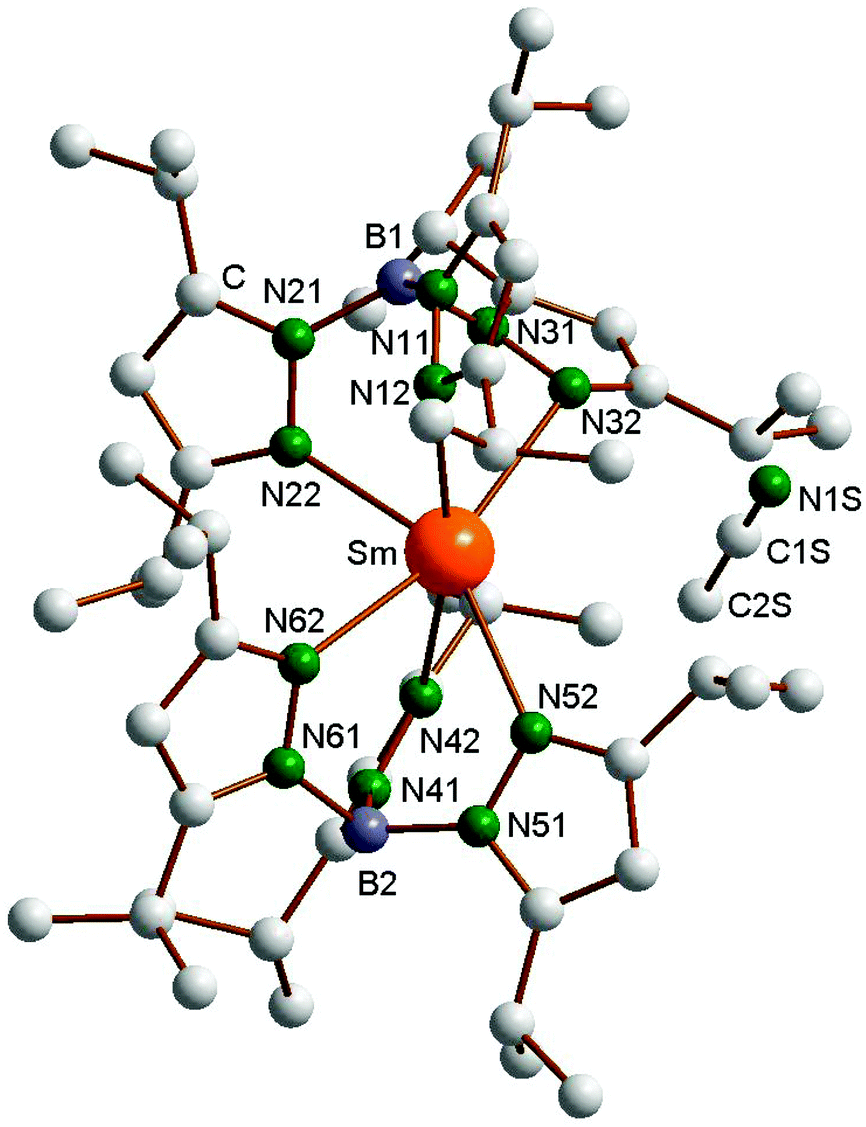
The new compounds 4–6 were structurally authenticated through single-crystal X-ray diffraction. Bright red X-ray quality single-crystals of 4 were obtained by cooling a very concentrated solution in n-pentane to −20 °C, whereas single crystals of both 5 (green) and 6 (colorless) were obtained from the reaction of 1 with acetonitrile in diethyl ether according to Scheme 3. The single-crystals of 4 were found to contain one molecule of n-pentane per formula unit. Crystallographic data of 4–6 are listed in Table 1. The molecular structure of the Yb complex, with numbering scheme, is shown in Fig. 1. Just like the Sm and Tm compounds 1 and 3, the ytterbium(II) complex Yb(TpiPr2)2 also exhibits the “bent sandwich-like” geometry, and indeed the compound is isomorphous with the Tm analogue and contains two independent molecules per asymmetric unit. The B–Yb–N angles in the two independent molecules are 151.1° and 153.9°, respectively. This can be favorably compared to the B–Ln–B angles of 150.1° in the samarium(II) analogue 1 and 152.2° in Tm(TpiPr2)2 (3).14 As expected from the nearly identically sized Yb(II) and Tm(II) ions,17 the Ln–N distances in 3 and 4 are nearly identical and the bond angles and torsional angles are similar as well (cf. Table S1 in the ESI‡).
Table 1 Crystallographic data for 4–6
| |
4
|
5
|
6
|
| Empirical formula |
C59H104B2N12Yb |
C56H95B2N13Sm |
C47H79BN11Sm |
|
a (Å) |
13.44670 (10) |
12.9942 (10) |
13.9188 (9) |
|
b (Å) |
20.6731 (2) |
13.6332 (10) |
16.8238 (11) |
|
c (Å) |
25.1841 (2) |
20.9744 (16) |
21.9614 (15) |
|
α (°) |
83.2050 (10) |
104.2666 (8) |
90 |
|
β (°) |
74.7430 (10) |
94.8690 (8) |
90.3416 (10) |
|
γ (°) |
77.6100 (10) |
115.6405 (7) |
90 |
|
V (Å3) |
6582.53 (10) |
3167.5 (4) |
5142.5 (6) |
|
Z
|
4 |
2 |
4 |
| Formula weight |
1176.20 |
1122.41 |
959.37 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/n |
|
T (°C) |
−173 |
−100 |
−80 |
|
λ (Å) |
0.71073 |
0.71073 |
0.71073 |
|
D
calcd (g cm−3) |
1.187 |
1.177 |
1.239 |
|
μ (mm−1) |
1.464 |
0.971 |
1.183 |
| Data/restraints/parameters |
39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 188/0/1345 188/0/1345 |
14330/0/658 |
12276/0/542 |
| Goodness-of-fit on F2 |
1.028 |
1.051 |
1.024 |
|
R (Fo or Fo2) |
0.0363 |
0.0271 |
0.0287 |
|
R
w (Fo or Fo2) |
0.0689 |
0.0678 |
0.0715 |
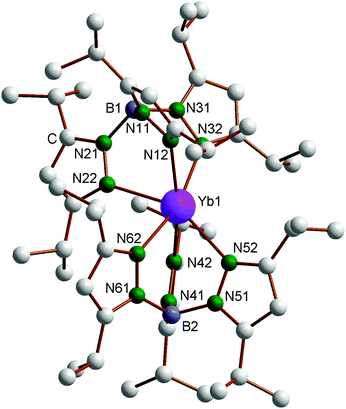 |
| | Fig. 1 Molecular structure of Yb(TpiPr2)2 (4). Selected bond lengths (Å) and angles (°), molecule 1: Yb1–N12 2.528(2), Yb1–N22 2.530(2), Yb1–N32 2.550(2), Yb1–N42 2.487(2), Yb1–N52 2.589(2), Yb1–N62 2.478(2), B1–Yb1–B2 151.1. Molecule 2: Yb2–N12′ 2.525(2), Yb2–N22′ 2.522(2), Yb2–N32′ 2.528(2), Yb2–N42′ 2.498(2), Yb2–N52′ 2.540(2), Yb2–N62′ 2.502(2), B3–Yb2–B4 153.9. | |
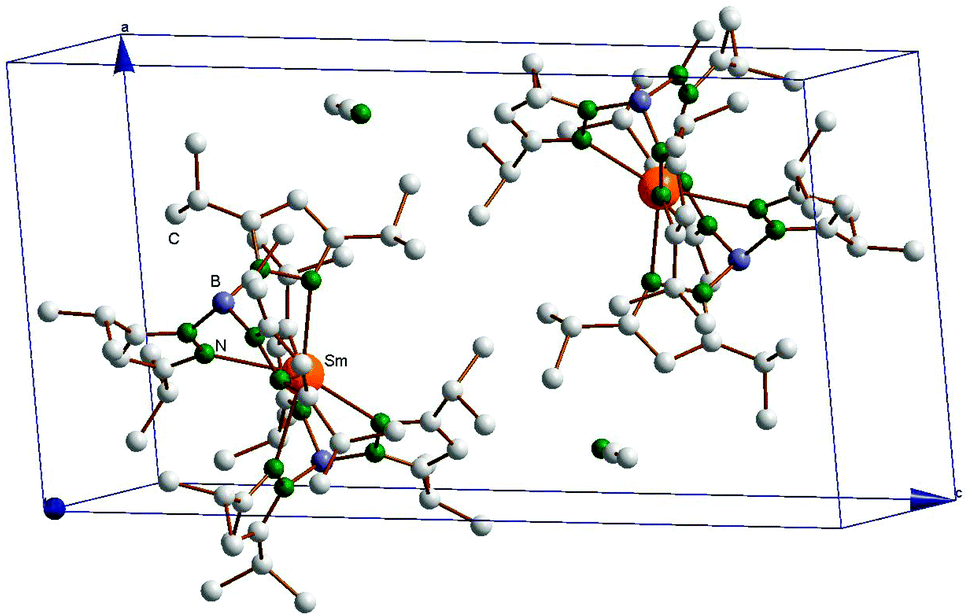
The structure of the Sm(II) compound SmII(TpiPr2)2·CH3CN (5), obtained by crystallization from CH3CN/Et2O, is shown in Fig. 2, and Fig. 3 shows the packing diagram. The lattice acetonitrile is just a solvate as the distance between Sm and N1S is over 6 Å, thus there is no bonding contact between Sm and NCCH3 molecule. As opposed to the crystals obtained from pentane, in this case there is only one molecule per asymmetric unit. Nevertheless, the geometry is still “bent sandwich-like” and the B1–Sm–B2 angle of 151.19(5)° is very similar to the 150.1° in the previously reported structure of SmII(TpiPr2)2,14 demonstrating once again that the bent geometry is an inherent molecular feature of all divalent Ln(TpiPr2)2 complexes and is not due to crystal packing effects. However, the latter may have some subtle effect since the Sm–N32 distance of 2.735(2) Å is longer that the 2.655(6) Å seen before and the torsion angle of this pyrazolyl moiety is also large, Sm–N32–N31–B1 = 62.4(2)°, as opposed to the 20° average observed before.14
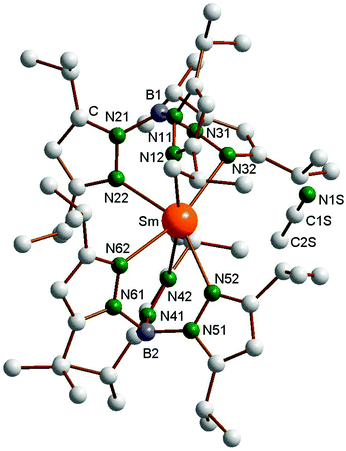 |
| | Fig. 2 Molecular structure of SmII(TpiPr2)2·CH3CN (5). Selected bond lengths (Å) and angle (°): Sm–N12 2.642(2), Sm–N22 2.638(2), Sm–N32 2.735(2), Sm–N42 2.631(2), Sm–N52 2.634(2), Sm–N62 2.661(2), B1–Sm–B2 151.2. | |
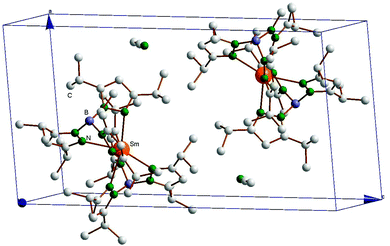 |
| | Fig. 3 Crystal packing diagram of SmII(TpiPr2)2·CH3CN (5). | |
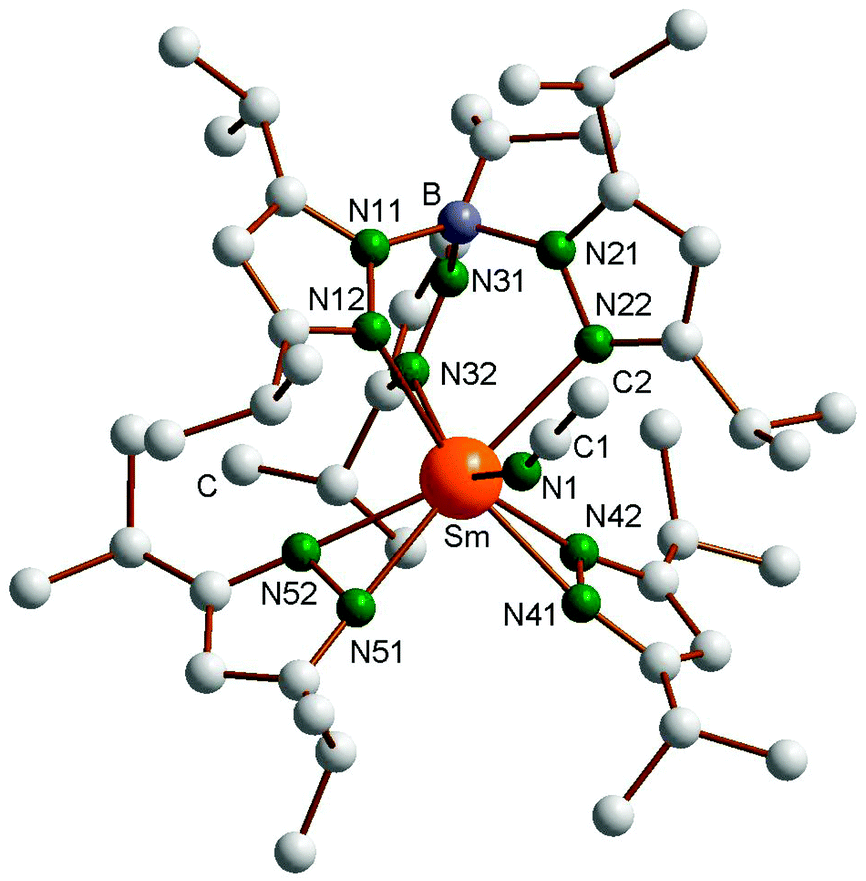
The molecular structure of the oxidized product (TpiPr2)Sm(3,5-iPr2Pz)2(NCCH3) (6) is shown in Fig. 4, with important bond distances and angles also listed in the figure caption. The coordination sphere of the Sm(III) center is defined by a classical κ3-TpiPr2 ligand, two almost symmetrically bonded κ2-pyrazolides and N1-bound acetonitrile. The coordination geometry can be roughly described as distorted octahedral, with N12, N22 and mid-points of N41N42 and N51N52 occupying the equatorial and N32 and N1 the axial positions (N32–Sm–N1 = 143.20(6)°). As expected, the Sm–N distances to the anionic κ2-pyrazolides (2.40 Å ave) are shorter than those to the κ3-TpiPr2 ligand (2.560 Å ave), which in turn is shorter than the Sm–NCCH3 distance of 2.601 Å. The Sm–N(TpiPr2) distances are shorter than those in Sm(TpiPr2)2 (1), reflecting the smaller size of Sm(III) compared to Sm(II) and also the more congested nature of the latter complex. That sterics still have an influence on the arrangement of the ligands in complex 6 is shown by the less than 180° of the Sm–N1–C1 angle (159.2(2)°), the bending being away from the iPr substituent of the two κ2-pyrazolide ligands.
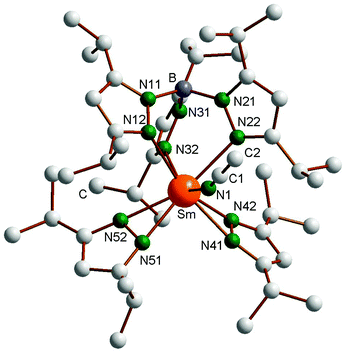 |
| | Fig. 4 Molecular structure of (TpiPr2)SmIII(3,5-iPr2pz)2(NCCH3) (6). Selected bond lengths (Å) and angles (°): Sm–N1 2.601(2), Sm–N12 2.574(2), Sm–N22 2.571(2), Sm–N32 2.536(2), Sm–N41 2.379(2), Sm–N42 2.420(2), Sm–N51 2.384(2), Sm–N52 2.409(2), N1–Sm–N32 143.20(6), Sm–N1–C1 159.2(2). | |
2.3 Luminescence study of Eu(TpiPr2)2 (2)
In general, the luminescence behaviour of divalent lanthanides is very different compared to that of the trivalent ones due to two main reasons. On the one hand, the position of the excited 4fn−15d1 states relative to the 4fn ground state is strongly influenced by the environment and, thus, variable over a wide spectral range. On the other hand, the respective 4fn ↔ 4fn−15d1 transitions are parity allowed leading to an intense emission in the most cases.18 Due to these advantages the most stable divalent ion, Eu2+, is mostly used in modern materials for applications, like LED phosphors, displays and medical markers.19 While the luminescence properties of Eu2+ ions doped in ionic compounds, especially the structure–luminescence-relationship, is well investigated,20 such investigations of molecular complexes are rather scarce. A notable example is the strong luminescence exhibited by Eu(CpBIG)2 (CpBIG = C5(C6H4nBu-4)5).21 In this contribution we present first results on the photoluminescence behavior of compound 2. It was found that Eu(TpiPr2)2 (2) shows extremely bright yellowish-green luminescence upon UV irradiation at room temperature in the solid state as well as in n-pentane solution, which can be also observed by daylight-excitation (Fig. 5). Fig. 6 shows the luminescence of solid 2 under UV light.
 |
| | Fig. 5 Bright yellow luminescence of Eu(TpiPr2)2 (2) in n-pentane solution excited by daylight. | |
 |
| | Fig. 6 Luminescence of solid Eu(TpiPr2)2 (2) under UV light. | |
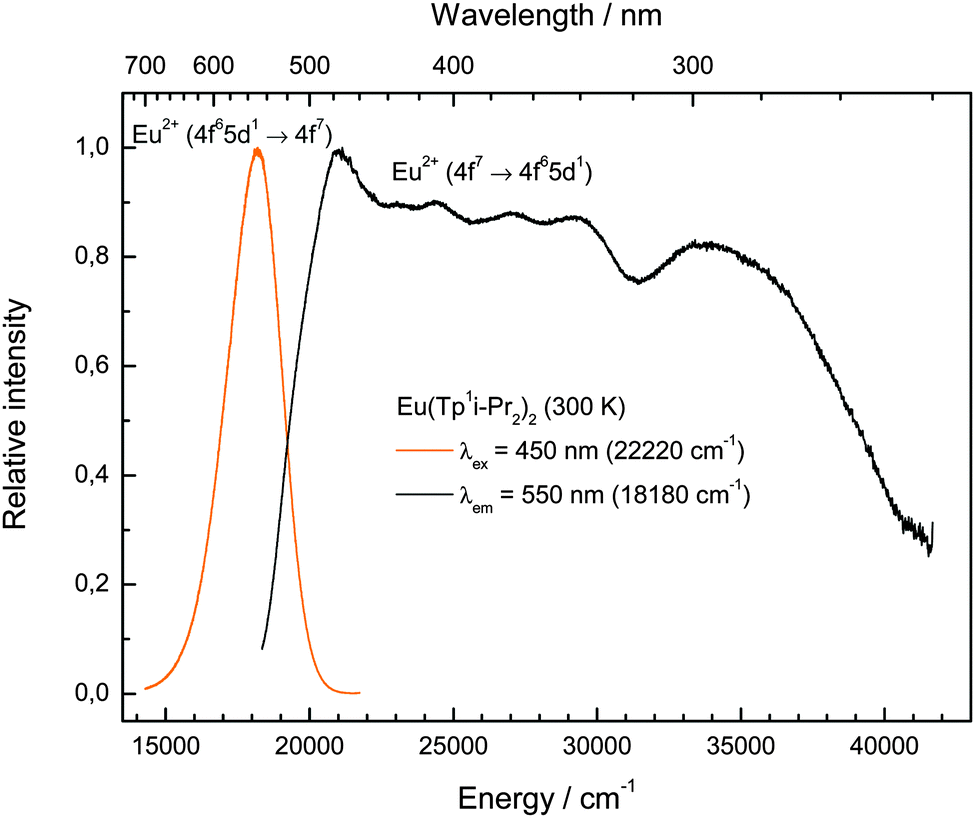
The photoluminescence emission and excitation spectra of this compound are depicted in Fig. 7. The shape and position are typical for Eu(II) photoluminescence, so that it is obvious that Eu2+ is the only emitting species. The broad (FWHM = 2245 cm−1) slightly asymmetric emission band peaking at 552 nm (18120 cm−1) can be assigned to the parity-allowed 4f65d1 → 4f7 transition of Eu(II). Its position is in very good agreement with the emissions reported for EuTp2,22 and Eu(II)-activated nitridosilicates, in which Eu(II) is also coordinated by N-based ligands in its first coordination sphere.23 A ligand-to-metal charge transfer (LMCT) induced luminescence as suggested for EuTp222 is excluded due to the high excitation wavelength of 450 nm (22220 cm−1) used for the detection of the emission spectrum of Eu(TpiPr2)2 (2). The π → π* transitions of the pyrazole units leading to a LMCT are typically located in the range of 220 nm and thus beyond the range of measureable wavelengths of the used spectrometer.24
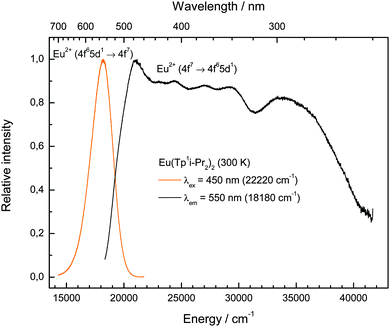 |
| | Fig. 7 Room temperature emission (left, yellow line, λex = 450 nm) and excitation (right, black line, λem = 550 nm) spectra of Eu(TpiPr2)2 (2). | |
The photoluminescence excitation spectrum reveals the presence of a raw fine structure that is characteristic for the 7FJ levels arising from the 4f6 core of the excited 4f65d1 configuration assuming a weak Coulomb interaction between the 4f and 5d electrons and rarely observed.25 This feature is another evidence for the presence of Eu(II) in the compound and justifies the assignment of the respective emissive transition. Moreover, the emission does not change with different excitation energies (320–480 nm), which is another evidence that the whole excitation band is originated by Eu(II). From the photoluminescence spectra of compound (2), the Stokes shift was estimated with 2830 cm−1. Both the Stokes shift and the FWHM of the emission band are relatively low for a molecular compound and close to values known for Eu(II)-activated ionic compounds.19 This can be explained by the rigidity of the very bulky tripodal TpiPr2 ligands that do not allow a large change in metal–ligand distances upon excitation of the complex. An indication that this argument is correct is the even smaller Stokes Shift of Eu(CpBIG)2 (2140 cm−1),21 where a large degree of rigidity can be assumed too.
3. Conclusions
In summarizing the work reported here, the series of homoleptic lanthanide(II) hydro-tris(pyrazolyl)borate complexes Ln(TpiPr2)2 (Ln = Sm (1), Eu (2), Tm (3), Yb (4); TpiPr2 = hydro-tris(3,5-diisopropylpyrazolyl)borate) has been completed by the high-yield synthesis of the hitherto unknown europium and ytterbium derivatives 2 and 4. A single crystal X-ray diffraction study of the ytterbium(II) derivative 4 revealed the same “bent sandwich-like” structure as was previously found for the Sm and Tm compounds 1 and 3. This finding confirmed that the bent geometry is an inherent structural feature of the Ln(TpiPr2)2 complexes. An initial reactivity study toward acetonitrile revealed a significant increase in reactivity in the sequence Eu ≈ Yb < Sm < Tm. The Eu (2) and Yb (4) compounds did not react with acetonitrile even upon heating. The Sm(II) complex 1 afforded the dark green acetonitrile solvate SmII(TpiPr2)2·CH3CN (5) in addition to the partially ligand fragmented Sm(III) complex, (TpiPr2)SmIII(3,5-iPr2pz)2(NCCH3) (6). The most reducing thulium(II) complex 3 only yielded ligand fragmented pyrazabole and unidentified Tm(III) species. Despite the opening of the coordination sphere in the “bent sandwich-like” Ln(TpiPr2)2 complexes, apparently not even rod-like donor ligands such as CH3CN are able to enter the coordination sphere of the central Ln2+ ions. Together with the previous studies on the “linear” homoleptic Ln(II) complexes Ln(TpMe2)2 these results clearly demonstrate that there is an intricate balance between stability and reactivity of such homoleptic lanthanide(II) tris(pyrazolyl)borate complexes. While the smaller TpMe2 provided versatile reactivity to the Sm(TpMe2)2 complex,8–11 it could only produce a thermally very sensitive Tm(TpMe2)2.14 In contrast, the very bulky TpiPr2 ligand stabilizes Tm(II), but hinders reactivity. Photoluminescence studies on Eu(TpiPr2)2 at room temperature revealed an exceptionally intense yellow emission at 552 nm (18120 cm−1, FWHM = 2245 cm−1, Stokes shift = 2830 cm−1) under excitation at 450 nm that can be assigned to a parity-allowed 4f65d1 → 4f7 transition of Eu(II) in the complex, supporting the sole presence of the divalent lanthanide. An intense emission is also observable at sunlight excitation. The luminescence characteristics are comparable to the values known from N-coordinated Eu(II) in ionic compounds, and indicates that the TpiPr2 ligands provide a rigid coordination environment to the Eu(II) center.
4. Experimental section
4.1 General procedures
All operations were performed with rigorous exclusion of air and water in oven-dried or flame-dried glassware under an inert atmosphere of dry argon, employing standard Schlenk, high-vacuum and glovebox techniques (MBraun MBLab; <1 ppm O2, <1 ppm H2O or Vacuum Atmosphere, model HE-553-2). THF, diethyl ether, and n-pentane were dried over sodium/benzophenone and freshly distilled under nitrogen atmosphere prior to use. Acetonitrile was dried over calcium hydride. All glassware was oven-dried at 120 °C for at least 24 h, assembled while hot and cooled under high vacuum prior to use. THF solvates of the three lanthanide diiodides, LnI2(THF)2, were prepared from the rare-earth metal powders and 1,2-diiodoethane in THF according to a well-established method by Kagan.26 The starting material KTpiPr2 was obtained through a melt reaction between KBH4 and 3 equiv. of 3,5-diisopropylpyrazole at 260 °C according to the method published by Kitajima et al.13 The NMR spectra were recorded in C6D6 or THF-d8 solutions on a Bruker DPX 600 (1H: 600.1 MHz; 13C: 150.9 MHz) or a Bruker AVANCE III 400 MHz (5 mm BB, 1H: 400.1 MHz; 13C: 100.6 MHz), 1H and 13C shifts are referenced to internal solvent resonances and reported in parts per million relative to TMS. IR (KBr) spectra were measured using a Perkin-Elmer FT-IR 2000 spectrometer. Mass spectra (EI, 70 eV) were run on a MAT 95 apparatus. Microanalyses of the compounds were performed using a Leco CHNS 923 apparatus. Photoluminescence measurements were performed at room temperature on a Fluorolog3 spectrofluorometer FL3-22 from Horiba JobinYvon equipped with double Czerny–Turner monochromators and a 450 W Xe lamp. The emission spectrum was corrected for the photomultiplier sensitivity and the excitation spectrum for the intensity of the excitation source. Measurements were made on crystalline compound 2, after the solid was sealed in silica ampoules under vacuum.
4.2 Preparation of Eu(TpiPr2)2 (2)
Solid EuI2(THF)2 (1.73 g, 3.16 mmol) was added to a stirred solution of KTpiPr2 (3.2 g; 6.33 mmol) in 150 ml of THF. Stirring at r.t. was continued for 24 h to give a “neon-yellow”, strongly fluorescent solution and a white precipitate (KI). After filtration and evaporation of the clear, yellow filtrate to dryness, the residue was extracted with n-pentane (80 ml) and filtered again. The filtrate was concentrated in vacuo to a total volume of ca. 25 ml. Cooling to −20 °C for 3d produced bright yellow crystals of 2. Yield: 2.84 g (83%). Decomposition range: 78–84 °C. Anal. calcd for C54H92B2EuN12 (1082.99 g mol−1): C, 59.89; H, 8.56; N, 15.52. Found: C, 59.39; H, 8.31; N, 14.98. IR (KBr): νmax = 3222m, 3103m, 2964vs, 2930s, 2871s, 2552w, 2471w, 2240w, 1959w, 1638m, 1567m, 1535m, 1468s, 1427m, 1381s, 1367s, 1300s, 1235m, 1174s, 1138m, 1105m, 1073m, 1050s, 1009m, 991m, 959w, 924w, 897w, 879w, 791s, 771m, 724m, 717m, 663m cm−1. MS (EI): m/z 988 (7%), 935 (6, [M − pziPr2]), 768 (100, [TpiPr2Eu + pziPr2]), 616 (88, [TpiPr2Eu]), 477 (40, [TpiPr2 + BH]), 302 (23, [pziPr2]2). 11B NMR (300 K, C6D6, 128 MHz): δ = −7 (v, br).
4.3 Preparation of Yb(TpiPr2)2 (4)
In a similar manner as described for 2, the reaction of YbI2(THF)2 (1.69 g, 2.96 mmol) with KTpiPr2 (3.0 g, 5.93 mmol) in 80 ml THF afforded, after crystallization from n-pentane and thorough drying to remove residual n-pentane, bright red crystalline 4 in 77% isolated yield (2.51 g). Mp. 99 °C (beginning dec.). Anal. calcd for C54H92B2N12Yb (1104.07 g mol−1): C, 58.75; H, 8.40; N, 15.22. Found: C, 58.33; H, 8.10; N, 14.88. IR (KBr): νmax = 3222m, 3092m, 2966vs, 2931s, 2870s, 2555m, 2237w, 1959w, 1638s, 1566m, 1538s, 1470s, 1426m, 1381s, 1368s, 1298s, 1237m, 1175s, 1138m, 1106m, 1053s, 1020m, 960w, 924w, 896w, 878w, 794s, 725m, 717m, 661m cm−1. MS (EI): m/z 996 (7%), 968 (8), 953 (6, [M − pziPr2]), 817 (28), 790 (100, [TpiPr2Yb + pziPr2]), 637 (6, [TpiPr2Eu]), 476 (9, [TpiPr2 + BH]), 321 (14, [pziPr2 + Yb]), 169 (19), 137 (25). 1H NMR (300 K, C6D6, 400 MHz): δ = 5.93 (s, 6H, pyrazolyl), 5.22 (s br, 2H, B–H), 3.73 (sept, 6H, C–HiPr, J = 6.8 Hz), 2.66 (sept, 6H, C–HiPr, J = 6.8 Hz), 1.22 (d, 36 H, CH3iPr, J = 6.8 Hz), 1.04 (d, 36 H, CH3iPr, J = 6.8 Hz). 13C NMR (300 K, C6D6, 100 MHz): δ = 160.6 (q-C pyrazolyl), 156.9 (q-C pyrazolyl), 97.5 (C–H pyrazolyl), 28.0 (C–H iPr), 27.1 (C–H iPr), 24.8 (CH3iPr), 23.8 (CH3iPr). 171Yb (300 K, C6D6, 70 MHz, relative to [Yb(η-C5Me5)2(THF)2]): δ = 619.1. 11B NMR (300 K, C6D6, 128 MHz): δ = −6.2 (s, br).
4.4 Behavior of Sm(TpiPr2)2 (1) toward acetonitrile
Addition of acetonitrile (2–3 ml) to ca. 200 mg of Sm(TpiPr2)2 (1) produced an almost colorless supernatant and a dark green, almost black solid (unchanged Sm(II)). The supernatant was pipetted off and the solid dissolved in Et2O to give a very dark green solution, to which acetonitrile was again added. An attempt to grow crystals by cooling at −30 °C was unsuccessful. The dark green solution was left to slowly evaporate at RT, and overnight deposited a mixture of dark green and colorless crystals. The supernatant was removed and the mixture of crystals briefly dried. From this mixture a dark green and a colorless crystal were selected and, by X-ray diffraction, were shown to be complexes 5 and 6, respectively.
4.5 X-ray crystallographic studies of 4–6
The intensity data of 4 were registered on an Oxford Diffraction Nova A diffractometer using mirror-focussed CuKα radiation. Absorption correction was applied using the multi-scan method. The structure was solved by direct methods (SHELXS-97)27a and refined by full matrix least-squares methods on F2 using SHELXL-97.27b Intensity data for 5 and 6 were collected on a Bruker D8/APEX II CCD diffractometer using graphite-monochromated MoKα radiation. Programs for diffractometer operation, data collection, data reduction and absorption correction were those supplied by Bruker. Absorption corrections were applied using the Gaussian integration (face-indexed) method. The structures were solved and refined using the programs SHELXT and SHELXL-2013.28 Data collection parameters for 4–6 are given in Table 1.
Acknowledgements
Financial support by the Otto-von-Guericke-Universität Magdeburg is gratefully acknowledged. JT thanks NSERC and the University of Alberta for financial support. Thanks are due to Ms Desirée Schneider and Dr Volker Lorenz for technical assistance in the preparation of the manuscript.
References
- Review:
F. T. Edelmann, Complexes of Scandium, Yttrium and Lanthanide Elements, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2006, p. 1 Search PubMed.
-
(a) W. J. Evans, L. A. Hughes and T. P. Hanusa, J. Am. Chem. Soc., 1984, 106, 4270 CrossRef CAS;
(b) W. J. Evans, J. W. Grate, H. W. Choi, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 941 CrossRef CAS;
(c) W. J. Evans, L. A. Hughes and T. P. Hanusa, Organometallics, 1986, 5, 1285 CrossRef CAS;
(d) C. J. Burns and R. A. Andersen, J. Am. Chem. Soc., 1987, 109, 5853 CrossRef CAS , and references cited therein;
(e) D. J. Berg, C. J. Burns, R. A. Andersen and A. Zalkin, Organometallics, 1989, 8, 1865 CrossRef CAS;
(f) W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Am. Chem. Soc., 1991, 113, 7423 CrossRef CAS , and references cited therein;
(g) M. Shultz, C. J. Burns, D. J. Schwartz and R. A. Andersen, Organometallics, 2000, 19, 781 CrossRef;
(h) J. A. Moore, A. H. Cowley and J. C. Gordon, Organometallics, 2006, 25, 5207 CrossRef CAS.
- W. J. Evans, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1988, 110, 6877 CrossRef CAS.
-
(a) S. N. Konchenko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, H. Schnöckel and P. W. Roesky, J. Am. Chem. Soc., 2009, 131, 5740 CrossRef CAS PubMed;
(b) T. Li, M. T. Gamer, M. Scheer, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2013, 49, 2183 RSC;
(c) N. Arlet, S. Bestgen, M. T. Gamer and P. W. Roesky, J. Am. Chem. Soc., 2014, 136, 14023 CrossRef PubMed.
-
(a) R. A. Andersen, J. M. Boncella, C. J. Burns, J. C. Green, D. Hohl and N. Rösch, J. Chem. Soc., Chem. Commun., 1986, 405 RSC;
(b) J. C. Green, D. Hohl and N. Rösch, Organometallics, 1987, 6, 712 CrossRef CAS;
(c) T. K. Hollis, J. K. Burdett and B. Bosnich, Organometallics, 1993, 12, 3385 CrossRef CAS;
(d) T. V. Timofeeva, J.-H. Lii and N. L. Allinger, J. Am. Chem. Soc., 1995, 117, 7452 CrossRef CAS;
(e) J. Marcalo, A. Pires de Matos and W. J. Evans, Organometallics, 1996, 15, 345 CrossRef CAS;
(f) L. Perrin, L. Maron, O. Eisenstein, D. J. Schwartz, C. J. Burns and R. A. Anderson, Organometallics, 2003, 22, 5447 CrossRef CAS;
(g) S. Labouille, C. Clavaguera and F. Nief, Organometallics, 2013, 32, 1265 CrossRef CAS;
(h) C. E. Kefalidis, S. Essafi, L. Perrin and L. Maron, Inorg. Chem., 2014, 53, 3427 CrossRef CAS PubMed;
(i) C. E. Kefalidis, L. Perrin, C. J. Burns, D. J. Berg, L. Maron and R. A. Andersen, Dalton Trans., 2015, 44, 2575 RSC.
-
S. Trofimenko, The Coordination Chemistry of Scorpionates – Pyrazolylborate Ligands, Imperial College Press, London, 1999 Search PubMed.
-
C. Pettinari, Scorpionates II: Chelating Borate Ligands, Imperial College Press, London, 2008 Search PubMed.
- J. Takats, X. W. Zhang, V. W. Day and T. A. Eberspacher, Organometallics, 1993, 12, 4286 CrossRef CAS.
- G. H. Maunder, A. Sella and D. A. Tocher, J. Chem. Soc., Chem. Commun., 1994, 885 RSC.
- A. C. Hillier, X. W. Zhang, G. H. Maunder, S. Y. Liu, T. A. Eberspacher, M. V. Metz, R. McDonald, Â. Domingos, N. Marques, V. W. Day, A. Sella and J. Takats, Inorg. Chem., 2001, 40, 5106 CrossRef CAS PubMed.
- Review: N. Marques, A. Sella and J. Takats, Chem. Rev., 2002, 102, 2137 CrossRef CAS PubMed.
- C. E. Davies, I. M. Gardiner, J. C. Green, M. L. H. Green, N. J. Hazel, P. D. Grebenik, V. S. B. Mtetva and K. Prout, J. Chem. Soc., Dalton Trans., 1985, 669 RSC.
- N. Kitajima, K. Fujisawa, C. Fujimoto, Y. Moro-oka, S. Hasimoto, T. Kitagawa, K. Toriumi, K. Tatsumi and A. Nakamura, J. Am. Chem. Soc., 1992, 114, 1277 CrossRef CAS.
- A. Momin, L. Carter, Y. Yang, R. McDonald, S. Essafi, F. Nief, I. Del Rosal, A. Sella, L. Maron and J. Takats, Inorg. Chem., 2014, 53, 12066 CrossRef CAS PubMed.
-
(a) A. G. Avent, M. A. Edelman, M. F. Lappert and G. A. Lawless, J. Am. Chem. Soc., 1989, 111, 3423 CrossRef CAS;
(b) J. M. Keates, G. A. Lawless and M. P. Waugh, Chem. Commun., 1996, 1627 RSC;
(c)
J. M. Keates and G. A. Lawless, 171Yb NMR Spectroscopy, in Advanced Applications of NMR to Organometallic Chemistry, ed. M. Gielen, R. Willem and B. Wrackmeyer and J. Wiley & Sons, Chichester, 1996, ch. 12, pp. 357–370 Search PubMed;
(d) J. M. Keates and G. A. Lawless, Organometallics, 1997, 16, 2842 CrossRef CAS.
-
(a) X. Y. Zhang, R. McDonald and J. Takats, New J. Chem., 1995, 19, 573 CAS;
(b) P. B. Hitchcock, M. F. Lappert and S. Tian, Organometallics, 2000, 19, 4320 CrossRef;
(c) K. Izod, P. O'Shaughnessy, J. M. Sheffield, W. Clegg and S. T. Liddle, Inorg. Chem., 2000, 39, 4741 CrossRef CAS;
(d) P. B. Hitchcock, M. F. Lappert and S. Prashar, J. Organomet. Chem., 2000, 613, 105 CrossRef CAS;
(e) M. Niemeyer, Eur. J. Inorg. Chem., 2001, 1969 CrossRef CAS;
(f) G. B. Deacon and C. M. Forsyth, Chem. – Eur. J., 2004, 10, 1798 CrossRef CAS PubMed;
(g) M. Niemeyer, Inorg. Chem., 2006, 45, 9085 CrossRef CAS PubMed;
(h) L. J. Bowman, K. Izod, W. Clegg and R. W. Harrington, Organometallics, 2007, 26, 2646 CrossRef CAS;
(i) A. Edelmann, S. Blaurock, V. Lorenz, L. Hilfert and F. T. Edelmann, Angew. Chem., Int. Ed., 2007, 46, 6732 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
-
C. Ronda, Luminescence, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
-
W. M. Yen, S. Shionoya and H. Yamamoto, Phosphor Handbook, CRC Press, 2007 Search PubMed.
- P. Dorenbos, J. Lumin., 2003, 104, 239 CrossRef CAS.
- S. Harder, D. Naglav, C. Ruspic, C. Wickleder, M. Adlung, W. Hermes, M. Eul, R. Pöttgen, D. B. Rego, F. Poineau, K. R. Czerwinski, R. H. Herber and I. Nowik, Chem. – Eur. J., 2013, 37, 12272 CrossRef PubMed.
- C. P. Shipley, S. Capecchi, O. V. Salata, M. Etchells, P. J. Dobson and V. Christou, Adv. Mater., 1999, 11, 533 CrossRef CAS.
-
(a) Y. Q. Li, J. E. J. V. Steen, J. W. H. V. Krevel, G. Botty, A. C. Delsing, F. J. DiSalvo, G. D. With and H. T. Hintzen, J. Alloys Compd., 2006, 417, 273 CrossRef CAS PubMed;
(b) H. A. Höppe, H. Lutz, P. Morys, W. Schnick and A. Seilmeier, J. Phys. Chem. Solids, 2000, 61, 2001 CrossRef;
(c) V. Bachmann, C. Ronda, O. Oeckler, W. Schnick and A. Meijerink, Chem. Mater., 2009, 21, 316 CrossRef CAS.
- D. S. Noyce, E. Ryder and B. H. Walker, J. Org. Chem., 1955, 20, 1681 CrossRef CAS.
-
(a) S. H. M. Poort, J. W. H. van Krevel, R. Stomphorst, A. P. Vink and G. Blasse, J. Solid State Chem., 1996, 122, 432 CrossRef CAS;
(b) A. Meijerink and G. Blasse, J. Lumin., 1989, 43, 283 CrossRef CAS;
(c) M. Suta, P. Larsen, F. Lavoie-Cardinal and C. Wickleder, J. Lumin., 2014, 149, 35 CrossRef CAS PubMed.
- J. L. Namy, H. B. Kagan and P. E. Caro, New J. Chem., 1981, 5, 479 CAS.
-
(a)
G. M. Sheldrick, SHELXL-97 Program for Crystal Structure Refinement, Universität Göttingen, Germany, 1997 Search PubMed;
(b)
G. M. Sheldrick, SHELXS-97 Program for Crystal Structure Solution, Universität Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of Professor Michael F. Lappert, a pioneer of organometallic chemistry. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1051399–1051401. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00568j |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a