
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In vitro characterization of a novel C,N-cyclometalated benzimidazole Ru(II) arene complex: stability, intracellular distribution and binding, effects on organic osmolyte homeostasis and induction of apoptosis†
Celina
Støving Dam
ab,
Sergio
Alejo Perez Henarejos
c,
Theodosia
Tsolakou
ab,
Christian
Alexander Segato
b,
Bente
Gammelgaard
a,
Gorakh S.
Yellol
c,
José
Ruiz
c,
Ian
Henry Lambert
b and
Stefan
Stürup
*a
aUniversity of Copenhagen, Department of Pharmacy, Universitetsparken 2, 2100 Copenhagen Ø, Denmark. E-mail: stefan.sturup@sund.ku.dk
bUniversity of Copenhagen, Department of Biology, Universitetsparken 13, 2100 Copenhagen Ø, Denmark
cDepartment of Inorganic Chemistry and Regional Campus of International Excellence “Campus Mare Nostrum”, University of Murcia and Institute for Bio-Health Research of Murcia IMIB-Arrixaca, E-30071 Murcia, Spain
First published on 25th March 2015
Abstract
In the present work a novel C,N-cyclometalated benzimidazole Ru(II) arene complex (GY34) was characterized by applying an alternative, diverse approach considering both chemical and biological aspects. RP-HPLC-ICP-MS and RP-HPLC-ESI-MS analysis proved that GY34 in both RPMI-1640 cell medium and ammonium acetate buffer was transformed into several subspecies and the importance of evaluating and controlling analyte stability throughout experiments was demonstrated. Applying a novel cell fractionation protocol GY34 was found to target cell nuclei and mitochondria in Ehrlich Lettré Ascites (ELA) cells, with the intracellular distribution depending on GY34 concentration in the cell medium during incubation. In ELA cells 96 ± 0.2% of cytosolic GY34 was bound to high-molecular species. Furthermore, using the tracer technique GY34 was found to reduce uptake and increase release of the organic osmolyte taurine in ELA cells, with innate resistance to Cisplatin and in A2780 human ovarian cancer cells, with acquired resistance to Cisplatin. Importantly, FACS analysis revealed that GY34 induced apoptosis in ELA cells. The present data suggest the potential of GY34 in overcoming Cisplatin resistance. The methodology applied can be used as a general protocol and an additional tool in the initial evaluation of novel metal-based drugs.
Introduction
Today cancer ranges among the principal causes of morbidity and mortality worldwide and the World Health Organization expects a 70% increase in the number of new cases over the next two decades.1 For many years Cisplatin has been the choice of treatment for a wide range of cancers, e.g. ovarian, testicular, head and neck, bladder and lung cancers.2 However, the efficiency of Cisplatin is limited by acquired or intrinsic resistance3 and by severe side effects such as ototoxicity, peripheral neuropathy, myelosuppression and nephrotoxicity.4Consequently, focus is shifting towards the discovery of novel improved anti-cancer drugs and numerous new compounds are synthesized in search of a promising drug candidate. In the initial work of selecting a candidate with an ideal profile, determination of IC50 values, apoptotic studies and cell cycle checkpoint assays are usually performed. However, knowledge on the stability of the compound and the mechanism of action is also very valuable when deciding the further fate of a new compound. Particularly, the latter is of significant importance as a mechanism of action dissimilar to that of Cisplatin may be able to overcome Cisplatin resistance, and hence improve anti-cancer therapy.
The purpose of the current work was to characterize the novel C,N-cyclometalated [(η6-p-cym)RuCl(κ2-N,C-L)] L = deprotonated 1-bytyl-2-phenyl-benzimidazole carboxylate complex GY34 (Fig. 1, published by Yellol et al.5) applying a more diverse approach than usually employed, addressing both biological and chemical aspects of the compound. GY34 has been shown to exert an increased cytotoxic activity compared to Cisplatin in colorectal adenocarcinoma HT29 cells (IC50 GY34: 2.2 ± 0.4 μM, IC50 Cisplatin: 9.5 ± 0.2 μM), in human breast cancer T47D cells (IC50 GY34: 5.5 ± 0.2 μM, IC50 Cisplatin: 38 ± 2 μM) and in Cisplatin-resistant human ovarian cancer A2780 cells (IC50 GY34: 6.4 ± 0.1 μM, IC50 Cisplatin: 15 ± 1 μM).5 Moreover, GY34 arrests cells in the S phase of the cell cycle and induces apoptosis of HT29 cells.5 Finally, the level of metal accumulation in T47D cells was increased after treatment with GY34 relative to treatment with Cisplatin.5 Altogether this presents GY34 as a potential future drug candidate for anti-cancer treatment and signifies the importance of further investigation of the compound.
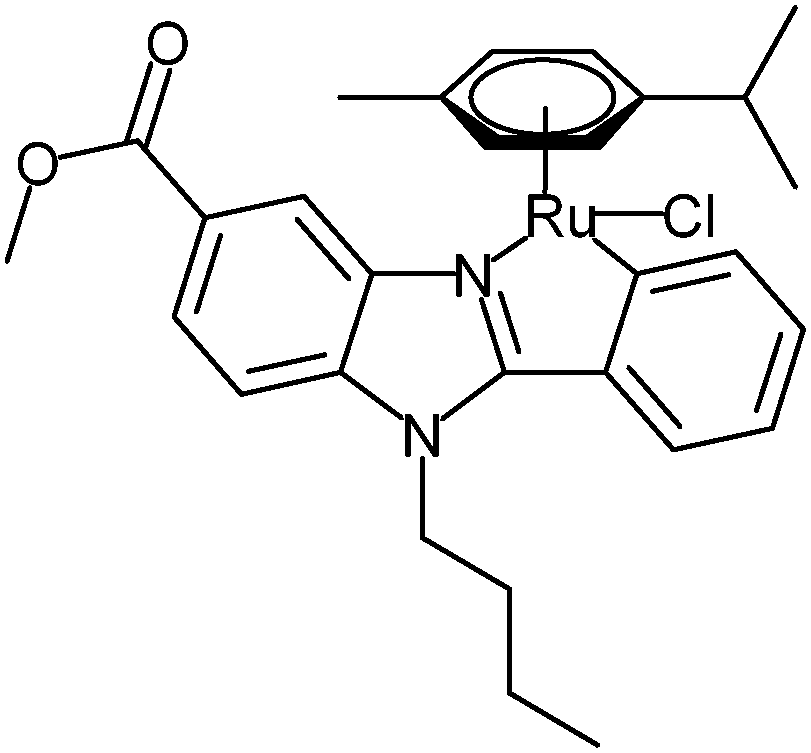 | ||
| Fig. 1 Structure of GY34.9 | ||
Experimental
Reagents
All reagents were of analytical grade and purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated.The Ru-based compound GY34 was synthesized as described elsewhere5 by the research group of Prof. José Ruiz, Department of Inorganic Chemistry, University of Murcia, Spain. 1 mM stock solutions of GY34 were prepared in DMSO. For cell experiments, volumes of 1 mM GY34 stock solutions in DMSO were added directly to the cell medium to obtain the preferred concentration. GY34 concentrations applied in the various experiments were selected on the basis of the IC50 value for human ovarian cancer A2780 cells reported by Yellol et al.5 (refer Introduction). The applied concentration of Cisplatin was based on previously performed caspase 3 activity assay.6
The mobile phase for RP-HPLC-ICP-MS and RP-HPLC-ESI-MS analysis consisted of 20 mM ammonium acetate in 65% v/v MeOH, pH 6.8.
PBS for cell culturing and cell fractionation consisted of 137 mM NaCl, 2.6 mM KCl, 6.5 mM Na2HPO4 and 1.5 mM KH2PO4, pH 7.4. KCl-Tris buffer used for cell fractionation consisted of 100 mM KCl, 50 mM Tris-HCl, 5 mM MgCl2, 1 mM Na2EDTA, pH 7.4. Lysis buffer consisted of 1% SDS, 150 mM NaCl, 20 mM HEPES, 1 mM EDTA, 0.5% Triton X-100, 1 mM NaVO3 and 1% protease inhibitor. Percoll (GE Healthcare, Wauwatosa, WI, USA) solutions were prepared in KCl-Tris buffer. The isotonic NaCl medium and MgCl2 solution used for taurine flux experiments contained 143 mM NaCl, 5 mM KCl, 1 mM Na2HPO4, 1 mM CaCl2, 1 mM MgSO4, 10 mM HEPES and 100 mM MgCl2, respectively.
SDS page gel electrophoresis and western blotting were performed using materials from Invitrogen Life Technologies (Thermo Fischer Scientific, Waltham, MA, USA): XCell SureLock Mini-Cell, 1.0 mm 10 well NuPAGE Novex 10% Bis-Tris gels, NuPAGE LDS Sample Buffer, BenchMark Protein Ladder, NuPAGE MOPS SDS Running Buffer, NuPAGE Antioxidant, XCell II Blot Module, 0.2 μm Nitrocellulose Pre-Cut Blotting Membranes and NuPAGE Transfer Buffer. Membranes were blocked with 5% non-fat dry milk (retail store) in TBST, i.e. 0.01 M Tris-HCl, 0.15 M NaCl, and 0.1% tween 20, pH 7.4. Primary rabbit mAB antibodies against histone H3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500, 18 kDa, Cell Signaling Technology, Danvers, MA, USA), malate dehydrogenase 2 (1:100, 36 kDa) and lactate dehydrogenase B (1:1000, 35 kDa, Novus Biologicals, Littleton, CO, USA) were used along with the secondary anti-rabbit IgG antibody (1:5000). Bands were developed using the BCIP/NBT Phosphatase Substrate 3-component System (Kirkegaard & Perry Laboratories Inc., Gaithersburg, MD, USA).
:500, 18 kDa, Cell Signaling Technology, Danvers, MA, USA), malate dehydrogenase 2 (1:100, 36 kDa) and lactate dehydrogenase B (1:1000, 35 kDa, Novus Biologicals, Littleton, CO, USA) were used along with the secondary anti-rabbit IgG antibody (1:5000). Bands were developed using the BCIP/NBT Phosphatase Substrate 3-component System (Kirkegaard & Perry Laboratories Inc., Gaithersburg, MD, USA).
The Ru standard used for the ICP-MS determinations was purchased from CPI International (Santa Rosa, CA, USA) and was a custom made multi-element standard containing Au, Ir, Pd, Pt, Os and Rh apart from Ru. The standards for external calibration were prepared in a diluted acid solution containing 0.1% HCl and 0.65% HNO3. The same mixture of diluted acids was used to dilute samples for ICP-MS analysis.
The incubation buffer used for the apoptosis assay consisted of 10 mM HEPES, 140 mM NaCl and 5 mM CaCl2, pH = 7.4. The labeling solution was prepared by diluting 20 μL of annexin-V-Fluos labeling reagent (Roche, Basel, Switzerland) in 1 mL of incubation buffer and adding 20 μL of 50 μg mL−1 propidium iodide solution.
Stability of GY34 in cell medium and the mobile phase
The stability of GY34 in dilute solution was initially examined by RP-HPLC-ICP-MS analysis applying an Agilent 1100 Series HPLC system consisting of a quaternary pump, a degasser and an autosampler (Agilent Technologies, Santa Clara, CA, USA). HPLC was performed using a Phenomenex (Torrance, CA, USA) Luna C18 column (3 μm, 100 Å, 100 × 2 mm), with a flow rate of 200 μL min−1 and an injection volume of 20 μL. The HPLC system was connected to a Perkin Elmer (Waltham, MA, USA) Sciex ELAN 6100 DRC-e ICP-MS instrument via a Cetac (Teledyne Technologies Inc., Omaha, NE, USA) Aridus II membrane desolvation system in order to remove organic solvent from the mobile phase prior to introduction into the ICP-MS instrument (refer Møller et al.7 for thorough description of the setup). The desolvation system was equipped with a Cetac 200 μL min−1 C-flow PFA concentric nebulizer, the spray chamber temperature was 110 °C, the desolvator temperature was 160 °C, the argon sweep gas flow was 5 L min−1 and the nitrogen gas flow was 6 mL min−1. The ICP-MS dwell time was 200 ms and 1 sweep/reading, 1 replicate and 500 readings/replicate were applied. Isotopes 99Ru+ and 101Ru+ were monitored. The nebulizer gas flow was 0.9 mL min−1 while RF power and lens voltage were optimized on a daily basis. A 10 ppb Ru standard prepared in 0.1% HCl and 0.65% HNO3 yielded a signal of ∼142000 counts. 1 μM solutions of GY34 (corresponding to 100 ppb Ru) in the mobile phase and Roswell Park Memorial Institute (RPMI-1640) cell medium, respectively, were analyzed immediately after preparation and after 24 h at room temperature.
In order to elucidate the structure of the various GY34 species indicated by the RP-HPLC-ICP-MS analysis, RP-HPLC-ESI-MS was performed using the same HPLC system and parameters as described above, but with a manual injector and with an injection volume of 5 μL. The HPLC system was connected to a Bruker (Billerica, MA, USA) Esquire ion trap equipped with an electrospray ionization source. ESI-MS was performed using the following parameters: positive ionization mode, 300–2200 m/z scan window, 50 ms max. accumulation time, 50000 ion charge control target, average 5, 10 L min−1 drying gas flow, 350 °C drying gas temperature, 45 psi nebulizer gas, 214 V RF amplitude, 42 trap drive, 53 V capillary exit, 18 V skimmer 1, and 5.7 V skimmer 2. A 1 mM GY34 solution prepared in DMSO and RPMI-1640 cell medium (20:80% v/v) was analyzed immediately after preparation and after 24 h of incubation at room temperature.
Cell culturing
Murine Ehrlich Lettré Ascites (ELA) cells, an adherent subtype of non-adherent wild type Ehrlich Ascites Tumor cells (EATC), were obtained from ATCC (Washington, DC, USA) and grown in RPMI-1640 cell medium supplied with 10% heat-inactivated fetal bovine serum and 1% antibiotics (penicillin, streptomycin). The cells were kept as a monolayer in 75 cm2 CellStar culture flasks at 37 °C, 5% CO2, 100% humidity and passaged every 3–4 days using 0.5% trypsin in PBS to detach cells.A2780 human ovarian carcinoma Cisplatin-sensitive wild-type (A2780 WT, provided by Dr Antonio Donaire, University of Murcia, Spain) and Cisplatin-resistant (A2780 RES, kindly donated by Dr Isolda Romero-Canelón, University of Warwick, United Kingdom) cells were grown and passaged under similar conditions, however with 1% L-glutamine added to the cell medium. Cisplatin resistance in A2780 RES cells was maintained by treatment with 1 μM Cisplatin between every three passages.
Cell fractionation
Prior to fractionation, ELA cells were grown to 80–90% confluence in four 175 cm2 culture flasks at 37 °C, 5% CO2, 100% humidity and incubated under the same conditions for 18 h with a nominal concentration of 10 μM GY34. Immediately before the fractionation was initiated a sample of the cell medium was taken out in order to measure the GY34 concentration in the medium and evaluate stability and solubility of the compound. An overview of the cell fractionation procedure is shown in Fig. 2. The fractionation was performed at room temperature; however in between each step in the process samples were kept on ice in order to minimize degradation. The roman numbers in the following provide overview of the different fractionation steps and should be related to Fig. 2.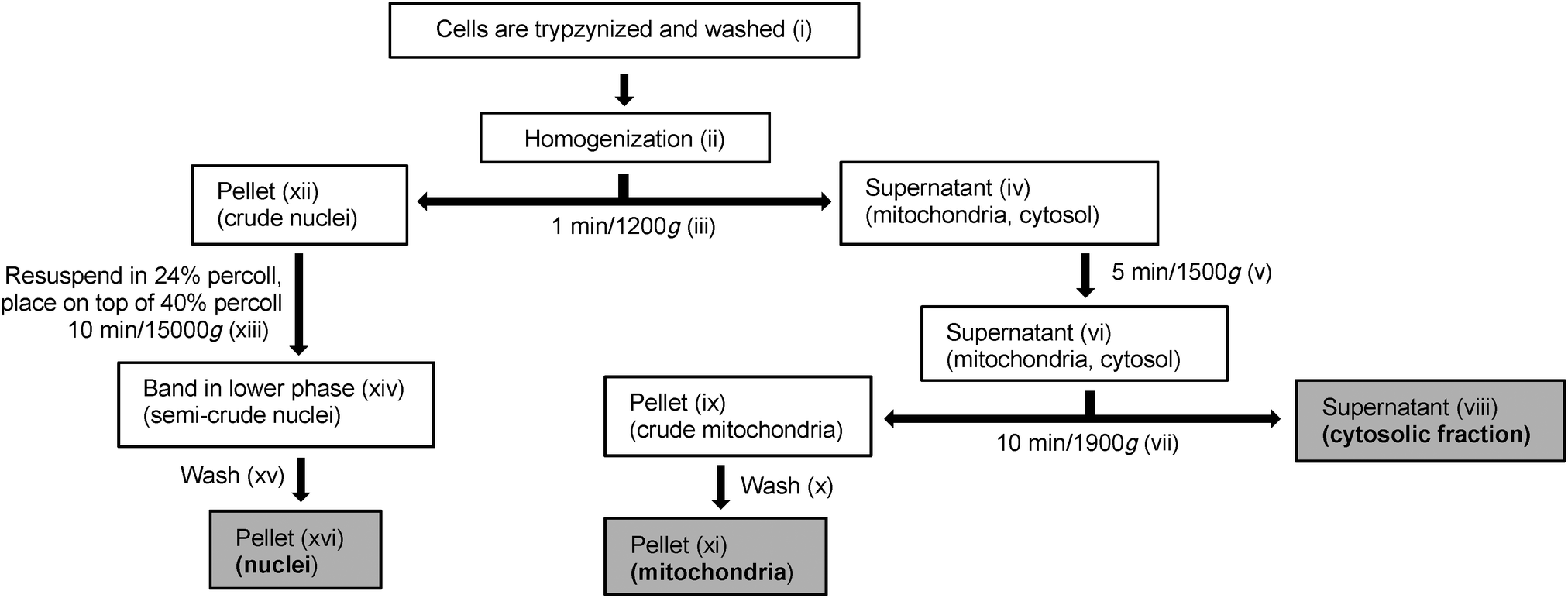 | ||
| Fig. 2 Overview of the procedure used to fractionate murine Ehrlich Lettré Ascites cells into nuclei, mitochondria and cytosol. Roman numbers in parentheses refer various steps in the process and are further specified in the experimental section Cell Fractionation. | ||
(i) Initially, the cell medium was removed and the cells in each of the four 175 cm2 culture flasks were washed in 5 mL of PBS. PBS was removed, 5 mL of 0.5% trypsin in PBS was added and the cells were maintained at 37 °C until loosened. The cells were then resuspended in 10 mL of RPMI-1640 medium and the cell suspensions in the four 175 cm2 culture flasks were transferred to four falcon tubes. The cells were washed twice in 5 mL of PBS (centrifuging 4 min/500g) and pooled in two falcon tubes. After removing PBS, 1 mL of KCl-Tris buffer was added to each falcon tube. KCl-Tris buffer was used to mimic cellular ion composition. The cells were resuspended and the cell suspension in each falcon tube was transferred to an eppendorf tube. From each tube 50 μL of cell suspension (unbroken cells) was collected. The remaining of the cell suspensions was then centrifuged (1 min/1200g/4 °C) and the supernatants were discarded. The remaining pellets (unbroken cells) were each resuspended in 100 μL of KCl-Tris buffer. (ii) The cells were homogenized using polypropylene pellet pestles (2 × 10 strokes, put on ice in between) and 700 μL of KCl-Tris buffer was added to each eppendorf tube. (iii) The homogenized cells were centrifuged (1 min/1200g/4 °C) and (iv) 700 μL of each of the supernatants (mitochondria and cytosol) were collected in new eppendorf tubes. (xii) The remaining pellets (crude nuclei) were stored on ice for later purification. (v) The collected supernatants were centrifuged (5 min/1500g/4 °C), (vi) the resulting supernatants were pooled and the pellet was discarded. (vii) The pooled supernatant was centrifuged (10 min/9000g/4 °C) and (viii) the resulting supernatant (cytosolic fraction) was collected. To 93 μL of the cytosol fraction 5 μL of 10% SDS, 1 μL of NaVO3 and 1 μL of protease inhibitor were added; this was used for protein determination and western blotting. (ix) The remaining pellet (crude mitochondria) (x) was washed three times in 500 μL of KCl-Tris buffer (centrifuging 10 min/9000g/4 °C) and (xi) the pellet (mitochondria) was collected and resuspended in 70 μL of lysis buffer (mitochondrial fraction).
(xiii) The crude nuclei were each resuspended in 600 μL of 24% percoll and each 24% suspension was carefully placed on top of 1 mL of 40% percoll in new eppendorf tubes creating two layers. The two-layer suspensions were centrifuged (10 min/15000g/4 °C) causing the nuclei to migrate through the 24% percoll layer to the bottom of the 40% percoll layer, leaving impurities in the 24% percoll layer. For each centrifuged two-layer suspension, most of the upper phase was removed and (xiv) 300 μL of the nuclei band (semi-crude nuclei) was collected and resuspended in 1 mL of KCl-Tris buffer followed by centrifugation (10 min/15000g/4 °C). (xv) The pellets were each washed in 500 μL of KCl-Tris buffer, centrifuged (10 min/15000g/4 °C) and pooled. The pooled pellet was washed in 500 μL of KCl-Tris buffer, centrifuged (10 min/15000g/4 °C) and (xvi) the resulting pellet (nuclei) was resuspended in 100 μL of lysis buffer (nuclear fraction) and sonicated.
Three replicates of the fractionation were made; i.e. cells from three different passages of the ELA cells were fractionated.
Protein determination
Protein content in cell fractions, used for SDS page gel electrophoresis, was determined by a Bradford colorimetric assay (bio-Rad, Hercules, CA), measuring absorbance at 600 nm (GeneQuant Pro spectrophotometer, GE Healthcare) and correlating values to a standard curve (0–30 μg μL−1). Protein content in influx experiments was determined by a standard Lowry method using standards in the range 0–1 mg mL−1.Evaluation of cell fractionation
The purity of the cell fractions was evaluated by western blotting. Samples containing the same amount of protein (15–20 μg) were proceeded for SDS page gel electrophoresis together with a crude total cell homogenate as control. Protein was then transferred to nitrocellulose membranes and the transfer was confirmed by Ponceau staining. The membranes were blocked for 1 h at 37 °C, incubated with the relevant primary antibody in a humid chamber overnight at 4 °C and washed with TBST. The membranes were then incubated in a humid chamber with the secondary antibody for 1 h at room temperature, washed with TBST and developed using the BCIP/NBT Phosphatase Substrate.Quantitation of GY34
For quantitation of GY34, a Perkin Elmer Sciex Elan 6000 ICP-MS instrument, equipped with a Perkin Elmer low-flow GemCone nebulizer and a Glass Expansion (West Melbourne Vic, Australia) cyclonic spray chamber, was applied. RF power, lens voltage and nebulizer gas flow were optimized on a daily basis. The remaining instrumental parameters applied were: 45 s sample flush, 30 s read delay, 90 s wash, 1 sweep/reading, 25 readings/replicate, 5 replicates, 50 ms dwell time and isotopes 99Ru+ and 101Ru+. Samples were delivered to the ICP-MS instrument with the aid of a Cetac ASX-110FR autosampler. Prior to analysis, samples were prepared according to the following procedure: exact volumes were evaporated to dryness using an Eppendorf (Hamburg, Germany) Concentrator Plus vacuum centrifuge. Subsequently, samples (excl. nuclei) were digested on a heat block with 200 μL of 65% HNO3 and 50 μL of 30% H2O2 at 60 °C for 6 h while nuclei were digested in a CEM (Matthews, NC, USA) MDS-81D microwave oven with 400 μL of 65% HNO3 and 100 μL of 30% H2O2 for 10 min at 60% capacity (corresponds to approximately 10 W). The digests were diluted to 5 mL, filtered (Q-max® RR Syringe Filters, 0.45 μm, Frisenette, Knebel, Denmark) and further diluted before analysis, if necessary. The cell medium was not digested, but diluted 200 times and filtered before analysis. The GY34 concentration was determined by external calibration (0–10 ppb Ru). LOD = 0.005 ppb (blank + 3*SD of blank).Binding of GY34 to cytosolic biomolecules
The distribution of GY34 between high- and low-molecular species in the cytosol was studied using centrifugal filtration. ELA cells were grown to 80–90% confluence in a 175 cm2 culture flask at 37 °C, 5% CO2, 100% humidity and were then incubated with 10 μM GY34 for 18 h under the same conditions. The cytosol was isolated by the following procedure: after removing the cell medium, the cells were washed twice in 10 mL of PBS and PBS was removed. 250 μL of lysis buffer was added, the cells were collected using a rubber policeman, sonicated and centrifuged (5 min/20000g/4 °C). The resulting supernatant (cytosol) was collected. An exact volume of the cytosol was then fractionated using 3 kDa Amicon Ultra 2 mL spin filters (EMD Millipore, Billerica, MA, USA). The cytosol was centrifuged (60 min/4500g/4 °C) and the filtrate (<3 kDa fraction) was collected. The filter was reversed, centrifuged (15 min/1500g/4 °C) and the concentrate (>3 kDa fraction) was collected. The content of GY34 in the <3 kDa and >3 kDa fractions and the unfractionated cytosol was determined as described above. The experiment was performed in triplo, i.e. on three different passages of ELA cells.
Taurine flux assays
Taurine uptake via the taurine transporter (TauT) as well as taurine release under isotonic conditions from A2780 WT, A2780 RES and ELA cells grown in the presence of either 5 μM Cisplatin or 5 μM GY34 for 18 h was determined by the tracer technique at room temperature as previously described.8,9 For influx experiments, cells were grown to 80% confluence in 6-well polyethylene culture plates (9.6 cm2 per well). Five wells were used to determine the taurine uptake and the residual well was used to determine the representative protein content in a well. Cells were washed three times with isotonic NaCl medium and left with 600 μL of isotonic NaCl medium. Influx was initiated by the addition of 50 μL of 3H-taurine stock solution containing 37000 Bq mL−1 (0.005 μM taurine) to wells 1–5 at time 0, 2, 4, 6 and 8 min. Influx was terminated by the removal of the extracellular medium at time 10 min, rapid rinse of cells by the addition/aspiration of 1 mL of ice-cold MgCl2 solution, followed by cell lysis with 96% ethanol. After evaporation of ethanol, 3H-activity was extracted with ddH2O and determined using a Perkin Elmer scintillation counter using Ultima Gold™. The cellular taurine content (nmol per g protein) in each well was determined from the 3H-taurine activity (cpm per well) using the extracellular specific activity (cpm nmol−1) and the protein content (mg protein per well). Taurine uptake (nmol g protein−1 min−1) was determined as the slope of a plot of cellular taurine content versus time using linear regression. At least three replicates of the experiment were made, i.e. on three different passages of each of the three cell lines.
Taurine efflux was estimated for cells grown to 80% confluence in 6-well polyethylene culture plates and loaded in cell media supplemented with 3H-taurine (18500 Bq per well) for 2 h (37 °C, 5% CO2, 100% humidity). Before initiation of the efflux, the loading medium and the cells were washed three times with 1 mL of isotonic NaCl medium. The efflux experiment was performed by transferring the NaCl medium from the well to vials and replacing it with the new medium at two minute intervals. After removal of the last sample, the isotope remaining inside the cells was determined by the addition of 1 mL of 1 M NaOH, gently shaking (1 h) and subsequently transferring NaOH and two times wash-outs (ddH2O) to vials. 3H-activity was determined using Ultima Gold™. The total 3H activity in the cell system was determined as the sum of 3H activity released during the efflux experiment and 3H activity detected in the NaOH/ddH2O wash-outs. The fractional rate constant (min−1) for taurine release under isotonic conditions was calculated from the equation:  , where X1 and X2 are the fractions remaining in the cell at times t1 and t2, divided by the time interval. At least three replicates of the experiment were made, i.e. on three different passages of each of the three cell lines.
, where X1 and X2 are the fractions remaining in the cell at times t1 and t2, divided by the time interval. At least three replicates of the experiment were made, i.e. on three different passages of each of the three cell lines.
Apoptosis assay
Apoptosis studies on cells exposed to Cisplatin and GY34 were performed by flow cytometry. 105 ELA cells were seeded in 2 mL of RPMI-1640 cell medium in 6-well plates and incubated for 24 h at 37 °C, 5% CO2. Cells were subsequently exposed to 5 μM GY34 or 5 μM Cisplatin for 18 h under the same conditions. One well remained untreated serving as a control. After incubation the cell medium was collected and the cells were detached by trypsination (0.25% trypsin/0.5 mM EDTA in 1 mL of PBS, 37 °C, 3 min). Trypsin was inactivated by the addition of 1 mL of cell medium. The resulting 2 mL of cell suspension was added to the collected cell medium hence permitting both floating and adherent cells to be considered in the assay. The cells were centrifuged (10 min/250g) and the precipitated cells were washed twice in 1 mL of PBS. PBS was removed after the final centrifugation and cells were resuspended in 160 μL of incubation buffer. Subsequently, 40 μL of labeling solution was added and the cells were protected from light at room temperature for 15 min. 200 μL of PBS was added immediately before detection of light emission at wavelengths of 620 nm (propidium iodide) and 525 nm (annexin-V) using a Becton-Dickinson FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). During registration 10000 events were acquired in each case. The experiment was performed in duplo, i.e. on two different passages of ELA cells.
Results and discussion
In this work, the novel Ru-based complex GY34 (Fig. 1) was characterized with respect to stability and transformation in solution, intracellular distribution and binding to cytosolic biomolecules, and organic osmolyte flux across cells along with its ability to induce apoptosis. Relevant comparisons to Cisplatin were made.Stability of GY34 in cell medium and the mobile phase
The stability of GY34 in dilute solution was evaluated by applying RP-HPLC-ICP-MS analysis. 1 μM solutions of GY34 in the mobile phase (20 mM ammonium acetate in 65% v/v MeOH, pH 6.8) and RPMI-1640 cell medium, respectively, were analyzed immediately after preparation and after 24 h at room temperature. The results are outlined in Fig. 3. The chromatograms obtained immediately after preparation of the GY34 solutions both contain one large peak (tr ∼ 4.5 min) that most likely can be assigned to GY34. After 24 h at room temperature a second large peak (tr ∼2 min) appears. This indicates that GY34 at room temperature is transformed to another Ru-containing species and that the transformation occurs in both the mobile phase and cell medium.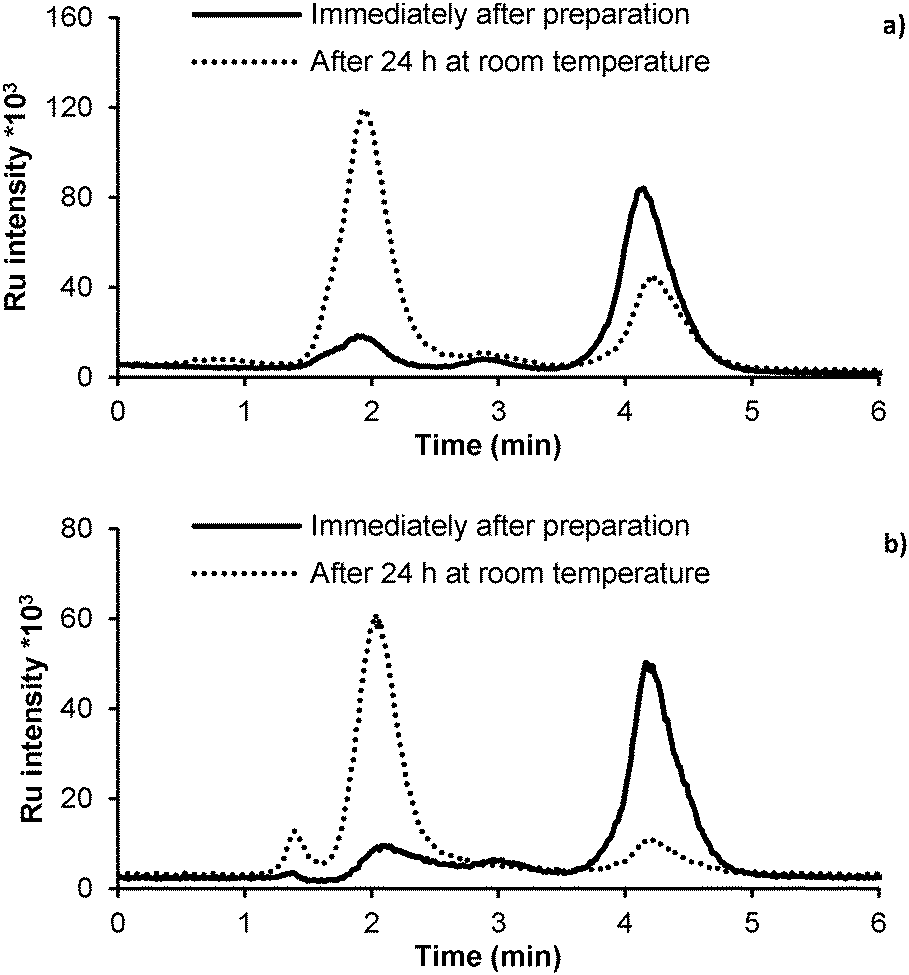 | ||
| Fig. 3 RP-HPLC-ICP-MS chromatograms of 1 μM GY34 solutions in (a) mobile phase (20 mM ammonium acetate in 65% MeOH, pH 6.8) and (b) RPMI-1640 cell medium. Solid curves were obtained immediately after preparation of the GY34 solutions and dotted curves after 24 h at room temperature. | ||
An attempt to elucidate the structures of the GY34 species was made using RP-HPLC-ESI-MS. A 1 mM GY34 solution prepared in DMSO and RPMI-1640 cell medium (20:80% v/v) was analyzed immediately after preparation (the total ion chromatogram (TIC) displays one large peak at tr ∼ 4.5 min and a smaller peak at tr ∼ 2 min, not shown) and after 24 h at room temperature (Fig. 4 and 5). Taking into account the slight difference in retention time caused by different dead volumes in the RP-HPLC-ICP-MS and RP-HPLC-ESI-MS systems, the three peaks shown in Fig. 4 most likely correspond to the peaks at tr ∼ 2, ∼3 and ∼4.5 min in the ICP-MS chromatograms shown in Fig. 3. The dissimilar peak intensity between the ICP-MS and ESI-MS chromatograms is probably due to the different sensitivity of the two methods. The respective mass spectra (Fig. 5) prove that the ∼3 min TIC peak contains species of m/z 529.3 and 607.2 and that the ∼4.5 min TIC peak corresponds to species of m/z 543.2 and 621.3. Verified by comparison to the theoretical mass spectrum (refer ESI†), the ∼3 min TIC peak can be assigned to the structure of GY34 where chlorine is removed and the methyl ester is hydrolyzed (m/z 529.3, refer Fig. 5 for structure) and to the same structure with DMSO being added (m/z 607.2). In the same manner, the ∼4.5 min TIC peak can be assigned to GY34 without chlorine (m/z 543.2, refer Fig. 5 for structure) and to the same structure with DMSO being added (m/z 621.3). The addition of DMSO to the GY34 species is not expected to occur in solution but rather as a result of an interaction between GY34 and DMSO in the ESI-MS interface. The mass spectrum of the ∼2 min TIC peak (not shown) did not allow structure elucidation of the related Ru species as the characteristic Ru isotopic pattern was absent. An explanation could be that the ∼2 min TIC peak corresponds to a GY34 subspecies that is fragmented during ESI leaving the Ru-containing fragment unionized and hence not detectable by ESI-MS, but still able to produce a signal in ICP-MS as it contains Ru.
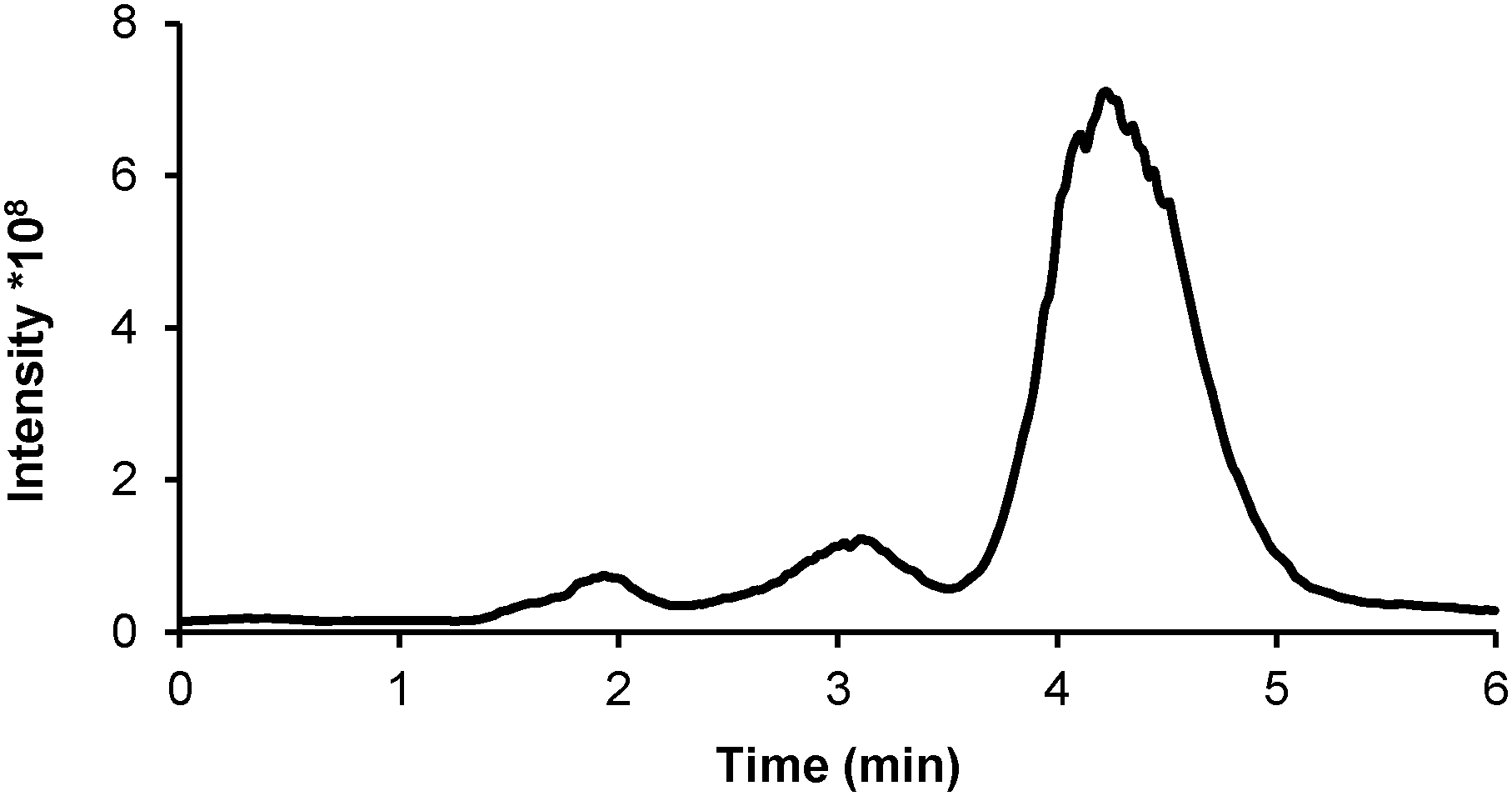 | ||
| Fig. 4 Total ion chromatogram obtained from RP-HPLC-ESI-MS analysis of 1 mM GY34 in RPMI-1640 cell medium 24 h after preparation. | ||
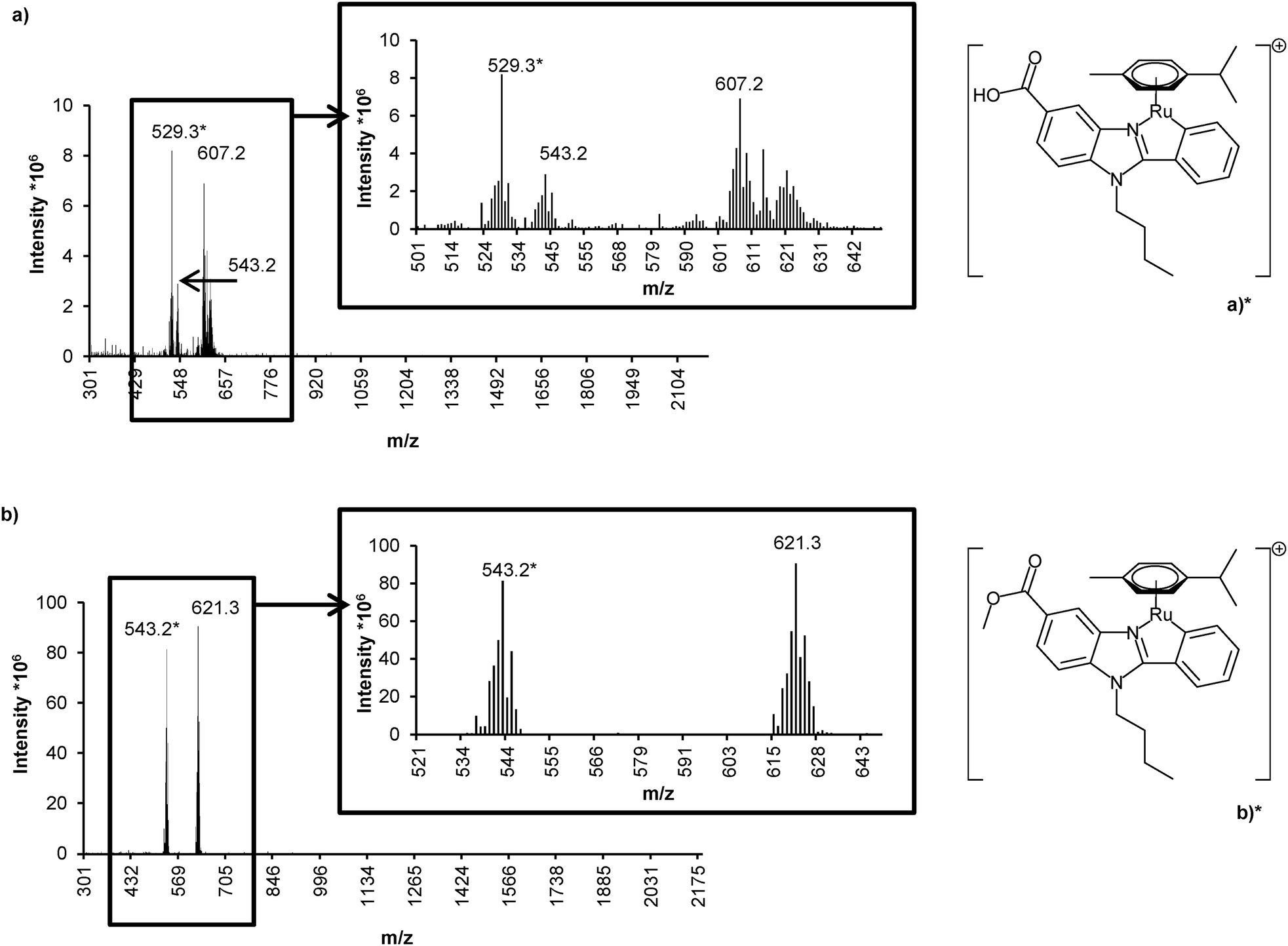 | ||
| Fig. 5 Mass spectra of (a) the ∼3 min and (b) the ∼4.5 min total ion chromatogram peaks in Fig. 4 with peaks of interest extracted. Structures of the m/z 529.3 and 543.2 species are depicted as (a)* and (b)*, respectively. | ||
Fig. 5 demonstrates that the chlorine of GY34 is removed immediately after dissolving the compound in cell medium or the mobile phase. In addition to this, GY34 is transformed to several different species as a function of time. Thus the cytotoxic activity of GY34 proposed by Yellol et al.5 is most likely not caused by GY34 itself but instead by a subspecies of the compound. This is in accordance with the knowledge that Cisplatin as well in solution is hydrolyzed, exchanging one or both chlorines with water.10 The possible transformation in solution of newly synthesized metallo-drugs should therefore be kept in mind when performing experiments.
Another important aspect of stability in solution is the solubility of the analyte. By NMR it has been shown that GY34 is soluble in pure DMSO at 20 mM concentration, and that 200 μM GY34 solutions prepared in 10% DMSO are stable up to 48 h.11 Slightly soluble compounds often precipitate over time and the actual concentration applied when performing experiments may not be consistent with the expected concentration. In the current work, the intracellular distribution of GY34 was investigated as described below. For these experiments, GY34 was added to the cell medium to obtain a nominal concentration of 10 μM. After incubation an aliquot of the cell medium was taken out and the GY34 concentration was determined by ICP-MS analysis. Interestingly, actual GY34 concentrations in the cell medium were found to range between ∼2–10 μM for different replicates. As discussed below the intracellular distribution of GY34 appears to be dependent on the GY34 concentration in the cell medium during incubation. Once more, this establishes analyte stability as a very critical parameter and stresses the significance of evaluating and controlling the actual analyte concentration throughout experiments.
The limited solubility of GY34 in the cell medium also signifies the importance of the procedure used to prepare samples for ICP-MS analysis. Simply filtering and diluting the samples will cause potentially precipitated GY34 to be lost and lead to erroneous results. In this work the samples were digested at 60 °C using 65% HNO3 and 30% H2O2, with nuclei even requiring microwave digestion. Thus, should some GY34 precipitate in the samples prior to ICP-MS analysis, the result will not be affected, as the sample preparation procedure will ensure that all GY34 is dissolved.
Intracellular distribution of GY34
In order to obtain knowledge on the mechanism of action of GY34, the intracellular distribution of the compound in ELA cells was investigated. The cells were incubated for 18 h with nominal 10 μM GY34 and afterwards the cell suspension was fractionated to a nuclear, a mitochondrial and a cytosolic fraction by applying a novel protocol developed for this work (refer Fig. 2 and the experimental section Cell Fractionation). The content of GY34 in each fraction was determined by ICP-MS analysis and the result is given in Table 1. According to Table 1 most of the GY34 taken up by the ELA cells is located in the cytosol and the mitochondria. This indicates that GY34 might target mitochondria in contrast to Cisplatin which is known to target DNA in the cell nucleus.12 ELA cells are known to be resistant to Cisplatin6 and the ability of GY34 to induce apoptosis in ELA cells (see below) might therefore arise from a different mechanism of action of GY34 compared to that of Cisplatin.| Fraction | % GY34 in fraction | |
|---|---|---|
| 1 | 2 | |
| Nuclei | 4 | 3 |
| Mitochondria | 18 | 21 |
| Cytosol | 78 | 76 |
As mentioned above, the concentration of GY34 in the cell medium during incubation varied between replicates. Interestingly, the concentration in the cell medium appeared to affect the intracellular distribution on GY34. For replicates 2–3 (∼10 μM GY34 detected in cell medium) more GY34 was located in the mitochondria compared to the nuclei (Table 1), on the other hand for replicate 1 (∼2 μM GY34 detected in cell medium) 32%, 10% and 58% of GY34 taken up by the cells were located in nuclei, mitochondria and cytosol, respectively. This indicates that GY34 at low concentrations is distributed primarily to the cell nuclei whereas the compound in higher concentrations also targets the mitochondria. Possibly, GY34 targets the cell nuclei until saturation is obtained, after which GY34 distribution to the mitochondria is initiated. It should be noted that the differing intracellular GY34 distribution between replicates is not owing to the applied cell fractionation procedure but rather related to the stability difficulties of GY34 in the cell medium. Applying the method on A2780 WT cells incubated with 10 μM Cisplatin yielded 1 ± 0.3% Cisplatin in the nuclei, 6 ± 0.9% in the mitochondria and 93 ± 0.6% in the cytosol (n = 3), demonstrating a satisfying reproducibility between replicates.
The importance of obtaining clean fractions when predicting the intracellular distribution of a compound is stressed by the elevated content of GY34 in the cytosol compared to nuclei and mitochondria. Should the nuclei or mitochondria be contaminated with cytosol a misleadingly higher nuclear or mitochondrial content of GY34 would appear leading to false deductions on the mechanism of action. In this work, SDS page and western blotting were performed to evaluate the purity of the obtained fractions. The result is shown in Fig. 6. From Fig. 6 it can be seen that the nuclei as expected contain histone H3 (HIST, a marker for nuclei) but do not contain malate dehydrogenase 2 (MDH, a marker for mitochondria) or lactate dehydrogenase B (LDH, a marker for cytosol). Thus, the nuclear fraction is not contaminated with either mitochondria or cytosol. In the same manner, the cytosol contains LDH but not HIST or MDH and the cytosol fraction is therefore not contaminated with histone or mitochondria. The mitochondria contain a substantial amount of MDH and no HIST however a very faded band for LDH is visible. This could indicate a minor contamination of the mitochondrial fraction with cytosol. Conversely, the observed LDH in the mitochondria could also very likely originate from LDH embedded in the inner mitochondrial membrane13 and would thus not be due to contamination with cytosol. Repeated wash of the mitochondria (refer step (x) in Fig. 2 and the experimental section Cell Fractionation) ought to have removed most of the cytosol from the surface of the mitochondria. Still, it cannot be ruled out that a small amount of cytosol has diffused into the inner membrane space of the mitochondria causing a minor contamination. Based on the above the authors believe that the purity of mitochondrial fractions obtained using the current fractionation protocol is sufficient and that a minor content of cytosol in the inner mitochondrial membrane is insignificant.
 | ||
| Fig. 6 Representative western blot of nuclei, mitochondria and cytosol obtained from fractionation of murine Ehrlich Lettré Ascites cells incubated for 18 h with nominal 10 μM GY34 (n = 3). The left band of each membrane piece is the control (total cell homogenate) and the right band is the sample, i.e. nuclei, mitochondria or cytosol as indicated. The histone H3 (HIST, 18 kDa, marker for nuclei), malate dehydrogenase 2 (MDH, 36 kDa, marker for mitochondria) and lactate dehydrogenase B (LDH, 35 kDa, marker for cytosol) bands are indicated as well. | ||
Several examples of subcellular fractionation and isolation of subcellular organelles are described in the literature.14–18 However, to the knowledge of the authors no procedures with features identical to the current presented fractionation method have been reported. In the work of Hornig-Do et al., superparamagnetic microbeads were used to isolate mitochondria from human 293 HEK, HeLa and osteosarcoma cells. Western blotting rebutted cytosolic contamination but showed a trace of nuclei in the mitochondrial fraction. Only mitochondria were isolated, not nuclei or cytosol.14 Applying gradient centrifugation with percoll, Wieckowski et al. were able to obtain a mitochondrial fraction from MEF cells that did not contain cytosol or nuclei.15 In the same manner, differential centrifugation allowed Dai et al. to isolate clean mitochondria from SKOV3 and A2780 cells.16 Neither Wieckowski et al. nor Dai et al. isolated nuclei or cytosol.15,16 Zayed et al. fractionated A549 human lung adenocarcinoma epithelial cells and human HT29 and HCA7 colorectal cancer cells into the cytosol, cell membranes, nuclei and the cytoskeleton using the Merck Millipore ProteoExtract® Subcellular Proteome Extraction Kit, but did not verify the purity of the obtained fractions.17 Various fractionation kits are commercially available however these are expensive and yield crude preparations that might need further purification.19,20 In the work of Groessl et al., the Thermo Scientific Mitochondria Isolation Kit for Cultured Cells and the Biovision FractionPREP™ Cell Fractionation Kit were used to isolate nuclei, mitochondria and cytosol i.a. from A2780 cells. The western blots provided in the ESI† of the paper show that the mitochondria contain LDH.18 Whether this is caused by a cytosolic contamination of the mitochondria or by LDH located in the inner mitochondrial membrane13 as discussed above remains unclear. The nuclear and cytosolic fractions are clean.18 In order to map the intercellular distribution of a compound obtaining clean fractions is of high importance. Also, the method must be able to fraction cells into nuclei, mitochondria and cytosol. The authors believe that the fractionation method presented in this work features both. In addition the method is less expensive than the commercially available fractionation kits.
A critical step in the fractionation procedure is the homogenization (step (ii), Fig. 2). Applying too much force will break the mitochondria resulting in a reduced yield of mitochondria along with mitochondrial contamination of the cytosol. Conversely, when a too little force is applied, only a minor part of the cells will be homogenized. This results in a decreased mitochondrial yield as well and will furthermore contribute to the contamination of the nuclei with unbroken cells. Step (iii) in the fractionation procedure serves to precipitate the nuclei which subsequently can be removed. However, should the homogenate contain small, unbroken cells with a density similar to nuclei the nuclear fraction will be contaminated with unbroken cells and western blot bands for LDH and MDH will appear. The probability of nuclear contamination with small, unbroken cells increases with the number of unbroken cells that again depends on the force applied during homogenization. To validate the quality of the homogenization, SDS page gel electrophoresis and western blotting should always be performed in connection with a fractionation.
In order to evaluate the amount of GY34 lost during the fractionation the mass balance was calculated. Prior to homogenization a volume of unbroken cells was sampled allowing the total amount of GY34 taken up by the cells to be determined by ICP-MS analysis. The mass balance was then obtained by relating the sum of the GY34 detected in the nuclear, mitochondrial and cytosolic fractions to the total GY34 taken up by the cells. 31 ± 9% (average ± SD, n = 3) of the total GY34 taken up by the cells was recovered in the fractions. A portion of GY34 was probably lost with the unbroken cells and some degree of loss is to be expected from the numerous washing and decanting steps in the procedure. Finally, the effort to avoid contamination of the fractions also causes a loss: e.g. when decanting a supernatant a part of the supernatant had to be left on top of the pellet to ensure no pellet was transmitted. Thus, a trade-off between fraction purity and metal recovery must be made. Although a relatively small amount of GY34 was recovered in the fractions the authors do not believe that it affects the obtained distribution of GY34 in the cells. In the work of Zayed et al. mentioned above a platinum recovery of >99% was obtained after subcellular fractionation was carried out using an extraction kit.17 Apparently, the attained fractions were not washed to the same extent as the current fractionation method requires, which most likely explains the improved recovery. In this work fraction purity was prioritized due to its significance when determining the intracellular distribution, as discussed earlier.
Binding of GY34 to cytosolic biomolecules
Another way to gain knowledge on the mechanism of action of a new compound is to study whether it binds to biomolecules in the cell. As reviewed by de Almeida et al.21 and Casini & Reedijk,22 there is every indication that the biodistribution, uptake and pharmacological action of metallo-drugs depend on interactions with proteins. In this work, ELA cells were incubated with 10 μM GY34 for 18 h and the cytosol was isolated and fractionated using 3 kDa spin filters. The GY34 content in the resulting two fractions was determined by ICP-MS analysis. 96 ± 0.2% (average ± SD, n = 3) of GY34 in the cytosol was found in the high-molecular fraction (>3 kDa). This indicates that in the cytosol the majority of GY34 is bound to high-molecular biomolecules, most likely proteins, and that only a minor part of the compound remains unbound or bound to small molecules. Thus the cytotoxicity of GY34 is most likely facilitated by interaction with protein targets.Similar experiments were carried out by Kasherman et al. with Cisplatin on A2780 cells suggesting that two-thirds of Cisplatin were associated with >3 kDa species.23 This supports the hypothesis that metallo-drugs bind to proteins once inside the cytosol instead of remaining unbound. The hypothesis is to some extent supported by the work of Heffeter et al. Performing SEC-ICP-MS on the cytosol of human cervical carcinoma-derived KB-3-1 cells treated with the Ru-based drug KP1019 and its sodium salt KP1339 they found that after 3 h the majority of Ru was found in the >150 kDa fraction and that Ru was redistributed to the <40 kDa fraction after 24 h.24
In order to validate that no GY34 was lost in the spin filters the GY34 content in the unfractionated cytosol was determined as well, allowing calculation of the mass balance. 91 ± 9% (average ± SD, n = 3) of the total cytosolic GY34 was located in the <3 kDa and >3 kDa fractions validating an insignificant loss of GY34 during the centrifugal filtration.
Effect of GY34 on the organic osmolyte flux
Cells exposed to physiological or cytotoxic stimuli are eliminated by apoptosis which is a genetically well-orchestrated cellular process. Apoptosis is characterized by an initial cell shrinkage (apoptotic volume decrease), which reflects the net loss of ions, organic osmolytes and water.25,26 It has been shown that the onset of apoptosis can be postponed/prevented by limitation of the activity of volume-sensitive and volume-insensitive leak pathways, that normally facilitate loss of the organic osmolyte taurine, and/or up-regulation of Na+-dependent transporters, e.g. TauT that facilitates taurine accumulation.27 Taurine accounts for about 0.1% of our total body weight28 and besides being an important organic osmolyte in mammalian cells taurine is recognized for its role in foetal development, lung functions, mitochondrial functions, antioxidative defense and as a modulator of the apoptotic response once it has been initiated.27 Cisplatin resistance in ELA cells, when compared to Cisplatin sensitivity in EATCs, has previously been demonstrated to correlate with less nuclear Cisplatin accumulation, decrease in the initial ion- and water loss as well as an increased TauT activity.6 In A2780 cells, acquirement of Cisplatin resistance correlates with up-regulation in TauT activity and a concomitant down-regulation in the volume-sensitive taurine leak pathway.29 In congruence, TauT activity has been shown to promote Cisplatin resistance in kidney cells30 and multidrug resistance in colorectal cancer.31In the present work, Cisplatin-sensitive A2780 WT cells (wild type), Cisplatin-resistant A2780 RES cells (acquired, extrinsic resistance) as well as Cisplatin-resistant ELA cells (innate, intrinsic resistance) were exposed to 5 μM Cisplatin or 5 μM GY34 for 18 h. Taurine influx and taurine release under isotonic conditions were determined by the tracer technique. From Fig. 7 it is seen that 18 h of Cisplatin exposure increases taurine uptake and concomitantly reduces taurine release in A2780 WT cells. It has recently been shown that Cisplatin resistance in A2780 RES cells correlates with an increased taurine accumulation, followed by an increased ability to accumulate taurine and a concomitant impairment of a volume-sensitive taurine release pathway,29i.e. the response to Cisplatin seen in A2780 WT cells (Fig. 7) most probably reflects initiation of a resistance phenotype within 18 h of exposure to Cisplatin. In contrast, 18 h of exposure to Cisplatin has no effect on taurine transport in A2780 RES or ELA cells. GY34 on the other hand reduces taurine uptake in all three cell lines and stimulates taurine release in the resistant cell lines. As down-regulation of taurine uptake and increase in taurine release following exposure to GY34 could reflect that GY34 induces cell death in the wild type as well as the Cisplatin-resistant cell lines, the progression of cell death and apoptosis in ELA cells following exposure to Cisplatin and GY34 was analyzed. Initiation of apoptosis by chemical drugs typically involves DNA-damage, activation of specific kinases (ATM/ATR) which through phosphorylation/activation of the transcription factor p53 provokes synthesis of pro-apoptotic proteins and subsequently activation of caspase 3.29 Exposure of phosphatidylserine on the surface of apoptotic cells is a clear signal of apoptotic progression and normally serves as a signal to phagocytic cells to engulf/degrade the apoptotic cell.32 From Fig. 8, which illustrates FACS analysis of ELA cells following exposure to 5 μM GY34 or 5 μM Cisplatin for 18 h, it can be seen that GY34 initiates cell death (increase in propidium iodide signaling) and apoptosis (increase in annexin-V staining). This may indicate that GY34 initiates cell death partly by apoptosis in Cisplatin-resistant cells and that GY34 therefore may be able to overcome Cisplatin resistance.
 | ||
| Fig. 7 Taurine influx and taurine release under isotonic conditions in Cisplatin-sensitive wild type (A2780 WT) and acquired Cisplatin-resistant (A2780 RES) human ovarian carcinoma cells along with innate Cisplatin-resistant murine Ehrlich Lettré Ascites (ELA) cells. Cells were grown in the absence (white bars) or presence of either 5 μM Cisplatin (light grey bars) or 5 μM GY34 (dark grey bars) for 18 h before determination of taurine influx and taurine release by the tracer technique as indicated in the experimental section Taurine Flux Assays. (a) Influx is given relative to control values, i.e. 0.009 ± 0.001 (A2780 WT), 0.018 ± 0.002 (A2780 RES) and 0.157 ± 0.016 (ELA) nmol g prot−1 min−1 and represents 8/3 (A2780 WT), 3/3 (A2780 RES) and 5/5 (ELA) sets of experiments with Cisplatin/GY34. (b) Efflux, determined as the fractional rate constant, is given relative to control values, i.e., 0.0044 ± 0.0003 (A2780 WT), 0.0028 ± 0.003 (A2780 RES) and 0.0022 ± 0.0003 (ELA) min−1 and represents 8/7 (A2780 WT), 8/6 (A2780 RES) and 9/5 (ELA) sets of experiments with Cisplatin/GY34. Values are given as mean value ±SEM. * indicates the significant difference from control values (Students t-test). | ||
 | ||
| Fig. 8 Representative FACS analysis dot plots of (a) control (untreated murine Ehrlich Lettré Ascites (ELA) cells). (b) ELA cells exposed to 5 μM Cisplatin for 18 h. (c) ELA cells exposed to 5 μM GY34 for 18 h (n = 3). Migration to quadrant Q2 indicates apoptosis. | ||
Conclusion
Interestingly, the potential of the novel Ru-based compound GY34 in overcoming Cisplatin resistance has been found in the present work. A novel fractionation procedure has been presented, able to obtain clean nuclei, mitochondria and cytosol for prediction of the intracellular metallo-drug distribution. Furthermore, the stability and transformation of the analyte during experiments have been found to significantly influence the outcome and the importance of monitoring these parameters has thus been demonstrated. As an alternate, more diverse approach than commonly practiced, considering both chemical and biological aspects, has been applied in this work, the experiments performed can be used as a general protocol and an additional tool in the initial evaluation of novel metal-based drugs.Acknowledgements
The authors kindly wish to thank Dorthe Nielsen who contributed significantly to the experimental work of this paper and Camilla Jensen for assistance in the laboratory. We also thank COST1105 for facilitating the collaboration between the Danish and Spanish contributors to this work. This work was supported by the Faculty of Health and Medical Sciences of University of Copenhagen, “Læge Sofus Carl Emil Friis og Olga Doris Friis's legat”, “Agnes og Poul Friis's Fond”, the Spanish Ministerio de Economía y Competitividad and FEDER (Project SAF2011-26611).References
- World Health Organization, http://www.who.int/mediacentre/factsheets/fs297/en/, accessed January 2015.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- L. Galuzzi, L. Senovilla, I. Vitale, J. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869–1883 CrossRef PubMed.
- C. A. Rabik and M. E. Dolan, Cancer Treat. Rev., 2007, 33, 9–23 CrossRef CAS PubMed.
- G. S. Yellol, A. Donaire, J. G. Yellol, V. Vasylyeva, C. Janiak and J. Ruiz, Chem. Commun., 2013, 49, 11533–11535 RSC.
- H. S. Tastesen, J. B. Holm, J. Møller, K. A. Poulsen, C. Møller, S. Stürup, E. K. Hoffmann and I. H. Lambert, Cell. Physiol. Biochem., 2010, 26, 809–820 CrossRef CAS PubMed.
- L. H. Møller, C. S. Jensen, T. T. T. N. Nguyen, S. Stürup and B. Gammelgaard, J. Anal. At. Spectrom., 2015, 30, 277–284 RSC.
- J. B. Holm, R. Grygorczyk and I. H. Lambert, Am. J. Physiol.: Cell Physiol., 2013, 305, C48–C60, DOI:10.1152/ajpcell.00412.2012.
- K. R. Villumsen, L. Duelund and I. H. Lambert, Amino Acids, 2010, 39, 1521–1536 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- J. Ruiz, Department of Inorganic Chemistry, University of Murcia.
- V. Cepeda, M. A. Fuertes, J. Castilla, C. Alonso, C. Quevo and J. M. Pérez, Anti-Cancer Agents Med. Chem., 2010, 10, 3–18 Search PubMed.
- E. E. Rojo, B. Guiard, W. Neupert and R. A. Stuart, J. Biol. Chem., 1998, 273, 8040–8047 CrossRef CAS PubMed.
- H. Hornig-Do, G. Günther, M. Bust, P. Lehnartz, A. Bosio and R. J. Wiesner, Anal. Biochem., 2009, 389, 1–5 CrossRef CAS PubMed.
- M. R. Wieckowski, C. Giorgi, M. Lebiedzinska, J. Duszynski and P. Pinton, Nat. Protoc., 2009, 4(11), 1582–1590 CrossRef CAS PubMed.
- Z. Dai, J. Yin, H. He, W. Li, C. Hou, X. Qian, N. Mao and L. Pan, Proteomics, 2010, 10, 3789–3799 CrossRef CAS PubMed.
- A. Zayed, T. Shoeb, S. E. Taylor, G. D. D. Jones, A. L. Thomas, J. P. Wood, H. J. Reid and B. L. Sharp, Int. J. Mass Spectrom., 2011, 307, 70–78 CrossRef CAS PubMed.
- M. Groessl, O. Zava and P. J. Dyson, Metallomics, 2011, 3, 591–599 RSC.
- Thermo Fisher Scientific Inc., http://www.piercenet.com/instructions/2161477.pdf, accessed January 2015.
- Sigma-Aldrich Co., http://https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Bulletin/mitoiso2bul.pdf, accessed January 2015.
- A. de Almeida, B. L. Oliveira, J. D. G. Correia, G. Soveral and A. Casini, Coord. Chem. Rev., 2013, 257, 2689–2704 CrossRef CAS PubMed.
- A. Casini and J. Reedijk, Chem. Sci., 2012, 3, 3135–3144 RSC.
- Y. Kasherman, S. Stürup and D. Gibson, J. Med. Chem., 2009, 52, 4319–4328 CrossRef CAS PubMed.
- P. Heffeter, K. Böck, B. Atil, M. A. R. Hoda, W. Körner, C. Bartel, U. Jungwirth, B. K. Keppler, M. Micksche, W. Berger and G. Koellensperger, JBIC, J. Biol. Inorg. Chem., 2010, 15, 737–748 CrossRef CAS PubMed.
- E. K. Hoffmann, I. H. Lambert and S. F. Pedersen, Physiol. Rev., 2009, 89, 193–277 CrossRef CAS PubMed.
- I. H. Lambert, E. K. Hoffmann and S. F. Pedersen, Acta Physiol. Scand., 2008, 194, 255–282 CrossRef CAS PubMed.
- I. H. Lambert, D. M. Kristensen, J. B. Holm and O. H. Mortensen, Acta Physiol., 2015, 213, 191–212 CrossRef CAS PubMed.
- R. J. Huxtable, Physiol. Rev., 1992, 72, 101–163 CAS.
- B. H. Sorensen, U. A. Thorsteinsdottir and I. H. Lambert, Am. J. Physiol.: Cell Physiol., 2014, 307, C1071–C1080, DOI:10.1152/ajpcell.00274.2014.
- X. Han, J. Yue and R. W. Chesney, J. Am. Soc. Nephrol., 2009, 20, 1323–1332 CrossRef CAS PubMed.
- M. Yasunaga and Y. Matsumura, Sci. Rep., 2014, 4, 4852, DOI:10.1038/srep04852.
- J. G. Kay and S. Grinstein, in Lipid-mediated Protein Signaling, ed. D. G. S. Caputello, Springer Science, ch. 10, 2013, pp. 177–193 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mt00056d |
| This journal is © The Royal Society of Chemistry 2015 |