
In situ solvent and counteranion-induced synthesis, structural characterization and photoluminescence properties of Pb-based MOFs†
Qi-Bing
Bo
*,
Juan-Juan
Pang
,
Hong-Yan
Wang
,
Chun-Hua
Fan
and
Zhen-Wei
Zhang
School of Chemistry and Chemical Engineering, University of Jinan, Jinan 250022, China. E-mail: chm_boqb@ujn.edu.cn
First published on 20th October 2014
Abstract
Based on an attempt to investigate the influence of solvents and counteranions on the structures and photoluminescence properties of Pb-based metal–organic frameworks, the hydrothermal reactions of the same amounts of Pb(NO3)2 and Pb(OAc)2 with the ligand 5-tert-butylisophthalic acid (H2tip) in the presence of water and a water–ethanol mixture have yielded three compounds [Pb(H2O)(tip)]n (1), [Pb3(μ4-O)(tip)2]n (2) and [Pb4(μ4-O)(tip)3]n (3) under similar reaction conditions (tip = 5-tert-butylisophthalate anion). These compounds represented the first examples of Pb(II) metal–organic frameworks with H2tip. All of them have been characterized by means of FT-IR spectra, elemental analysis, single-crystal X-ray diffraction, powder X-ray diffraction, thermogravimetric analysis, and photoluminescence spectra. The reaction processes revealed that 1 and 2 had selectivity for specific solvents. And the selectivity for specific counteranions was also found in 2 and 3. Single-crystal X-ray diffraction showed that 1 and 3 crystallized in the orthorhombic crystal system with space groups of Iba2 and Pna21, respectively, while 2 crystallized in the monoclinic P21/c space group. The structure of 1 featured a 2D bilayer structure containing a uninodal sql-type topological motif with a Schlafli symbol of (44). The 2D layer framework of 2 was constructed from a unique 8-connected hexanuclear cluster secondary building unit Pb6O2(COO)8, resulting in a uninodal hxl-type topological motif with a Schlafli symbol of (36·46·53). The structure of 3 could be described as a 3D microporous framework with a 6-connected tetranuclear cluster Pb4O(COO)6. Topological analysis revealed that 3 represented a uninodal dia-type topological motif with a Schlafli symbol of (66). All of the solid state compounds exhibited the photoluminescence properties at room temperature. Furthermore, taking the emissions of the free ligand into consideration, the emissions 1 and 2 could be assigned to metal-centered s → p transition transitions, while the emissions of 3 were due to ligand-to-metal charge transfers between the delocalized p bonds of carboxylate groups and p orbitals of Pb(II) centers. Especially, the in situ solvent and counteranion-induced synthesis strategy reported here could afford us new opportunities in the rapid design of new materials with interesting structures and properties.
Introduction
The design and construction of functional metal–organic frameworks (MOFs) has been one of the most active areas of materials research. The increasingly great interest in these materials has been not only due to their intriguing variety of structural topologies but also due to their promising properties.1 To date, many MOFs based on d-transition metals, f-lanthanide metals and 3d–4f heterometals have been extensively synthesized and investigated. However, less attention has been paid to the construction of the heavy p-block Pb(II) MOFs.2 As a heavy p-block Pb(II) ion, its toxic effects have increased with its increasing use in industry, such as in paints and batteries. As a result, the Pb(II) ion has polluted the environment and had damaging effects on human health.3 Therefore, a good knowledge of the Pb(II) ion properties, including aspects such as a large ion radius, the lone pair of electrons, flexible coordination environment, and variable stereochemical activity, would be of great significance to further understand the toxicological properties of Pb(II) ions. Furthermore, it would also provide unique opportunities for the construction of Pb-based MOFs with intriguing structural topologies and interesting properties. For the above-mentioned reasons, several types of Pb-based MOFs with organic ligands have been reported.2In our attempt to investigate the design and control of the self-assembly of d10- and f-based MOFs with organic ligands, we recently reported that the reaction of the Zn2+, Cd2+ and lanthanide salts with 5-methylisophthalic acid (H2mip) and 5-tert-butylisophthalic acid (H2tip) gave rise to the formation of various luminescent MOFs with novel structures.4 As part of our ongoing studies in the development of the coordination chemistry of the heavy main group elements, we chose typical Pb(II) centers with distinct coordination preferences to assemble with the H2tip ligand. To the best of our knowledge, no report for Pb(II)–tip MOFs has been presented though several other Pb(II) MOFs have been reported and studied by other groups. On the other hand, a solvent-induced synthesis of new MOFs has been of particular interest in materials chemistry.5 In this study, with a solvent or counteranion as the only variable, hydrothermal reactions with the same amounts of H2tip, NaOH, Pb(OAc)2·3H2O or Pb(NO3)2, yielded three new Pb-based MOFs [Pb(H2O)(tip)]n (1), [Pb3(μ4-O)(tip)2]n (2) and [Pb4(μ4-O)(tip)3]n (3) under the same reaction conditions. All of these MOFs were characterized by single-crystal X-ray diffraction, infrared spectroscopy, thermogravimetric analysis, elemental analysis, and PXRD measurements. The solvent and counteranion-induced synthesis and the photoluminescence properties of the products were also discussed in detail.
Results and discussion
Considering the solvent or counteranion as the only variable component, hydrothermal reactions of 5-tert-butyl isophthalic acid with the same amounts of H2tip, NaOH, Pb(OAc)2·3H2O or Pb(NO3)2 afforded three colorless crystals formulated by single-crystal X-ray diffraction as [Pb(H2O)(tip)]n (1), [Pb3(μ4-O)(tip)2]n (2) and [Pb4(μ4-O)(tip)3]n (3). Here, tip denotes the 5-tert-butylisophthalate dianions. The results of single-crystal X-ray diffraction analysis showed that all of the crystals have different structures, and all the 5-tert-butyl isophthalic acids were deprotonated to coordinate with the Pb2+ ions.Crystal structure of [Pb(H2O)(tip)]n (1)
Single-crystal X-ray diffraction analysis revealed that 1 crystallized in the space group Iba2 of the orthorhombic crystal system. Fig. 1 (represented in the “Ball-and-stick” model) showed that the independent crystallographic unit of 1 contained one Pb2+ ion, one coordinated water molecule and one tip anion.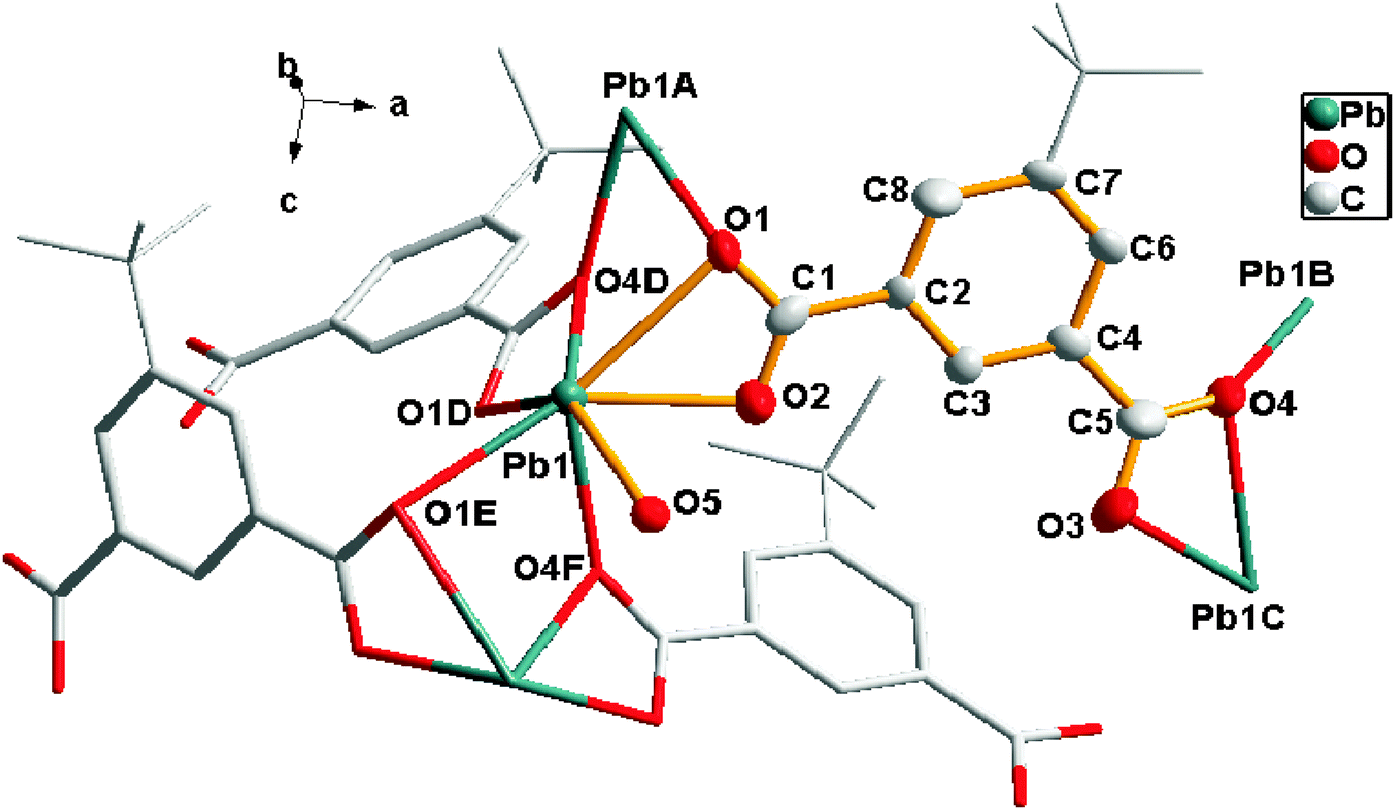 | ||
| Fig. 1 The coordination geometry of the tip ligand and Pb atoms in 1. Hydrogen atoms are omitted for clarity. Symmetry codes: A (−x, y, −0.5 + z), B (0.5 − x, 1.5 − y, −0.5 + z), C (0.5 + x, 1.5 − y, z), D (−0.5 + x, 1.5 − y, z), E (−x, y, 0.5 + z), F (0.5 − x, 1.5 − y, 0.5 + z). | ||
Each Pb center was connected to six carboxylate oxygen donors (O1, O2, O1D, O4D, O1E, O4F) from four different tip anions (Pb–O 2.384–2.817 Å). It is noticeable that the Pb1–O5 distance from the coordinated water molecule (Pb–O 2.938 Å) suggested a nonnegligible interaction,6 generating a seven coordination sphere for the lead atom. The coordination environment of tip for 1 revealed that each tip anion acting as bridge-linking ligand coordinated to four adjacent Pb atoms simultaneously with carboxylate groups. And each carboxylate group of tip adopted μ2-η2:η1-bridging modes (Chart 1a), forming a 2D bilayered structure (labeled with mauve and green parts in Fig. 2) in the ac plane.
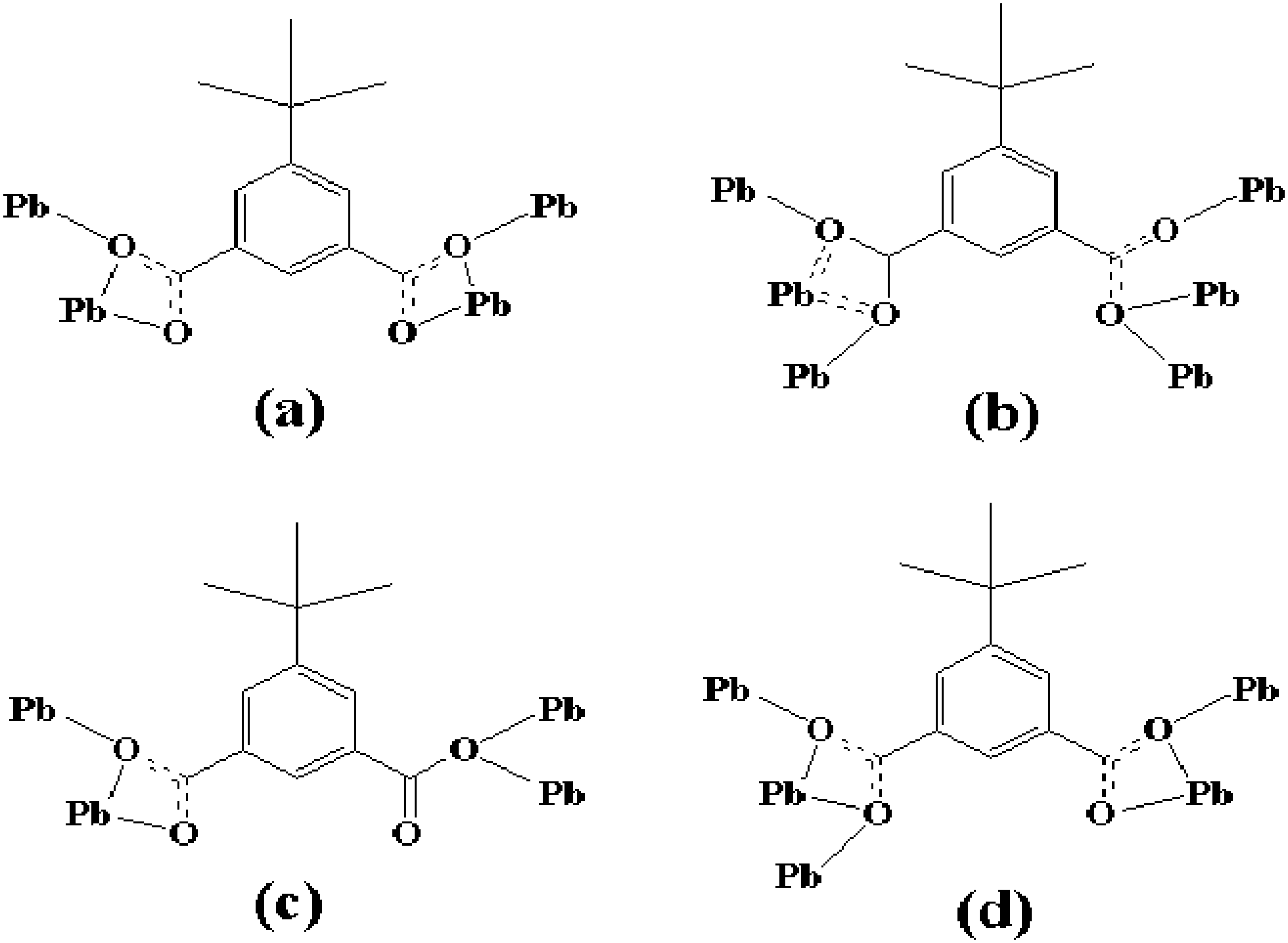 | ||
| Chart 1 Diverse coordination modes of the tip ligands. For each carboxylate group: μ2-η2:η1-bridging mode (a); μ3-η2:η2-bis-bridging mode and μ3-η1:η2-tri-monodentate mode (b); μ2-η1:η2-bridging mode and μ2-η0:η2-bis-monodentate mode (c); μ3-η2:η2-bis-bridging mode and μ2-η1:η2-bridging mode (d). | ||
 | ||
| Fig. 2 The 2D bilayer structure in ac plane for 1. | ||
Considering the tip ligand and the Pb center as nodes, the equivalent 2D topology framework for 1 could be viewed as a uninodal sql-type topological motif with a Schlafli symbol of (44), as shown in the ESI 1.†
Crystal structure of [Pb3(μ4-O)(tip)2]n (2)
Single crystal X-ray diffraction analysis indicated that 2 crystallized in the monoclinic crystal system with the space group P21/c. Fig. 3 showed that three Pb2+ ions were clustered by tip anions in multiple coordination modes and these three Pb2+ ions adopted six, five, and five coordination numbers, respectively. | ||
| Fig. 3 The coordination geometry of the tip ligand and Pb atoms in 2. Hydrogen atoms are omitted for clarity. Symmetry codes: A (−x, 1 − y, −z), B (−x, −0.5 + y, 0.5 − z), C (x, y, −1 + z), D (x, 1.5 − y, −0.5 + z), E (−x, 0.5 + y, 0.5 − z), F (x, 1.5 − y, 0.5 + z), G (x, y, 1 + z). | ||
The coordination environment of Pb1 was a six-coordinated geometry completed by one oxygen atom (O9) from μ4-O2−, five carboxylate oxygen atoms (O1, O2, O8B, O6A, O3C) from four tip ligands. Pb2 adopted a five-coordination geometry defined by two oxygen atoms (O9, O9A) from μ4-O2−, and three carboxylate oxygen atoms (O5, O6, O7B) from two tip ligands. Pb3 was five-coordinated by one oxygen atom (O9) from μ4-O2− and four carboxylate oxygen atoms (O1, O5, O3C, O7D) from four tip anions. The Pb–O bond lengths were in the range of 2.274–2.802 Å, which were comparable to those reported for other lead(II) carboxylates.7 The coordination environment of tip for 2 was shown in Chart 1b and c. It is evident that two kinds of the tip anions acting as bridge-linking ligands coordinated to the adjacent lead atoms with carboxylate groups. One adopted the μ3-η2:η2-bis-bridging mode and μ3-η1:η2-tri-monodentate mode (Chart 1b), while the other linked the lead atoms adopting the μ2-η1:η2-bridging mode and μ2-η0:η2-bis-monodentate mode (Chart 1c).
It is noteworthy that compound 2 displayed a two-dimensional network consisting of μ4-O2− anion-bridged Pb6O2 clusters. As shown in Fig. 4, six symmetry-related lead atoms (Pb1, Pb2, Pb3, Pb1A, Pb2A and Pb3A) were clustered by two μ4-O2− anions (O9 and O9A). Then, the cluster was edge-bridged by eight different carboxylate groups (O–C1–O, O–C1A–O, O–C5C–O, O–C5H–O, O–C13–O, O–C13A–O, O–C17B–O, and O–C17D–O) of the tip ligands to give an 8-connected centrosymmetric hexanuclear cluster SBU (secondary building unit) Pb6O2(COO)8. Each of the cluster SBUs was further interlinked by eight tip ligands, resulting in a 2D layer structure in bc plane, as shown in Fig. 5.
 | ||
| Fig. 4 Schematic representation of an 8-connected hexanuclear cluster SBU Pb6O2(COO)8 for 2. Symmetry codes: A (−x, 1 − y, −z), B (−x, −0.5 + y, 0.5 − z), C (x, y, −1 + z), D (x, 1.5 − y, −0.5 + z), H (−x, 1 − y, 1 − z). | ||
 | ||
| Fig. 5 The 2D layer structure constructed from 8-connected Pb6O2 clusters and tip ligands for 2. Hexanuclear clusters are represented in polyhedrons for clarity. | ||
Topologically, the tip ligands could be viewed as the linkers, and each Pb6O2 cluster served for a 6-connected node. The equivalent 2D topology framework for 2 can be viewed as a uninodal hxl-type topological motif with a Schlafli symbol of (36·46·53), as shown in the ESI 2.†
Crystal structure of [Pb4(μ4-O)(tip)3]n (3)
Compound 3 crystallized in the orthorhombic space group Pna21. The structure of 3 featured a complicated 3D network. The asymmetric unit of 3 contained four crystallographically unique Pb2+ ions, one μ4-O2− anion, and three tip anions. As shown in Fig. 6, Pb1, Pb2, and Pb3 were all six-coordinated by five carboxylate oxygen atoms (O1, O5, O6, O12, and O3A belong to Pb1; O1, O2, O8C, O9C, and O4A belong to Pb2; O5, O11, O7C, O8C, and O10C belong to Pb3) from four different tip anions and one oxygen atom from μ4-O2− anion (O13). Pb4 adopted another kind of coordination manner. It coordinated with five carboxylate oxygen atoms (O10C, O11, O12, O3A, and O4A belong to Pb4A) from three different tip anions and one oxygen atom from the μ4-O2− anion (O13). Distances of Pb–O in the range 2.283–2.818 Å were in good agreement with the reported values.8 Only the Pb–O bond distances for Pb4A–O10C [2.883 Å], Pb4A–O11 [2.932 Å], and Pb2–O2 [2.938 Å], were longer, but the values were still in the bonding range.9 At the same time, the subtle distinction among Pb1, Pb2, Pb3 and Pb4 originated from three kinds of tip ligands adopting four, five, and six coordination numbers, respectively. The detailed coordination modes for these three tip ligands are described in Chart 1a, b, and d. | ||
| Fig. 6 The coordination geometry of the tip ligand and Pb atoms in 3. Hydrogen atoms are omitted for clarity. Symmetry codes: A (2 − x, 1 − y, 0.5 + z), B (2 − x, 1 − y, −0.5 + z), C (0.5 + x, 1.5 − y, z), D (−0.5 + x, 1.5 − y, z), E (1.5 − x, 0.5 + y, 0.5 + z). | ||
It is worth noting that one μ4-oxygen atom (O13) connected the four crystallographically unique Pb atoms into an isolated tetrahedral cluster Pb4O, which was further shared with the six carboxylate groups (O–C1–O, O–C13–O, O–C30–O, O–C6A–O, O–C18C–O, and O–C25C–O), resulting in a 6-connected tetranuclear cluster SBU (secondary building unit) Pb4O(COO)6, as shown in Fig. 7. Each of the cluster SUBs was further linked to four neighboring ones by six tip ligands, reticulating into the final 3D porous framework (Fig. 8). By virtue of the PLATON analysis,10 approximately 41.5% of the crystal volume is occupied by the bulky tertbutyl groups (1657.5 out of the 3991.1 Å3 in each cell unit), which means that the compound 3 has a microporous framework located in the disordered tertbutyl groups.
 | ||
| Fig. 7 Schematic representation of a 6-connected tetranuclear cluster SBU Pb4O(COO)6 for 3. Symmetry codes: A (2 − x, 1 − y, 0.5 + z), C (0.5 + x, 1.5 − y, z). | ||
 | ||
| Fig. 8 The 3D microporous framework constructed from 6-connected Pb4O clusters and tip ligands for 3. Tetranuclear clusters are represented in polyhedrons for clarity. | ||
As shown in the ESI 3,† the tetrahedral Pb4O clusters acted as 4-connected nodes and the tip groups acted as the linkers. The equivalent 3D topology framework for 3 could be viewed as a uninodal dia-type topological motif with a Schlafli symbol of (66).
Solvent and counteranion-induced synthesis
The basic consideration of our synthetic strategy was to study the influence of solvents and counteranions on the structures and photoluminescence properties of Pb-based MOFs with the ligand 5-tert-butylisophthalic acid. Under the similar reaction conditions, we selected the same amounts of 5-tert-butylisophthalic acid, NaOH, lead salts of nitrate and acetate, water and water–ethanol mixture as the starting materials to carry out this investigation. As shown in Chart 2, when 5-tert-butylisophthalic acid, NaOH, and lead acetate were chosen as the reactants, the use of solvent water alone generated the crystal product of [Pb(H2O)(tip)]n (1), which showed a 2D bilayered structure. | ||
| Chart 2 Solvent and counteranion-induced synthesis. | ||
When the solvent was changed from pure water to a water/ethanol mixture (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, V/V) with everything else kept the same as in 1, the other crystal [Pb3(μ4-O)(tip)2]n (2) with a different 2D layer structure was formed. With the counteranion as the only variable, following the procedure adopted for 2, changing the lead acetate to nitrate afforded the crystal sample [Pb4(μ4-O)(tip)3]n (3) as a novel 3D porous framework. However, following the synthesis process of 3, when the solvent mixture (water–ethanol) was substituted with the pure solvent (water), only unspecific white precipitates were obtained, which was perhaps due to the quick hydrolysis of lead nitrate under the pure solvent conditions. In contrast to 1, no solvent was incorporated into 2 and 3, which may be likely due to the different solvent polarity or the configuration of solvent molecules. Furthermore, no nitrate or acetate ion was bonded to the lead center for the Pb-based MOFs mentioned above. At the same time, once isolated, all of the Pb-based MOFs were stable in air and insoluble in common organic solvents and water. From Chart 2, it is evident that MOFs 1 and 2 had selectivity for specific solvents during the synthetic process. And the selectivity for specific counteranions was also found in MOFs 2 and 3. The above observations suggested that both solvents and counteranions played crucial roles in the controllable synthesis of MOFs under the same amounts of starting materials and similar reaction conditions. The water–ethanol mixture and counteranions (acetate) might be highly efficient as structure-directing agents during the reaction processes, since the coordination environment around the building units or metal centers was always affected by the excellent H-bond formation abilities of the water–ethanol mixture and acetate. The findings of the in situ solvent and counteranion-induced synthesis strategy afforded us new opportunities in the rapid design of materials with interesting structures and properties.
:8, V/V) with everything else kept the same as in 1, the other crystal [Pb3(μ4-O)(tip)2]n (2) with a different 2D layer structure was formed. With the counteranion as the only variable, following the procedure adopted for 2, changing the lead acetate to nitrate afforded the crystal sample [Pb4(μ4-O)(tip)3]n (3) as a novel 3D porous framework. However, following the synthesis process of 3, when the solvent mixture (water–ethanol) was substituted with the pure solvent (water), only unspecific white precipitates were obtained, which was perhaps due to the quick hydrolysis of lead nitrate under the pure solvent conditions. In contrast to 1, no solvent was incorporated into 2 and 3, which may be likely due to the different solvent polarity or the configuration of solvent molecules. Furthermore, no nitrate or acetate ion was bonded to the lead center for the Pb-based MOFs mentioned above. At the same time, once isolated, all of the Pb-based MOFs were stable in air and insoluble in common organic solvents and water. From Chart 2, it is evident that MOFs 1 and 2 had selectivity for specific solvents during the synthetic process. And the selectivity for specific counteranions was also found in MOFs 2 and 3. The above observations suggested that both solvents and counteranions played crucial roles in the controllable synthesis of MOFs under the same amounts of starting materials and similar reaction conditions. The water–ethanol mixture and counteranions (acetate) might be highly efficient as structure-directing agents during the reaction processes, since the coordination environment around the building units or metal centers was always affected by the excellent H-bond formation abilities of the water–ethanol mixture and acetate. The findings of the in situ solvent and counteranion-induced synthesis strategy afforded us new opportunities in the rapid design of materials with interesting structures and properties.
FT-IR spectra, thermogravimetric analyses and PXRD patterns
All of the FT-IR spectra for 1, 2 and 3 exhibited the characteristic bands of the asymmetric stretching vibrations of the carboxylate groups at 1630–1520 cm−1, as well as the symmetric stretching vibrations between 1310 and 1480 cm−1 (ESI 4†). The results indicated the existence of a deprotonated carboxylate group coordinated to the metal ion, in agreement with the solid-state structures.In order to examine the thermal stability of the three compounds, thermogravimetric analyses (TGA) were performed on single-phase polycrystalline samples of these materials (ESI 5†). For 1, the first weight loss of 3.5% (calculated value 4.0%) occurred in the temperature range of 30–140 °C, which was equivalent to the release of the coordinated water molecules. And then there was a plateau of stability before 360 °C, indicating that the framework of 1 could be stable up to 360 °C. Upon heating to above 360 °C, a rapid collapse of the framework took place. This may be attributed to the decomposition of the ligand. 1 did not lose weight at higher temperatures up to 550 °C and the residue (50.6%, calculated value 50.1%) might be PbO. For 2 and 3, no weight loss was observed from room temperature up to 380 °C in the TG curves, which indicated that both of them were anhydrous. Evidently, the TGA results for 2 and 3 were also in accordance with the single crystal structure analysis. The weight losses of 59.2% and 62.1% from 380 to 580 °C should be ascribed to the decomposition of the organic frameworks for 2 and 3, respectively, which were both in good agreement with the calculated values (59.3% for 2, 62.0% for 3), considering the final product as PbO.
The powder X-ray diffraction patterns (PXRD) of 1, 2 and 3 were performed at room temperature (ESI 6†). Their different structures have also been indicated by their different XRPD patterns. Furthermore, all the XRPD patterns measured for the as-synthesized samples were all in good agreement with those simulated from single crystal structural data. Thus all the compounds 1, 2 and 3 were obtained as a single phase, which proved the purity of the bulk phases.
Photoluminescence properties
With regard to MOFs, studies have been essentially restricted to d10 and 4f metals, and little attention has been paid to the luminescence of MOFs of main group metals such as Pb.1,2 It is noteworthy that the Pb(II) complexes are a potential class of functional materials with interesting photic properties, due to the fact that complexes of heavy metals with the s2 electron configuration might reduce the radiative lifetime of triplets by increasing spin–orbit coupling and promote emission from the triplet state under ambient conditions.11 In this work, the photoluminescence properties of 1–3 have been explored in the solid state at room temperature. And in order to understand the nature of these emission bands, the photoluminescence properties of the free ligand were also examined. As shown in Fig. 9, the free ligand H2tip displayed ultraviolet emission at 345 nm (λex = 315 nm), which could probably be assigned to the π* → π transition. | ||
| Fig. 9 The emission spectra excited at 315, 360, 350 and 325 nm for H2tip, 1, 2 and 3, respectively. | ||
Three Pb-based MOFs displayed emission bands centered at 527 nm (λex = 360 nm) for 1, 502, 547 and 568 nm (λex = 350 nm) for 2, and 416 and 458 nm (λex = 325 nm) for 3. It was evident that the emission bands of the Pb-based MOFs were all red-shifted compared to the free ligand, and emitted luminescence in the range 400–600 nm at room temperature. However the free ligand H2tip exhibited no observable fluorescence emission in the range of 400–600 nm, which eliminated the ligand-centered (LC) and ligand-to-ligand charge transfer (LLCT) excited states. Therefore, taking the emission bands of the free ligand into consideration, the emission bands at 416 and 458 nm of 3 could be attributed to the ligand-to-metal charge transfer (LMCT) between delocalized p bonds of the aromatic carboxylate groups and p orbitals of Pb(II) centers, which is similar to those reported in the literature for the Pb-based MOFs.12 Compared with 3, the emission wavelengths of 1 and 2 became longer and should be different from that of LMCT. The low-energy emissions with large Stokes shifts, including 527 nm for 1, 502, 547 and 568 nm for 2 could be assigned to a metal-centered s → p transition as proposed by Vogler.13 These photoluminescence results imply that the coordination modes of the Pb(II) cations played an important role in influencing the emissive peak position of compounds, and had a significant influence on the emission mechanism of the Pb-based MOFs.
Conclusion
With solvents or counteranions as the only variable, hydrothermal reactions with the same amounts of 5-tert-butylisophthalic acid, NaOH, Pb(OAc)2·3H2O or Pb(NO3)2, afforded three new Pb-based metal–organic frameworks [Pb(H2O)(tip)]n (1), [Pb3(μ4-O)(tip)2]n (2) and [Pb4(μ4-O)(tip)3]n (3) under the same reaction conditions. Considering the synthetic process, the Pb-based compounds had selectivity for specific solvents and counterions in the self-assembly process. In this study, the water–ethanol mixture, nitrate and acetate ions could be highly efficient as structure-directing agents during the synthesis processes of Pb-based compounds. The structural analysis indicated that 1 featured a 2D bilayer structure containing a uninodal sql-type topological motif; the 2D layer framework of 2 was constructed from a unique 8-connected hexanuclear cluster secondary building unit Pb6O2(COO)8, with a uninodal hxl-type topological motif. The 3D microporous framework of 3 was formed by a 6-connected tetranuclear cluster Pb4O(COO)6 with a uninodal dia-type topological motif. Furthermore, strong luminescent emissions are observed for 1, 2 and 3, which might be good candidates for obtaining photoluminescent materials. The Pb-based metal–organic frameworks 1, 2 and 3, obtained under the same amounts of starting materials and similar synthetic procedures, were distinct from each other in properties, such as composition, topology, and photoluminescence properties, which could afford us new opportunities in the rapid design of materials with interesting structures and properties. A feasible strategy for controlling the synthesis of other framework architectures through this in situ solvent and counteranion-induced synthesis method is underway in our laboratory.Experimental section
Reagents and instrumentation
Reagents were purchased commercially and were used without further purification. Elemental analyses (C and H), IR spectra, TG analyses, single crystal X-ray diffraction data, PXRD, solid-state emission and excitation spectra were all performed on the corresponding instruments similarly to our recently published paper.4 And the crystal structures were solved using a direction method and refined using a full matrix least-squares technique based on F2 and the SHELXL 97 program.14 All of the non-hydrogen atoms were refined anisotropically. The organic hydrogen atoms were generated geometrically, and the aqua hydrogen atoms were located from difference maps and refined using isotropic temperature factors. CCDC 1019800 (1), 1019801 (2) and 1019802 (3). The crystallographic data and structure refinement for all the MOFs are summarized in Table 1.| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Empirical formula | C12H14O5Pb | C24H24O9Pb3 | C36H36O13Pb4 |
| Formula weight | 445.42 | 1078.00 | 1505.41 |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic |
| Space group | Iba2 | P21/c | Pna21 |
| a/Å | 16.2354(8) | 18.7933(9) | 18.4043(5) |
| b/Å | 19.1181(10) | 17.7755(6) | 30.9956(9) |
| c/Å | 8.2268(4) | 8.2471(3) | 6.9963(2) |
| α/deg | 90 | 90 | 90 |
| β/deg | 90 | 99.723(4) | 90 |
| γ/deg | 90 | 90 | 90 |
| Volume/Å3 | 2553.5(2) | 2715.46(19) | 3991.1(2) |
| Z | 8 | 4 | 4 |
| ρ cal/g cm−3 | 2.317 | 2.637 | 2.505 |
| μ/mm−1 | 13.224 | 18.602 | 16.884 |
| F (000) | 1664 | 1944 | 2736 |
| GOF | 1.023 | 1.029 | 1.055 |
| R 1 (I > 2σ(I)) | 0.0326 | 0.0599 | 0.0354 |
| wR2 (I > 2σ(I)) | 0.0678 | 0.1319 | 0.0749 |
| R 1 (all data) | 0.0557 | 0.0975 | 0.0410 |
| wR2 (all data) | 0.0799 | 0.1551 | 0.0778 |
| Flack parameter | 0.07(3) | 0.038(13) |
Preparation of MOFs
Elemental analysis (%) calcd for 1: C 32.36, H 3.17; found: C 32.29, H 3.22.
IR (KBr, cm−1) for 1: 3615 w, 3299 m, 3175 w, 2965 s, 2910 w, 2866 w, 1628 w, 1609 w, 1598 s, 1518 s, 1430 m, 1370 m, 1356 m, 1312 m, 1273 m, 1205 w, 1180 m, 1130 w, 1114 m, 927 w, 910 m, 822 s, 787 s, 754 s, 726 s, 696 m, 602 w, 578 w, 520 w, 481 w.
Elemental analysis (%) calcd for 2: C 26.74, H 2.24; found: C 26.65, H 2.16.
IR (KBr, cm−1) for 2: 3422 s, 2965 s, 2910 w, 2873 w, 1846 w, 1603 s, 1540 s, 1480 m, 1433 s, 1400 m, 1370 s, 1350 s, 1312 w, 1268 s, 1204 w, 1177 w, 1114 s, 1045 w, 1001 w, 927 m, 907 m, 820 s, 781 s, 751 s, 721 s, 696 w, 608 w, 578 w, 514 w, 484 w.
Elemental analysis (%) calcd for 3: C 28.72, H 2.41; found: C 28.68, H 2.38.
IR (KBr, cm−1) for 3: 3435 s, 2959 s, 2873 w, 1851 w, 1631 s, 1595 s, 1548 w, 1513 s, 1480 w, 1463 w, 1425 m, 1367 m, 1351 s, 1309 s, 1279 s, 1254 m, 1205 w, 1161 m, 1130 w, 1111 w, 1004 w, 910 m, 814 m, 776 s, 754 m, 721 s, 713 s, 600 w, 578 w, 512 w, 487 w.
Acknowledgements
We gratefully acknowledge financial support by the National Natural Science Foundation of China (grant no. 21171068) and the Shandong Provincial Natural Science Foundation of China (grant no. ZR2010BM036, ZR2013BQ009).References
- (a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (b) W. Lu, Z. Wei, Z. Y. Gu, T. F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H. C. Zhou, Chem. Soc. Rev., 2014, 43, 5561 RSC; (c) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC; (d) J. B. Decoste and G. W. Peterson, Chem. Rev., 2014, 114, 5695 CrossRef CAS PubMed; (e) C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222 CrossRef CAS PubMed.
- (a) A. Santra and P. K. Bharadwaj, Cryst. Growth Des., 2014, 14, 1476 CrossRef CAS; (b) X. M. Lin, T. T. Li, L. F. Chen, L. Zhang and C. Y. Su, Dalton Trans., 2012, 41, 10422 RSC; (c) S. C. Chen, Z. H. Zhang, Y. S. Zhou, W. Y. Zhou, Y. Z. Li, M. Y. He, Q. Chen and M. Du, Cryst. Growth Des., 2011, 11, 4190 CrossRef CAS.
- (a) A. P. Neal and T. R. Guilarte, Toxicol. Res., 2013, 2, 99 RSC; (b) C. V. Gherasim, J. Křivčík and P. Mikulášek, Chem. Eng. J., 2014, 256, 324 CrossRef CAS; (c) M. S. Tellis, M. M. Lauer, S. Nadella, A. Bianchini and C. M. Wood, Aquat. Toxicol., 2014, 146, 220 CrossRef CAS PubMed; (d) A. Üstünda, C. Behm, W. Föllmann, Y. Duydu and G. H. Degen, Arch. Toxicol., 2014, 88, 1281 CrossRef PubMed.
- (a) Q. B. Bo, H. Y. Wang, D. Q. Wang, Z. W. Zhang, J. L. Miao and G. X. Sun, Inorg. Chem., 2011, 50, 10163 CrossRef CAS PubMed; (b) Q. B. Bo, H. Y. Wang, J. L. Miao and D. Q. Wang, RSC Adv., 2012, 2, 11650 RSC; (c) Q. B. Bo, H. Y. Wang and D. Q. Wang, New J. Chem., 2013, 37, 380 RSC; (d) Q. B. Bo, H. T. Zhang, H. Y. Wang, J. L. Miao and Z. W. Zhang, Chem. – Eur. J., 2014, 20, 3712 CrossRef CAS PubMed.
- (a) Q. B. Bo, Z. X. Suna and W. Forsling, CrystEngComm, 2008, 10, 232 RSC; (b) X. Liu, P. Cen, H. Li, H. Ke, S. Zhang, Q. Wei, G. Xie, S. Chen and S. Gao, Inorg. Chem., 2014, 53, 8088 CrossRef CAS PubMed; (c) L. L. Qu, Y. L. Zhu, Y. Z. Li, H. B. Du and X. Z. You, Cryst. Growth Des., 2011, 11, 2444 CrossRef CAS; (d) J. Qian, J. Hu, J. Zhang, H. Yoshikawa, K. Awaga and C. Zhang, Cryst. Growth Des., 2013, 13, 5211 CrossRef CAS; (e) J. Li, S. Meng, J. Zhang, Y. Song, Z. Huang, H. Zhao, H. Wei, W. Huang, M. P. Cifuentes, M. G. Humphreyd and C. Zhang, CrystEngComm, 2012, 14, 2787 RSC.
- (a) H. T. Xiao and A. Morsali, Solid State Sci., 2007, 9, 155 CrossRef CAS; (b) J. L. Song, C. Lei, Y. Q. Sun and J. G. Mao, J. Solid State Chem., 2004, 177, 2557 CrossRef CAS; (c) J. L. Song, J. G. Mao, Y. Q. Sun and A. Clearfield, Eur. J. Inorg. Chem., 2003, 4218 CrossRef CAS; (d) M. R. S. J. Foreman, T. Gelbrich, M. B. Hursthouse and M. J. Plater, Inorg. Chem. Commun., 2000, 3, 234 CrossRef CAS.
- (a) X. Zhang, J. K. Cheng, F. Chen, M. L. Sun and Y. G. Yao, Inorg. Chem. Commun., 2011, 14, 358 CrossRef CAS; (b) K. L. Zhang, Y. Chang, C. T. Hou, G. W. Diao, R. T. Wu and S. W. Ng, CrystEngComm, 2010, 12, 1194 RSC; (c) X. L. Wang, Y. Q. Chen, Q. Gao, H. Y. Lin, G. C. Liu, J. X. Zhang and A. X. Tian, Cryst. Growth Des., 2010, 10, 2174 CrossRef CAS; (d) J. Yang, J. F. Ma, Y. Y. Liu, J. C. Ma and S. R. Batten, Cryst. Growth Des., 2009, 9, 1894 CrossRef CAS.
- (a) R. Abhinandan, K. J. Swapan, B. Madhusudan, H. Debdoot, S. C. Durga, Z. Ennio and D. Sudipta, J. Solid State Chem., 2013, 197, 46 CrossRef; (b) J. D. Lin, S. T. Wu, Z. H. Li and S. W. Du, CrystEngComm, 2010, 12, 4252 RSC; (c) L. Zhang, Z. J. Li, Q. P. Lin, Y. Y. Qin, J. Zhang, P. X. Yin, J. K. Cheng and Y. G. Yao, Inorg. Chem., 2009, 48, 6517 CrossRef CAS PubMed.
- (a) X. H. Lou, C. Xu, H. M. Li, Z. Q. Wang, H. Guo and D. X. Xue, CrystEngComm, 2013, 15, 4606 RSC; (b) D. Sunirban, K. Hyunuk and K. Kimoon, J. Am. Chem. Soc., 2009, 131, 3814 CrossRef PubMed; (c) P. Du, Y. Yang, J. Yang, Y. Y. Liu, W. Q. Kan and J. F. Ma, CrystEngComm, 2013, 15, 6986 RSC.
- A. L. Spek, PLATON 99, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1999 Search PubMed.
- S. K. Dutta and M. W. Perkovic, Inorg. Chem., 2002, 41, 6938 CrossRef CAS PubMed.
- (a) E. C. Yang, J. Li, B. Ding, Q. Q. Liang, X. G. Wang and X. J. Zhao, CrystEngComm, 2008, 10, 158 RSC; (b) X. Q. Li, H. B. Zhang, S. T. Wu, J. D. Lin, P. Lin, Z. H. Li and S. W. Du, CrystEngComm, 2012, 14, 936 RSC.
- (a) P. C. Ford and A. Vogler, Acc. Chem. Res., 1993, 26, 220 CrossRef CAS; (b) A. Vogler, A. Paukner and H. Kunkely, Coord. Chem. Rev., 1980, 33, 227 CrossRef; (c) H. Nikol, A. Becht and A. Vogler, Inorg. Chem., 1992, 31, 3277 CrossRef CAS; (d) G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer Verlag, Berlin, 1994 Search PubMed.
- (a) G. M. Sheldrick, SHELXL-97, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Topological framework, X-ray crystallographic data in CIF format, the experimental and simulated PXRD patterns, FT-IR spectra, TG curves, etc. CCDC 1019800–1019802. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01506a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |