
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of noncovalent interactions on the structures of metal–organic hybrids based on a [VO2(2,6-pydc)]− tecton with cations of imidazole, pyridine and its derivatives†
Tanja
Koleša-Dobravc
ab,
Anton
Meden
ab and
Franc
Perdih
*ab
aFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, P. O. Box 537, SI-1000 Ljubljana, Slovenia
bCO EN–FIST, Trg Osvobodilne fronte 13, SI-1000 Ljubljana, Slovenia. E-mail: franc.perdih@fkkt.uni-lj.si
First published on 17th March 2015
Abstract
Seven different dioxido(pyridine-2,6-dicarboxylato)vanadate(V) compounds with pyridinium (Hpy+) (1·2H2O and 1), 2-hydroxypyridinium (H2pyon+) (2·H2O), 4-aminopyridinium (H4apy+) (3·H2O and 3), 4-(dimethylamino)pyridinium (Hdmap+) (4·H2O) and imidazolium (Himd+) (5) cations have been prepared via different pathways starting either from pyridine-2,6-dicarboxylic acid or its esters, and were structurally characterized by single-crystal X-ray diffraction. The vanadium metal center in dioxido(pyridine-2,6-dicarboxylato)vanadate(V) anion is pentacoordinated in all of the compounds: having two oxido oxygen atoms in a mutual cis position and a tridentate pyridine-2,6-dicarboxylic ligand. Study of hydrogen bonds and weak interactions in the compounds revealed the relationship between the type of cation and the hydrogen bonding network in the compounds. While in 1·2H2O, 2·H2O and 4·H2O a one-dimensional (band, pillar or chain) hydrogen bonding network via N/O–H⋯O bonds is preferred, anhydrous 3 and 3·H2O favor a two-dimensional hydrogen-bonded framework, and the Himd+ cation facilitates a three-dimensional hydrogen bonding in 5. The unique vanadium coordination environment with two easily accessible oxido oxygen atoms of the VO2+ unit is suitable for the construction of non-covalent metal–organic hybrids. In 2·H2O, 3·H2O, 4·H2O and 5 both oxido oxygen atoms of the VO2+ unit participate as acceptors, however, in 1·2H2O and 3 only one oxido oxygen atom is involved in classical hydrogen bonding. Besides N/O–H⋯O hydrogen bonding, also other weak non-covalent interactions, such as C–H⋯O, π⋯π and C–H⋯π interactions, play an important role in stabilizing the crystal lattices.
Introduction
The self-assembly of tectons into extended network structures is the core of supramolecular chemistry and crystal engineering. Metal–organic hybrids and especially metal–organic frameworks are currently an extremely important topic and an active area of research because of their intriguing architectures and topologies,1 as well as due to their potential application in catalysis,2 chemical separation processes,3 gas storage,4 magnetism5 and as sensors.6Metal–organic hybrids can be assembled by a covalent approach using bridging ligands or by a non-covalent approach using hydrogen bonding and other weak interactions. The covalent approach is primarily based on strong coordinate bonds connecting metal cations and organic ligands into robust polymeric structures. Different kinds of these materials have been designed with special attention dedicated to the geometry of the metal ions as well as flexibility, bridging potential and coordination preferences of different organic linkers.1 In the non-covalent approach much weaker forces, such as hydrogen bonding, C–H⋯π/F interactions, π⋯π stacking, and halogen bonding, are employed. Although weak by nature multiple non-covalent forces can adjust the dimensionality and enable new topologies to arise. Therefore, the desired functions of supramolecular assemblies can be achieved.7–10 Among non-covalent interactions the hydrogen bonding is a particularly powerful building motif used in crystal engineering since it provides unique directionality and can be easily introduced into structures. There exists a great variety of hydrogen bonding donors–acceptors and their numbers can be varied through simple design, thus making them a particularly good choice for the construction of self-assemblies. With this approach a wide variety of mononuclear, dinuclear and polynuclear coordination compounds/ions can be assembled into desirable motifs. Multiple weak non-covalent interactions can even control the topology of metal–organic frameworks11 as well as the coordination geometry.12
Although great efforts have been made toward the understanding of the assembly process, rational control in the construction of supramolecular structures of complexes is still a challenging task. For the construction of a desired framework and functionality, it is important to control and understand the factors, such as counterions, solvents, temperature and pH value, that tend to influence the structural prediction on the assembly of the final coordination frameworks and govern the crystal growth and the stability of the overall crystals.1b,13 Among the above factors, it has been demonstrated that counterions have a significant effect on the formation of the product. Different coordination abilities, sizes, and geometries of anions have a great influence on the structural assembly and importantly influence the prediction of the overall supramolecular architectures of coordination compounds.1b,14 Cations can also have a significant influence on the crystal architecture. “Naked” alkali cations are useful tectons due to their different sizes and polarisability, while in the case of NH4+ and hydrated cations, besides their size and charge, the hydrogen bonding capacity can enable the formation of high-dimensional frameworks.15 Diverse effects can be introduced by organic cations, most commonly protonated cationic amines and pyridines. Organic cations can be varied through simple design and diverse functionalities can be combined. Coulombic interactions are a principal force for cation–anion arrangements in supramolecular structures.16 However, protonated organic cations can also act as multi-hydrogen bond donors as well as acceptors and thus easily adjust the topologies via additional non-covalent interactions. Charge-assisted or ionic hydrogen bonds are in general stronger hydrogen bonds since ionic charge on a donor or an acceptor enhances the hydrogen bond strength.17 Furthermore, employing organic cations enables introduction of additional weak non-covalent interactions, such as π⋯π stacking, C–H⋯π interactions and halogen bonding.18
Recently, the use of bioactive framework materials (BioMOFs) has gained considerable attention in biology and medicine. This has stimulated the search for new types of bioactive organic linkers capable of ligating biorelevant metal ions for the design of functional materials.19,20 Multidentate pyridinedicarboxylato ligands (pydc2−) have been widely used in recent years for the construction of organic–inorganic hybrid materials. Because they possess diverse coordination abilities, flexibilities and various bridging modes, supramolecular networks of high structural stability have been assembled either via coordination bonds, hydrogen bonds and/or aromatic interactions.21 Some biological activities of multidentate pyridinedicarboxylate have already been demonstrated, such as antimicrobial activity and DNA cleavage.22
The field of vanadium metal–organic hybrids started to grow considerably since the discovery of MIL-26 and MIL-47 in the early period of the last decade.23 We are especially interested in extending the knowledge of self-assembly of vanadium compounds because of their potential therapeutic application such as insulin-enhancing agents. An important advantage of vanadium compounds in the treatment of diabetes mellitus compared to insulin is the possibility of oral administration.24
In this study new supramolecular networks built by pyridine-2,6-dicarboxylic acid (2,6-H2pydc, dipicolinic acid) were generated containing various organic cations in order to evaluate the role of cations and anions in crystal engineering. Compared with other transition metal cations, vanadium ions possess distinctly different properties and coordination modes, which play key roles in the formation of both coordination structures and packing structures of complexes. For example, vanadium(V) compounds usually contain a typical cis-VO2+ moiety with an overall five- or six-coordination. The VO2+ as well as VO2+ group can also participate in weak V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯C interactions as pointed out recently.25 In the studied [VO2(2,6-pydc)]− systems the VO2+ moiety is additionally coordinated by one tridentate 2,6-pydc2− ligand forming pentacoordinated geometry. While oxygen atoms being part of the anion allow the formation of numerous hydrogen bonds, the aromatic pyridine ring of 2,6-pydc2− ligand enables additional π⋯π stacking interactions.
O⋯C interactions as pointed out recently.25 In the studied [VO2(2,6-pydc)]− systems the VO2+ moiety is additionally coordinated by one tridentate 2,6-pydc2− ligand forming pentacoordinated geometry. While oxygen atoms being part of the anion allow the formation of numerous hydrogen bonds, the aromatic pyridine ring of 2,6-pydc2− ligand enables additional π⋯π stacking interactions.
Experimental section
Materials and physical measurements
Reagents and chemicals were purchased as reagent grade from commercial sources and were used without any further purification. Diethyl pyridine-2,6-dicarboxylate ester was prepared according to a published procedure.26 Infrared (IR) spectra (4000–600 cm−1) of the samples were recorded using a Perkin-Elmer Spectrum 100, equipped with a Specac Golden Gate Diamond ATR as a solid sample support. Elemental (C, H, N) analysis was performed using a Perkin-Elmer 2400 Series II CHNS/O Elemental Analyzer. Powder X-ray diffraction patterns were collected on a Siemens D-5000 diffractometer with θ–2θ Bragg–Brentano geometry operating using Cu-Kα radiation.Preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 30 mL). To the solution KOH (151 mg, 2.69 mmol) in methanol (3 mL) was added in small portions. After stirring overnight at room temperature, water (30 mL) was added and volatile solvents were removed in vacuo. The water residue was extracted with dichloromethane (20 mL) and then acidified to pH 2 with 1 M HCl. The aqueous layer was extracted with dichloromethane (5 × 15 mL). Combined organic layers were washed with brine (15 mL) and dried with anhydrous MgSO4. Solvents were removed in vacuo yielding a white solid compound. Yield: m = 272 mg (46%). 1H NMR (CDCl3, 500 MHz): δ 8.54 (m, 1H, Py-H), 8.50 (d, J = 7 Hz, 2H, Ar-H 2,6-pydc), 8.20 (t, J = 7 Hz, 1H, Ar-H 2,6-pydc), 7.91 (m, 1H, Py-H), 7.35 (m, 1H, Py-H), 7.28 (m, 1H, Py-H).
:1, 30 mL). To the solution KOH (151 mg, 2.69 mmol) in methanol (3 mL) was added in small portions. After stirring overnight at room temperature, water (30 mL) was added and volatile solvents were removed in vacuo. The water residue was extracted with dichloromethane (20 mL) and then acidified to pH 2 with 1 M HCl. The aqueous layer was extracted with dichloromethane (5 × 15 mL). Combined organic layers were washed with brine (15 mL) and dried with anhydrous MgSO4. Solvents were removed in vacuo yielding a white solid compound. Yield: m = 272 mg (46%). 1H NMR (CDCl3, 500 MHz): δ 8.54 (m, 1H, Py-H), 8.50 (d, J = 7 Hz, 2H, Ar-H 2,6-pydc), 8.20 (t, J = 7 Hz, 1H, Ar-H 2,6-pydc), 7.91 (m, 1H, Py-H), 7.35 (m, 1H, Py-H), 7.28 (m, 1H, Py-H).
Hpy[VO2(2,6-pydc)]·2H2O (1·2H2O) was prepared according to procedure A. Yield: m = 53 mg (30%), CHN elemental analysis: calculated for monohydrate C12H11N2O7V (%): C 41.64, H 3.20, N 8.09; found: C 41.45, H 3.08, N 8.01. IR (ATR, cm−1): 3365m, 3097w, 3066w, 2663br, 2190w, 1685s, 1636m, 1545w, 1484m, 1435w, 1333s, 1252w, 1168m, 1148w, 1078m, 1033w, 946s, 918s, 881m, 850w, 749s, 690m, 677s.
Hpy[VO2(2,6-pydc)] (1) was prepared according to procedure C. Only a few crystals were taken out from the solution and used for X-ray analysis. Other crystals were left in the mother liquid and after a few days, probably due to the incorporation of air moisture, transformed into 1·2H2O crystals.
H2pyon[VO2(2,6-pydc)]·H2O (2·H2O) was prepared according to procedures A and D. Yield: m = 96 mg (53%), CHN elemental analysis: calculated for C12H11N2O8V (%): C 39.80, H 3.06, N 7.73; found: C 39.68, H 2.93, N 7.65. IR (ATR, cm−1): 3393m, 3106w, 3093w, 2496br, 2114w, 1695m, 1600s, 1592s, 1552m, 1476w, 1438w, 1386m, 1371m, 1342s, 1263w, 1169w, 1078m, 951s, 925s, 875m, 852w, 779m, 765m, 748s, 679m, 622w.
H4apy[VO2(2,6-pydc)]·H2O (3·H2O) was prepared according to procedure B. Yield: m = 132 mg (77%), CHN elemental analysis: calculated for anhydrous C12H10N3O6V (%): C 42.00, H 2.94, N 12.24; found: C 41.85, H 2.76, N 12.25. IR (ATR, cm−1): 3336w, 3160m, 3084m, 2941s, 2873w, 2071w, 1681m, 1656s, 1636m, 1530s, 1432w, 1337s, 1191w, 1164w, 1077m, 958m, 937w, 916s, 844w, 817m, 753m, 675m.
H4apy[VO2(2,6-pydc)] (3) was prepared according to procedure C. Yield: m = 25 mg (29%), CHN elemental analysis: calculated for C12H10N3O6V (%): C 42.00, H 2.94, N 12.24; found: C 42.15, H 2.61, N 12.22. IR (ATR, cm−1): 3336w, 3160m, 3084m, 2941s, 2873w, 2071w, 1681m, 1656s, 1636m, 1530s, 1432w, 1337s, 1191w, 1164w, 1077m, 958m, 937w, 916s, 844w, 817m, 753m, 675m.
Hdmap[VO2(2,6-pydc)]·H2O (4·H2O) was prepared according to procedures B and C. Yield: m = 140 mg (72%), CHN elemental analysis: calculated for C14H16N3O7V (%): C 43.31, H 3.89, N 10.82; found: C 43.10, H 3.87, N 10.78. IR (ATR, cm−1): 3508br, 3222w, 3093w, 2964w, 1681s, 1643m, 1632m, 1602w, 1568m, 1408w, 1334s, 1219m, 1066s, 944s, 922m, 818m, 752m, 744m, 674m.
Himd[VO2(2,6-pydc)] (5) was prepared according to procedures B and C. Yield: m = 111 mg (70%), CHN elemental analysis: calculated for C10H8N3O6V (%): C 37.87, H 2.54, N 13.25; found: C 37.92, H 2.35, N 13.22. IR (ATR, cm−1): 3160br, 3130m, 3075w, 3048w, 1680s, 1599w, 1563m, 1471w, 1424w, 1397w, 1337s, 1176m, 1150w, 1101w, 1069w, 1038w, 1029w, 923s, 907s, 803s, 768m, 745s, 670s, 627s.
Crystallography
Single-crystal X-ray diffraction data were collected at room temperature (1, 3, 4·H2O, 5) or at 150 K (1·2H2O, 2·H2O, 3·H2O) either on a Nonius Kappa CCD Diffractometer or an Agilent Technologies SuperNova Dual diffractometer with an Atlas detector using monochromated Mo-Kα radiation (λ = 0.71073 Å) (1, 3, 4·H2O, 5) or Cu-Kα radiation (λ = 1.54184 Å) (1·2H2O, 2·H2O, 3·H2O). The data were processed using DENZO28 or CrysAlis Pro.29 The structures were solved by direct methods using the program SHELXS-9730 (1·2H2O), SIR9731 (1, 2·H2O, 3, 4·H2O, 5) or SIR200432 (3·H2O), and refined on F2 using full-matrix least-squares procedures (SHELXL-97).30 All non-hydrogen atoms were refined anisotropically, except atoms of a minor part (34%) of disordered 4-aminopyridinium cation in 3·H2O. In 4·H2O the water molecule (O6) and the hydrogen atoms in the methyl groups of the Hdmap cation are disordered over the mirror plane in 0.50:0.50 ratio. Hydrogen atoms on aromatic rings, methyl, amino and hydroxyl groups were treated as riding atoms in geometrically idealized positions. Hydrogen atoms on water molecules were located from difference Fourier maps and refined by fixing the bond lengths and isotropic temperature factors as Uiso(H) = 1.5Ueq(O). Crystallographic data are summarized in Table 1.
| 1·2H2O | 1 | 2·H2O | 3·H2O | 3 | 4·H2O | 5 | |
|---|---|---|---|---|---|---|---|
| a R = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = {∑[w(Fo2 – Fc2)2]/∑[w(Fo2)2]}1/2. c S = {∑[(Fo2 – Fc2)2]/(n/p}1/2 where n is the number of reflections and p is the total number of parameters refined. | |||||||
| Formula | C12H13N2O8V | C12H9N2O6V | C12H11N2O8V | C12H12N3O7V | C12H10N3O6V | C14H16N3O7V | C10H8N3O6V |
| M r | 364.18 | 328.15 | 362.17 | 361.19 | 343.17 | 389.24 | 317.13 |
| T (K) | 150(2) | 293(2) | 150(2) | 150(2) | 293(2) | 293(2) | 293(2) |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/m | P21/m | P21/c | Pbcm | C2 |
| a (Å) | 6.7436(3) | 7.9144(3) | 7.8481(2) | 8.1855(2) | 14.4966(3) | 8.17150(10) | 9.3492(9) |
| b (Å) | 8.1400(3) | 12.5749(7) | 6.42100(10) | 6.5443(2) | 7.3713(2) | 29.4539(5) | 12.4260(5) |
| c (Å) | 13.7971(5) | 13.4045(6) | 13.8887(3) | 13.8669(3) | 14.6252(3) | 6.63980(10) | 6.4360(7) |
| α (°) | 103.904(4) | 88.379(3) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| β (°) | 97.539(3) | 89.985(2) | 95.055(2) | 97.755(2) | 118.6960(10) | 90.00 | 129.421(17) |
| γ (°) | 95.474(4) | 74.931(3) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| Volume (Å3) | 722.45(5) | 1287.63(10) | 697.17(3) | 736.03(3) | 1370.88(5) | 1598.08(4) | 577.59(9) |
| Z | 2 | 4 | 2 | 2 | 4 | 4 | 2 |
| D c (Mg m−3) | 1.674 | 1.693 | 1.725 | 1.630 | 1.663 | 1.618 | 1.823 |
| μ (mm−1) | 6.192 | 0.801 | 6.416 | 6.041 | 0.758 | 0.665 | 0.891 |
| F(000) | 372 | 664 | 368 | 368 | 696 | 800 | 320 |
| Crystal size (mm) | 0.50 × 0.20 × 0.17 | 0.13 × 0.08 × 0.08 | 0.40 × 0.20 × 0.05 | 0.40 × 0.25 × 0.07 | 0.50 × 0.22 × 0.12 | 0.25 × 0.18 × 0.03 | 0.30 × 0.30 × 0.30 |
| Reflections collected | 6961 | 10594 | 3489 | 3693 | 5849 | 3499 | 3063 |
| Reflections unique (Rint) | 2950 (0.0299) | 5902 (0.0598) | 1552 (0.0267) | 1627 (0.0267) | 3137 (0.0147) | 1987 (0.0177) | 1484 (0.0193) |
| Parameters | 220 | 379 | 140 | 159 | 199 | 160 | 93 |
| R, wR2 [I > 2σ(I)]a | 0.0395, 0.1105 | 0.0532, 0.1162 | 0.0333, 0.0913 | 0.0362, 0.1009 | 0.0320, 0.0866 | 0.0323, 0.0825 | 0.0247, 0.0560 |
| R, wR2 (all data)b | 0.0402, 0.1114 | 0.1365, 0.1504 | 0.0335, 0.0916 | 0.0367, 0.1018 | 0.0412, 0.0933 | 0.0429, 0.0879 | 0.0257, 0.0577 |
| GOF, Sc | 1.044, 1.043 | 0.964 | 1.089, 1.090 | 1.058 | 1.063 | 1.054 | 1.087 |
| Extinction coefficient | 0.0144(14) | 0.023(2) | |||||
| Flack parameter | 0.019(18) | ||||||
Results and discussion
Synthesis and spectroscopic properties
Seven structures of dioxido(pyridine-2,6-dicarboxylato)vanadate(V) salts with pyridinium (Hpy+), 2-hydroxypyridinium (H2pyon+), 4-aminopyridinium (H4apy+), 4-(dimethylamino)pyridinium (Hdmap+), and imidazolium (Himd+) counterions were determined. Colourless to pale yellow vanadium(V) pyridine-2,6-dicarboxylic complexes were isolated from aqueous or alcoholic mixtures of the ammonium or sodium metavanadate, pyridine-2,6-dicarboxylic acid (dipicolinic acid) and imidazole or pyridine analogues stirred at 70 °C for 15–30 minutes. After slow evaporation of solvents, crystals of 1·2H2O, 2·H2O, 3·H2O, 4·H2O and 5 were grown from the solutions. We have also attempted to prepare vanadium(IV) complexes with the pyridine-2,6-dicarboxylato ligand. However, when combining vanadyl sulphate with esters of pyridine-2,6-dicarboxylic acid and the corresponding nitrogen base only vanadium(V) complexes were obtained in rather low yields. In all these cases vanadium(IV) turned into vanadium(V) and the same structures, i.e.2·H2O and 4·H2O, 5 were obtained, except in the case of 4apy and py where the anhydrous 1 and 3 were formed. We have observed that monohydrate 3·H2O and dihydrate 1·2H2O are unstable outside the solution. When exposed to air, crystals of 3·H2O decompose, lose the crystal water, and, with the respect to the elemental analysis and IR spectra, transform into anhydrous compound 3. Crystals of dihydrate 1·2H2O also decompose, but lose only one equivalent of crystal water, as confirmed by elemental analysis.The symmetric and asymmetric stretching vibrations of the VO2+ moiety are observed in the range 951–907 cm−1 and have a shape of shoulder or are split into two or three bands. The position of these bands is similar to those in the NH4[VO2(2,6-pydc)] complex.33 Strong characteristic bands of the carboxyl groups are observed in the range 1695–1656 cm−1 for the asymmetric vibrations (νas) and 1342–1333 cm−1 for the symmetric vibrations (νs). The difference between asymmetric and symmetric stretching vibrations (Δ = νas − νs) of the carboxylate groups between 319 and 352 cm−1 is in accordance with monodentate coordination to the VO2+ moiety.34 The vibrational modes corresponding to aromatic C–H vibrations in all compounds appear in the range 3178–3050 cm−1. For compound 4·H2O also weak vibrations in the range 2964–2870 cm−1 are present, which could correspond to the vibrations of the methyl groups of the Hdmap cation. Compounds 1·2H2O, 2·H2O and 4·H2O show additional broad IR bands in the range 3506–3365 cm−1 corresponding to the O–H stretching vibrations of the crystal water.
Description of X-ray crystal structures
 | ||
| Scheme 1 Structure of the [VO2(2,6-pydc)]− anion with tridentate coordination of the pyridine-2,6-dicarboxylate ligand and the nitrogen bases used to form counterions. | ||
Selected bond distances and angles for individual complexes are summarized in Tables S1 and S2 (in ESI†), respectively. The bond distances between vanadium and pyridyl nitrogen atoms for all complexes are in the range 2.0824(19)–2.0993(13) Å, the distances between vanadium and carboxylate oxygen atoms are slightly shorter and are in the range 1.9746(17)–2.0109(17) Å. Double bonds between vanadium and oxido oxygen atoms of 1.6078(14)–1.6311(14) Å are as expected the shortest bonds in complexes.
Distortion of the pentacoordinated structure due to the chelation of the pyridine-2,6-dicarboxylato ligand is even more evident from the observed bond angles around vanadium. Angles between carboxylate oxygen atoms and pyridyl nitrogen atom are 73.86(5)–75.02(4)°, and angles between carboxylate oxygen atoms are 147.97(5)–150.04(7)°. Angles between carboxylate oxygen atoms and oxido oxygen atoms are 97.92(15)–100.15(15)°, and angles between two oxido oxygen atoms are in the range of 107.92(7)–110.49(13)°. These bond distances and angles are in the same range as previously reported for ammonium, guanidinium33 and 2,9-dimethyl-1,10-phenanthrolinium35 compounds. The distortion of a square-pyramid can be best described by the structural parameter τ (0 for an ideal square pyramid and 1 for an ideal trigonal bipyramid),36 which in these cases is in the range 0.38–0.42, except for 3 and 1·2H2O where it is 0.16 and 0.35, respectively. This difference will be discussed later.
The vanadium atom in 2·H2O, 3·H2O, 4·H2O and 5 lies in the plane with the pyridine-2,6-dicarboxylate O and N binding atoms; however, in other compounds vanadium is positioned above this plane for 0.047 Å (1·2H2O), 0.036 and 0.030 Å (1), and 0.157 Å (3).
. One asymmetric unit of 1·2H2O contains one complex anion, one cation and two molecules of crystal water (Fig. S1, ESI†). Hydrogen bonds and weak C–H⋯O interactions found in 1·2H2O are listed in Table S3 (in ESI†). In 1·2H2O only the protonated pyridinium group would act as the hydrogen bond donor, but due to the additional water molecules more diverse hydrogen bonding is possible. Water molecules act as hydrogen bond donors and hydrogen bond acceptors, and exhibit linkage between adjacent anions and cations. Infinite chains of anions parallel to vector [110] with the C22(12) graph set37 motif are formed via O–H⋯O (dD⋯A = 2.80–2.88 Å) hydrogen bonds between anions and water molecules and via O–H⋯O (dD⋯A = ∼2.67 Å) hydrogen bonds between two water molecules. These chains are further connected into parallel pairs via O–H⋯O (dD⋯A = ∼2.81 Å) hydrogen bonds forming flat bands with R66(24) and R44(16) graph set motifs. To the water molecules on the sides of the band pyridinium cations are hydrogen-bonded via charge-assisted N–H⋯O hydrogen bonds (dD⋯A = ∼2.66 Å) (Fig. 1). Additional stabilization of the crystal lattice is provided by weak C–H⋯O and π⋯π stacking interactions. Weak π⋯π stacking interactions connect pairs of parallel pyridinium rings with a centroid-to-centroid distance of 4.1093(13) Å that belong to the adjacent hydrogen-bonded bands and stabilize packing of hydrogen-bonded bands along [101] (Table S8, ESI†). Packing is facilitated in all three dimensions by C–H⋯O interactions (dD⋯A = 3.15–3.27 Å) (Fig. 1).
 | ||
| Fig. 1 Hydrogen bonding network in 1·2H2O. (a) A fragment of hydrogen-bonded band with R66(24) and R44(16) graph set motifs. (b) Packing of adjacent bands by C–H⋯O interactions. (c) Space filled representation of the three-dimensional packing along the [110] direction. (d) Packing diagram with π⋯π stacking interactions between cation rings. | ||
Anhydrous pyridinium salt 1 also crystallizes in the triclinic space group P. One asymmetric unit of anhydrous 1 contains two complex anions and two cations (Fig. S2, ESI†). Hydrogen bonds and weak C–H⋯O interactions found in anhydrous 1 are listed in Table S3 (in ESI†). Due to the absence of crystal water, only two hydrogen bonds could be formed in one asymmetric unit of the anhydrous compound 1. Furthermore, both cations in the asymmetric unit are connected to the same anion via N–H⋯O hydrogen bonds with the D11(2) graph set motifs. The first pyridinium cation is hydrogen-bonded to carboxylate O7 atom (dD⋯A = ∼3.13 Å), and the second cation is hydrogen-bonded to the oxido O11 atom (dD⋯A = ∼2.88 Å). The second vanadium anion is not involved in the hydrogen bonding (Fig. 2).
 | ||
| Fig. 2 Hydrogen bonding network in anhydrous 1. (a) Hydrogen bonds and π⋯π stacking interaction in the asymmetric unit of anhydrous 1. (b) Packing diagram along the a axis facilitated by weak interactions. (c) Space filled representation of packing along the [110] direction. (d) Packing diagram emphasizing the π⋯π stacking interactions. | ||
Interestingly, due to the lack of strong non-covalent intermolecular interactions such as hydrogen bonds anions are involved to a larger extent in weak interactions. For instance, weak VO⋯C interactions previously described by Stilinović et al.25 have been observed between the adjacent V1 and V2 complex anions of the asymmetric unit. The VO⋯C interactions are formed between oxido groups and carboxyl carbon atoms with O⋯C distances of 2.968(5) and 3.039(5) Å and VO⋯C angles of 139.1(2) and 141.0(2)°. Ions of different asymmetric units are further connected only by weak C–H⋯O and π⋯π stacking interactions. π⋯π stacking interactions were observed along the a axis between pairs of cations that belong to the same asymmetric unit with a centroid-to-centroid distance of 3.878(4) Å and an inter-ring dihedral angle of 6.3(3)°, and between cations of the adjacent asymmetric units with a centroid-to-centroid distance of 4.230(4) Å (Fig. 2, Table S8, ESI†). Supramolecular architecture is therefore controlled by additional weak hydrogen bonds extending in all three dimensions between aromatic CH groups of Hpy+ and V2 anions, oxido and carbonyl oxygen atoms of V1 anions, and oxido and carboxyl oxygen atoms of V2 anions (Fig. 2).
The three-dimensional framework is achieved by connecting the pillars to each other by several C–H⋯O interactions (dD⋯A = 3.14–3.48 Å) (Fig. 3). However, no significant π⋯π interactions have been observed. View of the packing along the b-axis reveals formation of canals between anions and cations that are parallel to the b-axis and are occupied by water molecules (Fig. 4).
 | ||
| Fig. 3 Hydrogen bonding network in 2·H2O. (a) A fragment of the crystal structure of 2·H2O emphasizing the connection of ionic pairs through a water molecule via the R66(24) graph set motif. (b) Square pillar formation due to the hydrogen bonding. (c) Space filled representation of the packing. (d) Space filled representation of the packing along the b-axis. | ||
 | ||
| Fig. 4 Packing of 2·H2O along the b-axis showing the canals filled with water molecules. Water molecules are shown as space-filling models for clarity. | ||
:0.34. The centers of both rings lie almost at the same position, but the cations are rotated for approximately 135°. Both possible cation positions enable similar hydrogen bonding as shown in Fig. 5. Hydrogen bonds found in 3·H2O are listed in Table S5 (in ESI†).
 | ||
| Fig. 5 The possibility of the formation of hydrogen bonds for both positions of a disordered 4-aminopyridinium cation in 3·H2O. Dashed lines indicate hydrogen bonds (symmetry codes: (i) x, y, z − 1; (ii) −x, y + 1/2, −z + 1; (iii) −x + 1, y + 1/2, −z + 1). | ||
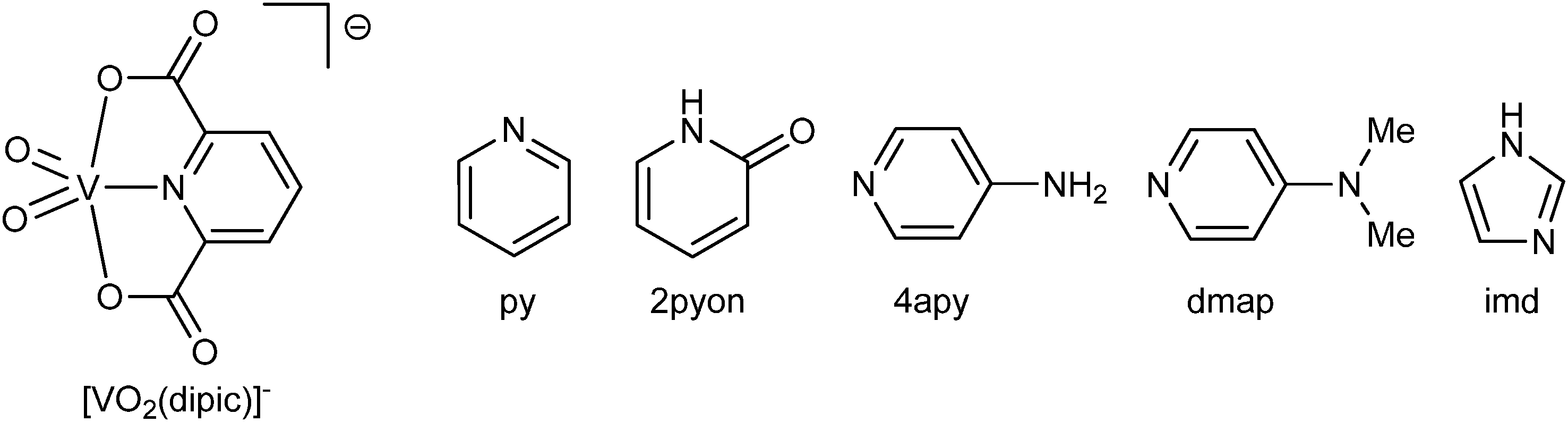
The Coulomb interactions between H4apy+ cations and [VO2(2,6-pydc)]− anions supported by charge-assisted hydrogen bonding between them organize chain formation. Cations and anions on the symmetry plane are connected via N–H⋯O hydrogen bonds (dD⋯A = 2.77–2.84 Å for the main component) along the c axis with the C22(12) graph set motif involving NH and NH2 moieties of H4apy (donors) and the carboxyl and carbonyl oxygen atoms of the 2,6-pydc2− ligand (acceptors). This chain is further connected to adjacent chains through water molecules forming an R66(26) motif via O–H⋯O (dD⋯A = ∼2.76 Å) and N–H⋯O (dD⋯A = ∼2.82 Å for the main component) hydrogen bonds connecting the NH2 moiety through a water molecule to the oxido oxygen atom of the VO2+ moiety (Fig. 6). Such two-dimensional frameworks are packed into crystal structure; however, no significant C–H⋯O or π⋯π interactions have been observed among them. View of the packing along the b-axis reveals a similar structure to that in 2·H2O. Between anions and cations canals parallel to the b-axis filled with water are formed (Fig. 7).
 | ||
| Fig. 6 Hydrogen bonding network in 3·H2O for the major part of a disordered 4-aminopyridinium cation. (a) A fragment of the crystal structure of 3·H2O emphasizing the connection of adjacent chains through a water molecule via the R66(26) graph set motif. (b) Chains connected into a two-dimensional framework. (c) Packing diagram along the b-axis. (d) Space filled representation of the packing along the c-axis. | ||
 | ||
| Fig. 7 Packing of 3·H2O along the b-axis showing the canals filled with water molecules. Water molecules are shown as space-filling models for clarity. | ||
Anhydrous 4-aminopyridinium salt 3 crystallizes in the monoclinic space group P21/c. One asymmetric unit of anhydrous 3 contains one complex anion and one H4apy+ cation (Fig. S5, ESI†). Similar to the 3·H2O structure the amino group and the protonated pyridine group in H4apy+ act as hydrogen bond donors, but only one oxido and two carboxyl oxygen atoms act as hydrogen bond acceptors. Hydrogen bonds found in anhydrous 3 are listed in Table S5 (ESI†).
As mentioned earlier the structural parameter τ describing the distortion of a square-pyramid is noticeably smaller for anhydrous 3 (τ = 0.16) than for all the other complex anions (τ = 0.35–0.42). The parameter τ is based on the difference between the largest two X–M–X angles α and β in the complex according to the equation τ = (α − β)/60°.33 We have investigated the reasons for such unexpected difference in τ values in the same type of complexes and observed that the largest angle α is Ocarboxyl–V–Ocarboxyl, and has similar values in all compounds (∼148°); however, the second largest angle β is N–V–Ooxido and is close to 125° in all cases, except in the anhydrous 3 being 138.42(7)°. This larger angle β (N1–V1–O6) in anhydrous 3 causes a small tilt of a VO2+ group, as can be observed also by a smaller N1–V1–O5 angle (113.63(7)°). The distortion of the VO2+ moiety probably happens due to the involvement of only one oxido oxygen atom of the VO2+ moiety in the hydrogen bonding. This has also an effect on the difference in VO bond lengths of the VO2+ moiety (1.6311(14) vs. 1.6078(14) Å), which is the largest difference in all compounds.
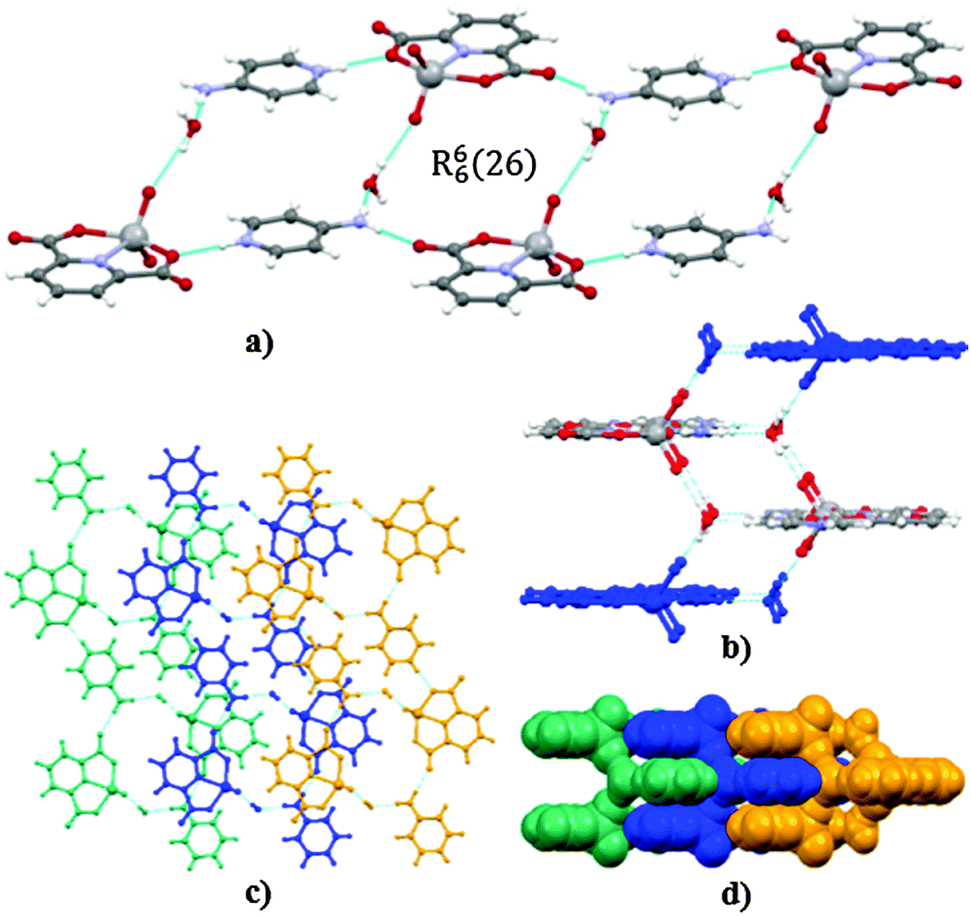
Coulomb interactions between H4apy+ cations and [VO2(2,6-pydc)]− anions supported by charge-assisted hydrogen bonding between them organize the packing of cations and anions. Cations and anions are connected via N–H⋯O hydrogen bonds (dD⋯A = 2.78–3.00 Å) into infinite chains with the C66(30) graph set motif involving NH and NH2 moieties of H4apy and one oxido oxygen of the VO2+ moiety as well as the carboxyl and carbonyl oxygen atoms of the 2,6-pydc2− ligand. Chains are further connected into infinite double layers perpendicular to the c axis by R44(16) and R88(44) graph set motifs. Further stabilization of the crystal lattice is enabled by weak C–H⋯O interactions between cations and anions (dD⋯A = 3.08–3.23 Å) or between adjacent anions (dD⋯A = ∼3.48 Å) that help to connect two-dimensional layers into a three-dimensional framework (Fig. 8). No significant π⋯π stacking has been observed in this crystal structure.
 | ||
| Fig. 8 Hydrogen bonding network in anhydrous 3. (a) A fragment of the crystal structure of 3 emphasizing the connection of cations and anions via the R44(16) graph set motif. (b) View along the c axis on the two-dimensional framework emphasizing the R88(44) motif. (c) Space filled representation of the packing of layers facilitated by weak C–H⋯O interactions. (d) Packing diagram along the b axis. | ||
:0.50 due to the mirror plane. Hydrogen bonds found in 4·H2O are listed in Table S6 (in ESI†). In 4·H2O, beside a water molecule, only a protonated pyridine group of the Hdmap+ cation acts as a hydrogen bond donor, while the two oxido and two carboxyl oxygen atoms of the anion act as hydrogen bond acceptors. Adjacent anions are connected through water molecules via O–H⋯O hydrogen bonds (dD⋯A = 3.07–3.14 Å) into twisted infinite chains. Chains are parallel to the c axis and (with the omission of one part of the disordered water molecule) possess the C44(6) graph set motif involving water molecules as donors, and carbonyl and oxido oxygen atoms as acceptors. To the water molecules belonging to these chains also cations are connected via charge-assisted N–H⋯O hydrogen bonds (dD⋯A = ∼2.82 Å). Stabilization of the crystal structure is further controlled by weak hydrogen bonds, as well as π⋯π stacking interactions listed in Table S8 (ESI†). The π⋯π stacking spreading along the c axis has been observed between parallel 2,6-pydc2− and Hdmap+ rings with a centroid-to-centroid distance of 3.87 Å that are part of two different hydrogen-bonded chains, and helps to stabilize packing along the a axis. Weak C–H⋯O interactions (dD⋯A = 3.22–3.46 Å) formed between methyl and aromatic hydrogen atoms of cations and oxygen atoms of anions spread along a and b axes, and stabilize packing of 4·H2O in all three dimensions (Fig. 9). View of the packing diagram along the c-axis reveals formation of canals between anions and cations parallel to the c-axis. Canals are wider than in 2·H2O and 3·H2O, and contain two rows of water molecules (Fig. 10).
 | ||
| Fig. 9 Hydrogen bonding network in 4·H2O. (a) A hydrogen-bonded chain parallel to the c axis with the C44(12) motif. (b) π⋯π stacking interactions between parallel anions and cations. (c) Space filled representation and packing diagram along the b axis indicating π⋯π stacking interactions. (d) Packing diagram along the c axis (facilitated by weak interactions). | ||
 | ||
| Fig. 10 Packing of 4·H2O along the c-axis showing the canals filled with water molecules. Water molecules are shown as space-filling models for clarity. | ||
 | ||
| Fig. 11 Hydrogen bonding network in 5. (a) A fragment of the hydrogen bonding in 5 with the R65(28) graph set motif. (b) Packing along the [1,0,−1] direction. (c) Packing diagram with the R65(24) graph set motif. (d) π⋯π stacking interactions between anions and cations. | ||
Structural comparison
The vanadium center in the dioxido(pyridine-2,6-dicarboxylato)vanadate(V) anion has a distorted square-pyramidal structure. Distortion also depends on the presence of hydrogen bonding. In most cases both oxido oxygen atoms are involved in the hydrogen bonding and both VO bond lengths are of the same range. The value of trigonality parameter τ in these compounds is usually 0.38–0.42. However, in 1·2H2O and 3 only one oxido oxygen atom is involved in the hydrogen bonding and this VO distance is elongated compared to the others. Interestingly, in these two compounds the distortion of the vanadium coordination sphere is the least pronounced, as can be seen by the τ values (in 1·2H2O is 0.35 and in 3 is 0.16).
In the imidazolium salt, in which each of the two NH groups binds to two acceptors, three-dimensional hydrogen bonding is possible. In 4-aminopyridinium salts with NH and NH2 donating groups two-dimensional systems were observed due to the classical hydrogen bonding. On the other hand, in pyridinium salts with only one donating NH group only isolated or one-dimensional hydrogen bonded systems could be formed and additional packing is stabilized by weak C–H⋯O and π⋯π interactions. We have also noticed that the water molecules in the crystal lattice acting as hydrogen bond donors and acceptors additionally increase the number and diversity of hydrogen bonds, but do not necessarily increase the stability of the structures. Powder X-ray diffraction (PXRD) experiments were carried out in order to confirm the phase purity of the bulk materials. We have observed that 1·2H2O is unstable outside the solution. When exposed to air, crystals of 1·2H2O decompose and lose one equivalent of crystal water, as confirmed by elemental analysis. Due to this partial dehydration the experimental PXRD pattern of monohydrate 1·H2O does not correspond with the one computer-simulated from the single crystal data of dihydrate 1·2H2O (Fig. S8 in ESI†). We have also observed that 3·H2O is not stable outside the solution. When exposed to air, crystals of 3·H2O decompose, lose the crystal water, and, with respect to the elemental analysis and IR spectra, transform into anhydrous 3 compound. The experimental PXRD patterns of 2·H2O, 3, 4·H2O and 5 correspond well with the ones computer-simulated from the single crystal data, indicating the high purity of the synthesized samples (Fig. S9 in ESI†). The differences in reflection intensities between the simulated and the experimental pattern are due to the variation in the preferred orientation of the powder samples as well as due to the fact that crystal 2·H2O was measured at 150 K while all PXRD data were collected at room temperature.
It has to be stressed that vanadium possesses a unique coordination environment in comparison to the other first row transition metals. In addition to the carboxylic group of the 2,6-pydc2− ligand, the [VO2(2,6-pydc)]− moiety has two easily accessible oxido oxygen atoms of the VO2+ unit. The availability of H-bond donating sites is also reflected in the number of participating acceptors in the [VO2(2,6-pydc)]− coordination anion. In 5 with a three-dimensional hydrogen-bonded framework and in 3·H2O with a two-dimensional hydrogen-bonded framework four out of six oxygen atoms of the [VO2(2,6-pydc)]− anion participate as N/O–H⋯O hydrogen bond acceptors including both oxido oxygen atoms of the VO2+ unit. The number of the acceptor sites of [VO2(2,6-pydc)]− anions is reduced to three out of six in 1·2H2O, 2·H2O and 4·H2O with a one-dimensional (band, pillar or chain) hydrogen bonding network and in anhydrous 3 with a two-dimensional hydrogen-bonded framework. However, in 2·H2O and 4·H2O both oxido oxygen atoms of the VO2+ unit participate as acceptors of classical hydrogen bonds while in 1·2H2O and 3 only one oxido oxygen atom acts as the acceptor.
Conclusion
We have prepared and structurally characterized seven different dioxido(pyridine-2,6-dicarboxylato)vanadate(V) compounds with aromatic nitrogen cations: pyridinium (1·2H2O and 1), 2-hydroxypyridinium (2·H2O), 4-aminopyridinium (3·H2O and 3), 4-(dimethylamino)pyridinium (4·H2O) and imidazolium cation (5). The compounds were synthesized via different pathways starting either from pyridine-2,6-dicarboxylic acid or its esters. Pyridinium and 4-aminopyridinium salts crystallized depending on the type of preparation either in their hydrous or anhydrous form, while 2-hydroxypyridinium, 4-(dimethylamino)pyridinium and imidazolium salts crystallized in the same way regardless of the synthetic route. The interactions between hydrogen-bonded organic cations and the [VO2(2,6-pydc)]− anion has been examined for the formation of self-assembly systems. The unique vanadium coordination environment with two easily accessible oxido oxygen atoms of the VO2+ unit is suitable for the construction of non-covalent metal–organic hybrids. In 2·H2O, 3·H2O, 4·H2O and 5 both oxido oxygen atoms of the VO2+ unit participate as acceptors, however, in 1·2H2O and 3 only one oxido oxygen atom is involved in classical hydrogen bonding. The ability of hydrogen bond formation and especially the number of H-bond donating sites on the cation has a significant impact on the supramolecular structure. In the case of 1·2H2O and 4·H2O with one H-bond donating Hpy+ and Hdmap+ cations one-dimensional (band or chain) hydrogen bonding networks via N/O–H⋯O bonds are formed. In 2·H2O with two H-bond donating H2pyon+ cation a hydrogen-bonded pillar structure with a hydrophilic interior is formed. On the other hand in the case of the Himd+ cation also with two H-bond donating sites a three-dimensional hydrogen bonding network was observed in 5. In anhydrous 3 and 3·H2O a two-dimensional hydrogen-bonded framework is formed even though the H4apy+ cation possesses the highest number of H-bond donating sites. Besides N/O–H⋯O hydrogen bonding, also other weak non-covalent interactions, such as C–H⋯O, π⋯π, C–H⋯π and VO⋯C interactions play an important role in stabilizing the crystal lattices. Thus, the rational choice of the cation may be an effective way to construct novel metal–organic hybrids with desired structures and properties. This research can be of great help in the field of emerging vanadium hybrid materials particularly from the viewpoint of being aware of the existing building blocks and their binding potentials according to the concepts of crystal engineering.
Acknowledgements
The authors thank the Ministry of Education, Science and Sport of the Republic of Slovenia and the Slovenian Research Agency for financial support (P1–0230–0175) as well as the EN–FIST Centre of Excellence, Trg Osvobodilne fronte 13, 1000 Ljubljana, Slovenia for use of the Supernova diffractometer.References
- (a) C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22–43 RSC; (b) M. G. Goesten, F. Kapteijn and J. Gascon, CrystEngComm, 2013, 15, 9249–9257 RSC; (c) M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619–1636 RSC; (d) M. W. Hosseini, Coord. Chem. Rev., 2003, 240, 157–166 CrossRef CAS; (e) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed; (f) K. Biradha, C.-Y. Su and J. J. Vittal, Cryst. Growth Des., 2011, 11, 875–886 CrossRef CAS; (g) E. R. Parnham and R. E. Morris, Acc. Chem. Res., 2007, 40, 1005–1013 CrossRef CAS PubMed; (h) G. Férey, Chem. Soc. Rev., 2007, 37, 191–214 RSC.
- (a) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC; (b) A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2014, 43, 5750–5765 RSC; (c) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- (a) B. V. de Voorde, B. Bueken, J. Denayer and D. D. Vos, Chem. Soc. Rev., 2014, 43, 5766–5788 RSC; (b) E. Barea, C. Montoro and J. A. R. Navarro, Chem. Soc. Rev., 2014, 43, 5419–5430 RSC; (c) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- (a) Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC; (b) M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed; (c) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- (a) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC; (b) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed; (c) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- (a) W. Zhang and R.-G. Xiong, Chem. Rev., 2012, 112, 1163–1195 CrossRef CAS PubMed; (b) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC.
- (a) L. Brammer, Dalton Trans., 2003, 3145–3157 RSC; (b) M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757–1788 RSC.
- (a) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC; (b) S. M. Malathy Sony and M. N. Ponnuswamy, Cryst. Growth Des., 2006, 6, 736–742 CrossRef.
- (a) K. Rissanen, CrystEngComm, 2008, 10, 1107–1113 RSC; (b) L. Brammer, G. M. Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 RSC; (c) R. W. Troff, T. Mäkelä, F. Topić, A. Valkonen, K. Raatikainen and K. Rissanen, Eur. J. Org. Chem., 2013, 1617–1637 CrossRef CAS; (d) V. G. Saraswatula and B. K. Saha, New J. Chem., 2014, 38, 897–901 RSC.
- (a) S. Petrosyants, Z. Dobrokhotova, A. Ilyukhin and V. Novotortsev, New J. Chem., 2014, 38, 3803–3812 RSC; (b) H. S. Jena, New J. Chem., 2014, 38, 2486–2499 RSC; (c) M. T. Johnson, Z. Džolić, M. Cetina, O. F. Wendt, L. Öhrström and K. Rissanen, Cryst. Growth Des., 2012, 12, 362–368 CrossRef CAS PubMed; (d) F. Perdih, Monatsh. Chem., 2012, 143, 1011–1017 CrossRef CAS; (e) F. Perdih, J. Coord. Chem., 2012, 65, 1580–1591 CrossRef CAS; (f) F. Perdih, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, M64–M68 CAS; (g) F. Perdih, Struct. Chem., 2014, 25, 809–819 CrossRef CAS.
- (a) H. W. Roesky and M. Andruh, Coord. Chem. Rev., 2003, 236, 91–119 CrossRef CAS; (b) L. Brammer, Chem. Soc. Rev., 2004, 33, 476–489 RSC.
- I. Cvrtila, V. Stilinović and B. Kaitner, CrystEngComm, 2013, 15, 6585–6593 RSC.
- (a) S. S. Sunkari, B. Kharediya, S. Saha, B. Elrezcd and J.-P. Sutter, New J. Chem., 2014, 38, 3529–3539 RSC; (b) J. Chen, S.-H. Wang, Z.-F. Liu, M.-F. Wu, Y. Xiao, F.-K. Zheng, G.-C. Guo and J.-S. Huang, New J. Chem., 2014, 38, 269–276 RSC; (c) X.-M. Lin, H.-C. Fang, Z.-Y. Zhou, L. Chen, J.-W. Zhao, S.-Z. Zhu and Y.-P. Cai, CrystEngComm, 2009, 11, 847–854 RSC; (d) S. Macksasitorn, Y. Hu and J. R. Stork, CrystEngComm, 2013, 15, 1698–1705 RSC; (e) F. Jin, H.-Z. Wang, Y. Zhang, Y. Wang, J. Zhang, L. Kong, F.-Y. Hao, J.-X. Yang, J.-Y. Wu, Y.-P. Tian and H.-P. Zhou, CrystEngComm, 2013, 15, 3687–3695 RSC; (f) C.-Q. Wan, A.-M. Li, S. A. Al-Thabaiti and T. C. W. Mak, Cryst. Growth Des., 2013, 13, 1926–1936 CrossRef CAS.
- (a) Y. Wen, T. Sheng, S. Hu, Y. Wang, C. Tan, X. Ma, Z. Xue, Y. Wang and X. Wu, CrystEngComm, 2013, 15, 2714–2721 RSC; (b) Z. Chen, L. Zhang, F. Liu, R. Wang and D. Sun, CrystEngComm, 2013, 15, 8877–8880 RSC; (c) R. K. Prajapati, J. Kumar and S. Verma, CrystEngComm, 2013, 15, 9316–9319 RSC; (d) A. M. Owczarzak, N. Kourkoumelis, S. K. Hadjikakou and M. Kubicki, CrystEngComm, 2013, 15, 3607–3614 RSC; (e) M. Mirzaei, H. Eshtiagh-Hosseini, M. M. Abadeh, M. Chahkandi, A. Frontera and A. Hassanpoor, CrystEngComm, 2013, 15, 1404–1413 RSC; (f) V. J. Argyle, L. M. Woods, M. Roxburgh and L. R. Hanton, CrystEngComm, 2012, 15, 120–134 RSC; (g) A. Singh, R. P. Sharma, T. Aree and P. Venugopalan, CrystEngComm, 2013, 15, 1153–1163 RSC.
- (a) P. Thuéry, CrystEngComm, 2014, 16, 1724–1734 RSC; (b) K. Molčanov, B. Kojić-Prodić, D. Babić and J. Stare, CrystEngComm, 2013, 15, 135–143 RSC; (c) K. Molčanov, B. Kojić-Prodić, D. Babić, D. Pajić, N. Novosel and K. Zadro, CrystEngComm, 2012, 14, 7958–7964 RSC; (d) K. Molčanov, B. Kojić-Prodić, D. Babić, D. Žilić and B. Rakvin, CrystEngComm, 2011, 13, 5170–5178 RSC; (e) K. Molčanov, I. Sabljić and B. Kojić-Prodić, CrystEngComm, 2011, 13, 4211–4217 RSC; (f) K. Molčanov, B. Kojić-Prodić and A. Meden, CrystEngComm, 2009, 11, 1407–1415 RSC.
- F. Zhuge, B. Wu, J. Liang, J. Yang, Y. Liu, C. Jia, C. Janiak, N. Tang and X.-J. Yang, Inorg. Chem., 2009, 48, 10249–10256 CrossRef CAS PubMed.
- (a) M. Meot-Ner, Chem. Rev., 2012, 112, PR22–PR103 Search PubMed; (b) J. K. Maclaren and C. Janiak, Inorg. Chim. Acta, 2012, 389, 183–190 CrossRef CAS PubMed; (c) D. Mekhatria, S. Rigolet, C. Janiak, A. Simon-Masseron, M. A. Hasnaoui and A. Bengueddach, Cryst. Growth Des., 2011, 11, 396–404 CrossRef CAS; (d) B. M. Drašković, G. A. Bogdanović, M. A. Neelakantan, A.-C. Chamayou, S. Thalamuthu, Y. S. Avadhut, J. Schmedt auf der Günne, S. Banerjee and C. Janiak, Cryst. Growth Des., 2010, 10, 1665–1676 CrossRef; (e) B. Gil-Hernández, H. A. Höppe, J. K. Vieth, J. Sanchiz and C. Janiak, Chem. Commun., 2009, 8270–8272 Search PubMed.
- (a) M. B. Andrews and C. L. Cahill, CrystEngComm, 2013, 15, 3082–3086 RSC; (b) K. Molčanov and B. Kojić-Prodić, CrystEngComm, 2010, 12, 925–939 RSC; (c) A. Pevec and A. Demšar, J. Fluorine Chem., 2008, 129, 707–712 CrossRef CAS PubMed; (d) A. Pevec, M. Tekavec and A. Demšar, Polyhedron, 2011, 30, 549–555 CrossRef CAS PubMed.
- (a) A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef CAS PubMed; (b) P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- (a) S. W. Jaros, P. Smoleński, M. F. C. G. da Silva, M. Florek, J. Król, Z. Staroniewicz, A. J. L. Pombeiro and A. M. Kirillov, CrystEngComm, 2013, 15, 8060–8064 RSC; (b) S. R. Miller, P. Horcajada and C. Serre, CrystEngComm, 2011, 13, 1894–1898 RSC; (c) S. R. Miller, D. Heurtaux, T. Baati, P. Horcajada, J.-M. Grenèche and C. Serre, Chem. Commun., 2010, 46, 4526–4528 RSC.
- (a) F. Semerci, O. Z. Yeşilel, S. Keskin, C. Darcan, M. Taş and H. Dal, CrystEngComm, 2013, 15, 1244–1256 RSC; (b) H. Eshtiagh-Hosseini, M. Mirzaei, M. Biabani, V. Lippolis, M. Chahkandi and C. Bazzicalupi, CrystEngComm, 2013, 15, 6752–6768 RSC; (c) M. Mirzaei, H. Eshtiagh-Hosseini, Z. Karrabi, K. Molčanov, E. Eydizadeh, J. T. Mague, A. Bauzá and A. Frontera, CrystEngComm, 2014, 16, 5352–5363 RSC; (d) G. Hundal, M. S. Hundal, Y. K. Hwang and J. S. Chang, Cryst. Growth Des., 2014, 14, 172–176 CrossRef CAS; (e) A. Husain and C. L. Oliver, CrystEngComm, 2014, 16, 3749–3757 RSC; (f) T.-W. Tseng, T.-T. Luo, C.-C. Su, H.-H. Hsu, C.-I. Yang and K.-L. Lu, CrystEngComm, 2014, 16, 2626–2633 RSC; (g) B.-M. Kukovec, G. A. Venter and C. L. Oliver, Cryst. Growth Des., 2012, 12, 456–465 CrossRef CAS; (h) M. A. Sharif and G. R. Najafi, Acta Chim. Slov., 2013, 60, 138–143 CAS; (i) L. B. Hamdy, P. R. Raithby, L. H. Thomas and C. C. Wilson, New J. Chem., 2014, 38, 2135 RSC.
- (a) A. T. Çolak, F. Çolak, O. Z. Yeşilel, D. Akduman, F. Yılmaz and M. Tümer, Inorg. Chim. Acta, 2010, 363, 2149–2162 CrossRef PubMed; (b) E.-J. Gao, M.-C. Zhu, Y. Huang, L. Liu, H.-Y. Liu, F.-C. Liu, S. Ma and C.-Y. Shi, Eur. J. Med. Chem., 2010, 45, 1034–1041 CrossRef CAS PubMed.
- (a) P. Van Der Voort, K. Leus, Y.-Y. Liu, M. Vandichel, V. Van Speybroeck, M. Waroquierb and S. Biswasa, New J. Chem., 2014, 38, 1853–1867 RSC; (b) M. Riou-Cavellec, M. Sanselme and G. Férey, J. Mater. Chem., 2000, 10, 745–748 RSC; (c) K. Barthelet, J. Marrot, D. Riou and G. Férey, Angew. Chem., Int. Ed., 2002, 41, 281–284 CrossRef CAS.
- (a) G. R. Willsky, L.-H. Chi, M. Godzala III, P. J. Kostyniak, J. J. Smee, A. M. Trujillo, J. A. Alfano, W. Ding, Z. Hu and D. C. Crans, Coord. Chem. Rev., 2011, 255, 2258–2269 CrossRef CAS PubMed; (b) T. Kiss, T. Jakusch, B. Gyurcsik, A. Lakatos, É. A. Enyedy and É. Sija, Coord. Chem. Rev., 2012, 256, 125–132 CrossRef CAS PubMed; (c) H. Sakurai, Y. Yoshikawa and H. Yasui, Chem. Soc. Rev., 2008, 37, 2383–2392 RSC; (d) T. Koleša-Dobravc, E. Lodyga-Chruscinska, M. Symonowicz, D. Sanna, A. Meden, F. Perdih and E. Garribba, Inorg. Chem., 2014, 53, 7960–7976 CrossRef PubMed; (e) T. Koleša-Dobravc, A. Meden and F. Perdih, Monatsh. Chem., 2014, 145, 1263–1275 CrossRef PubMed.
- V. Stilinović, D.-K. Bučar, I. Halasz and E. Meštrović, New J. Chem., 2013, 37, 619–623 RSC.
- G. Labbé, A. P. Krismanich, S. de Groot, T. Rasmusson, M. Shang, M. D. R. Brown, G. I. Dmitrienko and J. G. Guillemette, J. Inorg. Biochem., 2012, 112, 49–58 CrossRef PubMed.
- W. Rasshofer, W. M. Mueller and F. Voegtle, Chem. Ber., 1979, 112, 2095–2119 CrossRef CAS.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- CrysAlis PRO, Agilent Technologies, Yarnton, England, 2011 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381–388 CrossRef CAS.
- B. S. Parajón-Costa, O. E. Piro, R. Pis-Diez, E. E. Castellano and A. C. González-Baró, Polyhedron, 2006, 25, 2920–2928 CrossRef PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Theory and Applications in Inorganic Chemistry, John Wiley & Sons, New York, 1997 Search PubMed.
- H. Aghabozorg and E. Sadr-khanlou, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m1753 CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- (a) J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed., 1995, 34, 1555–1573 CrossRef CAS; (b) M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- (a) T. Dorn, A.-C. Chamayou and C. Janiak, New J. Chem., 2006, 30, 156–167 RSC; (b) J. K. Maclaren, J. Sanchiz, P. Gili and C. Janiak, New J. Chem., 2012, 36, 1596–1609 RSC; (c) M. Enamullah, V. Vasylyeva and C. Janiak, Inorg. Chim. Acta, 2013, 408, 109–119 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1030256–1030262. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00164a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |