
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot preparation of cationic charged Pt nanoparticles by the autocatalytic hydrolysis of acetylthiocholine†
Yohei
Ishida
,
Thatchanont
Jirasupangkul‡
and
Tetsu
Yonezawa
*
Division of Material Science and Engineering, Faculty of Engineering, Hokkaido University, Kita 13, Nishi 8, Kita-ku, Sapporo, Hokkaido 060-8628, Japan. E-mail: tetsu@eng.hokudai.ac.jp
First published on 8th April 2015
Abstract
We propose a novel synthetic approach for cationic charged platinum nanoparticles via the autocatalytic hydrolysis of acetylthiocholine. This method can be extended to the platinum group of metals.
In recent years, significant advances have been made toward the development of metallic nanoparticles (NPs)1 having a controlled size, shape, and composition for applications in photonics,2,3 electronics,4 medicine,5 and catalysis.6,7 For these applications, it is imperative to control the size of the metal NPs, and several molecules such as amines, mercaptans, and polymers have been used as stabilizers due to their good coordination properties.1
Recently, the self-organization of monodisperse metal NPs has been extensively investigated to considerably broaden the spectrum of their applications.8 Several methods have been reported for assembling NPs into superstructures, including the interaction between NPs9,10 as well as that between NPs and other host materials such as inorganic nanosheets,11,12 cavitands,13 and DNA.14,15 Such organization between NPs and/or hosts can be achieved through van der Waals, dipole–dipole, and electrostatic interactions as well as other interactions. Moreover, self-assembly can also be achieved by the effects of an external electrical16 field, irradiation,17 and a magnetic18 field. The regular superstructures of NPs thus obtained can offer unique possibilities such as the control of the coupling between excitons and plasmons19 and the transfer of excitation energies.20
A major approach for the formation of NP assemblies is by exploiting electrostatic interactions,9,10,21 whereby the charged NPs attach to oppositely charged NPs or hosts via electrostatic interactions and form superstructures. This self-assembly requires the use of charged NPs; however, reports of the synthesis of cationic charged metal NPs22 are quite limited as compared with those of the synthesis of anionic charged NPs, such as those stabilized by mercaptoundecanoic acid,23 mercaptopropionic acid,24 or glutathione.25 The scarcity of the reports on cationic charged NPs possibly originates from the low availability and selectability of cationic charged organic stabilizers containing mercapto groups. Meanwhile, cationic charged ligands such as cetyltrimethylammonium bromide have been used as the stabilizing reagent for metal NPs. The mechanism for stabilization involves the formation of a micelle; hence, as compared to thiol-stabilized systems, the resultant structure is not as stable.26 Moreover, if an easy and facile approach can be developed for the synthesis of thiol-stabilized cationic NPs, the chemistry of supramolecular NP assemblies will accelerate, and the synthetic strategies for such structures will considerably broaden.
Herein, we report the synthesis of cationic charged Pt NPs by a novel approach based on organic chemistry. Typically, the synthesis of metal NPs only utilizes metal ions as the metal source, while in our new approach, the Pt ion is used as the metal source and the Pt NPs formed initially act as autocatalysts for the hydrolysis of the mercaptan precursor, namely, acetylthiocholine. By employing this strategy, we can eliminate the chemical synthesis of mercaptan; furthermore, this approach can be widely applied to a wide variety of metals in the platinum group. To the best of our knowledge, this is the first report of the synthesis of metal NPs by the autocatalytic reaction of a ligand precursor by the formed NPs (Scheme 1).
 | ||
| Scheme 1 Hydrolysis from acetylthiocholine to thiocholine. | ||
Cationic charged platinum NPs (Pt NPs) were obtained by the chemical reduction of H2PtCl6 with acetylthiocholine (ATC) and NaBH4. The one-pot synthesis of cationic Pt NPs was performed as follows. First, 5 mL of an H2PtCl6 solution (10 mM in methanol) was added to 5 mL of an aqueous ATC solution (10, 20, or 30 mM). Next, the solution mixture immediately turned colloidal, and it was stirred for 30 min. To evaluate transparency, absorption spectra of the solutions were recorded without dilution using a 1 mm quartz cell (Fig. 1). By the addition of ATC, the absorption of platinum ions at 456 nm (black line) almost disappeared, and large scattering was observed in the whole wavelength region (blue line), which is attributed to the colloidal species formed by the ion complex of PtCl42− and ATC+. To the colloidal suspension, 1.7 mL of a freshly prepared NaBH4 aqueous solution (0.4 M) was slowly added at the rate of 1 drop per minute, and the stirring of the mixture was continued for another 3 h. With the addition of several drops, the colour of the suspension started to turn black (red line; Fig. 1), indicating the reduction of Pt ions and the formation of Pt NPs. Notably, NaBH4 does not catalyze the hydrolysis of ATC. Typically, as catalytic hydrolysis by Pt proceeds under alkaline conditions, the slow addition of NaBH4 would simultaneously suppress the rapid formation of NPs and enhance the catalytic hydrolysis of ATC to thiocholine (TC).
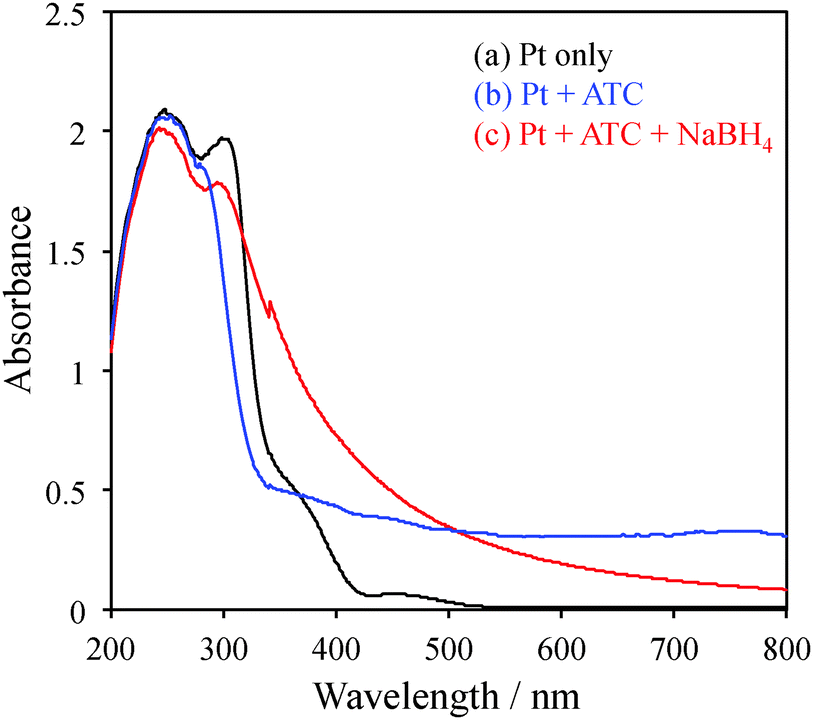 | ||
| Fig. 1 Absorption spectra of (a) H2PtCl4 solution in methanol, (b) after the addition of an aqueous acetylthiocholine (ATC) solution to (a), (c) after addition of the NaBH4 aqueous solution to (b). All measurements were performed using a 1 mm quartz cell without dilution. | ||
For transmission electron microscopy (TEM) analysis, the obtained suspension of the Pt NPs was purified by ultracentrifugation. After centrifugation at 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 3 h, the impurities remained in the supernatant and Pt NPs precipitated out. Furthermore, after repeated ultracentrifugation (3 times) with the addition of pure water to each cycle, purified Pt NPs were obtained. The Pt NPs were then redispersed in water and methanol, and the TEM sample was prepared by dropping an aqueous dispersion of Pt NPs onto a carbon-coated Cu grid. Fig. 2 shows the TEM images and the size distribution of the obtained Pt NPs at a 1:3 mol ratio of Pt:ATC. Highly monodisperse spherical NPs were observed, and the distribution histogram was well described by the Gaussian distribution. The average diameter of the obtained NPs was 2.3 nm. Moreover, at a lower ATC concentration (Pt:ATC = 1:1 and 1:2), slightly larger NPs were obtained (both 2.4 nm). In this study, the change in diameter with different ligand concentrations was not significant, and it was within the error. This small change is attributed to the synthetic mechanism, whereby the ATC ligands attached to Pt ions (as an ion complex) can only act as a stabilizer immediately after the addition of NaBH4, suggesting that the actual concentration of ATC near Pt ions is the same under different ATC concentrations. Thus, the ATC–Pt ion complex is the actual precursor for the synthesis of Pt NPs in this system, and the obtained size of Pt NPs is thus almost constant under different ATC concentrations.
000 rpm for 3 h, the impurities remained in the supernatant and Pt NPs precipitated out. Furthermore, after repeated ultracentrifugation (3 times) with the addition of pure water to each cycle, purified Pt NPs were obtained. The Pt NPs were then redispersed in water and methanol, and the TEM sample was prepared by dropping an aqueous dispersion of Pt NPs onto a carbon-coated Cu grid. Fig. 2 shows the TEM images and the size distribution of the obtained Pt NPs at a 1:3 mol ratio of Pt:ATC. Highly monodisperse spherical NPs were observed, and the distribution histogram was well described by the Gaussian distribution. The average diameter of the obtained NPs was 2.3 nm. Moreover, at a lower ATC concentration (Pt:ATC = 1:1 and 1:2), slightly larger NPs were obtained (both 2.4 nm). In this study, the change in diameter with different ligand concentrations was not significant, and it was within the error. This small change is attributed to the synthetic mechanism, whereby the ATC ligands attached to Pt ions (as an ion complex) can only act as a stabilizer immediately after the addition of NaBH4, suggesting that the actual concentration of ATC near Pt ions is the same under different ATC concentrations. Thus, the ATC–Pt ion complex is the actual precursor for the synthesis of Pt NPs in this system, and the obtained size of Pt NPs is thus almost constant under different ATC concentrations.
 | ||
| Fig. 2 TEM images and size distribution histogram of Pt nanoparticles prepared at a 1:3 mol ratio of Pt:acetylthiocholine (ATC). | ||
To clarify whether the catalytic hydrolysis of ATC to TC proceeded as we expected, 1H-NMR (400 MHz) measurements were performed. All spectra were recorded in D2O. Fig. 3a shows the 1H-NMR spectrum of ATC. A sharp singlet peak was observed at 2.4 ppm, corresponding to the methyl group of ACT. Under several relaxation time conditions, no peaks were observed for the purified Pt NPs, caused by the attachment of organic ligands onto the NP surfaces. Typically, treatment by I2 is employed to extract mercaptans from metal NPs via the formation of disulfides.27 Hence, Pt NPs were treated with I2. The treatment involved the addition of 1 mg of I2 powder to 5 mL of an aqueous suspension containing 1 mg of Pt NPs. A black precipitate formed instantly. The supernatant was dried under vacuum, and the powder obtained was then dissolved in D2O for 1H-NMR measurement (Fig. 3b). The characteristic singlet of ATC disappeared, indicating the hydrolysis of ATC to TC. Moreover, we chemically synthesized TC by the hydrolysis of ATC in the presence of HBr according to a previous paper.28 The synthetic TC thus exhibited an NMR spectrum (Fig. 3c) similar to that of the TC extracted by I2 treatment (Fig. 3b). Thus, we can conclude that the synthesis of cationic Pt NPs was successfully achieved by the hydrolysis of ATC. FT-IR spectra of ATC, synthetic TC and Pt NPs also support the discussion (see Fig. S1 in ESI†). A characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of ATC observed at 1680 cm−1 disappeared in Pt NPs.
O stretching vibration of ATC observed at 1680 cm−1 disappeared in Pt NPs.
 | ||
| Fig. 3 1H-NMR (400 MHz) spectra of (a) acetylthiocholine (ATC), (b) ligand of Pt NPs that was extracted by I2 treatment, and (c) synthesized thiocholine (TC) treated with I2. All measurements were performed in D2O. | ||
A question that still remained was whether Pt ions or the Pt NPs that are formed initially catalyze the hydrolysis of ATC. To answer this question, a solution mixture containing H2PtCl6 and ATC was stirred with NaOH instead of NaBH4. This experiment aimed to clarify whether Pt ions can act as a catalyst for the hydrolysis of ATC to TC, as NaOH does not reduce Pt ions but only increases the solution pH, similar to that observed for NaBH4 systems. After stirring for 3 h, the solution mixture containing H2PtCl6, ATC, and NaOH was dried, and the residue was dissolved in DMSO-d6 for 1H-NMR measurement. All peaks for ATC were observed in the 1H-NMR spectrum (Fig. S2, ESI†). Hence, the results from this experiment clearly indicate that the Pt NPs formed initially, not Pt ions, catalyze the hydrolysis of ATC.
As this synthesis depends on the catalytic ability of Pt, it can be extended to other metals in the platinum group, such as Pd. On the other hand, this synthetic route cannot be applied to metals that do not catalyze hydrolysis reactions. To examine the generality of this method for other metals, we examined the synthesis of Pd and Au NPs using H2PdCl4 and HAuCl4, respectively, as the metal sources in the presence of ATC. Fig. 4a–c show TEM images and size distribution histograms of the Pd and Au NPs thus prepared at a 1:3 mol ratio of metal:ATC. The Pd NPs thus obtained had a small size, with the average NP size being 2.0 nm ± 0.6 nm. On the other hand, Au NPs exhibited large aggregates with sizes ranging from 30 to 200 nm. These results clearly indicate that Pd, not Au, can catalyze the hydrolysis of ATC, indicating that only metals in the platinum group can be used for the autocatalysis of thioacetyl compounds. Our previous study on the synthesis of Au NPs having a diameter of 2.7 nm by using TC as a stabilizer also supports this conclusion.28
 | ||
| Fig. 4 (a) TEM image of Pd NPs, (b) size distribution histogram of Pd NPs, and (c) TEM image of Au NPs. Pd and Au NPs were prepared at a 1:3 mol ratio of metal:acetylthiocholine (ATC). | ||
In conclusion, we described a novel approach for preparing cationic charged Pt NPs by the autocatalytic hydrolysis of acetylthiocholine. Typically, the synthesis of metal NPs uses only a metal ion as the metal source, while in our proposed approach, the Pt ion is used as the metal source and the Pt NPs formed initially serve as autocatalysts for the hydrolysis of the mercaptan precursor. This strategy eliminates the chemical synthesis of mercaptan; furthermore, it can be applied to a wide variety of metals in the platinum group. Charged, especially cationic charged NPs, thiol-stabilized metal NPs are fascinating as building blocks for supramolecular NP assemblies. Regular NP superstructures can offer unique possibilities, such as exciton–plasmon coupling, magnetic enhancement, as well as the utilization of excitation energy; hence, this method presented in this study can serve as a novel, simple synthetic approach for broadening the scope of NPs.
Experimental section
The details of the synthesis of platinum nanoparticles (Pt NPs) are provided in the results and discussion section. H2PtCl6, PdCl2, and HAuCl4 were purchased from Kojima Chemicals (Japan), and acetylthiocholine iodide was purchased from Aldrich. NaBH4 was obtained from Kanto Chemical. The counter ion of acetylthiocholine iodide was converted to the corresponding chloride by using an ion-exchange column (Organo Amberlite IRA400JCL) before the use.Acknowledgements
This work is partially supported from Grant-in-Aid for Scientific Research (A) (to TY, 24241041) and from Hokkaido University (to YI and TY). Authors acknowledge Mr Hideo Genda and Prof. Kunihito Koumoto (Nagoya Univ.) for fruitful discussions. A part of this work is supported by Building of Consortia for the Development of Human Resources in Science and Technology from Hokkaido University (to YI), and by Hokkaido University, microstructural characterization platform as a program of Nanotechnology Platform of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.Notes and references
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389 CrossRef CAS PubMed.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568 CrossRef CAS PubMed.
- X. Yuan, Z. Luo, Q. Zhang, X. Zhang, Y. Zheng, J. Y. Lee and J. Xie, ACS Nano, 2011, 5, 8800 CrossRef CAS PubMed.
- H. C. Choi, S. Kundaria, D. Wang, A. Javey, Q. Wang, M. Rolandi and H. Dai, Nano Lett., 2003, 3, 157 CrossRef CAS.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740 RSC.
- A. Taketoshi and M. Haruta, Chem. Lett., 2014, 43, 380 CrossRef CAS.
- N. Toshima and T. Yonezawa, New J. Chem., 1988, 1179 Search PubMed.
- L. Xu, W. Ma, L. Wang, C. Xu, H. Kuang and N. A. Kotov, Chem. Soc. Rev., 2013, 42, 3114 RSC.
- R. Klajn, K. J. M. Bishop and B. A. Grzybowski, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10305 CrossRef CAS PubMed.
- Q. Yao, Z. Luo, X. Yuan, Y. Yu, C. Zhang, J. Xie and J. Y. Lee, Sci. Rep., 2014, 4, 3848 Search PubMed.
- L. Zhu, S. Letaief, Y. Liu, F. Gervais and C. Detellier, Appl. Clay Sci., 2009, 43, 439 CrossRef CAS PubMed.
- P. Podsiadlo, M. Michel, K. Critchley, S. Srivastava, M. Qin, J. W. Lee, E. Verploegen, A. J. Hart, Y. Qi and N. A. Kotov, Angew. Chem., Int. Ed., 2009, 48, 7073 CrossRef CAS PubMed.
- B. Mondal, N. Kamatham, S. R. Samanta, P. Jagadesan, J. He and V. Ramamurthy, Langmuir, 2013, 29, 12703 CrossRef CAS PubMed.
- T. Yonezawa, S. Onoue and T. Kunitake, Kobunshi Ronbunshu, 1999, 56, 855 CrossRef CAS.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935 CrossRef CAS PubMed.
- S. Acharya, I. Patla, J. Kost, S. Efrima and Y. Golan, J. Am. Chem. Soc., 2006, 128, 9294 CrossRef CAS PubMed.
- S. Srivastava, A. Santos, K. Critchley, K. S. Kim, P. Podsiadlo, K. Sun, J. Lee, C. Xu, G. D. Lilly, S. C. Glotzer and N. A. Kotov, Science, 2010, 327, 1355 CrossRef CAS PubMed.
- A. Ahniyaz, Y. Sakamoto and L. Bergström, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17570 CrossRef CAS PubMed.
- P. Lodahl, A. Floris Van Driel, I. S. Nikolaev, A. Irman, K. Overgaag, D. Vanmaekelbergh and W. L. Vos, Nature, 2004, 430, 654 CrossRef CAS PubMed.
- E. V. Shevchenko, M. Ringler, A. Schwemer, D. V. Talapin, T. A. Klar, A. L. Rogach, J. Feldmann and A. P. Alivisatos, J. Am. Chem. Soc., 2008, 130, 3274 CrossRef CAS PubMed.
- F. Aldeek, M. A. H. Muhammed, G. Palui, N. Zhan and H. Mattoussi, ACS Nano, 2013, 7, 2509 CrossRef CAS PubMed.
- A. M. Kalsin, B. Kowalczyk, S. K. Smoukov, R. Klajn and B. A. Grzybowski, J. Am. Chem. Soc., 2006, 128, 15046 CrossRef CAS PubMed.
- Z. Zhang, L. Xu, H. Li and J. Kong, RSC Adv., 2012, 3, 59 RSC.
- M. Kondon, J. Kim, N. Udawatte and D. Lee, J. Phys. Chem. C, 2008, 112, 6695 CAS.
- S. Link, A. Beeby, S. FitzGerald, M. A. El-Sayed, T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2002, 106, 3410 CrossRef CAS.
- L. Vigderman, P. Manna and E. R. Zubarev, Angew. Chem., Int. Ed., 2011, 51, 636–641 CrossRef PubMed.
- A. C. Templeton, M. J. Hostetler, E. K. Warmoth, S. Chen, C. M. Hartshorn, V. M. Krishnamurthy, M. D. Forbes and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 4845 CrossRef CAS.
- T. Yonezawa, S. Onoue and N. Kimizuka, Chem. Lett., 2002, 1172 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental conditions, FT-IR spectra, additional 1H-NMR spectra, and magnified TEM images. See DOI: 10.1039/c5nj00420a |
| ‡ Present address: Undergraduate Student, International School of Engineering, Chulalongkorn University, 254 Phyathai Road, Patumwan, Bangkok 10330, Thailand. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |