
Vesicular hydrogen silsesquioxane-mediated synthesis of nanocrystalline silicon dispersed in a mesoporous silica/suboxide matrix, with potential for electrochemical applications†
Ferenc
Somodi
,
Chang Sun
Kong
,
Jerome C.
Santos
and
Daniel E.
Morse
*
Institute for Collaborative Biotechnologies, California NanoSystems Institute, Materials Research Laboratory and Department of Molecular, Cellular, and Developmental Biology, University of California, Santa Barbara, CA 93106, USA. E-mail: d_morse@lifesci.ucsb.edu; Fax: +1 805 893 8062; Tel: +1 805 893 7442
First published on 3rd November 2014
Abstract
Room temperature hydrolysis of triethoxysilane (TES) in the presence of pluronic P-123 (PEO20–PPO70–PEO20) triblock copolymer under acidic conditions resulted in the formation of soft nanoscale vesicles with thin (<10 nm) hydrogen silsesquioxane (HSiO1.5)n gel walls. FTIR and 29Si MAS NMR showed that pyrolysis of this material at 1000 °C in H2/Ar atmosphere led to decomposition of the hydrogen silsesquioxane with the resulting formation of a silicon suboxide surrounding Si nanocrystals, all embedded in a SiO2 matrix. SEM, TEM, BET and SAXS measurements showed that this vesicular Si@SiOx–SiO2 composite material had a high surface area and interconnected mesoporous structure. Nanocrystalline silicon was confirmed by XRD after the high temperature pyrolysis. An optimum in Li-ion battery half-cell performance was observed after the pyrolysis at 1000 °C, apparently attributable to the mesoporous structure and silicon nanocrystals. The effect of the polymer binders sodium carboxymethyl cellulose (CMC) and polyacrylic acid (PAA) on the cyclic performance was investigated by cyclic voltammetry and electrochemical impedance spectroscopy. Results suggest that the formation of the solid electrolyte interface layer is slower in the case of PAA, resulting in lower resistance to lithium diffusion at the interface.
Introduction
Silicon has the highest lithium storage capacity amongst the metals and metalloids in the p-block of elements, frequently investigated as anode materials for Li-ion batteries due to their alloy formation with lithium.1,2During lithium insertion, however, silicon undergoes volume expansion – including the fourfold increase in unit cell volume observed in the case of Li22Si5.3 This reversible expansion and contraction with repeated cycling results in crack formation and disintegration of the electrode leading to loss of conductivity, decrease in specific capacity and short cycle life of the battery.4–6 Several solutions have been explored to overcome this problem,6 with the use of Si nanoparticles and nanostructures being one of the most important directions of current research. But simply decreasing the size of the Si particles is not sufficient to obtain stable, high capacity anodes; the structure of the electrode is an equally important factor.7 The anode must meet the following structural requirements:8 large surface area accessible to the electrolyte; short diffusion length for Li ions; large free space available to accommodate the volume change; and high electron conductivity. It has been shown that porous doped Si nanowires match these criteria and that half-cells made using this anode material exhibit long cycle life at high rates of charge and discharge.8 Others presented a clever design of core–shell composite anode structure made by coating commercially available Si nanoparticles with SiO2 first, then with polydopamine. After pyrolysis, the SiO2 was dissolved and the void space between the Si and carbon shell was able to accommodate the volume change of Si nanoparticles during lithium insertion over 1000 cycles.9 Despite the very high specific capacity of Si anodes, their widespread application has been hindered by the high cost and difficulty of large-scale production of silicon nanostructures and/or the relatively low areal capacity of these electrodes.10
Numerous studies have investigated SiO as anode in Li-ion batteries11–13 because of the natural occurrence of Si nanoclusters in this material. Silicon oxide is considered to be a mixture of Si and SiO2 domains at room temperature,14 rather than a random mixture of Si–Si and Si–O–Si bonds. Detailed spectroscopic investigation suggested that amorphous SiO is built up by clusters of silicon surrounded by a sub-oxide in a silicon dioxide matrix.15 The presence of the surrounding oxide matrix of amorphous SiO2 and suboxide around Si nanoclusters was shown to increase the cycling stability of Li-ion battery anodes.16 Oxygen incorporation into silicon thin film anodes and subsequent low temperature annealing resulted in increased cycling stability and rate capability with minimal capacity fading in the first 120 cycles.17 The major disadvantage of such SiO anodes is the difficulty and cost of their structural design; for instance, the size of the particles can only be modified by high energy ball milling and/or high temperature treatments.18,19
Sol–gel synthesis routes are low-cost and scalable processes that might provide structural control and open a new path towards the realization of high capacity Si containing anodes. Acidic hydrolysis of triethoxysilane or trichlorosilane and subsequent pyrolysis of the resulting hydrogensilsesquioxane (HSiO1.5)n gel product leads to the formation of Si nanoparticles embedded in a SiO2 matrix.20–23 Extending earlier work using the self-assembling amphiphilic P-123 triblock copolymer as a template to produce mesoporous silica from tetraethylorthosilicate,24 it was shown recently that a so-called “impossible material”, an ordered mesoporous (HSiO1.5)n gel, can be synthesized using that template with triethoxysilane as precursor in the presence of that sodium chloride and n-butanol in hydrochloric acid solution.25
Here we show that similar vesicular gels can be prepared by changing the reaction components for the triethoxysilane hydrolysis, using only pluronic P-123 (PEO20–PPO70–PEO20) triblock copolymer in HCl solution at room temperature, without any other additives. The subsequently pyrolyzed vesicular gel has been characterized and tested for its electrochemical performance vs. metallic lithium, investigating the effect of pyrolysis temperature and different binders on the product's structure, reversible electrochemical storage capacity and cyclic stability in Li-ion battery half-cells.
Experimental
Synthesis
3.5 g P-123 triblock copolymer (PEO20–PPO70–PEO20, MW ∼5800 g mol−1) was dissolved in 120 ml 0.1 M hydrochloric acid, after which 9 ml triethoxysilane (TES) (Sigma-Aldrich) was injected at room temperature. The clear transparent solution immediately turned white upon addition of TES, with no precipitate observed. After stirring for 24 h in a closed round bottom flask, the solvent was evaporated in 5 h at 70 °C on a hot plate under a fume hood. The structure-directing polymer was removed via extraction with ethanol in a Soxhlet apparatus for 3 × 8 h, leaving the material under ethanol overnight between extraction runs. Finally, the material was dried overnight in an oven at 60 °C. The resulting white solid was pyrolyzed in a horizontal tube furnace at 700 or 1000 °C in 5% H2–95% Ar mixture for 1 h, at a heating rate of 20 °C min−1. Another pyrolysis at 1400 °C was performed in a mini hot-press under otherwise identical conditions.Materials characterization
Structural characterization of the materials by transmission electron microscopy (TEM) was performed using a FEI XL40 Sirion FEG digital scanning electron microscope and FEI Tecnai G2 Sphera transmission electron microscope. Dynamic light scattering (DLS) measurements were done using a Zetasizer Nano ZS (Malvern) instrument.Nitrogen adsorption measurements were conducted with a MicroMetrics TriStar Porosimeter. Before measurement at liquid nitrogen temperature, the samples were degassed at 200 °C in flowing nitrogen atmosphere for 4 h. Infrared spectra before and after pyrolysis were recorded on a Nicolet Magna 850 FTIR spectrometer. For sample preparation, 10 mg of the material were suspended in 0.1 ml acetone and dropped onto disposable polyethylene IR sample cards. Spectra were collected after evaporation of the acetone.
Solid state 29Si MAS NMR measurements were performed on a Bruker AVANCE III HD 400 MHz (9.4 T) wide bore (89 mm) spectrometer, using a 4 mm zirconia rotor system at a spinning frequency of 10 kHz. Spectra were collected with a 30° 29Si excitation pulse of 1.67 μs using a 16 s relaxation delay, an acquisition size of 1024 points, and a 18 ms acquisition time where 1H decoupling of 82 kHz was applied. 4848, 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 931, and 21200 scans were collected for the (HSiO1.5)n gel samples after extraction with ethanol, after pyrolysis in H2/Ar at 700 °C, and at 1000 °C, respectively.
931, and 21200 scans were collected for the (HSiO1.5)n gel samples after extraction with ethanol, after pyrolysis in H2/Ar at 700 °C, and at 1000 °C, respectively.
Small angle X-ray diffraction and XRD measurements were obtained with a Rigaku Smartlab high-resolution diffractometer and Philips X'PERT MPD respectively, using Cu Kα radiation. For small angle X-ray diffraction measurements, samples were placed in a Kapton sample holder; for powder XRD, ∼10 mg of the sample was dispersed in acetone and this suspension was dropped and dried onto a glass microscope slide, which was placed on the diffractometer sample stage.
Electrochemical characterization
To prepare electrodes, the composite material (100 mg, 80 wt%) was mixed with 500 μl of a 12.5 mg ml−1 solution of sodium carboxymethyl cellulose (CMC, MW ∼ 250000; DS = 0.7) in water or polyacrylic acid (PAA, Mw ∼ 450000) in N-methyl-2-pyrrolidone (6.25 mg, 5 wt%); carbon black (Super P Li; 18.75 mg, 15 wt%) was added to these suspensions in a mortar to form slurries which were then dispersed on a 15 μm thick copper foil by using a 150 μm notch bar.26 The working electrodes (0.64 cm2) were cut and pressed (∼3.5 MPa, 2 min) after drying the coated copper foils in a vacuum oven overnight at 85 °C. Half-cells were assembled in an argon filled glove box, using lithium foil counter electrodes and porous separators (Celgrad 3501). The electrolyte was 1 M LiPF6 in a mixture of ethylene carbonate and dimethyl carbonate (1:1 by volume). The cells were tested on a BT2000 battery tester from Arbin Instruments at 50 mA g−1 current density between 0.005 and 2.0 V versus Li/Li+. The specific capacity was calculated using the weight of the active material which was 80 wt% of the total weight of the electrode (excluding the current collector foil).
Cyclic voltammetry (CV) measurements were conducted with a CHI 660C Electrochemical Analyzer/Workstation over the potential range of 0.005–2.0 V versus Li/Li+ at a scan rate of 0.1 mV s−1 up to 10 cycles. Electron Impedance Spectroscopy (EIS) was measured over the frequency ranges from 100 kHz to 10 MHz with an alternative current (AC) amplitude of 2 mV using a Solariton SI 1287 electrochemical interface and a SI 1260 impedance/gain-phase analyzer. EIS data were collected with ZPlot®, and the resulting data analyzed with the ZView® electrochemical software package. All measurements were carried out at room temperature.
Results and discussion
Synthesis of mesoporous hydrogen silsesquioxane
Previous investigators synthesized ordered mesoporous material using triethoxysilane (TES) as a precursor, with NaCl and n-butanol as swelling agents to enhance micelle formation of P-123.25 To prepare silicon-based anode materials, all of the additives must be removed from the (HSiO1.5)n gel prior to pyrolysis, but the removal process can reduce the concentration of the labile Si–H bonds that are essential for Si nanocrystal formation. While the templating P-123 polymer and n-butanol can readily be extracted with ethanol, the removal of NaCl requires washing of the silsesquioxane gel with water or dilute HCl. We observed that such washing resulted in gas evolution, indicating the premature cleavage of Si–H bonds to evolve hydrogen, whereas without washing, such cleavage and gas evolution can be minimized.27As reported previously, at a P-123 polymer template concentration of ∼0.03% w/v, micelles can be formed without sodium chloride.28 Likewise, our DLS measurements revealed that n-butanol had no effect on the micelle size (data not shown). Our synthesis conditions thus resemble those of Stucky and colleagues, who showed that ordered mesoporous silicas (such as SBA-15) and other metal oxides can be synthesized in the absence of any salt and swelling agents by using tetraethyl orthosilicate.24 Based on this information, the synthesis was carried out in the absence salts and swelling agents.
Surprisingly, instead of the precipitation of a gel with ordered mesopores, we obtained a stable colloidal suspension (Fig. 1). In good agreement with previous data,29,30 DLS measurements showed the presence of P-123 polymer micelles with 17.7 nm average hydrodynamic diameter and a polydispersity index of 0.039 in solution before addition of triethoxysilane (Fig. 1D). After injection of TES, the particle size detected by DLS increased, with vesicles identified by TEM (Fig. 1).
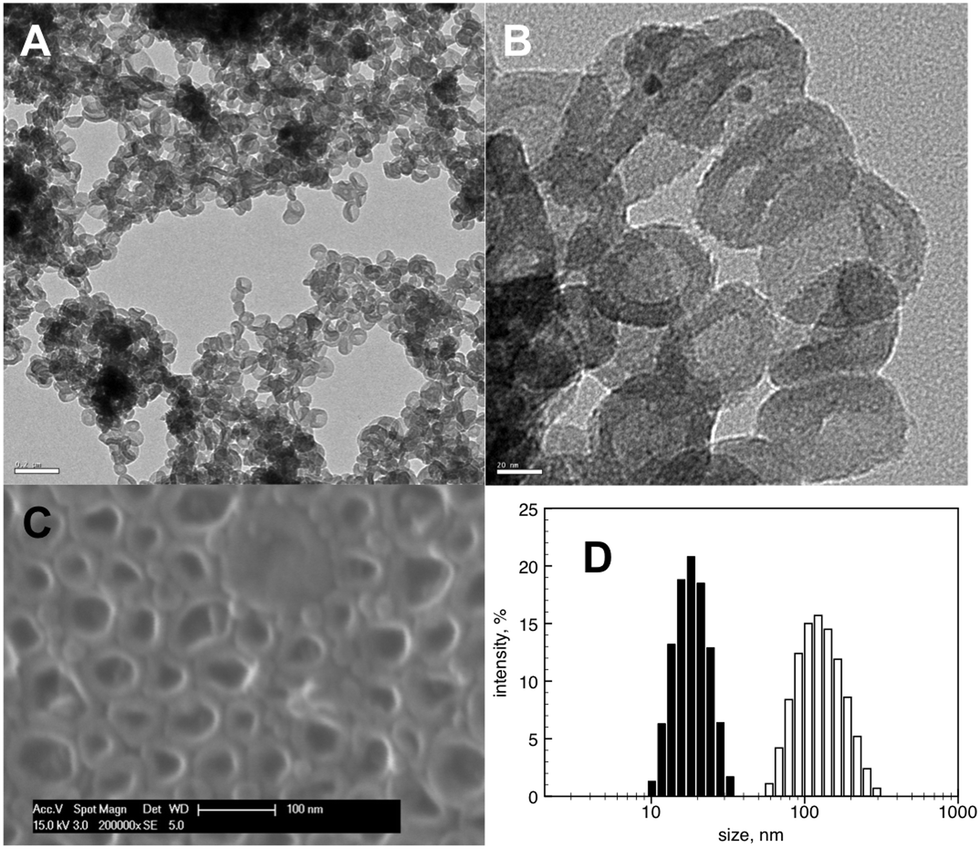 | ||
| Fig. 1 (A, B) TEM images of vesicles, sample was taken from reaction suspension after 24 h. Scale bars represent 200 nm and 20 nm respectively. (C) SEM image of mesoporous sample after solvent evaporation. (D) Size distribution by DLS. Black bars: P-123 micelles in 0.1 M HCl before addition of TES. White bars: (HSiO1.5)n vesicles 3 h after addition of TES to the P-123 solution. | ||
The observed vesicle formation can only be explained by the cooperative self-assembly of the structure-directing agent (P-123) and triethoxysilane and its hydrolysis products. It is plausible that multiple processes took place simultaneously when the TES was injected into the solution of the micellar P-123; these could include: (1) diffusion of TES into the PPO-core of the micelles; (2) transformation of the micelles to vesicles driven by the changed polarity of the reaction environment (theoretically possible,31 this transformation has not been observed experimentally in a PPO/PEO–water–oil system32); and (3) hydrolysis of TES to form the hydrogen silsesquioxane. The absence of a detectable precipitate or irregularly shaped gel product during synthesis suggests that hydrolysis and polycondensation of the triethoxysilane occurs at the hydrophilic–hydrophobic interface within the PPO–PEO, whether in a developing micelle or in a pre-formed vesicle.
When the hydrolysis occurs in the micelle-stabilized TES, the release of ethanol and polycondensation of the (HSiO1.5)n gel will cause a volume expansion of the hydrophobic PPO polymer chains, increasing the critical packing parameter as the system stabilizes in the final vesicular form. Similarly, other investigators prepared large vesicles by the conventional chloroform film method, mixing P-123 with a non-ionic surfactant (Span 65); they concluded that the surfactant increased the volume of the PPO core, thereby increasing the critical packing parameter and driving vesicle formation.33
We note that a stable colloid was not observed when NaCl (6 g/under our reaction conditions) was present. Instead, a precipitate was formed after one hour following the injection of TES. It is known that sodium chloride enhances vesicle formation by decreasing the critical micelle concentration, critical micelle temperature and cloud point.34 While the mechanism for this effect remains controversial, it is generally accepted that the presence of such a structure-enhancing salt leads to an increase in self-hydration of water through hydrogen bonding, hence reducing the solubility of the copolymer (i.e., strengthening the hydrophobic interactions in solution and salting out the polymer).35 In parallel to these considerations, the presence of structure-enhancing salts in block copolymer solutions results in increased solubility of hydrophobic compounds.36,37 These findings suggest that the diffusion of TES into the PEO core of the micelles is more favoured in the presence of NaCl, while stabilization of the system by micelle transformation is hindered. In this case, the hydrolysis is slower, leading to precipitation of large particles with ordered mesopores, as observed previously.25
Characterization of mesoporous hydrogen silsesquioxane before and after pyrolysis
After our synthesis as described above, the P-123 was extracted with ethanol in a Soxhlet apparatus. The absorption bands corresponding to the P-123 vibrations, including the C–H stretching (2800–3000 cm−1), C–H bending (1350–1500 cm−1) and C–O–C stretching (1000–1300 cm−1), were not visible after extraction indicating that the polymer was successfully removed (Fig. 2A). | ||
| Fig. 2 FTIR spectra of products after different processes. (HSiO1.5)n gel vesicles after extraction of P-123 with ethanol (A). (HSiO1.5)n gel vesicles after high temperature pyrolysis at 700 °C (B), 1000 °C (C) and 1400 °C (D) for 1 h in H2/Ar. | ||
The temperature induced changes of the chemical structure can be clearly seen in the FTIR spectra B and C in Fig. 2: (i) the intensity of the Si–H band at 2240 cm−1 significantly decreased and shifted to 2262 cm−1 and 2282 cm−1 after pyrolysis at 700 °C and 1000 °C, respectively; (ii) the intensity of the absorption band for the asymmetric Si–O–Si stretching (1200–1000 cm−1) and Si–O–Si bending (below 500 cm−1) vibrations increased together with the Si–O stretching vibrations at around 800 cm−1, while the bands for the O–Si–H bending vibrations (800–960 cm−1) disappeared. The position of the observed bands is in good agreement with the values published previously.38–40
Assignment of the new absorption band at ∼875 cm−1, observed after pyrolysis, is problematic (Fig. 2B and C). This might simply be attributed to O–Si–H bending vibrations, but its intensity does not decrease together with the Si–H stretching band following pyrolysis; in fact, a slight increase in intensity is observed. This band was seen on the FTIR spectrum of pyrolyzed hydrogen silsesquioxane and was attributed to Si–O vibrations affected by the presence of Si–Si bonds.27 The appearance of this band also was reported in thin layers of Si2O3 prepared by evaporation of SiO in an oxidizing atmosphere.41 The FTIR spectrum of Si2O3 prepared by hydrolysis of Si2Cl3 had the same absorption band at 875 cm−1 together with a weak band (2265 cm−1) in the Si–H stretching region, but no signal related to Si–H bonds was observed in the 29Si MAS NMR spectrum of that material.42 Our NMR measurements provided similar results. Fig. 3 shows the 29Si MAS NMR spectrum of the vesicular (HSiO1.5)n gel after extraction and pyrolysis at different temperatures. The peak positions after peak fitting can be seen in Table 1.
 | ||
| Fig. 3 29Si MAS NMR spectrum of the vesicular (HSiO1.5)n gel after extraction of P-123 with ethanol and after pyrolysis at different temperatures in H2/Ar. | ||
| Position | Area | (%) | ||
|---|---|---|---|---|
| After extraction | Si–(OSi)4 | −111.5 | 3967824 |
16.3 |
| Si–(OSi)3–OH | −101.5 | 1050182 |
4.3 | |
| H–Si–(OSi)3 | −85.1 | 17848710 |
73.4 | |
| H–Si–(OSi)2–OH | −75.9 | 1459205 |
6.0 | |
| 700 °C | Si–(OSi)4 | −110.7 | 72248640 |
81.5 |
| H–Si–(OSi)3 | −83.9 | 6171210 |
7.0 | |
| H–Si–(OSi)2–OH | −74.3 | 6573256 |
7.4 | |
| a-Si, Si3Si(OSi) | −65.1 | 3607594 |
4.1 | |
| 1000 °C | Si–(OSi)4 | −109.2 | 65684620 |
90.3 |
| c-Si | −81.7 | 747572 |
1.0 | |
| SiSi(OSi)3 | −72.2 | 2191690 |
3.0 | |
| a-Si, Si3Si(OSi) | −66.2 | 4136490 |
5.7 | |
The peaks at −75.9 and −85.1 ppm (Fig. 3) correspond to the T2 HSi(OSi)2(OH) and T3 HSi(OSi)3 centers, the units with Si–H bonding. The other two peaks at lower chemical shifts −101.5 ppm and −111.5 ppm – represent the Q3 (OH)Si(OSi)3 and Q4 Si(OSi)4 units, respectively. Their appearance is the consequence of the partial hydrolysis of Si–H bonds.25 In agreement with the FTIR results, after pyrolysis at 700 °C in H2/Ar, the intensity of T2 and T3 peaks decreased while the intensity of Q4 units significantly increased. The new peak observed in the NMR spectrum at −65.1 ppm can be assigned to amorphous silicon and/or silicon suboxide Si3Si(OSi) in which only one O atom is in the coordination sphere of a Si atom.15
After pyrolysis at 1000 °C, the relative intensity of this band slightly increased and new peaks appeared in the spectrum at −81.7 and −72.2 ppm. The former peak can be assigned to crystalline Si and the latter to Si2O3, in which three oxygen atoms are situated in the coordination sphere of a silicon atom Si2(OSi)3.15,42
The formation and growth of silicon particles upon pyrolysis is supported by XRD measurements as well. Fig. 4 shows the XRD patterns of the vesicular hydrogen silsesquioxane gels after pyrolysis at different temperatures. In accord with the 29Si MAS NMR results, crystalline Si nanoparticles were not observed when the pyrolysis temperature was lower than 1000 °C. The average crystallite size of the Si particles was 5.2 nm, calculated by the Scherrer-equation using the peak broadening of (111) line. The average crystallite size increased to 17.6 nm when the material was pyrolyzed at 1400 °C.
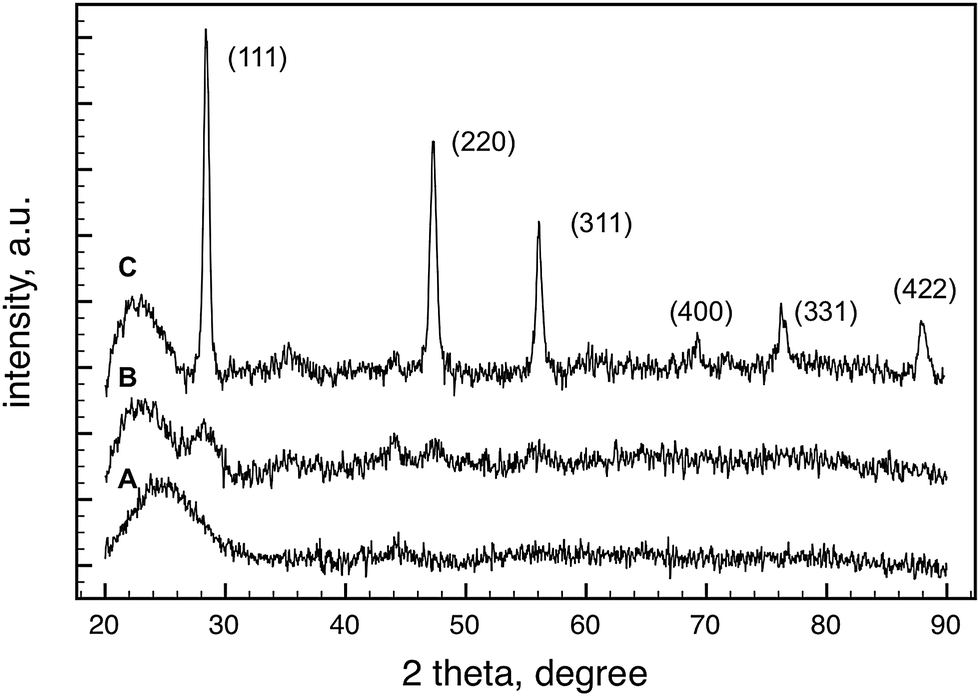 | ||
| Fig. 4 XRD patterns of the (HSiO1.5)n gel samples after pyrolysis at different temperatures in flowing H2/Ar atmosphere for 1 h. (A) 700 °C; (B) 1000 °C; (C) 1400 °C. | ||
Based on the FTIR, 29Si MAS NMR and powder XRD results and previously published data,26 we suggest that the transformation of (HSiO1.5)n gel to Si and silicon oxides occurs in two stages. Below 700 °C, more precisely at around 350–400 °C,26 silane forms through the redistribution of Si–H and Si–O bonds, and promptly decomposes to Si nanocrystallites:
| 4HSiO1.5 → SiH4 + ySiO2 + 3 − y(SiOx) | (1) |
| SiH4 → Si + 2H2 | (2) |
As the temperature increases, the remaining hydrogen silsesquioxane further decomposes to silsesquioxide, which together with other suboxides, disproportionates to silicon and silicon oxide:
| 2HSiO1.5 → Si2O3 + H2 | (3) |
| Si2O3 → 0.5Si + 1.5SiO2 | (4) |
This is why the band at 875 cm−1 cannot be seen after pyrolysis at 1400 °C. Based on these findings, we can conclude that the absorption band observed at ∼875 cm−1 in our FTIR spectra after pyrolysis is attributed to Si–O stretching vibrations influenced by the neighbouring Si–Si bonds, as suggested previously.27 According to the stoichiometry of reactions (1)–(4) the theoretical metallic Si content after 1000 °C pyrolysis would be 13.5 wt%. Based on solid state NMR results, the metallic Si content of this sample is around 6.7 wt%. The discrepancy might be explained by the partial decomposition of the material during evaporation of the solvent after synthesis or extraction of the templating polymer.
It is noteworthy that during pyrolysis, material transitions from white to orange-brown. Although this color change had previously been postulated as a characteristic of Si2O3 formation,43 we believe, in accordance with others,44 that this is not the case. Instead, this color change is the consequence of Si nanocrystal formation, as pure Si2O3 is white.42,45
Pyrolysis leads to changes, not only in the chemical but also in the physical structure of the material. To investigate the pore structure of the material, nitrogen adsorption measurements were conducted. As seen in Fig. 5, the vesicular (HSiO1.5)n gel shows a type IV adsorption isotherm with a H3 hysteresis loop, which is characteristic of mesoporous materials with disordered pore structure. Results of the nitrogen adsorption measurements are summarized in Table 2. The rapid decrease of surface area upon heating to high temperatures, which is not commonly observed in the case of mesoporous silicas, can be explained by the chemical instability of the amorphous hydrogen silsesquioxane gel walls. As shown above, during pyrolysis the hydrogen silsesquioxane decomposes to silane, silica and/or silicon suboxides which further disproportionate to silicon and silica; i.e., Si–O bonds break and reform during the processes. As a result of these transformations, the material lacks the solid covalent network of SiO4 units. The presence of oxygen vacancies significantly reduces the glass transition temperature and hence the thermal stability of the material.
 | ||
| Fig. 5 Nitrogen adsorption isotherms of vesicular (HSiO1.5)n gel (1) after extraction of P-123 with ethanol and after pyrolysis at 700 °C (2) and at 1000 °C (3) in flowing H2/Ar atmosphere for 1 h. | ||
| Pyrolysis temp. [°C] | BET surface area [m2 g−1] | Pore volumeb [cm3 g−1] | Average pore sizeb [nm] |
|---|---|---|---|
| a Sample was calcined in air for 6 h at 550 °C. Heating rate was 10 °C min−1. b Cumulative pore volume and average pore size were calculated based on the adsorption branch of the isotherms. | |||
| After extraction | 600.9 | 0.604 | 6.5 |
| 550a | 130.8 | 0.095 | 6.5 |
| 700 | 119.9 | 0.12 | 6.4 |
| 1000 | 19.1 | 0.025 | 6.3 |
In spite of the significant decrease in surface area and pore volume after high temperature pyrolysis, the type of the adsorption isotherm and the hysteresis loop did not change (Fig. 5) and we conclude that the material likely retained some of the original pore structure (Fig. 5). In ordered mesoporous materials, the average pore diameter calculated using the adsorption and desorption branches of the isotherm are typically the same. However, in the case of materials with H3 type hysteresis, desorption of nitrogen is delayed because of the presence of ink-bottle like pores with constricted necks. Previous investigators showed that if the diameter of the neck is smaller than a critical value (4 nm for nitrogen), desorption does not depend on neck size,46,47 and hence the average pore size calculated from the desorption branch will give unrealistic results; for this reason, the adsorption branch was used.
Small angle X-ray diffraction measurements were performed to provide more information about the structural changes during pyrolysis. The results show that the final material contains periodic building blocks, but their distribution lacks any symmetry (Fig. 6). The d-spacing calculated from the XRD pattern can be related to the distance between periodically repeated scattering centers, in other words, to the vesicle size. This value is 26 nm before and 24.5 nm after pyrolysis at 1000 °C, which is smaller than the DLS and sizes observed by SEM and TEM, probably as a result of the random distribution, and the probable collapse and rupture of the condensed vesicles after solvent evaporation, as the TEM images in Fig. 1 suggest.
 | ||
| Fig. 6 Small-angle X-ray diffraction from the vesicular (HSiO1.5)n gel after extraction of P-123 (solid line) and after pyrolysis at 1000 °C in H2/Ar (dashed line). | ||
The decrease in surface area after pyrolysis can be explained by the densification of the material that accompanies the increase in the number of Si(OSi)4 units (as shown by the NMR and FTIR results), leading to the closing or disappearance of pores situated in the vesicle walls or between the vesicles. The collapse of the mesoporous structure is even more pronounced after pyrolysis at 1400 °C, which also leads to the aggregation of small Si nanocrystals, as seen in the results of the XRD (Fig. 4) and TEM (Fig. 7) analyses.
 | ||
| Fig. 7 (A) TEM image of vesicular (HSiO1.5)n after pyrolysis at 1000 °C. (B) after pyrolysis at 1400 °C. The Si particles embedded in the SiO2 matrix can be clearly seen. The scale bars in A and B represent 100 nm. (C, D) Bright field and dark field images, respectively, of the material after pyrolysis at 1000 °C. The Si nanoparticles appear as bright spots on the dark field image. The scale bar represents 20 nm. | ||
Electrochemical characterization
Analyses of electrochemical performance as anode in lithium-ion battery half-cells show that the specific capacity of the material was strongly dependent on the pyrolysis temperature (Fig. 8). The lowest first cycle charge capacity (71 mA h g−1) was observed for the vesicular (HSiO1.5)n material that had been pyrolyzed at 1400 °C. This was initially surprising, as Si nanocrystals which were detected by XRD and their presence was further supported by TEM. It is plausible that the low specific capacity is the consequence of the dense SiO2 matrix, hindering access of lithium ions to the more deeply embedded Si nanoparticles; if so, only the outer surface of the composite may contain Si nanocrystals capable of reacting reversibly with lithium ions, as suggested for the proposed mechanism for lithiation of a Si–SiO2 composite.48 The authors of that study observed a five-fold capacity increase after 200 charge and discharge cycles, which was related to the increased fraction of Si phase formed at the interface. Our results show a very minor increase in the discharge capacity after 50 cycles, but the peaks at ∼0.2 V vs. Li/Li+ on the lithiation curve and at ∼0.3 and ∼0.45 V on the delithiation curve on the differential capacity plots (Fig. 9) indicate the phase transformations of amorphous silicon and lithium silicide49,50 during the electrode reactions, which were undetected during the first ten cycles. | ||
| Fig. 8 Top: specific charge (empty symbols) and discharge (filled symbols) capacities of the lithium ion battery half-cells upon cycling at 60 mA g−1 current density. Active material:carbon black:binder ratio was 80:15:5 using sodium carboxymethyl cellulose (CMC) as binder. The vesicular (HSiO1.5)n gel pyrolyzed at 1400 °C (□), 700 °C (○), 1000 °C (△). Curve (⋄) shows the specific capacities when polyacrylic acid (PAA) was used as binder with the vesicular (HSiO1.5)n gel pyrolyzed at 1000 °C. Bottom: rate capability performance of the latter (PAA) cell. | ||
 | ||
| Fig. 9 Galvanostatic voltage profiles (left) and the corresponding differential capacity (dQdV) analyses (right) of cells using vesicular (HSiO1.5)n pyrolyzed at 1000 °C (upper row) and at 1400 °C (bottom row). | ||
To investigate the role of the silicon in the cycling process, XRD measurements of the anode coating before and after 50 cycles was undertaken (see ESI†). The intensity of the small Si(111) peak in the XRD pattern did not change, confirming the suggestion that the contribution of the embedded Si nanoparticles in the lithiation process is indeed very small or negligible.
Higher first cycle charge capacities were observed when the vesicular (HSiO1.5)n gel was pyrolyzed at lower temperatures: 278 mA h g−1 and 483 mA h g−1 for 700 °C and 1000 °C, respectively (Fig. 8). The initial difference in the charge capacities between these cells decreased upon cycling and stabilized at around 200 mA h g−1 after 50 cycles. There is only limited information in the literature about the electrochemical performance of pyrolyzed hydrogen silsesquioxane. It was reported that an electrode containing non-mesoporous hydrogen silsesquioxane exhibited first cycle specific capacity of 516 mA h g−1, but that this value decreased to 99 mA h g−1 after 33 cycles.51 When that pyrolyzed hydrogen silsesquioxane was coated with carbon, the first cycle capacity increased to 905 mA h g−1 with a 60% capacity retention after the first 33 cycles.51 Others reported excellent cycling stability and high specific capacity when carbon coated non-mesoporous pyrolyzed hydrogen silsesquioxane electrode was used versus metallic lithium. The initial specific capacity of the electrode was around 925 mA h g−1 which only decreased to 740 mA h g−1 after 100 cycles.52 It appears that the mesoporous structure itself increases cycling stability, and that carbon coating is needed to increase both conductivity and capacity retention.
The higher specific capacity of the cells made with the material pyrolyzed at 1000 °C can be related to the higher concentration of accessible Si nanoparticles, which counterbalances the lower surface area due to the higher pyrolysis temperature. The voltage capacity plots and differential capacity (dQ/dV) curves of the cell using the anode pyrolyzed at 1000 °C are shown in Fig. 9. The contribution of Si in the electrochemical processes can be clearly seen. The data suggest that during the first cycle, at very low potential, ∼0.065 V, irreversible lithiation of the highly active sub-oxide regions takes place. During this process, as previously suggested from DFT calculations,48 the size of the Si nanoparticles increases; this conclusion is supported by the appearance of additional peaks at ∼0.1 and 0.2 V in the second lithiation curve of the dQ/dV plot, and commonly attributed to the formation of amorphous LixSi alloys.49,50,53
Note that when the vesicular (HSiO1.5)n gel calcined in air at 550 °C was also tested as anode, it exhibited very low lithium storage capacity, which did not change upon cycling (see ESI†). The material had almost the same surface area as the sample pyrolyzed at 700 °C (Table 2), but did not contain any Si or suboxide phase, only SiO2. This observation confirms our identification of silicon and its suboxide as the lithium acceptors.
Our results clearly show that the content of Si is more important than the surface area of the matrix. Higher temperatures produce larger and more abundant Si nanocrystals at the cost of collapsed structure and reduced surface area. At some critical temperature between 1000 and 1400 °C, the pore structure disappears and the redistribution of Si–O bonds results in the formation of a dense matrix, which occludes the embedded Si particles from lithiation and the electrochemical reactions.
Optimizing the composition and the nature of the electrode coating can further increase the capacity and cycling stability of the cells. As seen in Fig. 8, changing the binder from sodium carboxymethyl cellulose (CMC) to polyacrylic acid (PAA) leads to a ca. 30% increase in specific capacity. To further investigate the effect of the binder on the electrochemical performance of the cell made by using vesicular (HSiO1.5)n pyrolyzed at 1000 °C, cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were conducted. CV test data of the first 10 cycles were used for this analysis, since it is believed that the initial stage of the cell operation is significantly limited by the formation of the solid-electrolyte interphase (SEI) layer and the consequent rapid decay of the cell capacity. The CV profiles of Si@SiOx–SiO2-based anodes in which the two different polymer binders were used are compared in Fig. 10. When CMC was used as the binder (Fig. 10A, C, and E), the intensities of the cathodic current peaks progressively decreased while those of the anodic peaks slightly increased as the CV cycling proceeded. For the anode in which the PAA binder was used (Fig. 10B, D, and F), however, it was observed that both cathodic and anodic current peaks gradually increased as the cycling continued. These results suggest that the lithiation and delithiation processes in the anode are accelerated more readily in the PAA binder environment, and specifically that the insertion and extraction of Li ions occurred in a more reversible manner when PAA was used as binder.
 | ||
| Fig. 10 Cyclic voltammograms of Si@SiOx–SiO2 anodes in which two different polymer binders were used: CMC (A, C, and E); PAA (B, D, and F). Fig. (C) and (E) are enlarged sections of figure (A); (D) and (F) are enlarged sections of figure (B). | ||
These results can be explained by effects on the formation of the SEI layer. In the CMC-bonded anode, a cathodic current peak at the potential of 1.0 V is shown in the first cycle, which corresponds to the formation of the SEI layer at the interface between the electrode and electrolyte. In case of the PAA-based counterpart, however, no peak corresponding to SEI formation is observed during the first or subsequent cycles. This indicates that the SEI formation occurred at a slower rate with regard to the PAA-bonded composite when compared to the composite bonded with CMC.
In the anodic currents of both samples, two consecutive peaks are observed at the potentials of approximately 0.35 V and 0.48 V, corresponding to the extraction of Li ions from the anode. The progressive shift of these peaks toward higher positive potentials indicates the progressive loss of mobile Li ions as more are retained irreversibly at each cycle, while the gradual increase of peak intensity reflects the progressive expansion of the lithiated domains throughout the anode.3,54
Fig. 11 shows the results of the EIS measurements taken before and after each CV cycle shown in Fig. 10. Results of the automated nonlinear curve fitting are summarized in the Table 3. R0 is the combined resistance of the electrolyte and all other ohmic components of the system. In both cases, we see that the interface resistance (R1) contributed by the SEI layer is increased after the first cycle.
 | ||
| Fig. 11 Electrochemical impedance spectroscopy data of samples in which two different binders are used (A, B) PAA; (C, D) CMC; (E) corresponds to the equivalent circuit model for the Si@SiOx–SiO2 anode/electrolyte interface. Zre and Zim are real and imaginary components of the complex impedance. W is the Warburg impedance. CPE1 and CPE2 are constant phase elements of the SEI and electrode, respectively. R0, R1, and R2 are defined in Table 3. | ||
| Cycle 1 | Cycle 2 | Cycle 5 | Cycle 10 | |||||
|---|---|---|---|---|---|---|---|---|
| PAA | CMC | PAA | CMC | PAA | CMC | PAA | CMC | |
| R 0: combined internal resistance of the electrolyte and other ohmic component of the cell, R1: interface resistance of the SEI between the electrolyte and electrode, R2: charge-transfer resistance in the electrode. | ||||||||
| R 0 | 0.92 | 1.59 | 1.23 | 1.13 | 1.36 | 1.15 | 2.13 | 1.1 |
| R 1 | 3.22 | 1 × 1020 | 70.76 | 1 × 1020 | 142.3 | 1 × 1020 | 154.1 | 1 × 1020 |
| R 2 | 48.73 | 0.23 | 1.87 × 10−6 | 48.13 | 0.01 | 84.73 | 1.91 × 10−8 | 229.5 |
The electrolyte resistance was generally lower when the PAA binder was used. R1, which corresponds to the electric resistance of SEI layer, was already very high during the first CV cycle for the CMC sample, while in the case of the PAA sample, this resistance increased slowly. Similarly, electrode charge transfer resistance also increased gradually during cycling.
Conclusions
In summary, we have shown that vesicles of hydrogen silsesquioxane gel with uniform size distribution can be synthesized by the hydrolysis of triethoxysilane in hydrochloric acid solution containing P-123 triblock copolymer in the absence of added salt or swelling agent. We suggest that vesicle formation is the result of a cooperative self-assembly rather than a templated synthesis, since vesicles of the polymer were not observed before the addition of triethoxysilane.Evaporation of the solvent and extraction of the structure-directing polymer yields a condensed mesoporous structure with bimodal pore size distribution. Pyrolysis of this material in inert atmosphere leads to the formation of silicon nanocrystals embedded in a SiOx–SiO2 matrix. The size and concentration of silicon nanoparticles and the surrounding suboxide region strongly depends on the pyrolysis temperature. We found that the mesoporous vesicular gel pyrolyzed at 1000 °C has the highest specific capacity when used as an anode in Li-ion half-cells. At lower pyrolysis temperature (700 °C) the size and/or concentration of Si nanocrystals formed is insufficient for high electrochemical capacity, while pyrolysis at higher temperature (1400 °C) caused the mesoporous structure to collapse, making the silicon nanocrystals largely inaccessible to the lithium ions.
Further optimization of the electrode's carbon-based coating, both internally and externally, may further enhance stability with cycling. Use of polyacrylic acid instead of sodium carboxymethyl cellulose binder increased the specific capacity by ca. 30%; electrochemical analyses suggest this result may be explained by slower build-up of the SEI layer in those cells.
Acknowledgements
This research was supported by the U.S. Department of Energy through award no. DE-FG02-02ER46006 (for support of F.S., D.E.M., and all other research costs) and award no. DE-SC0001009 to the Center for Energy Efficiency, an Energy Frontier Research Center (for support of C.S.K.), and by the Institute for Collaborative Biotechnologies through grant W9111NF-09-0001 from the U.S. Army Research Office (for support of J.C.S.). Instrumentation for material characterization was provided by UCSB's Material Research Laboratory Shared Experimental Facilities, a member of the NSF-funded Materials Research Facilities Network, supported by the MRSEC Program of the NSF under Award No. DMR 1121053. The authors are thankful to Lyle Kaplan-Reinig and to Dr Krisztián Niesz for their helpful comments and valuable suggestions during the preparation of the manuscript.Notes and references
- J. O. Besenhard, J. Yang and M. Winter, J. Power Sources, 1997, 68, 87 CrossRef CAS.
- R. Nesper, Prog. Solid State Chem., 1990, 20, 1–45 CrossRef CAS.
- B. A. Boukamp, G. C. Lesh and R. A. Huggins, J. Electrochem. Soc., 1981, 128, 725–729 CrossRef CAS PubMed.
- H. Li, X. Huang, L. Chen, Z. Wu and Y. Liang, Electrochem. Solid-State Lett., 1999, 2, 547 CrossRef CAS PubMed.
- X. Yang, Z. Wen, X. Xu, B. Lin and Z. Lin, J. Electrochem. Soc., 2006, 153, A1341 CrossRef CAS PubMed.
- U. Kasavajjula, C. Wang and A. J. Appleby, J. Power Sources, 2007, 163, 1003 CrossRef CAS PubMed.
- R. Liu, J. Duay and S. B. Lee, Chem. Commun., 2011, 47, 1384 RSC.
- M. Ge, J. Rong, X. Fang and C. Zhou, Nano Lett., 2012, 12, 2318 CrossRef CAS PubMed.
- N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. Wang and Y. Cui, Nano Lett., 2012, 12, 3315 CrossRef CAS PubMed.
- L. Hu, H. Wu, S. S. Hong, L. Cui, J. R. McDonough, S. Bohy and Y. Cui, Chem. Commun., 2010, 47, 367 RSC.
- J. Yang, Y. Takeda, N. Imanishi, C. Capiglia, J. Xie and O. Yamamoto, Solid State Ionics, 2002, 152–153, 125 CrossRef CAS.
- Y. Nagao, H. Sakaguchi, H. Honda, T. Fukunaga and T. Esaka, J. Electrochem. Soc., 2004, 151, A1572 CrossRef CAS PubMed.
- T. Kim, S. Park and S. M. Oh, J. Electrochem. Soc., 2007, 154, A1112 CrossRef CAS PubMed.
- F. Bechstedt and K. Hübner, J. Non-Cryst. Solids, 1987, 93, 125 CrossRef CAS.
- A. Hohl, T. Wieder, P. van Aken, T. Weirich, G. Denninger, M. Vidal, S. Oswald, C. Deneke, J. Mayer and H. Fuess, J. Non-Cryst. Solids, 2003, 320, 255 CrossRef CAS.
- T. Zhang, J. Gao, H. P. Zhang, L. C. Yang, Y. P. Wu and H. Q. Wu, Electrochem. Commun., 2007, 9, 886 CrossRef CAS PubMed.
- P. R. Abel, Y.-M. Lin, H. Celio, A. Heller and C. B. Mullins, ACS Nano, 2012, 6, 2506 CrossRef CAS PubMed.
- Y. Hwa, C.-M. Park and H.-J. Sohn, J. Power Sources, 2013, 222, 129 CrossRef CAS PubMed.
- C.-M. Park, W. Choi, Y. Hwa, J.-H. Kim, G. Jeong and H.-J. Sohn, J. Mater. Chem., 2010, 20, 4854 RSC.
- M. Pauthe, E. Bernstein, J. Dumas, L. Saviot, A. Pradel and M. Ribes, J. Mater. Chem., 1999, 9, 187 RSC.
- E. J. Henderson, J. A. Kelly and J. G. C. Veinot, Chem. Mater., 2009, 21, 5426 CrossRef CAS.
- E. J. Henderson, A. J. Shuhendler, P. Prasad, V. Baumann, F. Maier-Flaig, D. O. Faulkner, U. Lemmer, X. Y. Wu and G. A. Ozin, Small, 2011, 7, 2507 CAS.
- J. A. Kelly, E. J. Henderson and J. G. C. Veinot, Chem. Commun., 2010, 46, 8704 RSC.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS.
- Z. Xie, E. J. Henderson, O. Dag, W. Wang, J. E. Lofgreen, C. Kübel, T. Scherer, P. M. Brodersen, Z.-Z. Gu and G. A. Ozin, J. Am. Chem. Soc., 2011, 133, 5094 CrossRef CAS PubMed.
- T. Marks, S. Trussler, A. J. Smith, D. Xiong and J. R. Dahn, J. Electrochem. Soc., 2011, 158, A51 CrossRef CAS PubMed.
- V. Belot, R. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, Chem. Mater., 1991, 3, 127 CrossRef CAS.
- P. Alexandridis and T. Alan Hatton, Colloids Surf., A, 1995, 96, 1 CrossRef CAS.
- P. Petrov, J. Yuan, K. Yoncheva, A. H. E. Müller and C. B. Tsvetanov, J. Phys. Chem. B, 2008, 112, 8879 CrossRef CAS PubMed.
- B. Yang, C. Guo, S. Chen, J. Ma, J. Wang, X. Liang, L. Zheng and H. Liu, J. Phys. Chem. B, 2006, 110, 23068 CrossRef CAS PubMed.
- N. Dan and S. A. Safran, Macromolecules, 1994, 27, 5766 CrossRef CAS.
- P. Alexandridis, U. Olsson and B. Lindman, Langmuir, 1998, 14, 2627 CrossRef CAS.
- T. Sakai, H. Kurosawa, T. Okada and S. Mishima, Colloids Surf., A, 2011, 389, 82 CrossRef CAS PubMed.
- P. Alexandridis and J. F. Holzwarth, Langmuir, 1997, 13, 6074 CrossRef CAS.
- K. Nakashima and P. Bahadur, Adv. Colloid Interface Sci., 2006, 123–126, 75 CrossRef CAS PubMed.
- P. Bahadur, K. Pandya, M. Almgren, P. Li and P. Stilbs, Colloid Polym. Sci., 1993, 271, 657 CAS.
- N. Pandit, T. Trygstad, S. Croy, M. Bohorquez and C. Koch, J. Colloid Interface Sci., 2000, 222, 213 CrossRef CAS PubMed.
- M. G. Albrecht and C. Blanchette, J. Electrochem. Soc., 1998, 145, 4019 CrossRef CAS PubMed.
- P. Bornhauser and G. Calzaferri, J. Phys. Chem., 1996, 100, 2035 CrossRef CAS.
- R. Al-Oweini and H. El-Rassy, J. Mol. Struct., 2009, 919, 140 CrossRef CAS PubMed.
- E. Ritter, Opt. Acta, 1962, 9, 197 CrossRef CAS.
- R. M. Hagenmayer, B. Friede and M. Jansen, J. Non-Cryst. Solids, 1998, 226, 225 CrossRef CAS.
- G. H. Wagner and A. N. Pines, Ind. Eng. Chem. Res., 1952, 44, 321 CrossRef CAS.
- C. M. Hessel, E. J. Henderson and J. G. C. Veinot, Chem. Mater., 2006, 18, 6139 CrossRef CAS.
- B. S. Suresh and D. K. Padma, J. Chem. Soc., Dalton Trans., 1984, 1779 RSC.
- P. I. Ravikovitch and A. V. Neimark, Langmuir, 2002, 18, 1550 CrossRef CAS.
- P. I. Ravikovitch and A. V. Neimark, Langmuir, 2002, 18, 9830 CrossRef CAS.
- C. Ban, B. B. Kappes, Q. Xu, C. Engtrakul, C. V. Ciobanu, A. C. Dillon and Y. Zhao, Appl. Phys. Lett., 2012, 100, 243905 CrossRef PubMed.
- K. Kang, K. Song, H. Heo, S. Yoo, G.-S. Kim, G. Lee, Y.-M. Kang and M.-H. Jo, Chem. Sci., 2011, 2, 1090 RSC.
- A. M. Chockla, M. G. Panthani, V. C. Holmberg, C. M. Hessel, D. K. Reid, T. D. Bogart, J. T. Harris, C. B. Mullins and B. A. Korgel, J. Phys. Chem. C, 2012, 116, 11917 CAS.
- H.-s. Kim and S.-k. Mah, US Pat., US8309252 B2, 2012 Search PubMed.
- M.-S. Park, E. Park, J. Lee, G. Jeong, K. J. Kim, J. H. Kim, Y.-J. Kim and H. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 9608 CAS.
- K. Song, S. Yoo, K. Kang, H. Heo, Y.-M. Kang and M.-H. Jo, J. Power Sources, 2013, 229, 229 CrossRef CAS PubMed.
- W. J. Weydanz, M. Wohlfahrt-Mehrens and R. A. Huggins, J. Power Sources, 1999, 81–82, 237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nj01762e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |