
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of enantiomerically pure δ-benzylproline derivatives†
Isabel
Rodríguez
,
M. Isabel
Calaza
,
Ana I.
Jiménez
and
Carlos
Cativiela
*
Departamento de Química Orgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC–Universidad de Zaragoza, 50009 Zaragoza, Spain. E-mail: cativiela@unizar.es; Tel: +34 976761210
First published on 5th February 2015
Abstract
The (2S,5R) stereoisomer of 5-benzylproline, i.e. the L-proline analogue that bears a δ-benzyl substituent cis to the carbonyl function, has been prepared in enantiomerically pure form and excellent global yield. The procedure involves the construction of the pyrrolidine ring through intramolecular cyclization and uses as starting material the enantiopure β-amino acid obtained by homologation of L-phenylalanine. The generation of an intermediate vinyl triflate with full regiochemical control followed by a stereoselective hydrogenation reaction allowed the isolation of the target δ-substituted L-proline analogue in optically pure form and 43% overall yield from the initial β-amino acid. The trans stereoisomer of (2R,5R) configuration is obtained as a minor product through a less stereoselective hydrogenation reaction.
Introduction
One of the most efficient approaches to control peptide conformation is the incorporation of amino acids with restricted conformational flexibility into the peptide chain. Among proteinogenic residues, only proline exhibits clearly defined conformational propensities.1 This is due to the presence of the five-membered pyrrolidine ring that includes the α carbon and the amino function, thus precluding rotation around the N–Cα bond. As a consequence, proline cannot adopt the fully extended conformation and acts as a turn inductor in peptide chains.1,2 Another effect associated to the cyclic nature of proline is the relative high probability of the so-called prolyl peptide bond (that formed by the pyrrolidine nitrogen and the carbonyl group of the preceding residue) to accommodate a cis arrangement.1,3The singular structure of proline may serve as a scaffold for the construction of new amino acids with well-defined conformational tendencies and, indeed, extensive research has been conducted on the synthesis of proline analogues with different types of modifications.4,5 In particular, the attachment of substituents to the pyrrolidine cycle gives rise to non-proteinogenic residues that should retain the conformational propensities of the natural amino acid while incorporating new side-chain functionalities. Such a combination of structural and functional properties makes substituted prolines very helpful to stabilize structural motifs in peptides and is particularly appealing in the case of proline analogues that bear side chains belonging to other proteinogenic amino acids. Moreover, by adequately choosing the position, nature, and steric bulk of the new substituent incorporated, the conformational propensities of the parent amino acid may be adjusted.5,6
In this regard, attachment of a substituent to the δ position of the pyrrolidine cycle (C-5) affects the cis–trans isomerization state of the prolyl peptide bond,5,6 which is governed by steric interactions of the acyl moiety linked to the pyrrolidine nitrogen with the contiguous α and δ positions of the pyrrolidine cycle. Thus, proline analogues with considerable steric hindrance at Cδ such as δ,δ-dimethylproline and δ-tert-butylproline have been shown to stabilize the cis state of this peptide bond,7 which, in turn, affects the overall conformation of the peptide backbone. The possibility to modulate amide geometry by using δ-substituted prolines has been applied, for example, to investigate the folding pathways of therapeutic proteins,3a,8 to elucidate the receptor-bound conformation of bioactive peptides,7c,d,9 or to establish the role of amide cis–trans isomerization of a single proline residue in switching the open/closed states of a neurotransmitter-gated ion channel.10 Moreover, several proline analogues containing aromatic substituents at the δ position have been used in the design of peptide-based pharmacologically active compounds.11 Among these analogues is δ-benzylproline,11d,e which combines the conformational constraints of proline with the side chain of phenylalanine. This proline–phenylalanine hybrid amino acid may not only provide fundamental insights into the precise conformational requirements for optimal receptor binding affinity in bioactive peptides, but can also be very useful to understand the influence of aromatic interactions in peptide folding and prolyl amide isomerization.
The exploitation of δ-benzylproline requires ready access to the different stereoisomers of this amino acid in optically pure form. The most general methods for the synthesis of enantiopure or highly enantioenriched δ-substituted prolines make use of substrates with a pre-formed proline skeleton in which the desired substituent at Cδ is further installed4d,e,5 (Scheme 1, routes a, b). In particular, L-pyroglutamic acid derivatives have been used as precursors for the synthesis of two δ-benzylproline stereoisomers.12 Alternatively, the five-membered pyrrolidine ring can be formed by intramolecular cyclization of adequately functionalized acyclic precursors.5 Among the cyclization strategies giving rise to highly enantioenriched or enantiomerically pure δ-substituted prolines, the approach described by Davis,13 which involves the use of chiral sulfinimines (>96% ee) derived from aldehydes (Scheme 1, route c), is the most efficient and versatile one. However, the synthesis of enantiopure δ-benzylproline by this method seems unfeasible because it would require the use of a sulfinimine that undergoes facile isomerization to the tautomeric enamine,14 thereby compromising preservation of optical purity. A few reports specifically oriented to the synthesis of δ-benzylproline through stereoselective cyclization reactions have been described and entail the laborious preparation and transformation of a chiral oxaziridine15a or acetylene-containing amino acid.15b Herein, we present a practical procedure for the synthesis of an enantiopure δ-benzylproline stereoisomer that makes use of a homochiral β-amino acid as the starting material. This methodology can be viewed as complementary to Davis' approach13 since it is amenable to extension to other readily available enantiopure β-amino acids, thus giving access to δ-substituted prolines that are not accessible by Davis' procedure.
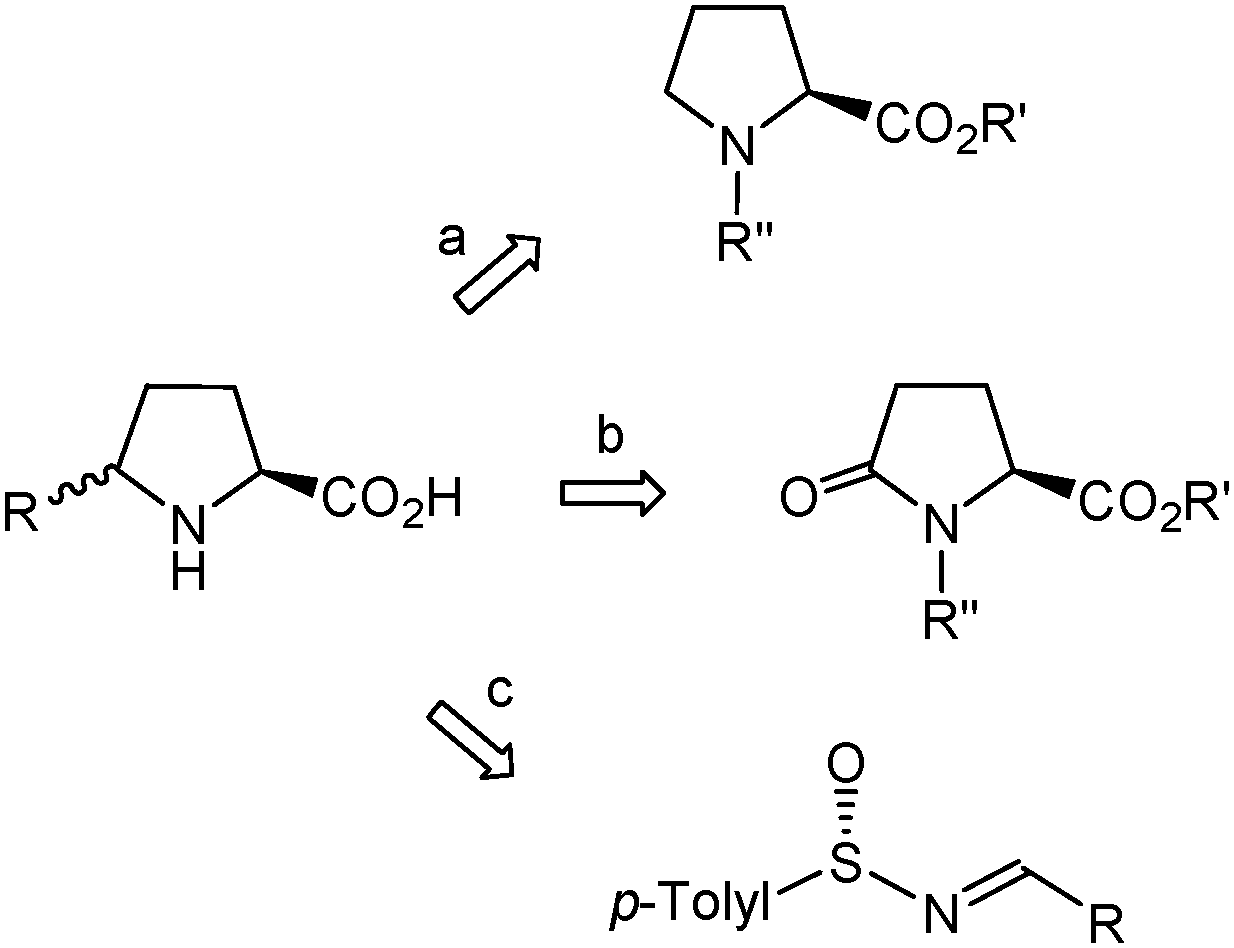 | ||
| Scheme 1 Most general strategies used for the synthesis of enantiopure or highly enantioenriched δ-substituted prolines. | ||
Results and discussion
We have recently shown that β-amino acids are suitable precursors for the synthesis of racemic proline analogues substituted at the β and/or δ positions of the pyrrolidine ring.16 Thus, we carried out the preparation of racemic β-phenylproline16a by cyclization of an α-diazo-β-ketoester derived from β-alanine through an intramolecular N–H insertion reaction mediated by a metal carbenoid17,18 and further functionalization of the intermediate β-ketoproline formed to incorporate the aromatic substituent. We subsequently showed that the procedure is applicable to the β-alanine derivative possessing two geminal methyl groups at C-3, which enabled the synthesis of δ,δ-dimethyl-β-phenylproline and δ,δ-dimethylproline.16b In both cases, non-chiral β-amino acids were used as substrates and the substituted proline analogues were obtained in racemic form.16 Yet the procedure should be adequate for the preparation of enantiomerically pure prolines with different substitution patterns by using homochiral β-amino acids as starting materials. In particular, we envisioned that the enantiopure β-amino acids readily accessible from proteinogenic L-amino acids through well-known racemization-free homologation procedures19 (many of them are also commercially available) could allow the synthesis of optically pure δ-substituted prolines that incorporate the side chain of the coded amino acid at the δ position (Scheme 2). The validity of this hypothesis was checked by addressing the preparation of enantiomerically pure δ-benzylproline starting from the β-amino acid derived from L-phenylalanine. To the best of our knowledge, no β-amino acid of this type has been used to date for the synthesis of δ-substituted proline derivatives.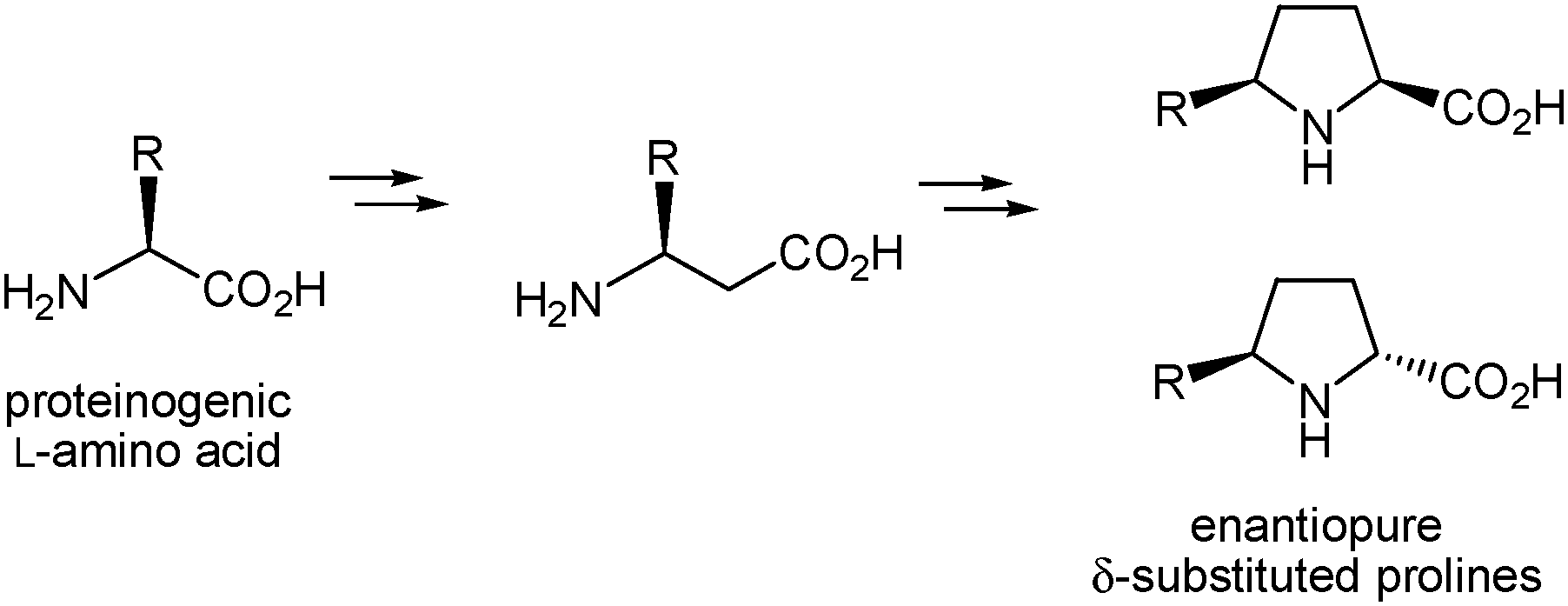 | ||
| Scheme 2 Proposed access to enantiopure δ-substituted prolines from β-amino acids derived from proteinogenic amino acids. | ||
The synthetic route began with the preparation of the desired β-amino acid bearing a tert-butoxycarbonyl (Boc) group at the amino function as this type of protection is widely used in standard peptide synthesis and is compatible with the synthetic transformations to be carried out. This compound, Boc-L-β-homophenylalanine or (S)-3-(N-tert-butoxycarbonyl)amino-4-phenylbutanoic acid, is available from commercial sources albeit at a very high price. Alternatively, it can also be obtained in multigram quantities and optically pure form by Arndt–Eistert homologation of non-expensive Boc-L-phenylalanine following the well-established literature procedure.20 We applied a slightly modified version of this method. Thus, the mixed anhydride generated by reaction of Boc-L-phenylalanine with ethyl chloroformate20 was treated with trimethylsilyldiazomethane (Scheme 3) instead of the very hazardous diazomethane20 to form the diazoketone 1. This replacement proved satisfactory even if it was discarded in the reported procedure.20 Our assay was based on the described use of trimethylsilyldiazomethane as a convenient substitute for diazomethane in the Arndt–Eistert homologation of acyl chlorides21 and its successful application to a carboxylic acid activated as a mixed anhydride in a more complex molecule.22 The conditions described in the latter work22 proved adequate in our case, when applied to Boc-L-phenylalanine, and the corresponding diazoketone (1) was obtained in good yield using the stable and less hazardous trimethylsilyldiazomethane (Scheme 3). Next, the diazoketone underwent a rearrangement process20 promoted by silver benzoate under sonication conditions22,23 to provide the desired N-Boc-protected β-amino acid (2) in enantiomerically pure form with retention of configuration.19,24
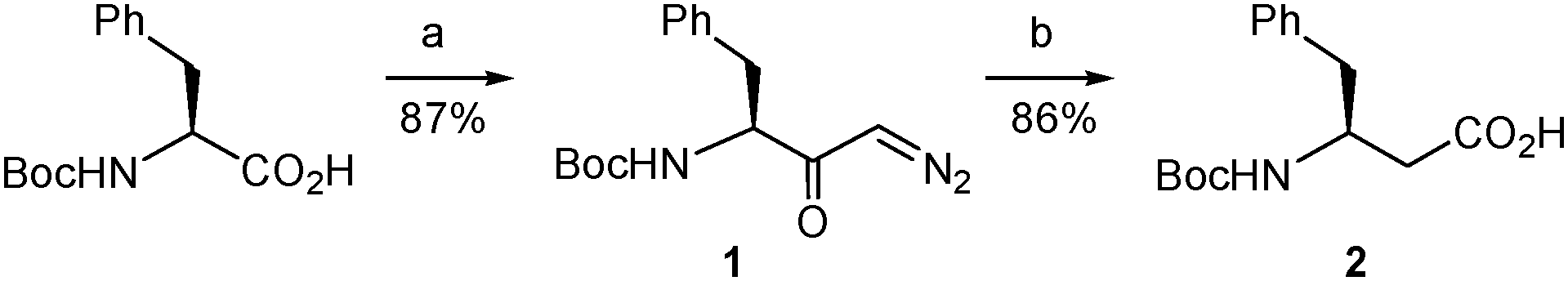 | ||
| Scheme 3 Synthesis of the β-amino acid 2. Reagents and conditions: (a) (i) EtOCOCl, Et3N, THF, −15 to −5 °C; (ii). Me3SiCHN2, MeCN, 4 °C; (b) Na2CO3, PhCO2Ag, dioxane/water, sonication, rt. | ||
Construction of the β-ketoproline skeleton was then addressed (Scheme 4). To this end, we followed the experimental procedure applied by Sorensen to β-alanine18 with slight variations.16 Thus, the carboxylic acid in 2 was activated with N,N′-carbonyldiimidazole and the imidazolide formed reacted with a malonate-derived soft enolate to afford the β-ketoester 3 after spontaneous decarboxylation. The crude compound was subjected to a diazo-transfer reaction by treatment with 4-(acetamido)benzenesulfonyl azide25 in the presence of triethylamine. The resulting α-diazo-β-ketoester (4) underwent decomposition promoted by rhodium diacetate catalysis under heating conditions. The intramolecular N–H insertion reaction of the metal carbenoid generated in situ delivered the δ-benzyl-β-ketoproline derivative 5 as an inseparable mixture of cis/trans stereoisomers.26 This mixture was obtained in 72% overall yield from the starting β-amino acid (2) with a single purification being necessary.
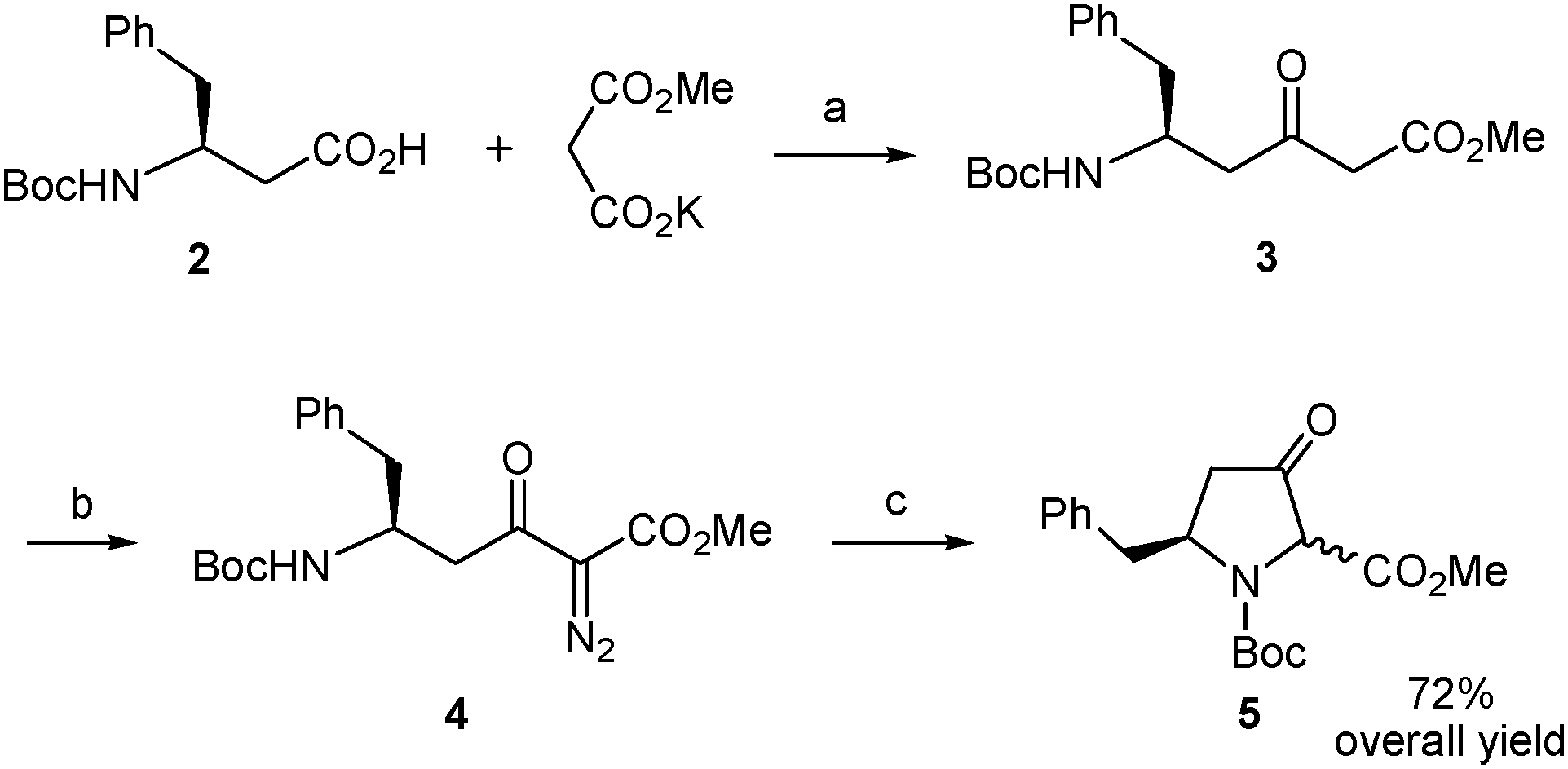 | ||
| Scheme 4 Synthesis of the δ-benzyl-β-ketoproline 5 from the β-amino acid 2. Reagents and conditions: (a) N,N′-carbonyldiimidazole, MgCl2, THF, rt; (b) 4-(acetamido)benzenesulfonyl azide, Et3N, MeCN, rt; (c) Rh2(OAc)4, toluene, 85–90 °C. | ||
The final step in the route to the target amino acid involved reduction of the ketone function at the β position (C-3) in 5. In our previous syntheses of substituted proline analogues starting from β-amino acids,16 we carried out this transformation through an intermediate vinyl triflate (triflate: trifluoromethanesulfonate, OTf). By using the adequate combination of base and triflating agent, we have established the conditions for the formation of this intermediate triflate in a completely regioselective manner so that only the regioisomer with a double bond connecting the α and β carbons (C-2, C-3) is obtained.16 Accordingly, there are two reasons to prefer the formation of this intermediate vinyl triflate over other methodologies for the reduction of the carbonyl moiety at C-3 in compound 5. First, as the intramolecular cyclization reaction giving rise to 5 (Scheme 4) proceeds with low diastereoselectivity,26 hydrogenation of the double bond in this triflate provides a further opportunity to obtain the desired δ-benzylproline with a higher degree of stereocontrol at the α carbon. Second, if desired, the triflate moiety at the β carbon may serve for the incorporation of additional substituents at this position, as demonstrated in our previous work.16
Therefore, we undertook the generation of the vinyl triflate 6 (Scheme 5) by applying the combination of base and triflating agent that proved essential to achieve full regiocontrol when dealing with other 3-ketoproline derivatives previously investigated.16 Thus, deprotonation of 5 (mixture of cis/trans diastereoisomers) with potassium bis(trimethylsilyl)amide (KHMDS) gave an enolate that was trapped with N-(5-chloro-2-pyridyl)triflimide (Comins' reagent27) to yield 6 as a single regioisomer.28 Compound 6 proved stable enough for chromatographic purification and when stored at −20 °C.
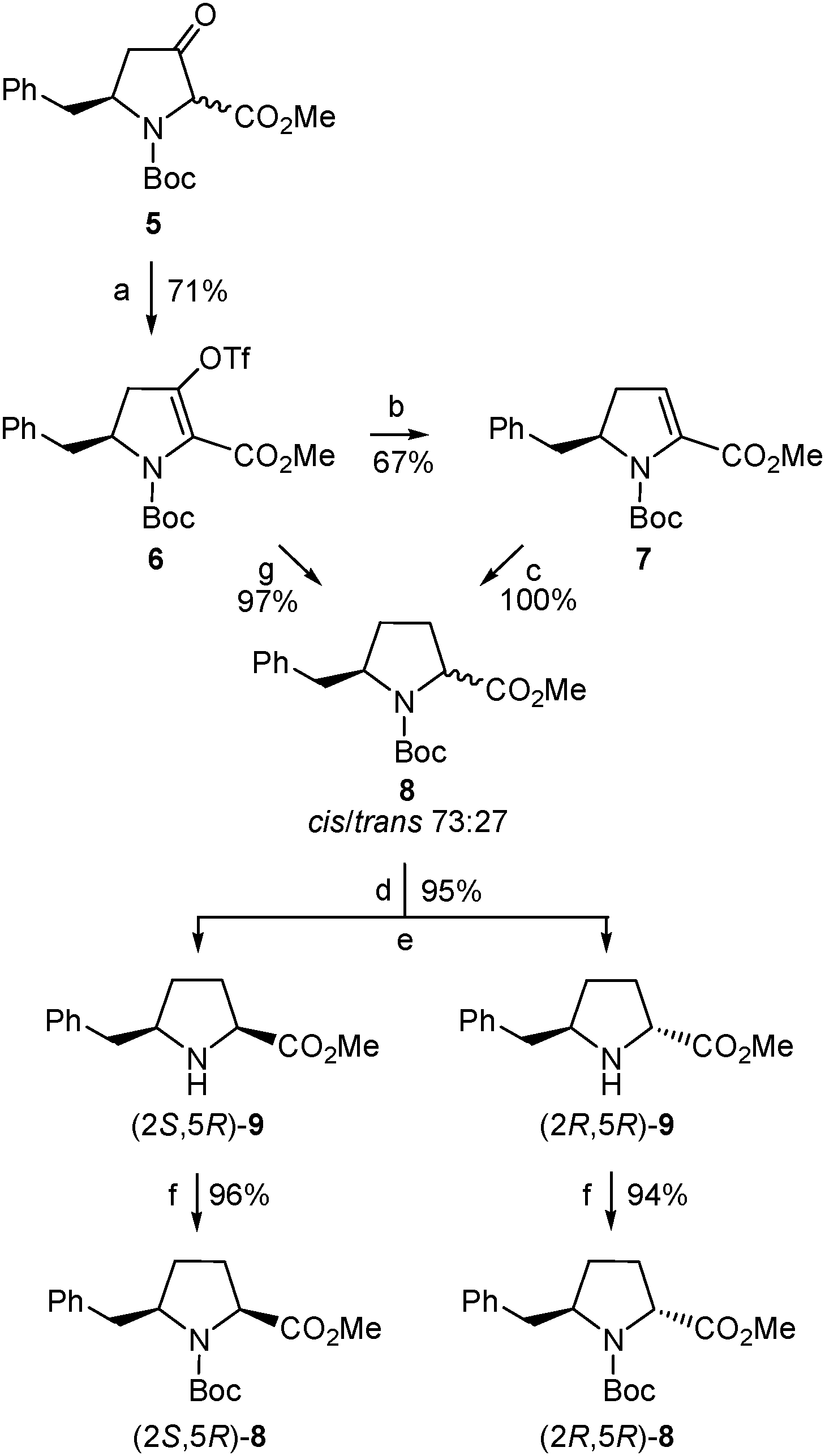 | ||
| Scheme 5 Synthesis of enantiomerically pure δ-benzylprolinates 8 and 9. Reagents and conditions: (a) KHMDS, N-(5-chloro-2-pyridyl)triflimide, THF, rt; (b) Et3SiH, Pd(PPh3)4, LiCl, Et3N, DMF, 75 °C; (c) H2, Pd/C, MeOH, rt; (d) TFA, CH2Cl2, rt; (e) separation of diastereoisomers by column chromatography; (f) Boc2O, CH2Cl2, rt; (g) H2, Pd/C, Na2CO3, MeOH, rt. | ||
With the aim to obtain the desired δ-benzylproline, triflate 6 was first subjected to a palladium-catalyzed reductive removal of the triflate moiety using triethylsilane as the hydride source (Scheme 5). The resulting α,β-dehydroproline derivative (7) was then submitted to hydrogenation to generate the saturated five-membered ring. Treatment of 7 with hydrogen gas at atmospheric pressure in the presence of palladium on carbon as a catalyst proceeded with quantitative yield but only moderate diastereoselectivity, so that a 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :27 mixture of δ-benzylprolinates 8 was formed (Scheme 5), as deduced from the 1H-NMR spectrum. The 8 diastereoisomers could not be separated by column chromatography and, therefore, their cis/trans relative configuration [corresponding, respectively, to a (2S,5R)/(2R,5R) absolute stereochemistry29] was not established at this stage. Yet a cis relative disposition of substituents around the pyrrolidine ring, and hence a (2S,5R) absolute configuration, was tentatively assigned to the major diastereoisomer assuming that hydrogenation had preferentially taken place on the less hindered face of the molecule, that is, opposite the benzyl substituent. Luckily enough, chromatographic separation of diastereoisomers was possible after removal of the Boc group of the (2RS,5R)-8 mixture under standard acidic conditions (Scheme 5). For characterization purposes, samples of the pure amino esters thus isolated, (2S,5R)-9 and (2R,5R)-9, were transformed into the corresponding N-Boc-protected precursors, (2S,5R)-8 and (2R,5R)-8, respectively (Scheme 5). In this way, the (2S,5R) and (2R,5R) stereoisomers of 8 and 9 were obtained in optically pure form.29
:27 mixture of δ-benzylprolinates 8 was formed (Scheme 5), as deduced from the 1H-NMR spectrum. The 8 diastereoisomers could not be separated by column chromatography and, therefore, their cis/trans relative configuration [corresponding, respectively, to a (2S,5R)/(2R,5R) absolute stereochemistry29] was not established at this stage. Yet a cis relative disposition of substituents around the pyrrolidine ring, and hence a (2S,5R) absolute configuration, was tentatively assigned to the major diastereoisomer assuming that hydrogenation had preferentially taken place on the less hindered face of the molecule, that is, opposite the benzyl substituent. Luckily enough, chromatographic separation of diastereoisomers was possible after removal of the Boc group of the (2RS,5R)-8 mixture under standard acidic conditions (Scheme 5). For characterization purposes, samples of the pure amino esters thus isolated, (2S,5R)-9 and (2R,5R)-9, were transformed into the corresponding N-Boc-protected precursors, (2S,5R)-8 and (2R,5R)-8, respectively (Scheme 5). In this way, the (2S,5R) and (2R,5R) stereoisomers of 8 and 9 were obtained in optically pure form.29
The stereochemistry of the enantiomerically pure 8 and 9 δ-benzylprolinates obtained (Scheme 5) was first established by 2D NOESY experiments performed on the amino esters. A nOe correlation was observed between the α and δ protons only for the major 9 stereoisomer, indicating a cis relative disposition of the benzyl and carboxylate substituents in this compound, that is, a (2S,5R) absolute configuration. This result is in agreement with the stereoselectivity expected for the hydrogenation process. The stereochemical assignment was corroborated by X-ray diffraction analysis of single crystals of the N-acetyl derivative obtained by reaction of the minor amino ester with acetic anhydride (Fig. 1). The R configuration at C-5, given by the starting β-amino acid derived from L-phenylalanine, was taken as a reference in these assignments.
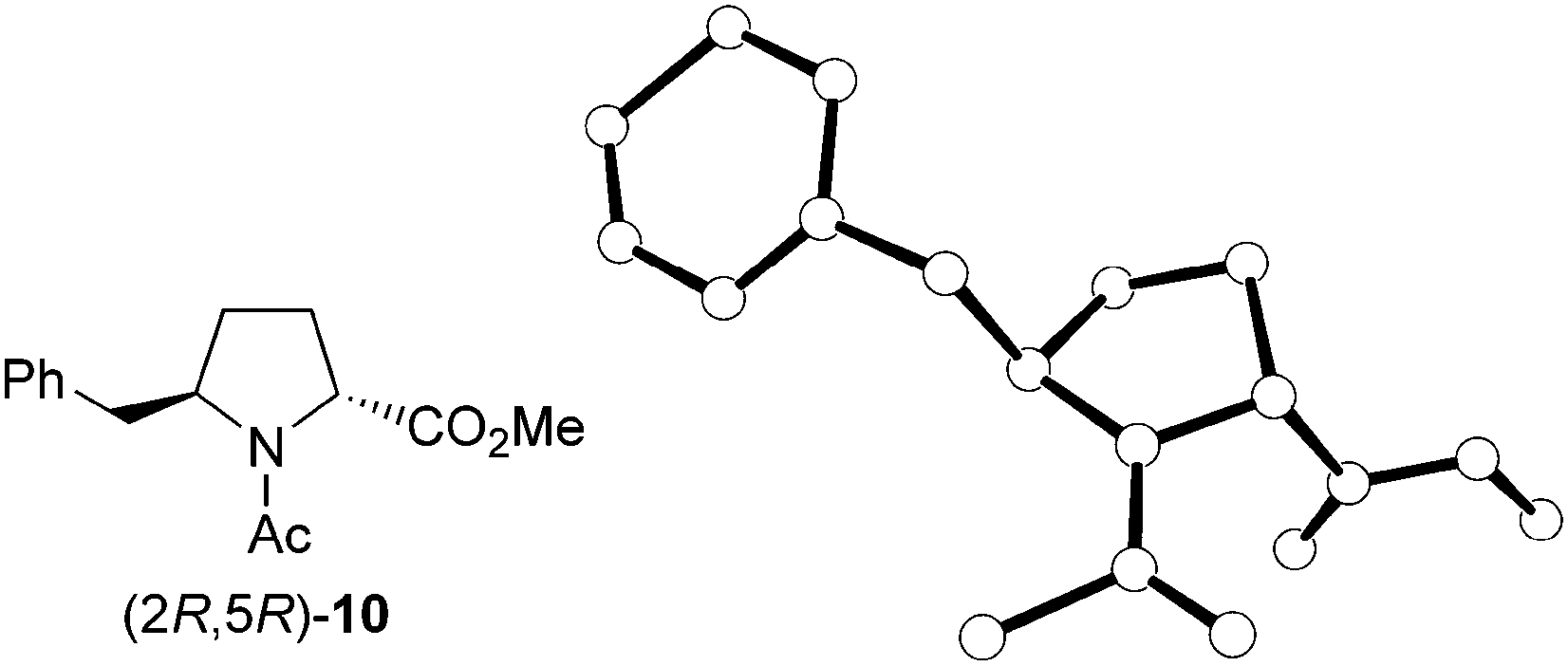 | ||
| Fig. 1 X-ray crystal structure of the acetyl derivative (2R,5R)-10 obtained from the minor amino ester, (2R,5R)-9. Hydrogen atoms have been omitted for clarity. | ||
In the search for a more selective, and hence efficient, procedure for the synthesis of (2S,5R)-δ-benzylproline, compound 6 was treated directly with hydrogen gas at atmospheric pressure using palladium on carbon as a catalyst. Under these conditions, removal of the triflate group and reduction of the olefin moiety occurred to cleanly afford a mixture of δ-benzylprolinates 8 (Scheme 5). The combined yield of (2S,5R)-/(2R,5R)-8 isolated was substantially higher (97% vs. 67% when obtained through the intermediate Δ2-pyrroline 7) but, unfortunately, the same 73:27 ratio of cis/trans diastereoisomers was determined in the 1H-NMR spectrum of the crude product.30 Therefore, the direct 6 to 8 transformation allowed the isolation of the major δ-benzylprolinate (2S,5R)-9 [as well as the minor (2R,5R)-9 stereoisomer] with significantly improved yield and avoiding one synthetic step. Accordingly, the synthesis of the intermediate 7 had no advantage in the route to the target δ-benzylprolinates 8/9. Even so, it should be noted that compound 7 is an enantiomerically pure α,β-dehydroproline derivative that may be useful as such for different types of studies. Moreover, 7 is also an interesting intermediate for the preparation of δ-benzylprolinates with further functionalization at the β (and α) position(s) taking advantage of the reactivity of the endocyclic conjugated double bond. The same holds true for 6, whose triflate moiety could enable the incorporation of additional substituents at the β carbon by means of cross-coupling reactions.16
In a further attempt to increase the diastereoselectivity of the process, the catalytic hydrogenation of triflate 6 was explored under different conditions. When palladium on carbon was replaced by platinum oxide, the cis8 diastereoisomer, (2S,5R)-8, was formed with higher diastereoselectivity, with only a small amount of the trans stereoisomer being detected in the 1H-NMR spectrum of the crude product.31 In this way, after removal of the Boc group, the pure cis amino ester, (2S,5R)-9, was obtained in 85% yield from 6 (Scheme 6). This modification therefore allowed the synthesis of enantiomerically pure δ-benzylprolinate (2S,5R)-9 in 43% overall yield from the initial β-amino acid (2). This δ-benzylproline derivative is ready for functionalization at the amino terminus while the corresponding N-Boc-protected amino acid suitable for standard peptide synthesis could be easily obtained following the procedure used for other proline analogues.32
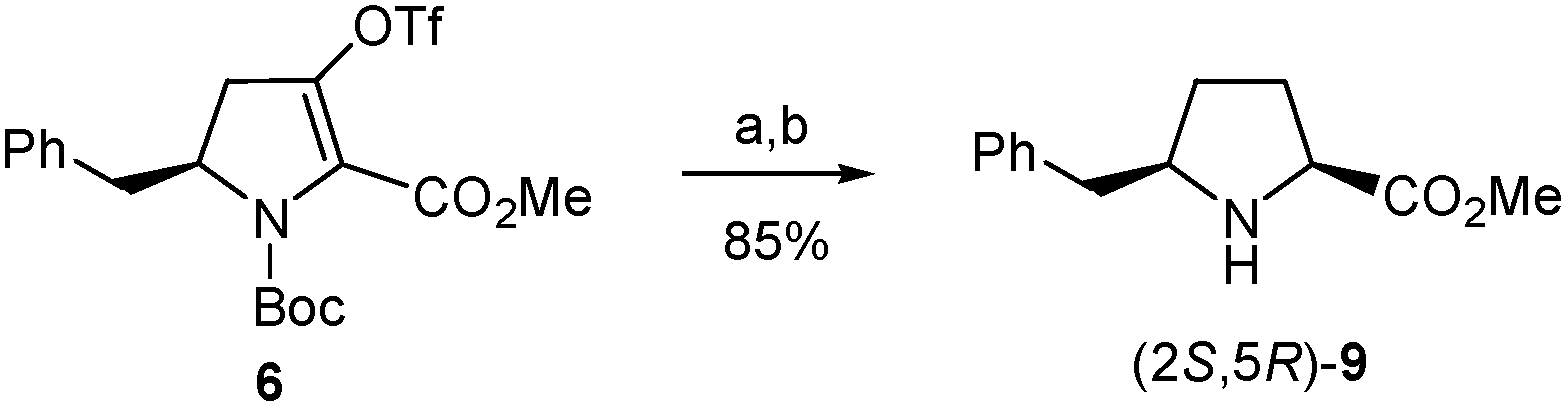 | ||
| Scheme 6 Optimized synthesis of (2S,5R)-9 from 6. Reagents and conditions: (a) H2, PtO2, Na2CO3, MeOH, rt; (b) TFA, CH2Cl2, rt. | ||
The results described above show that the β-amino acid derived from L-phenylalanine allows the efficient synthesis of the L-proline analogue (S configuration at Cα or C-2) that bears a δ-benzyl substituent cis to the carboxylate moiety [(2S,5R)-8/9 in Schemes 5 and 6]. In principle, this procedure should be extensible to β-amino acids derived from other proteinogenic amino acids (available from commercial sources or by well-known stereocontrolled homologation procedures19) thus providing access to enantiomerically pure cis δ-substituted L-proline analogues that contain the side chain of such coded amino acids at the δ position (note that the diastereoselectivity of the hydrogenation reaction may depend, to a great extent, on the nature of the substituent at Cδ and therefore differ from that observed in this work for the L-phenylalanine derivative). The present methodology therefore complements that developed by Davis,13 which has proven successful for the preparation of enantiopure δ-substituted prolines of the same configuration (cis δ-substituted L-prolines) with other types of substituents, namely aryl, vinyl, or tert-butyl (in general, those derived from aldehydes with no hydrogen at the α position or possessing a non-enolizable α hydrogen) but would not be adequate when the substituent at Cδ is the side chain of a proteinogenic amino acid. In all such cases the aldehyde to be used as the starting material has an enolizable α hydrogen and the chiral sulfinimine formed would be susceptible to the imine–enamine equilibrium leading to epimerization at the stereogenic centre14 that corresponds to C-5 in the final δ-substituted proline, thus hampering the isolation of enantiopure compounds.
Conclusions
A convenient procedure has been developed for the preparation of an enantiomerically pure L-proline analogue characterized by a δ-benzyl substituent cis to the carboxylate moiety. The fragment of the pyrrolidine ring bearing the benzyl group derives from L-phenylalanine. Thus, the synthetic route starts with the β-amino acid obtained by homologation of L-phenylalanine and involves the construction of a 3-oxopyrrolidine through an intramolecular cyclization reaction. The formation of an intermediate vinyl triflate with complete regioselectivity and the good diastereoselectivity attained in the subsequent hydrogenation of this compound using platinum oxide as a catalyst have allowed the isolation of the final cis δ-substituted L-proline derivative, methyl (2S,5R)-5-benzylprolinate, in enantiomerically pure form and 43% overall yield. If desired, the corresponding trans stereoisomer, methyl (2R,5R)-5-benzylprolinate, can be isolated as a minor product through a less diastereoselective palladium-catalyzed hydrogenation reaction. Clearly, starting from D-phenylalanine, the (2R,5S) and (2S,5S) stereoisomers would be obtained as major and minor products, respectively. The procedure could be adequate for extension to β-amino acids derived from proteinogenic residues other than L-phenylalanine, thus allowing the synthesis of enantiomerically pure δ-substituted proline analogues bearing the side chain of different coded amino acids at the δ carbon. This methodology complements other previously described methods for the synthesis of enantiopure or highly enantioenriched δ-substituted prolines.Experimental
Single-crystal X-ray diffraction
Colourless single crystals of (2R,5R)-10 were obtained by slow evaporation of a solution in dichloromethane–hexanes. The X-ray diffraction data were collected at 295 K on an Oxford Diffraction Xcalibur diffractometer provided with a Sapphire CCD detector, using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The structure was solved by direct methods using SHELXS-9733a and refinement was performed using SHELXL-9733b by the full-matrix least-squares technique with anisotropic thermal factors for heavy atoms. Hydrogen atoms were located by calculation and affected by an isotropic thermal factor fixed to 1.2 times the Ueq of the carrier atom (1.5 for the methyl protons). CCDC 1024381. Summary of crystallographic data for (2R,5R)-10 (C15H19NO3): monoclinic, space group P21; a = 9.7435(8) Å, b = 6.5523(4) Å, c = 11.9590(12) Å, β = 105.802(10)°; Z = 2; dcalcd = 1.181 g cm−3; 2659 reflections collected, 2297 unique (Rint = 0.020); data/parameters: 2297/175; final R indices (I > 2σ(I)): R1 = 0.059, wR2 = 0.128; final R indices (all data): R1 = 0.085, wR2 = 0.147; goodness of fit: 1.025; highest residual electron density: 0.12 e Å−3.Syntheses
:3) to give diazoketone 1 as a slightly yellow solid (2.37 g, 8.19 mmol, 87% yield). Spectroscopic data were identical to those reported.20
:1 v/v, 100 mL), sodium carbonate (769 mg, 7.26 mmol) and silver benzoate (42 mg, 0.18 mmol) were added. The resulting suspension was sonicated for 45 min in an ultrasound bath. The silver salts were filtered off and the reaction mixture was diluted with water and washed with diethyl ether (2 × 60 mL). The aqueous phase was acidified to pH 2 with 1 M HCl and extracted with ethyl acetate (3 × 80 mL). The combined organic layers were dried over anhydrous MgSO4, filtered and evaporated under reduced pressure. The resulting residue was purified by column chromatography (eluent: hexanes/ethyl acetate 4:1 containing 1% acetic acid) to afford 2 as a white solid (1.75 g, 6.26 mmol, 86% yield). [α]21D − 16.3 (c 1.84 in MeOH); lit.20 [α]20D − 15.7 (c 1.84 in MeOH) for 99% ee. Spectroscopic data were identical to those reported.20
:1) to afford 5 as a 2.2:1 mixture of diastereoisomers26 (sticky oil, 1.29 g, 3.87 mmol, 72% overall yield). 1H NMR (400 MHz, DMSO-d6, 373 K) (only signals of the major diastereoisomer are listed): δ 1.40 (9H, s), 2.33 (1H, m), 2.65 (1H, m), 2.94 (1H, m), 3.25 (1H, m), 3.74 (3H, s), 4.43 (1H, m), 4.70 (1H, s), 7.00–7.42 (5H, m). 13C NMR (100 MHz, DMSO-d6, 373 K) (only signals of the major diastereoisomer are listed): δ 27.4, 40.6, 41.6, 51.9, 55.0, 65.3, 79.7, 125.8, 127.9, 128.6, 137.5, 152.5, 166.6, 203.5. HRMS (ESI) C18H23NNaO5 [M + Na]+: calcd 356.1529, found 356.1510.
:1) to afford pure 6 as an oil (991 mg, 2.13 mmol, 71% yield). [α]21D + 42.9 (c 0.97 in CHCl3). IR (neat) νmax 1749, 1715, 1430, 1394, 1369 cm−1. 1H NMR (400 MHz, CDCl3): δ 1.37 (9H, s), 2.41 (1H, dd, J = 17.1 Hz, J = 2.5 Hz), 2.71 (1H, dd, J = 13.4 Hz, J = 9.2 Hz), 3.03 (1H, dd, J = 17.1 Hz, J = 9.6 Hz), 3.09 (1H, dd, J = 13.4 Hz, J = 4.9 Hz), 3.80 (3H, s), 4.45 (1H, m), 7.12–7.30 (5H, m). 13C NMR (100 MHz, CDCl3): δ 28.1, 33.8, 40.8, 52.7, 59.8, 82.6, 118.4 (q, J = 320.8 Hz), 127.0, 127.1, 128.8, 129.8, 136.1, 138.0, 152.6, 159.6. HRMS (ESI) C19H22F3NNaO7S [M + Na]+: calcd 488.0961, found 488.1043.
:1) to afford 7 as an oil (196 mg, 0.62 mmol, 67% yield). [α]23D + 20.5 (c 0.39 in CHCl3). IR (neat) νmax 1742, 1704, 1629 cm−1. 1H NMR (300 MHz, CDCl3): δ 1.34 (9H, s), 2.13 (1H, ddd, J = 17.9 Hz, J = 3.6 Hz, J = 2.3 Hz), 2.51–2.72 (2H, m), 2.99 (1H, dd, J = 13.2 Hz, J = 5.1 Hz), 3.71 (3H, s), 4.44 (1H, m), 5.62 (1H, m), 7.04–7.28 (5H, m). 13C NMR (75 MHz, CDCl3): δ 28.1, 33.4, 40.8, 51.9, 61.4, 80.9, 118.8, 126.4, 128.3, 129.7, 135.7, 137.3, 152.8, 162.6. HRMS (ESI) C18H23NNaO4 [M + Na]+: calcd 340.1519, found 340.1528.
From 7. A solution of 7 (300 mg, 0.95 mmol) in methanol (30 mL) was stirred at room temperature under hydrogen gas at atmospheric pressure in the presence of 10% Pd/C (30 mg). After 16 h, the catalyst was filtered off and washed with methanol. The solvent was concentrated in vacuo to furnish a 73
:27 mixture of (2S,5R)-8/(2R,5R)-8 (302 mg, 0.95 mmol, 100% yield). The crude oil was dissolved in dichloromethane (6 mL) and trifluoroacetic acid (3 mL) was added. After stirring at room temperature for 2 h, the mixture was washed with saturated aqueous NaHCO3 (3 × 5 mL) and brine (2 × 5 mL). The organic layer was dried over MgSO4, filtered and evaporated under reduced pressure. The residue was purified by column chromatography (eluent: ethyl acetate/dichloromethane 7:3) to afford 110 mg of (2S,5R)-9, 32 mg of (2R,5R)-9, and 55 mg of (2S,5R)-9/(2R,5R)-9 mixture (0.90 mmol, 95% combined yield).
From 6. A mixture of 6 (330 mg, 0.71 mmol), sodium carbonate (23 mg, 0.21 mmol) and 10% Pd/C (33.0 mg) in methanol (20 mL) was stirred for 1 h at room temperature under hydrogen gas at atmospheric pressure. The catalyst was filtered off and washed with methanol. The solvent was concentrated in vacuo to furnish a 73
:27 mixture of (2S,5R)-8/(2R,5R)-8 (220 mg, 0.69 mmol, 97% yield) that was treated as described above.
(2S,5R)-9. Oil. [α]27D − 33.5 (c 0.50 in MeOH). IR (neat) νmax 3350, 1736 cm−1. 1H NMR (500 MHz, CDCl3): δ 1.43 (1H, m), 1.84 (1H, m), 1.95 (1H, m), 2.09 (1H, m), 2.23 (1H, br s), 2.75 (1H, dd, J = 13.4 Hz, J = 6.9 Hz), 2.88 (1H, dd, J = 13.4 Hz, J = 6.9 Hz), 3.33 (1H, m), 3.70 (3H, s), 3.77 (dd, J = 8.9 Hz, J = 5.9 Hz), 7.14–7.29 (5H, m). 13C NMR (75 MHz, CDCl3): δ 29.9, 31.2, 42.3, 52.2, 59.8, 61.3, 126.3, 128.5, 129.0, 139.8, 175.7. HRMS (ESI) C13H18NO2 [M + H]+: calcd 220.1332, found 220.1334.
(2R,5R)-9. Oil. [α]27D + 11.8 (c 0.29 in MeOH). IR (neat) νmax 3349, 1737 cm−1. 1H NMR (500 MHz, CDCl3): δ 1.48 (1H, m), 1.86 (2H, m), 2.24 (1H, m), 2.31 (1H, br s), 2.66–2.80 (2H, m), 3.49 (1H, m), 3.69 (3H, s), 3.88 (1H, dd, J = 8.7 Hz, J = 5.3 Hz), 7.15–7.24 (3H, m), 7.25–7.32 (2H, m). 13C NMR (75 MHz, CDCl3): δ 29.4, 31.2, 42.9, 52.1, 59.2, 59.9, 126.2, 128.5, 129.1, 139.9, 176.4. HRMS (ESI) C13H17NNaO2 [M + Na]+: calcd 242.1151, found 242.1150.
:3) to afford (2S,5R)-9 (40 mg, 0.18 mmol, 85% yield). Spectroscopic data were identical to those described above.
:1) to afford pure (2S,5R)-8 as an oil (70 mg, 0.22 mmol, 96% yield). [α]24D − 55.9 (c 0.32 in CHCl3). IR (neat) νmax 1752, 1697 cm−1. 1H NMR (400 MHz, CDCl3) (mixture of rotamers, asterisks indicate signals corresponding to the minor rotamer): δ 1.36 and 1.39* (9H, two br s), 1.57–1.83 (2H, m), 1.95 (1H, m), 2.13 (1H, m), 2.51 (1H, m), 3.20* and 3.34 (1H, two dd, J = 13.3 Hz, J = 4.6 Hz, and J = 13.3 Hz, J = 4.0 Hz, respectively), 3.69 and 3.70* (3H, two s), 3.96* and 4.05 (1H, two m), 4.17 and 4.29* (1H, two m), 7.06–7.29 (5H, m). 13C NMR (100 MHz, CDCl3) (duplicate signals are observed for almost all carbon atoms): δ 28.2/28.3, 28.46/28.5, 28.9/29.6, 39.8/41.0, 52.1/52.3, 60.0, 60.3, 60.4, 80.16/80.18, 126.2/126.3, 128.5/128.6, 129.5/129.6, 139.3/139.4, 153.7/154.4, 174.0/174.1. HRMS (ESI) C18H25NNaO4 [M + Na]+: calcd 342.1676, found 342.1648.
:1) to afford (2R,5R)-8 as an oil (89 mg, 0.28 mmol, 94% yield). [α]24D + 54.1 (c 0.36 in CHCl3). IR (neat) νmax 1747, 1697 cm−1. 1H NMR (400 MHz, CDCl3) (mixture of rotamers, asterisks indicate signals corresponding to the minor rotamer): δ 1.37 and 1.47* (9H, two s), 1.62 (1H, m), 1.73 (1H, m), 1.78–1.98 (2H, m), 2.50* and 2.55 (1H, two overlapped dd, J = 13.1 Hz, J = 9.5 Hz, and J = 13.3 Hz, J = 9.5 Hz, respectively, 1H), 2.98* and 3.06 (1H, two dd, J = 13.2 Hz, J = 3.3 Hz, and J = 13.1 Hz, J = 3.3 Hz, respectively), 3.62 and 3.63* (3H, two s), 4.01–4.16 (1H, m), 4.17–4.34 (1H, m), 7.04–7.28 (5H, m). 13C NMR (100 MHz, CDCl3) (duplicate signals are observed for almost all carbon atoms, asterisks indicate signals corresponding to the minor rotamer): δ 26.8/27.4*, 27.6*/28.3, 28.5/28.6*, 39.6/40.5*, 52.0/52.2*, 59.5, 59.7, 60.1, 80.1/80.3*, 126.4/126.5*, 128.5/128.6*, 129.5*/129.7, 138.9*/139.0, 153.8/154.4*, 173.4*/173.8. HRMS (ESI) C18H25NNaO4 [M + Na]+: calcd 342.1676, found 342.1671.
:4) to afford (2R,5R)-10 as a white solid (26 mg, 0.10 mmol, 69% yield). mp 89–90 °C. [α]23D + 23.3 (c 0.18 in CHCl3). IR (nujol) νmax 1747, 1635 cm−1. 1H NMR (400 MHz, CDCl3) (mixture of rotamers, asterisks indicate signals corresponding to the minor rotamer): δ 1.66–2.23 (4H, m) overlapped with 1.99* and 2.14 (3H, two s), 2.55–2.76 (1H, m), 2.93 and 3.20* (1H, two dd, J = 13.7 Hz, J = 4.3 Hz and J = 13.2 Hz, J = 3.1 Hz, respectively), 3.71 and 3.74* (3H, two s), 4.17 and 4.41* (1H, two m), 4.26* and 4.38 (1H, two m), 7.11–7.37 (5H, m). 13C NMR (100 MHz, CDCl3) (duplicate signals are observed for all carbon atoms, asterisks indicate signals corresponding to the minor rotamer): δ 22.1/23.0*, 26.5*/26.9, 28.5/29.3*, 38.6*/41.2, 52.2/52.7*, 59.2/61.1*, 59.6*/60.5, 126.4*/127.0, 128.5*/128.9, 129.3/129.6*, 137.7/139.1*, 169.77/169.8*, 172.86*/172.9. HRMS (ESI) C15H19NNaO3 [M + Na]+: calcd 284.1257, found 284.1249.
Acknowledgements
The authors thank the Ministerio de Economía y Competitividad-FEDER (grants CTQ2010-17436, CTQ2013-40855-R, FPI fellowship to I.R.) and Gobierno de Aragón–FSE (research group E40) for financial support.Notes and references
- (a) M. W. MacArthur and J. M. Thornton, J. Mol. Biol., 1991, 218, 397–412 CrossRef CAS; (b) P. Chakrabarti and D. Pal, Prog. Biophys. Mol. Biol., 2001, 76, 1–102 CrossRef CAS.
- G. D. Rose, L. M. Gierasch and J. A. Smith, Adv. Protein Chem., 1985, 37, 1–109 CrossRef CAS.
- (a) K. P. Lu, G. Finn, T. H. Lee and L. K. Nicholson, Nat. Chem. Biol., 2007, 3, 619–629 CrossRef CAS PubMed; (b) C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475–2532 CrossRef CAS PubMed; (c) D. E. Stewart, A. Sarkar and J. E. Wampler, J. Mol. Biol., 1990, 214, 253–260 CrossRef CAS.
- (a) M. I. Calaza, F. J. Sayago, P. Laborda and C. Cativiela, Eur. J. Org. Chem. DOI:10.1002/ejoc.201403121; (b) F. J. Sayago, P. Laborda, M. I. Calaza, A. I. Jiménez and C. Cativiela, Eur. J. Org. Chem., 2011, 2011–2028 CrossRef CAS; (c) M. I. Calaza and C. Cativiela, Eur. J. Org. Chem., 2008, 3427–3448 CrossRef CAS PubMed; (d) S. K. Panday, J. Prasad and D. K. Dikshit, Tetrahedron: Asymmetry, 2009, 20, 1581–1632 CrossRef CAS PubMed; (e) C. Nájera and M. Yus, Tetrahedron: Asymmetry, 1999, 10, 2245–2303 CrossRef.
- P. Karoyan, S. Sagan, O. Lequin, J. Quancard, S. Lavielle and G. Chassaing, in Targets in Heterocyclic Systems. Chemistry and Properties, ed. O. A. Attanasi and D. Spinelli, Royal Society of Chemistry, Cambridge, 2005, vol. 8, pp. 216–273 Search PubMed.
- L. Moroder, C. Renner, J. J. Lopez, M. Mutter and G. Tuchscherer, in Cis-Trans Isomerization in Biochemistry, ed. C. Dugave, Wiley-VCH, Weinheim, 2006, ch. 11, pp. 225–259 Search PubMed.
- (a) L. Halab and W. D. Lubell, J. Am. Chem. Soc., 2002, 124, 2474–2484 CrossRef CAS PubMed; (b) L. Halab and W. D. Lubell, J. Org. Chem., 1999, 64, 3312–3321 CrossRef CAS PubMed; (c) S. S. A. An, C. C. Lester, J.-L. Peng, Y.-J. Li, D. M. Rothwarf, E. Welker, T. W. Thannhauser, L. S. Zhang, J. P. Tam and H. A. Scheraga, J. Am. Chem. Soc., 1999, 121, 11558–11566 CrossRef CAS; (d) C. Aubry, H. Oulyadi, G. Dutheuil, J. Leprince, H. Vaudry, X. Pannecoucke and J.-C. Quirion, J. Pept. Sci., 2006, 12, 154–160 CrossRef CAS PubMed.
- U. Arnold, M. P. Hinderaker, J. Koeditz, R. Golbik, R. Ulbrich-Hofmann and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 7500–7501 CrossRef CAS PubMed.
- L. Bélec, J. Slaninova and W. D. Lubell, J. Med. Chem., 2000, 43, 1448–1455 CrossRef PubMed.
- S. C. R. Lummis, D. L. Beene, L. W. Lee, H. A. Lester, R. W. Broadhurst and D. A. Dougherty, Nature, 2005, 438, 248–252 CrossRef CAS PubMed.
- (a) H. Hoveyda, D. Schils, L. Zoute and J. Parcq, PCT Int. Patent Appl., WO2011073376, 2011 Search PubMed; (b) F. Manfré and J. P. Pulicani, Tetrahedron: Asymmetry, 1994, 5, 235–238 CrossRef; (c) M.-C. Fournie-Zaluski, P. Coric, V. Thery, W. Gonzalez, H. Meudal, S. Turcaud, J.-B. Michel and B. P. Roques, J. Med. Chem., 1996, 39, 2594–2608 CrossRef CAS PubMed; (d) H. Ryder, D. A. Kendrick, G. Semple, K. Miyata, A. R. Batt, E. A. Mathews, D. P. Rooker, A. Nishida, S. Azuzawa and M. Szelke, PCT Int. Patent Appl., WO9320099, 1993 Search PubMed; (e) J. Das, S. D. Kimball and W. F. Lau, US Pat., 5 691 356, 1997 Search PubMed; (f) Z. Pei, X. Li, K. Longenecker, T. W. von Geldern, P. E. Wiedeman, T. H. Lubben, B. A. Zinker, K. Stewart, S. J. Ballaron, M. A. Stashko, A. K. Mika, D. W. A. Beno, M. Long, H. Wells, A. J. Kempf-Grote, D. J. Madar, T. S. McDermott, L. Bhagavatula, M. G. Fickes, D. Pireh, L. R. Solomon, M. R. Lake, R. Edalji, E. H. Fry, H. L. Sham and J. M. Trevillyan, J. Med. Chem., 2006, 49, 3520–3535 CrossRef CAS PubMed.
- (a) S.-G. Kim, E. J. Ahn and T.-H. Park, Bull. Korean Chem. Soc., 2007, 28, 1665–1669 CrossRef CAS; (b) I. Collado, J. Ezquerra and C. Pedregal, J. Org. Chem., 1995, 60, 5011–5015 CrossRef CAS.
- (a) F. A. Davis, T. Fang and R. Goswami, Org. Lett., 2002, 4, 1599–1602 CrossRef CAS PubMed; (b) F. A. Davis, B. Yang and J. Deng, J. Org. Chem., 2003, 68, 5147–5152 CrossRef CAS PubMed; (c) F. A. Davis, K. A. Bowen, H. Xu and V. Velvadapu, Tetrahedron, 2008, 64, 4174–4182 CrossRef CAS PubMed.
- F. A. Davis, R. E. Reddy, J. M. Szewczyk, G. V. Reddy, P. S. Portonovo, H. Zhang, D. Fanelli, R. T. Reddy, P. Zhou and P. J Carroll, J. Org. Chem., 1997, 62, 2555–2563 CrossRef CAS PubMed.
- (a) J. Aubé, B. Gülgeze and X. Peng, Bioorg. Med. Chem. Lett., 1994, 4, 2461–2464 CrossRef; (b) L. B. Wolf, K. C. M. F. Tjen, H. T. ten Brink, R. H. Blaauw, H. Hiemstra, H. E. Schoemaker and F. P. J. T. Rutjes, Adv. Synth. Catal., 2002, 344, 70–82 CrossRef CAS.
- (a) I. Rodríguez, M. I. Calaza, A. I. Jiménez and C. Cativiela, Tetrahedron, 2012, 68, 9578–9582 CrossRef PubMed; (b) I. Rodríguez, M. I. Calaza and C. Cativiela, Eur. J. Org. Chem., 2013, 1093–1099 CrossRef.
- M. P. Moyer, P. L. Feldman and H. Rapoport, J. Org. Chem., 1985, 50, 5223–5230 CrossRef CAS.
- R. J. Moreau and E. J. Sorensen, Tetrahedron, 2007, 63, 6446–6453 CrossRef CAS PubMed.
- Enantioselective Synthesis of β-Amino Acids, ed. E. Juaristy and V. A. Soloshonok, Wiley, Hoboken, 2005 Search PubMed.
- M. R. Linder, S. Steurer and J. Podlech, Org. Synth., Coll. Vol., 2004, 10, 194–199 Search PubMed.
- T. Aoyama and T. Shioiri, Tetrahedron Lett., 1980, 21, 4461–4462 CrossRef CAS.
- J. Cesar and M. S. Dolenc, Tetrahedron Lett., 2001, 42, 7099–7102 CrossRef CAS.
- A. Müller, C. Vogt and N. Sewald, Synthesis, 1998, 837–841 CrossRef PubMed.
- The specific rotation was in agreement with that reported in ref. 20: see Experimental section.
- J. S. Baum, D. A. Shook, H. M. L. Davies and H. D. Smith, Synth. Commun., 1987, 17, 1709–1716 CrossRef CAS.
- The 1H-NMR spectrum of the purified (2RS,5S)-5 mixture showed broadened, partially overlapped signals even when registered at high temperature. Under the best conditions (1H-NMR in DMSO-d6 at 373 K), the ratio of 5 diastereoisomers was roughly estimated to be ≈2.2:1. A more precise determination of this ratio was not considered essential, and therefore was not addressed, since this mixture of epimers at the α carbon would be transformed into the same compound subsequently in the synthesis. For the same reasons, identification of the cis/trans diastereoisomers in the mixture was not made.
- (a) D. L. Comins and A. Dehghani, Tetrahedron Lett., 1992, 33, 6299–6302 CrossRef CAS; (b) D. L. Comins, A. Dehghani, C. J. Foti and S. P. Joseph, Org. Synth., Coll. Vol., 1998, 9, 165–168 Search PubMed.
- Formation of the regioisomer with a double bond connecting C-3, C-4 was not observed. In our previous work (ref. 16a), the use of triflic anhydride or N-phenyltriflimide with this or other bases (as LiHMDS) led to mixtures of the desired vinyl triflate and the regioisomer exhibiting a double bond at C-3, C-4.
- Note that the configuration at C-5 (Cδ) is given by the starting homochiral β-amino acid while that at the α carbon (C-2) is generated in the hydrogenation process. In this regard, it is important to emphasize that the reaction conditions in Schemes 4 and 5 should not compromise the configurational integrity of the chiral carbon attached to the benzyl substituent in the initial β-amino acid (C-5 in the final δ-benzylprolinates), thus leading to enantiomerically pure compounds after separation of diastereoisomers. Even so, the enantiomeric purity of (2S,5R)-8 and (2R,5R)-8 was checked by HPLC analysis on six different chiral columns (Chiralpak® IA, IB, IC, ID, IE, and IF) eluting with n-hexane–2-propanol mixtures and a single peak was observed in all assays. Given the high enantiodiscrimination ability typically shown by these chiral stationary phases, the absence of a peak attributable to the other enantiomer in all of the chromatograms registered strongly supports the enantiomeric purity of the compounds analyzed, (2S,5R)-8 and (2R,5R)-8, and their precursors, (2S,5R)-9 and (2R,5R)-9. As a matter of fact, we have tested a great number of N- and C-protected non-proteinogenic amino acids on these columns (particularly, the IA, IB, and IC types, which have been on the market for some years) and never found a case in which separation of enantiomers was not observed on at least one of them. For some examples of enantioseparation of proline analogues on Chiralpak® IA, IB, or IC columns, see ref. 4b and 16b and (a) P. Fatás, A. M. Gil, M. I. Calaza, A. I. Jiménez and C. Cativiela, Chirality, 2012, 24, 1082–1091 CrossRef PubMed; (b) F. J. Sayago, A. I. Jiménez and C. Cativiela, Tetrahedron: Asymmetry, 2007, 18, 2358–2364 CrossRef CAS PubMed; (c) F. J. Sayago, M. J. Pueyo, M. I. Calaza, A. I. Jiménez and C. Cativiela, Chirality, 2011, 23, 507–513 CrossRef CAS PubMed; (d) A. M. Gil, E. Buñuel, P. López and C. Cativiela, Tetrahedron: Asymmetry, 2004, 15, 811–819 CrossRef CAS PubMed.
- The identical diastereoselectivity observed is not completely unexpected since this transformation has been proposed to occur through hydrogenolysis of the C–O bond followed by hydrogenation of the alkene: A. García Martínez, R. Martínez Álvarez, J. Arranz Aguirre and L. R. Subramanian, J. Chem. Soc., Perkin Trans. 1, 1986, 1595–1598 RSC.
- The presence of some impurities hampered precise determination of the diastereomeric ratio.
- If desired, the N-Boc-protected amino acid could be obtained by treatment of (2S,5R)-8 [generated by introduction of the Boc group on (2S,5R)-9 as shown in Scheme 5] with lithium hydroxide in methanol–water. These conditions have proven efficient for deprotection of the methyl ester function in other substituted proline analogues while being mild enough to preserve the optical purity of the substrate: see ref. 29a.
- (a) G. M. Sheldrick, SHELXS-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray structure of (2R,5R)-10 with atom numbering; 1H-NMR and 13C-NMR spectra of compounds 6–10. CCDC 1024381. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01894j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |