
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The functional roles of the three copper sites associated with the methionine-rich insert in the multicopper oxidase CueO from E. coli†
Laura
Cortes
,
Anthony G.
Wedd
and
Zhiguang
Xiao
*
School of Chemistry and The Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: z.xiao@unimelb.edu.au; Fax: +61 3 9347 5180; Tel: +61 3 9035 6072
First published on 28th January 2015
Abstract
CueO from Escherichia coli is a multicopper oxidase (MCO) involved in copper tolerance under aerobic conditions. It features the four typical copper atoms that act as electron transfer (T1) and dioxygen reduction (T2, T3; trinuclear) sites. In addition, it displays a methionine- and histidine-rich insert that includes a helix that blocks physical access to the T1 site. In crystalline form, the insert provides at least three additional Met-rich Cu(I) binding sites Cu5 (sCu), Cu6 and Cu7 that are proposed to facilitate rapid oxidation of bound Cu(I) to Cu(II) (S. K. Singh, et al., J. Biol. Chem., 2011, 43, 37849–37857). The activities of variants featuring mutations at sites Cu5 (D360M, M355LD360N), Cu6 (M358,362S), Cu7 (M364,368S) and Cu6,7 (M358,362,364,368S) were compared to that of the wild type form using three different air-stable model substrates (2,6-dimethoxyphenol, [CuI(Bca)2]3− and CuICuII-PcoC, a periplasmic Cu(I) binding protein from E. coli). The results demonstrate that the three copper sites play related but distinct roles in CueO oxidase activities. The internal Cu5 site is part of the essential electron transfer pathway connecting surface-exposed sites Cu6 and Cu7 to site T1. Both Cu6 and Cu7 are dominant substrate-docking-oxidation (SDO) sites on the protein surface. However, under physiologically relevant conditions, the SDO function of Cu6 relies largely on an electron transfer pathway via Cu7 to Cu5. These Met-rich sites in CueO provide a robust cuprous oxidase function for control of Cu(I) toxicity.
Introduction
The E. coli copper efflux system Cue confers copper tolerance under aerobic conditions.1,2 It codes for a P1B-type ATPase CopA (a Cu(I) membrane pump) and for a multicopper oxidase CueO (a soluble protein expressed to the periplasmic space). Expression of both enzymes is up-regulated in response to copper stress by the cytosolic metallo-regulatory protein CueR.3 Like all multicopper oxidases (MCOs), CueO features the three copper sites T1, T2, and binuclear T3 (Fig. 1a).4–6 This machinery couples one-electron substrate oxidation steps (at the T1 site in simpler MCOs) to the four-electron reduction of dioxygen to water (at the T2, T3 trinuclear copper cluster).4,7,8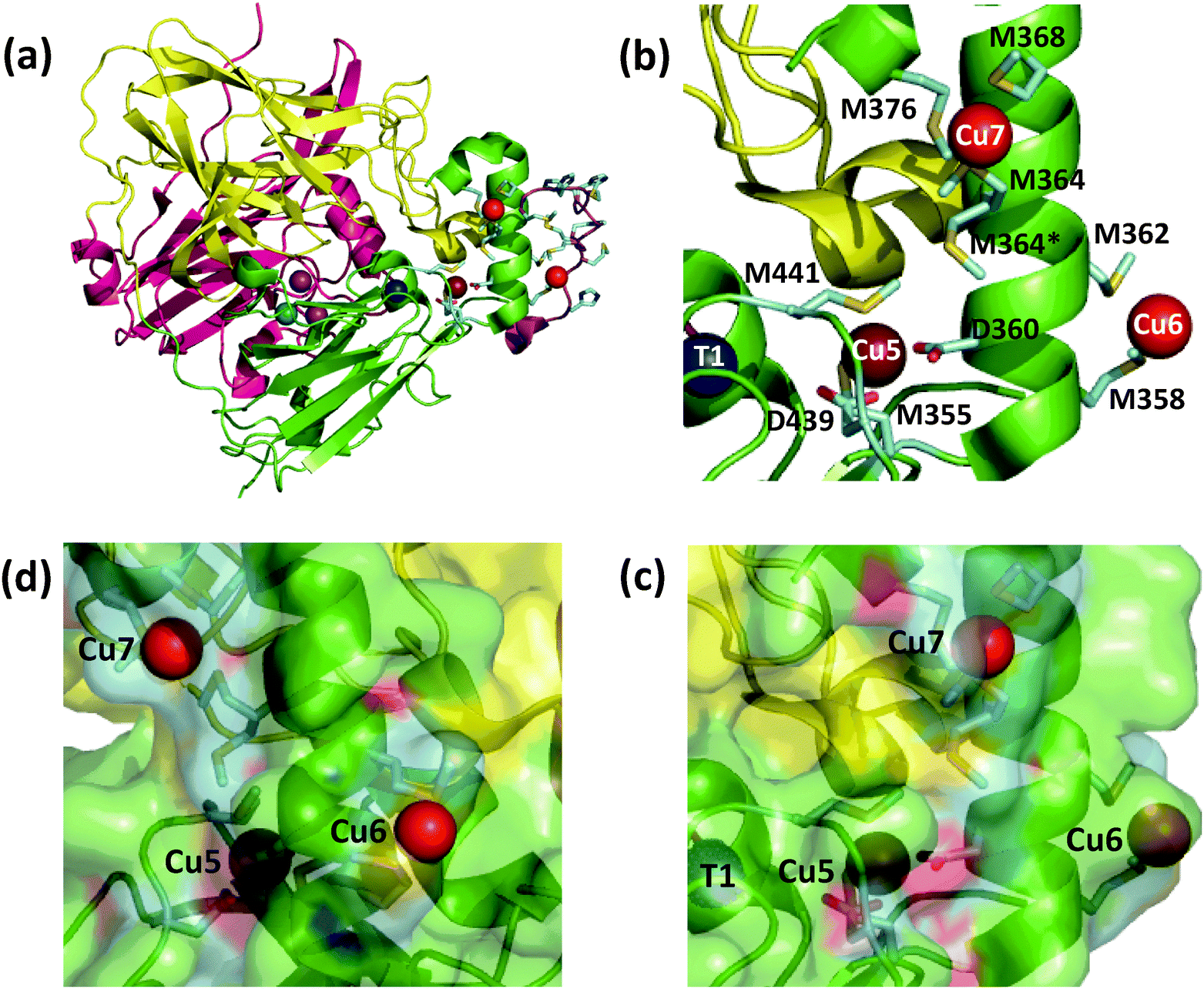 | ||
| Fig. 1 Ribbon model of fully copper-loaded CueO variant C500S (PDB: 3NT0). (a) Overall structure highlighting three structural domains (D1, red; D2, gold; D3, green), copper centres associated with MCO activity (T1, blue; T2, teal; T3 (binuclear), purple;) and additional Cu(I) centres associated with the methionine-rich insert (Cu5, chocolate; Cu6 & Cu7, red). Note: (i) T1 Cu is not present in the C500S structure but is added to the figure for illustration; (ii) the disordered Met- and His-rich loop (383–400; shown in purple) was not resolved but is added to the figure by superimposing the wild type CueO structure (PDB: 3OD3) to show potential alternative ligands for Cu6 and Cu7, especially in the Cu(II) form. (b) Molecular structure highlighting the Met-rich α-helix (in green) and the associated metal ligands (in sticks) for the Cu5, Cu6 and Cu7 sites (note: the Met- and His-rich loop added in (a) is not shown). (c) View as for (b) with a space-occupying transparent surface highlighting the buried site of Cu5 and the surface exposure Cu6 and Cu7. (d) Clockwise rotation of structure (c) around the vertical axis by 90 degrees. | ||
CueO displays robust cuprous oxidase and/or phenol oxidase activities under different conditions.9 In contrast to simpler MCOs, it features an additional Met- and His-rich insert (residues 355–402; 14 Met; 5 His) that includes a helix that blocks physical access to the T1 centre and a flexible surface loop that is frequently not resolved in the crystal structures (Fig. 1a).10 Crystals soaked with Cu(II) revealed an extra labile copper binding site adjacent to the helix (labelled Cu5 in Fig. 1b).11 This site is only 7.5 Å away from the T1 centre to which it is linked via a hydrogen bond involving a ligand from each centre. Ligand mutation at Cu5 led to loss of the oxidase activity and compromised protection against elevated copper levels in variant strains.11
Site Cu5 was labelled initially as rCu to suggest its regulatory role in the enzyme function and later as sCu to emphasise its putative role as a site for binding of substrate Cu(I).11,12 Complementary work from our group provided supporting evidence for Cu5 as the site of outer sphere phenol oxidation (when occupied by copper) and as the site of inner sphere cuprous oxidation.9 The latter role was inferred on the basis that the enzyme could extract Cu(I) from air-stable pseudo-substrate [CuI(Bca)2]3− (Bca = bicinchoninic acid anion; β2 = 1.6 × 1017) and from CuICuII-PcoC (KD = 2.0 × 10−13 M) efficiently for the oxidation. PcoC is a metallo-chaperone protein released to the periplasm of E. coli under copper stress conditions.13,14 These Cu(I) transfer reactions led to an estimate of KD = 1.3 × 10−13 M for the affinity of the Cu5 site of CueO for Cu(I). A weaker affinity was derived for Cu(II) (KD = 5.5 × 10−9 M), consistent with extraction of that product and regeneration of the apo form of the Cu5 site.
The intrinsic instability of unprotected Cu(I) ions in aqueous solution means that substrate Cu(I) must be transferred to the reaction site(s) in the enzyme in a protected form. This can be achieved by direct transfer of Cu(I) from the donor site in the substrate complex to the acceptor site(s) in the oxidase enzyme. Support for this requirement is provided by the observation that another bacterial periplasmic protein CopK binds Cu(I) with affinity indistinguishable to that of PcoC,15 but the air-stable complex CuICuII-CopK is not a Cu(I) substrate for CueO in 3-(N-morpholino)propanesulfonate (Mops) buffer whereas the air-stable CuICuII-PcoC is oxidised rapidly under the same conditions.9 The Cu(I) atom in CopK is bound close to an hydrophobic core15–17 and appears to be protected against direct molecular contact with CueO. On the other hand, the Cu(I) site in PcoC lies on a flexible Met-rich metal binding loop exposed to the protein surface14,18 and is likely to interact directly with CueO for efficient Cu(I) transfer and oxidation. However, examination of the available structures indicates that the Cu5 site in CueO is buried underneath the protein surface, raising the question of whether it is actually the direct Cu(I) substrate-docking-oxidation (SDO) site for the cuprous oxidase function.
Soaking of crystals of inactive C500S-CueO (disabled T1 site) with Cu(I) identified two additional high-occupancy copper binding sites that are associated with the Met-rich helix and that are solvent-exposed (labelled Cu6 and Cu7 in Fig. 1).12 Mutation of the Met ligands of these putative copper sites led to a four-fold reduction in kcat for the cuprous oxidase activity for model substrate [CuI(MeCN)4]+, suggesting a potential role as direct Cu(I) docking sites.12 However, uncertainty remains regarding their relationship to the previously identified Cu5 site and whether they act as Cu(I) SDO sites or solely as Cu(I) docking sites with the ion being transferred subsequently to the Cu5 site for oxidation. The functional nature of Cu5 site becomes uncertain too, since double site mutation of both Cu6 and Cu7 sites reduces, but does not disable the cuprous oxidase activity on the model substrate [CuI(MeCN)4]+.12 However, the latter Cu(I) substrate is itself dioxygen-sensitive and subject to auto-oxidation. In addition, it may be oxidised at the enzyme reaction site(s) via both inner and outer sphere mechanisms.9
The specific roles of sites Cu5, Cu6 and Cu7 in cuprous oxidase and phenol oxidase reactivities are examined in this work. Variants of the wild-type (wt) protein with mutations at these sites were tested using three different air-stable model substrates: 2,6-dimethoxyphenol (DMP), [CuI(Bca)2]3− and CuICuII-PcoC, a possible native substrate. It is demonstrated that the three copper sites play related but distinct roles in CueO oxidase activity: (i) the internal Cu5 site is part of the essential electron transfer pathway connecting surface-exposed sites Cu6 and Cu7 to site T1, but it is not a direct substrate docking site; (ii) both sites Cu6 and Cu7 are dominant SDO sites. However, under physiologically relevant conditions, the SDO function of Cu6 relies largely on an electron transfer pathway via Cu7 to Cu5. These distinct but correlated properties of different copper sites are discussed in relationship to the X-ray crystal structures and to the sophisticated requirements for specific metal oxidase enzymes.19
Experimental section
Materials and general methods
Chemicals and reagents (analytical grade) were purchased from normal commercial sources and were used as received. Cu(II) fluorescence probe DP3 was synthesised as reported.20 Stock solutions of [CuI(Bca)2]3− were prepared anaerobically in a glove box ([O2] < 2 ppm) by addition of [CuI(CH3CN)4]ClO4 in CH3CN into solutions of Na2Bca in Mops buffer (50 mM; pH 7.0) with varying molar ratios of Cu(I)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Bca. The Cu(I) concentration was calibrated by the absorbance of this anion while Bca concentrations were calibrated by titration with a Cu2+ standard in the presence of weak reductant NH2OH, as detailed previously.9
:Bca. The Cu(I) concentration was calibrated by the absorbance of this anion while Bca concentrations were calibrated by titration with a Cu2+ standard in the presence of weak reductant NH2OH, as detailed previously.9
UV-visible spectra and/or absorbance were recorded on a Varian Cary 300 spectrophotometer in dual beam mode with quartz cuvettes of 1.0 cm path length. Fluorescence emission spectra were recorded on a Varian Cary Eclipse spectrophotometer with a band pass of 20 nm for both excitation and emission. EPR spectra were recorded on Bruker Elexsys E 500 EPR spectrometer. The samples were 0.1 mM in concentration in either Mops or 2-[bis(2-hydroxyethyl)amino]-2(hydroxymethyl)-l,3-propanediol (BisTris) buffer (50 mM, pH 7.0) containing ∼10% glycerol and were run at 77 K in a liquid-nitrogen finger Dewar.
Generation of protein variants
wt-CueO was expressed and isolated as reported previously.9 Site-directed mutagenesis was performed by overlapping PCR reactions. The primer sequences used to generate variant CueO gene sequences are listed in Table S1 (ESI†). After confirming the correct insert of each expression plasmid by DNA sequencing, protein expression and purification followed the previous protocols.9 Yields of purified apo proteins were ca. 50 mg l−1 of culture in each case. Purity and identity were confirmed by SDS-PAGE and ESI-QTOF mass spectrometry (Fig. S1 and Table S2, ESI†). The periplasmic protein PcoC from E. coli that binds both Cu(I) and Cu(II) at separate sites was isolated and prepared as reported previously.14Copper incorporation and analysis
Incubation of apo-CueO with excess CuSO4 (∼10 equiv.) in Mops buffer (20 mM, pH 7) in the presence of reductant glutathione (GSH; 1 mM) at 4 °C for 1–2 h led to bright blue solutions. Excess reagents were removed using a Bio-Del P-6 DG gel desalting column (Bio-Rad) in Mops buffer (20 mM, pH 7.0) to generate a fully copper-loaded enzyme form designated as CueOMops. The concentrations of copper-incorporated wt-CueO and all its protein variants were calculated based upon the stable molar absorptivity for an intact T1 centre in CueO (ε610 = 5000 M−1 cm−1).9BisTris possesses modest affinity for Cu(II) (KD = 10−5.1 M at pH 7.0)21 and BisTris buffer at millimolar concentration may compete with certain labile Cu sites in CueO.9 CueOMops samples were converted to BisTris-treated forms CueOBT and analysed for labile copper content via the following experiments. The samples were incubated in BisTris buffer (20 mM; pH 7.0) for 10 min, followed by buffer change back to Mops buffer via a P-6 desalting column or a Vivaspin centrifugal concentrator. Both samples were characterised and assessed via their solution spectra (Fig. S2, ESI†), phenol oxidase activities (with DMP as model substrate in Mops buffer; vide infra) and removable labile Cu(II) contents. The last aspect was conducted with our recently-developed fluorescence probe DP3 that binds Cu(II) with KD = 10−12.3 M at pH 7.4. The probe emits intense fluorescence at λmax = 550 nm with λex = 330 nm and Cu(II) binding quenches the fluorescence intensities sensitively.20 Briefly, CueOMOPS or CueOBT (∼100 μM) was titrated into a solution of probe DP3 (2.0 μM) in Mops buffer (50 mM, pH 7.4). Cu(II) transfer from CueO to probe was monitored by fluorescence quenching at 550 nm. The difference in detectable Cu(II) content between the samples CueOMops and CueOBT was assigned to the ‘BisTris-removable Cu(II) content’ of the CueOMops samples.
Phenol oxidase activity
The activities of CueOMops and its variants as phenol oxidases were assessed via catalytic aerobic oxidation of model substrate DMP to brown dimeric 3,5,3′,5′-tetramethoxydiphenoquinone compound (TMPQ) that exhibits an absorption maximum at 469 nm with ε = 14800 M−1 cm−1.22 The reactions were performed in air-saturated Mops buffer (50 mM, pH 7.0) without or with addition of BisTris buffer (5.0 mM). The [O2] concentration in the buffers was assumed to be 285 μM.23 The reactions were conducted in a mixing cell with a total path length of ∼0.9 cm. The two half-cells were pre-loaded with the enzyme (varying concentrations in the range 0.04–0.4 μM) and the substrate DMP (10 mM), respectively, both in Mops buffer (50 mM, pH 7.0). After recording the initial absorbance, the reactions were started by a rapid mixing of the solutions (note that the concentrations of reactants are halved by the mixing) and followed by the increase in absorbance at 469 nm. The enzyme activity expressed as reaction molar velocity (i.e., the turnover number per min) was calculated from the initial steady-state reaction rate.
Cuprous oxidase activity
Two different cuprous substrates were employed. The assay approaches varied with the substrate properties. Air-stable model substrate [CuI(Bca)2]3− was employed as described previously.9 The two half-cells were loaded, respectively, with the enzyme (0.1–2.5 μM; BisTris buffer, 50 mM, pH 7.0) and the substrate [CuI(Bca)2]3− (∼240 μM with initial Bca:Cu(I) = 3.0 in the same buffer). After recording the initial absorbance at 562 nm for [CuI(Bca)2]3− (ε = 7900 M−1 cm−1), the reactions were started by rapid mixing and followed for about 20 min by the change in absorbance at 562 nm.
The concentration of ligand Bca was varied to alter its apparent affinity for Cu(I) and hence the initial availability of the ion for oxidation. On the other hand, free Bca is also a co-product of this catalysis and its generation is inhibiting. As a result, the apparent cuprous oxidase activity on [CuI(Bca)2]3− varied considerably, not only with the initial substrate conditions (both Cu(I):Bca ratio and concentration) but also with the reaction course. However, starting from the same initial substrate composition and concentration, the apparent cuprous oxidase activity (molar velocity in units min−1) was independent of the enzyme concentration under the condition of the same substrate concentration of [CuI(Bca)2]3− in solution. Consequently, meaningful activity comparisons between different variant enzymes must be conducted with the same starting substrate solution and the activity calculated from the solution with the same substrate absorbance at 562 nm along the reaction time course. However, the enzyme concentration may vary to account for the different activities of the variants.
The metallo-chaperone CuICuII-PcoC from E. coli was also employed as a Cu(I) substrate. The reactions were conducted under catalytic conditions (enzyme:substrate = 1:150–300) in air-saturated Mops buffer (20 mM, pH 7.0). Generation of product CuII-PcoC was followed semi-quantitatively via rapid separation of reactants and products on a CIM SO3 cation-exchange disk (0.1 mL; BIA Separations) connected to a Bio-Rad FPLC system. The substrate CuICuII-PcoC and the product CuII-PcoC eluted at ∼115 and ∼105 mM, respectively, under a NaCl gradient of 0–200 mM at a flow rate of 4.0 mL min−1. Briefly, to start the reaction, varying quantities of the variant enzymes in Mops buffer (20 mM, pH 7.0) were added to an air-saturated solution of CuICuII-PcoC (10 μM) in Mops buffer. The reaction solution (∼1 mL) was loaded immediately into a syringe and injected, in small aliquots (∼0.15 mL) as a function of time, into the FPLC system for product separation. The reaction rates were calculated by integration of the areas under the two elution profiles. Cu(II)-coordinating buffers such as BisTris accelerate the catalytic reaction considerably and prevent meaningful kinetic analysis with this approach. They must be excluded carefully from the whole assay procedure.
Results and discussion
Generation and characterisation of CueO samples
Five different protein variants of CueO were generated (Table 1). They were designed to perturb the Cu5, Cu6 and Cu7 sites by substituting ligand side chains with alternatives of differing properties (see Fig. 1b): Cu5-m1 (D360M), Cu5-m2 (M355LD360N), Cu6-m (M358,362S), Cu7-m (M364,368S), Cu6,7-m (M358,362,364,368S). They were each isolated in metal-free apo-forms but were reconstituted readily to produce copper-loaded forms with intact T1 and T2/3 copper centres, as indicated by their characteristic absorbance and EPR spectra (Fig. S2 and S3, ESI†). All copper incorporation procedures were conducted in Mops buffer only, to take advantage of its weak affinity for both Cu(I) and Cu(II). Incubation of the fully copper-loaded samples of CueOMops in Mops buffer containing BisTris (∼20 mM; KD, 10−5.1 M for Cu(II)21), followed by buffer change back to the same Mops buffer generated a new copper-loaded form CueOBT. The EPR signals for T1 and T2 Cu centre remained unchanged and so did the solution spectrum for the T1 centre at 610 nm (Fig. S2 and S3, ESI†). However, the absorbance at ∼330 nm was decreased due to removal of certain bound labile copper ions by BisTris buffer (Fig. S2, ESI†), as has been observed previously.9,24| Protein | Site variation | As DMP oxidase | As [CuI(Bca)2]3− oxidase | As CuICuII-PcoC oxidase | |||||
|---|---|---|---|---|---|---|---|---|---|
| (min−1)a | (wt%) | At A562 ∼ 0.7 | At A562 ∼ 0.6 | (min−1)e | (wt%) | ||||
| (min−1)c | (wt%) | (min−1)c | (wt%) | ||||||
| a Unless otherwise indicated, determined in real time via change in solution absorbance at 469 nm with an initial DMP concentration 5.0 mM in Mops buffer (50 mM, pH 7.0) (see Fig. 2). b Determined similarly but with BisTris (5.0 mM) added to the Mops buffer (see Fig. S3a, ESI). c Determined in real time via change in solution absorbance at 562 nm at the two reaction times when A562 = 0.70 ± 0.01 and 0.60 ± 0.01; initial [Cu(I)] = 120 μM and [Bca] = 360 μM in BisTris-HCl buffer (25 mM, pH 7.0; unless otherwise indicated) (see Fig. 3 and 4). d Determined as for (c) but with [Bca] = 600 μM (see Fig. 3b(i)). e Determined semi-quantitatively via ion-exchange separation in Mops buffer (20 mM, pH 7.0; Fig. 6); the reaction is too fast to be followed in buffers containing BisTris. | |||||||||
| wt-CueO | 13.0b | 2.4 | 16.2d | 12 | 10.6d | 13 | ∼312 | 100 | |
| 541 | 100 | 136 | 100 | 80.7 | 100 | ||||
| Cu5-m1 | D360M | ∼0 | ∼0 | 2.1 | 1.6 | 1.1 | 1.3 | ∼0 | ∼0 |
| Cu5-m2 | M355L,D360N | ∼0 | ∼0 | ∼0 | ∼0 | ∼0 | ∼0 | ∼0 | ∼0 |
| Cu6-m | M358,362S | 139 | 25.7 | 34.9 | 25.8 | 20.5 | 25.4 | ∼110 | ∼35 |
| Cu7-m | M364,368S | 32.7 | 6.0 | 9.3 | 6.9 | 3.9 | 4.8 | ∼72 | ∼23 |
| Cu6,7-m | M355,362,364,368S | 5.5 | 1.0 | ∼0 | ∼0 | ∼0 | ∼0 | ∼0 | ∼0 |
Contents of labile Cu(II) in various samples were analysed with the fluorescent peptide probe DP3 (KD = 10−12.3 M at pH 7.4).20 Titration of wt-CueOMops into a solution of DP3 (2.0 μM) in Mops buffer quenched the probe fluorescence at 550 nm (λex = 330 nm) with an apparent end point at ∼0.7 [CueO]tot/[DP3]tot, corresponding to transfer of ∼1.4 equiv. Cu from the wt-CueOMops sample to DP3 (Fig. S4, green titration curve with filled circles, ESI†). However, titration with wt-CueOBT quenched the fluorescence much more slowly with an apparent endpoint at >2 [CueO]tot/[DP3]tot, corresponding to transfer of <0.5 equiv. Cu from wt-CueOBT (Fig. S4, green titration curve with empty circles, ESI†). This implies the presence of ∼1.0 equiv. ‘BisTris-removable labile Cu(II)’ in wt-CueOMops samples.
Equivalent experiments with Cu5-m2Mops and Cu5-m2BT samples produced about the same results (Fig. S4, two red titration curves with filled and empty circles, ESI†), also indicating the presence of ∼1.4 and ∼1.0 equiv. of ‘DP3- and BisTris-removable labile Cu(II)’ in Cu5-m2Mops, respectively. However, the Cu5 site in Cu5-m2 is disabled via double mutation (M355L,D360N) and is not expected to bind Cu. On the other hand, the equivalent experiments on variants Cu6,7-mMops and Cu6,7-mBT detected removal of <0.3 equiv. ‘BisTris-removable labile Cu(II)’ (Fig. S4, two blue titration curves with filled and empty circles, ESI†). It may be concluded from these experiments that the labile Cu in wt-CueOMops is associated with sites Cu6,7, not with site Cu5. This is consistent with the buried position of Cu5 and the solvent-exposed positions of Cu6 and Cu7. Further experiments demonstrate the impact of these labile Cu sites on the oxidase functions of CueO on different substrates (vide infra).
Phenol oxidase activity
This is characteristic of simpler MCOs such as laccases and ascorbate oxidases that feature open T1 sites as outer sphere reaction centres.4,19 Initial experiments with DMP as a model substrate (5.0 mM) verified that the observed reaction rate of wt-CueOMops was dependent on the affinity of the buffer (pH 7.0) for Cu2+ (Fig. S5, ESI†): optimal rate in Mops (low affinity) and inactive in BisTris (higher affinity). Previous work was interpreted in terms of mandatory occupation of the Cu5 site by a copper ion in the active enzyme.9 The present results demonstrate that wt-CueOMops samples contain at least one equivalent of labile Cu(II) that is essential for phenol oxidase activity and which is associated with sites Cu6,7, rather than with site Cu5 (vide supra). Notably, wt-CueOBT with its labile Cu(II) removed is inactive in Mops buffer (Fig. S5a(iii), ESI†).The relative activities of wt-CueOMops and its variants were examined in Mops buffer (20 mM, pH 7.0) with the enzyme concentration varying in the range 0.1–0.4 μM (Fig. 2). The wild type form is the most active and the relative activities decrease in the following order (Table 1):
| wt (1.00) > Cu6-m (0.26) > Cu7-m (0.06) ≫ Cu6,7-m (∼0.01) > Cu5-m1 ∼ Cu5-m2 (∼0) | (1) |
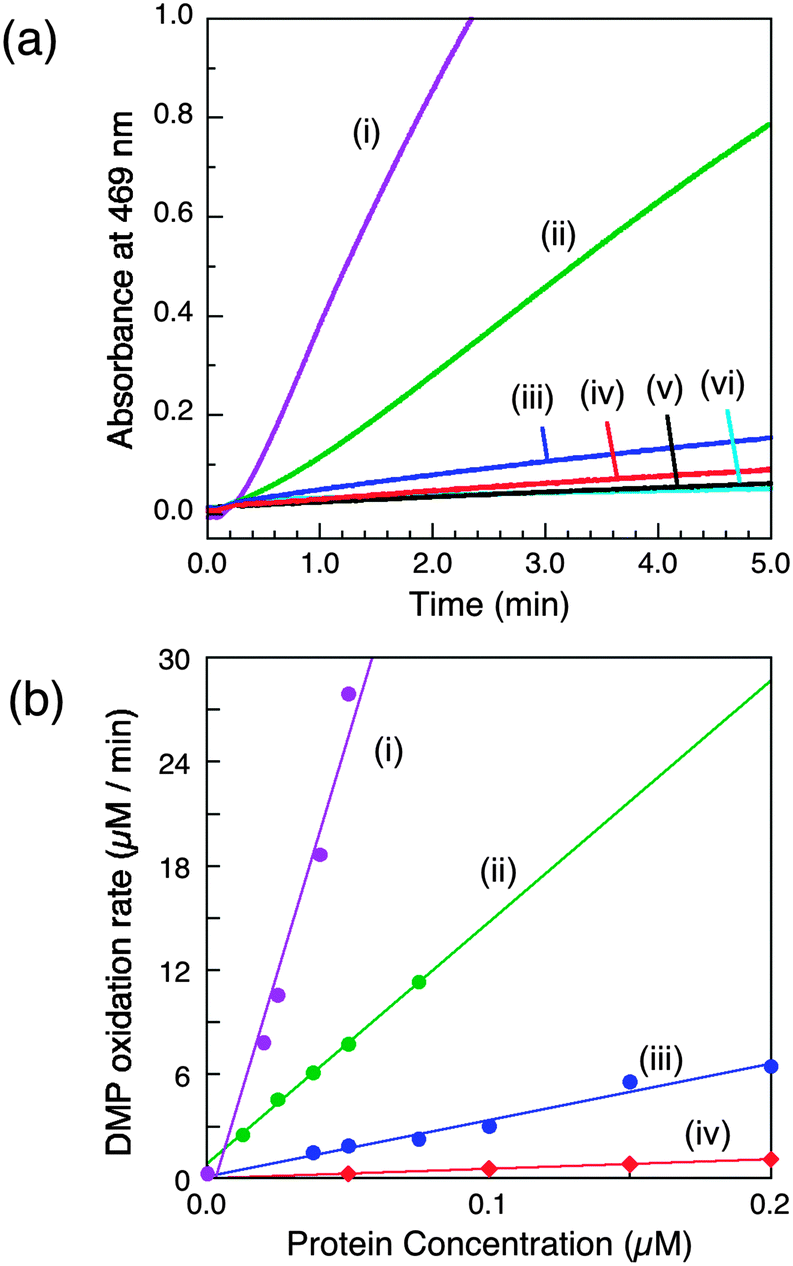 | ||
| Fig. 2 Phenol oxidase activity of wt-CueO and its variants on model substrate DMP (5.0 mM) in air-saturated Mops buffer (25 mM, pH 7.0). (a) Increase in absorbance at 469 nm due to oxidation of DMP catalysed by the same concentration of each enzyme (0.05 μM) and (b) molar activity determined via linear plots of DMP oxidation rate versus enzyme concentration: (i) wt-CueO; (ii) Cu6-m; (iii) Cu7-m; (iv) Cu6,7-m; (v) Cu5-m1 (indistinguishable with Cu5-m2); (vi) control with no enzyme. The derived data are given in Table 1. | ||
The following conclusions can be drawn from these experiments:
(i) The Cu5 site is essential as both Cu5-m1 and Cu5-m2 are inactive, an observation that matches previous in vivo assays.25 It has been assigned previously as the DMP SDO site.9 However, Cu5 is fully buried beneath the protein surface (Fig. 1b–d), suggesting it to be more likely to act as an electron transfer site connecting surface SDO site(s) to the T1 centre.
(ii) Variants Cu6-m and Cu7-m exhibit ∼26% and ∼6%, respectively, of the activity of wt-CueO while the double site variant Cu6,7-m is essentially inactive. It is apparent that the roles of solvent-exposed sites Cu6 and Cu7 are cooperative and that they both appear to be active as SDO sites for DMP. A plausible model (Scheme 1) suggests that activity due to occupation of Cu7 by copper is dependent upon electron transfer to Cu5 (separation 11.2 Å; Fig. 1b–d). Cu6 is separated from Cu5 (14.3 Å) and Cu7 (15.9 Å). Loss of Cu7 induces a loss of activity of ∼94%, consistent with Cu7 acting as the major electron transfer acceptor site for Cu6 oxidase activity. Cu6 is separated from both Cu7 and Cu5 by the body of the Met-rich α-helix that may play a direct role in electron transfer via its multiple Met and nearby His residues (Fig. 1a). It may also supply alternative transient copper-binding sites to expedite SDO activity and/or electron transfer to Cu5 via Cu7 (Fig. 1). The structure of T1-disabled CueO identified Cu6 and Cu7 as Cu(I), rather than Cu(II), binding sites, consistent with the Met-rich nature of these sites.12 However, in the absence of Cu(I), the sites are likely to bind Cu(II) weakly with possible participation of nearby His sidechain(s) as Cu(II) ligand(s) (Fig. 1a).26 This model is consistent with the experiments with probe DP3 that concluded that the labile Cu in wt-CueOMops is associated with sites Cu6 and 7, not with site Cu5 (Fig. S4, ESI†).
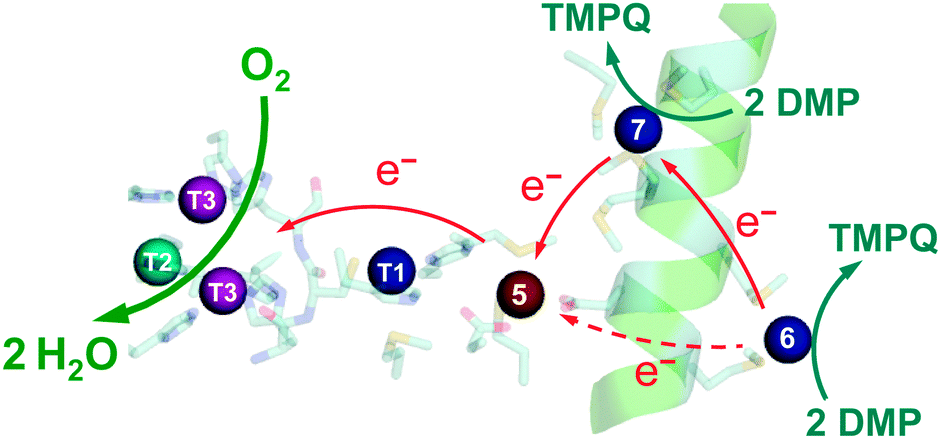 | ||
| Scheme 1 Proposed reaction mechanism for CueO as a DMP oxidase. The occupied sites Cu5,6,7 are essential for full activity. The Cu5 site functions as an electron-transfer mediator only. Both Cu6 and Cu7 are substrate oxidation sites. The activity at Cu6 depends primarily on the electron transfer via Cu7. | ||
Occupation of all seven Cu sites in CueO by copper appears to be essential for full phenol oxidase activity. They play distinct but related roles in the substrate oxidation. The function of DMP SDO sites Cu6 and Cu7 relies on the electron-transfer route Cu6 → Cu7 → Cu5 → T1 → T2,3 → O2 under Cu(II) limiting conditions. However, sites Cu6,7 can only bind Cu(II) weakly and are not likely to function under the physiological conditions of limited Cu availability. Thus, as emphasised previously,9 the conclusion is that the primary function of CueO in vivo is not that of a phenol oxidase.
Cuprous oxidase activity based on model substrate [CuI(Bca)2]3−
This Cu(I) substrate is stable in air-saturated buffers on the experimental time scale (Fig. 3a(i)), but is oxidised in the presence of catalyst wt-CueO:| [CuI(Bca)2]3− → Cu2+ + 2Bca2− + e− | (2) |
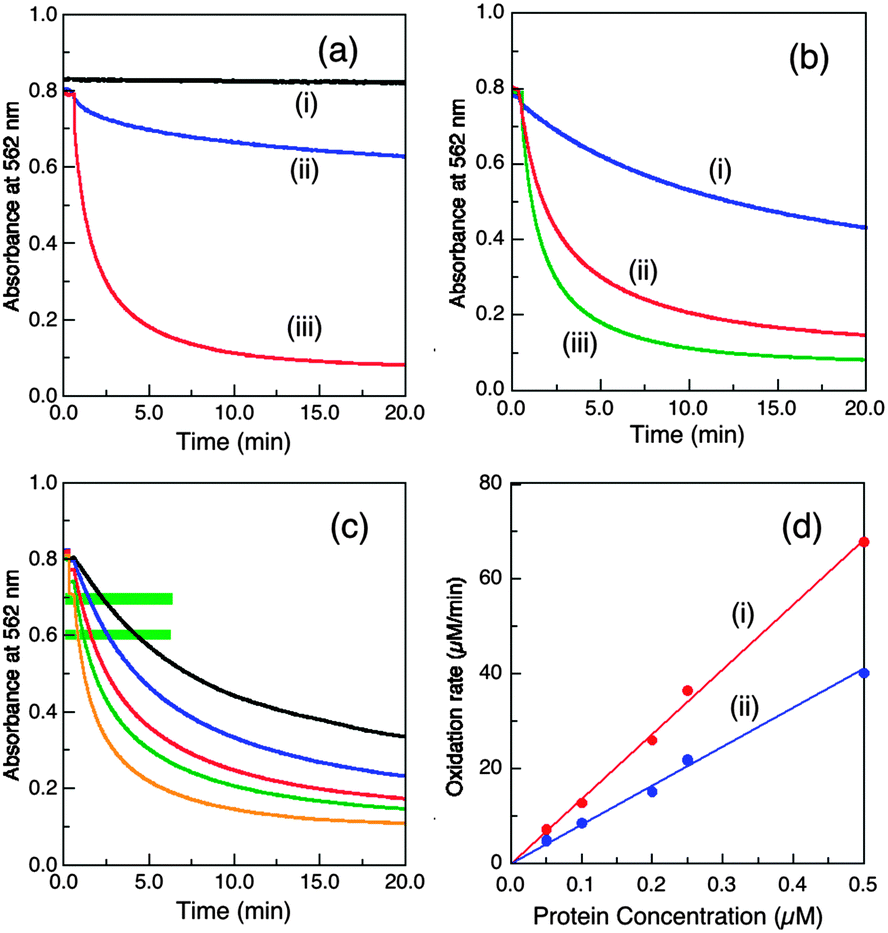 | ||
| Fig. 3 Conditions that modulate the cuprous oxidase activity of wt-CueO (0.25 μM in (a, b) and varying in (c, d)) on model substrate [CuI(Bca)2]3− (∼120 μM) in air-saturated buffer (25 mM, pH 7.0). (a) Buffer effect with initial substrate composition Bca/Cu(I) = 2.1: (i) no enzyme in BisTris buffer (ii) Mops buffer; (iii) BisTris buffer. (b) Effect of variation of initial Bca/Cu(I) ratio in BisTris buffer: (i) 5.0, (ii) 3.0, (iii) 2.1. (c, d) Effect of variation of enzyme concentrations: (c) reaction profiles under the condition of b(ii); from the top: 0.05, 0.1, 0.2, 0.25, 0.5 μM; (d) molar activity determined via linear plots of [CuI(Bca)2]3− oxidation rate versus enzyme concentration at reaction points defined by A562 = (i) 0.70 ± 0.01; (ii) 0.60 ± 0.01 (see green-highlighting in c). The derived apparent molar activities are 136 and 80.7 min−1, respectively, and are listed in Table 1. | ||
Buffers with a modest affinity for Cu(II) were required to abstract the oxidation product Cu2+ from the enzyme to regenerate the empty active site(s) and so the initial form of the enzyme (i.e., either CueOMops or CueOBT), is now not important. Catalysis is ineffective in Mops buffer (KD > 10−2 M) but effective in BisTris buffer (KD, 10−5.1 M; Fig. 3a).
In BisTris buffer (20 mM, pH 7.0) with the same initial substrate concentration of [CuI(Bca)23−] = 120 μM (after mixing), the apparent CueO activity varies considerably with the initial molar ratio of [Cu(I)]tot/[Bca]tot (Fig. 3b). The excess free ligand Bca limits Cu(I) availability and suppresses Cu(I) oxidation. On the other hand, oxidation product Cu2+ may also compete for the Cu(I) SDO site(s) and inhibit enzyme activity. Such inhibition is minimal in BisTris buffer at concentrations >10 mM as it can limit free Cuaq2+ to the sub-nanomolar range.9 However, free Bca is released during the catalysis (eqn (2)) and consequently, the apparent enzyme activity decreases continually along the reaction time course. For example, for a substrate solution defined by [CuI(Bca)23−] = 120 μM and initial [Cu(I)]tot/[Bca]tot = 1.0/3.0, the average catalytic molar velocity of wt-CueO for reaction 2 at reaction points defined by A(562) = 0.70 ± 0.01 and 0.60 ± 0.01 were calculated to be 136 min−1 and 80.7 min−1, respectively. The latter is an apparent decrease by more than 40% over the former (Fig. 4c and d). However, the above apparent enzyme activities determined at a fixed absorbance value for A(562) (which defines the solution composition at a given stage of the reaction) are insensitive to the enzyme concentration (Fig. 3c and d) and so can be used to compare the relative activities of the various CueO proteins quantitatively and reliably.
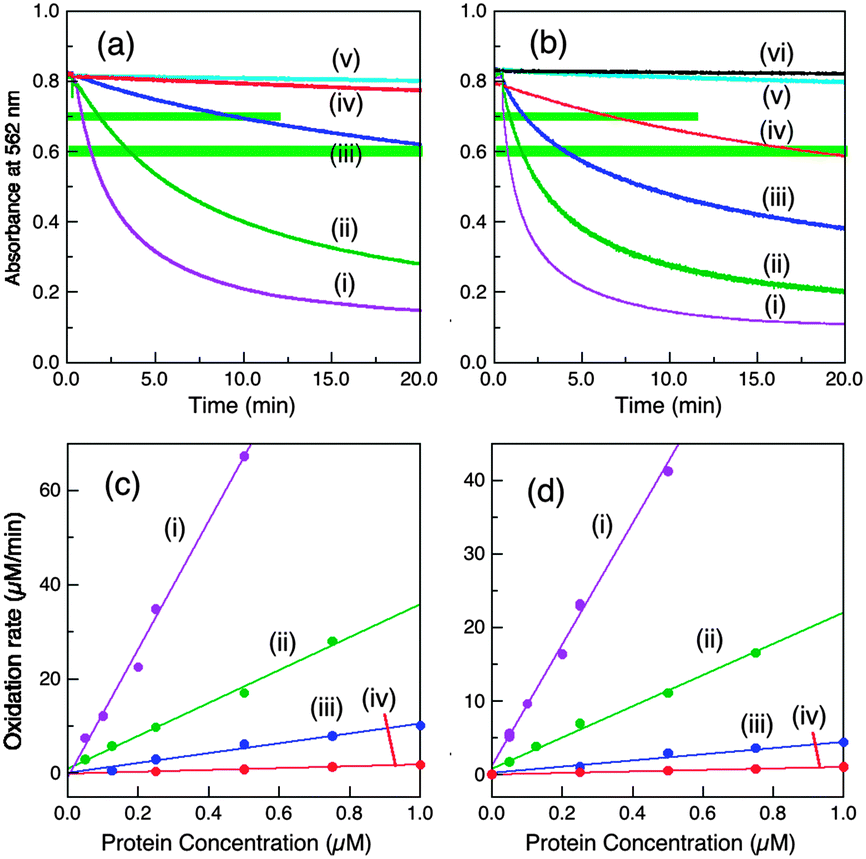 | ||
| Fig. 4 Cuprous oxidase activity of various CueO variants on model substrate [CuI(Bca)2]3− (∼120 μM with initial Cu(I):Bca = 1.0:3.0) in air saturated BisTris buffer (25 mM, pH 7.0). (a, b) Reaction profiles with a fixed amount of each enzyme (0.25 μM in (a) and 0.75 μM in (b)) and (c, d) molar activity determined via linear plots of the reaction rates versus each enzyme concentration under the solution conditions of A562 = 0.70 ± 0.01 (c) and 0.60 ± 0.01 (d) (both highlighted in green in a, b): (i) wt-CueO; (ii) Cu6-m; (iii) Cu7-m; (iv) Cu5-m1; (v) Cu6,7-m; (vi) Cu5-m2 (indistinguishable to that of control with no enzyme). Note: CueO variant Cu5-m1 at 0.75 μM catalysed the Cu(I) oxidation to a solution condition of A562 = 0.70 and 0.60 in ∼7 and ∼17 min, respectively, but this activity is <2% relative to that of wt-CueO. Consequently, all other CueO variants with lower activities than that of Cu5-m1 are counted as inactive. The data are summarised in Table 1. | ||
Two representative reaction points defined by A(562) = 0.70 ± 0.01 and 0.60 ± 0.01 for the substrate solution were chosen to define and compare relative cuprous oxidase activities for enzyme concentrations between 0.05 and 1.25 μM. The two sets of data, although numerically quite different, are essentially identical in terms of activity relative to that of the wild type protein, demonstrating the reliability of the approach (Table 1). This led to the following relative order of decreasing activity:
| wt (1.00) > Cu6-m (0.26) > Cu7-m (∼0.06) ≫ Cu5-m1 (<0.02) > Cu6,7-m ∼ Cu5-m2 (∼0) | (3) |
Interestingly, this order echoes the order of phenol oxidase activity (eqn (1)) despite the differences in reaction media (BisTris vs. Mops), substrates ([CuI(Bca)2]3−vs. DMP) and apparent reaction mechanisms (inner sphere vs. outer sphere).
Again, Cu5 is essential, consistent with a role as a fully-buried electron transfer site (Fig. 1c, d and Scheme 2) occupied by a copper ion. Disruption of both surface sites in Cu6,7-m rendered the enzyme essentially inactive while both Cu6 and Cu7 contribute to the Cu(I) SDO activity. The result again suggests very weak, if any, direct electron transfer between Cu6 and Cu5 under the Cuaq2+ limiting conditions imposed by the BisTris buffer. The data consolidate the conclusion from the phenol oxidase study that a facile electron transfer route of Cu6 → Cu7 → Cu5 → T1 → T2,3 → O2 exists for both oxidase functions. However, the cuprous oxidase activity in this case relies on the capability of the surface sites Cu6 and Cu7 to extract Cu(I) from its carrier and thus is optimised in weak Cu(II) binding buffer BisTris that promotes removal of inhibiting product Cu(II) from the reaction sites, as sketched in Scheme 2. Direct transfer of Cu(I) from site Cu6 to site Cu7 cannot be ruled out completely and may be promoted by the Met and His sidechains between the two sites (Fig. 1a). The same facile electron transfer route is utilised for outer sphere phenol oxidation and for inner sphere cuprous oxidation (Schemes 1 and 2).
 | ||
| Scheme 2 Proposed reaction mechanism for CueO as a cuprous oxidase. The Cu5 site is an essential electron-transfer mediator, not a substrate reaction site. Cu6 and Cu7 are the two reaction sites for Cu(I) loading and oxidation. The function of Cu6 depends primarily on electron transfer to Cu7. | ||
The thermodynamic affinities of the reaction site(s) for Cu(I) and Cu(II) were estimated previously to be around 0.13 pM and 5.5 nM, respectively, based on the analysis of the enzyme activity as cuprous oxidase and phenol oxidase under various conditions and were assigned to the proposed reaction site Cu5.9 However, it is now apparently that these affinities should be assigned more appropriately to the affinities of the labile SDO sites Cu6 and Cu7. Both sites are rich in methionine and should be able to bind Cu(I) with much higher affinity than they can Cu(II). The experiments with probe DP3 discussed above indicate that the copper centre in Cu5 in wt-CueO remains intact in BisTris buffer (Fig. S4, ESI†).
Cuprous oxidase activity based upon substrate CuICuII-PcoC
The cue operon is expressed by E. coli under normal aerobic nutrient conditions and the pco operon is induced at higher concentrations of environmental copper.1 PcoC is a copper metallo-chaperone with separated binding sites specific for Cu(I) and Cu(II).14 PcoA is a MCO with also a Met-rich insert in its sequence. It is able to oxidise PcoC-bound Cu(I) to less toxic Cu(II) in vitro.27,28 Metallo-chaperone PcoC is proposed to interact with PcoA as part of the pco resistance response but may also interact with the MCO CueO.9In air-saturated BisTris buffer, stable CuICuII-PcoC is oxidised rapidly and quantitatively upon addition of a catalytic amount of wt-CueO enzyme.9 The process is too fast to be followed by the available chromatographic separation approach and so the reactions were conducted in inhibiting Mops buffer to allow for semi-quantitative comparison of the relative activities of wt-CueO and its variants.
In air-saturated Mops buffer (20 mM, pH 7.0), the reaction of wt-CueO with air-stable CuICuII-PcoC under catalytic conditions (1:150–300) led to generation of products Cuaq2+ and CuII-PcoC that were separated from the original substrate by a cation-exchange column (Fig. 5a):
| CuICuII-PcoC → CuII-PcoC + Cuaq2+ + e− | (4) |
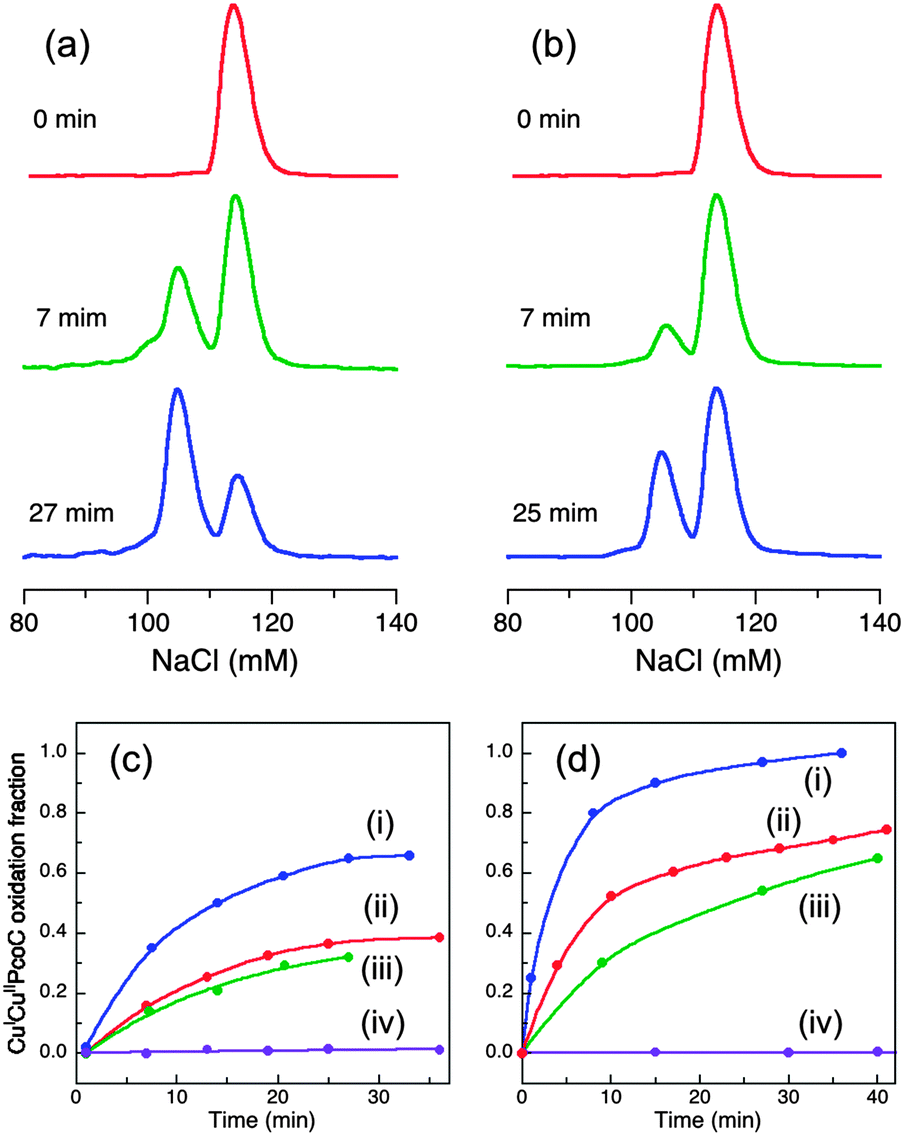 | ||
| Fig. 5 (a, b) Oxidation of CuICuII-PcoC (10 μM) catalyzed by wt-CueO (0.03 μM; (a)) or Cu6-m (0.03 μM; (b)) in air-saturated Mops buffer (20 mM, pH 7.0). The oxidation progress was monitored via chromatographic separation on a CIM SO3 cation-exchange column at the indicated reaction time. The elution peak intensities are normalized for clarity. (c, d) Plots of the fraction of oxidized product CuII-PcoC relative to total PcoC content versus reaction time for CueO enzymes at 0.03 μM in (c) and 0.06 μM in (d): (i) wt-CueO; (ii) Cu6-m; (iii) Cu7-m; (iv) Cu5-m1 (indistinguishable for Cu5-m2, Cu6,7-m or control with no enzyme and no appreciable oxidation for >10 h). | ||
The catalysis was suppressed to varying extents by different CueO variants. Activity decreased in the following order (Fig. 5b–d):
| wt (1.00) > Cu6-m (∼0.35) > Cu7-m (∼0.23) ≫ Cu6,7-m, Cu5-m1, Cu5-m2 (∼0) | (5) |
Both Cu5 site mutants Cu5-m1 and Cu5-m2 are again totally inactive, as is the double site variant Cu6,7-m, re-enforcing the above conclusions that Cu5 provides the essential electron-transfer pathway and that Cu6 and Cu7 are two Cu(I) SDO sites. Again, the apparently relative lower activity at Cu6 site (i.e., the Cu7-m variant) may reflect its persistent requirement of employing Cu7 as an electron-transfer pathway to Cu5. But such demand appears somewhat diminished due to possible establishment of a direct electron-transfer pathway from Cu6 to Cu5 in the presence of elevated free Cuaq2+ concentration under the catalytic condition. There are multiple Met and His residues associated with the helix and the nearby flexible loop (Fig. 1a). The individual activities of the two reaction sites are again not additive relative to the activity of the wt enzyme, suggesting some interdependency or cooperativity. Further speculation will be resisted due to the shortcomings of the present approach: (i) the semi-quantitative nature of the current analytical approach for reaction 4 prevents quantitative kinetic analysis and (ii) the activity in Mops buffer is inhibited by product Cuaq2+. This accumulation of free Cuaq2+ in non-coordinating buffer is physiologically irrelevant.
In conclusion, the present work confirmed that CuICuII-PcoC is a more robust cuprous substrate for wt-CueO than is [CuI(Bca)2]3−,9 presumably due to facile Cu(I) transfer from the substrate to the Cu(I) SDO sites Cu6 and Cu7 that is promoted by the surface-exposed Met-rich Cu(I) donating and receiving sites in the two molecules, as depicted in Fig. 6.
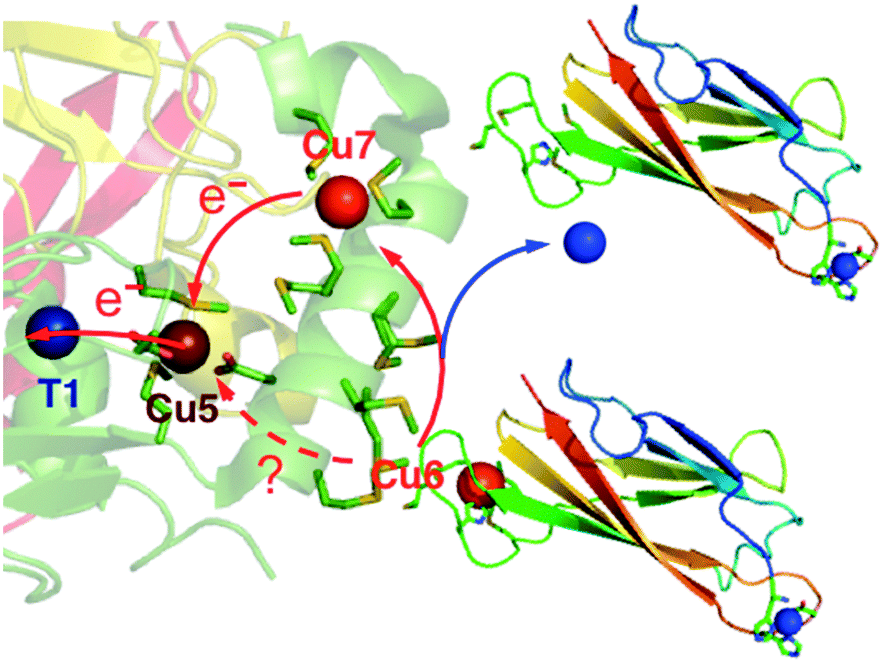 | ||
| Fig. 6 Reaction model of aerobic oxidation of CuICuII-PcoC catalyzed by cuprous oxidase enzyme CueO. The direct electron pathway between Cu5 and Cu6 may be promoted by adventitious binding of excess Cuaq2+ in Mops buffer. | ||
Summary and concluding remarks
CueO catalyses both phenol oxidation and cuprous oxidation via different reaction mechanisms.9 As a phenol oxidase, the reactivity depends on occupation of the reaction sites by Cu(II) ions that provide necessary oxidation and electron transfer sites for the substrate via an outer sphere mechanism. As a cuprous oxidase, the reactivity relies on the capability of the reaction sites to extract Cu(I) from its carriers for the oxidation via an inner sphere mechanism and on the subsequent release of the product Cu(II) from the reaction sites, as proposed for other metallo-oxidases.6,19 Thus the reaction sites must possess higher affinities for Cu(I) than for Cu(II). This work demonstrates, with three air-stable substrates, that the two surface-exposed sites Cu6 and Cu7 are the direct SDO sites for both oxidase functions and that buried site Cu5 channels electrons from the oxidations to the Type 1 site of the MCO machinery. Both sites Cu6 and Cu7 are Met-rich and expected to extract Cu(I) with sub-picomolar affinity.The pathway was demonstrated in BisTris buffer that is able to sequester product Cuaq2+ in a manner reminiscent of the physiologically relevant condition of limiting copper levels. Under such conditions, the phenol oxidase function is suppressed and the cuprous oxidase function is promoted. Consequently, the seven copper sites in CueO have evolved with specific roles to control copper toxicity in vivo as a robust cuprous oxidase.
Met- and His-rich regions have been identified in other systems and, in particular, in the N-terminal domains of the high affinity copper pumps Ctr1 from various sources.29,30 Such regions have been linked to sequestration and transport of Cu(I), rather than its oxidation, as discussed here in the context of control of its toxicity.
Acknowledgements
We thank the Australian Research Council for financial support under Grant DP130100728. Franziska Boneberg (an occupational trainee from Ludwig-Maximilian University, Munich, Germany) is thanked for assistance in DNA mutagenesis.Notes and references
- C. Rensing and G. Grass, Escherichia coli mechanisms of copper homeostasis in a changing environment, FEMS Microbiol. Rev., 2003, 27, 197–213 CrossRef CAS.
- E.-H. Kim, D. H. Nies, M. M. McEvoy and C. Rensing, Switch or Funnel: How RND-Type Transport Systems Control Periplasmic Metal Homeostasis, J. Bacteriol., 2011, 193, 2381–2387 CrossRef CAS PubMed.
- F. W. Outten, C. E. Outten, J. Hale and T. V. O'Halloran, Transcriptional activation of an Escherichia coli copper efflux regulon by the chromosomal MerR homologue, CueR, J. Biol. Chem., 2000, 275, 31024–31029 CrossRef CAS PubMed.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Multicopper Oxidases and Oxygenases, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed.
- J. M. Castagnetto, S. W. Hennessy, V. A. Roberts, E. D. Getzoff, J. A. Tainer and M. E. Pique, MDB: the Metalloprotein Database and Browser at The Scripps Research Institute, Nucleic Acids Res., 2002, 30, 379–382 CrossRef CAS PubMed.
- L. Quintanar, C. Stoj, A. B. Taylor, P. J. Hart, D. J. Kosman and E. I. Solomon, Shall we dance? How a multicopper oxidase chooses its electron transfer partner, Acc. Chem. Res., 2007, 40, 445–452 CrossRef CAS PubMed.
- D. A. Cooksey, Molecular mechanisms of copper resistance and accumulation in bacteria, FEMS Microbiol. Rev., 1994, 14, 381–386 CrossRef CAS PubMed.
- E. I. Solomon, A. J. Augustine and J. Yoon, O2 reduction to H2O by the multicopper oxidases, Dalton Trans., 2008, 3921–3932 RSC.
- K. Y. Djoko, L. X. Chong, A. G. Wedd and Z. Xiao, Reaction Mechanisms of the Multicopper Oxidase CueO from Escherichia coli Support Its Functional Role as a Cuprous Oxidase, J. Am. Chem. Soc., 2010, 132, 2005–2015 CrossRef CAS PubMed.
- S. A. Roberts, A. Weichsel, G. Grass, K. Thakali, J. T. Hazzard, G. Tollin, C. Rensing and W. R. Montfort, Crystal structure and electron transfer kinetics of CueO, a multicopper oxidase required for copper homeostasis in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2766–2771 CrossRef CAS PubMed.
- S. A. Roberts, G. F. Wildner, G. Grass, A. Weichsel, A. Ambrus, C. Rensing and W. R. Montfort, A labile regulatory copper ion lies near the T1 copper site in the multicopper oxidase CueO, J. Biol. Chem., 2003, 278, 31958–31963 CrossRef CAS PubMed.
- S. K. Singh, S. A. Roberts, S. F. McDevitt, A. Weichsel, G. F. Wildner, G. B. Grass, C. Rensing and W. R. Montfort, Crystal Structures of Multicopper Oxidase CueO Bound to Copper(I) and Silver(I): Functional Role of a Methionine-rich Sequence, J. Biol. Chem., 2011, 286, 37849–37857 CrossRef CAS PubMed.
- D. A. Rouch and N. L. Brown, Copper-inducible transcriptional regulation at two promoters in the Escherichia coli copper resistance determinant pco, Microbiology, 1997, 143(Pt 4), 1191–1202 CrossRef CAS PubMed.
- K. Y. Djoko, Z. Xiao, D. L. Huffman and A. G. Wedd, Conserved Mechanism of Copper Binding and Transfer. A Comparison of the Copper-Resistance Proteins PcoC from Escherichia coli and CopC from Pseudomonas syringae, Inorg. Chem., 2007, 46, 4560–4568 CrossRef CAS PubMed.
- L. X. Chong, M. R. Ash, M. J. Maher, M. G. Hinds, Z. Xiao and A. G. Wedd, Unprecedented binding cooperativity between Cu(I) and Cu(II) in the copper resistance protein CopK from Cupriavidus metallidurans CH34: implications from structural studies by NMR spectroscopy and X-ray crystallography, J. Am. Chem. Soc., 2009, 131, 3549–3564 CrossRef CAS PubMed.
- G. Sarret, A. Favier, J. Coves, J. L. Hazemann, M. Mergeay and B. Bersch, CopK from Cupriavidus metallidurans CH34 Binds Cu(I) in a Tetrathioether Site: Characterization by X-ray Absorption and NMR Spectroscopy, J. Am. Chem. Soc., 2010, 132, 3770–3777 CrossRef CAS PubMed.
- M.-R. Ash, L. X. Chong, M. J. Maher, M. G. Hinds, Z. Xiao and A. G. Wedd, Molecular Basis of the Cooperative Binding of Cu(I) and Cu(II) to the CopK Protein from Cupriavidus metallidurans CH34, Biochemistry, 2011, 50, 9237–9247 CrossRef CAS PubMed.
- A. K. Wernimont, D. L. Huffman, L. A. Finney, B. Demeler, T. V. O'Halloran and A. C. Rosenzweig, Crystal structure and dimerization equilibria of PcoC, a methionine-rich copper resistance protein from Escherichia coli, JBIC, J. Biol. Inorg. Chem., 2003, 8, 185–194 CrossRef CAS PubMed.
- Z. Xiao and A. G. Wedd, Metallo-Oxidase Enzymes: Design of their Active Sites, Aust. J. Chem., 2011, 64, 231–238 CrossRef CAS.
- T. R. Young, C. J. K. Wijekoon, B. Spyrou, P. S. Donnelly, A. G. Wedd and Z. Xiao, A Set of Robust Fluorescent Peptide Probes for Quantification of Cu(II) Binding Affinities in the Micromolar to Femtomolar Range, Metallomics, 2015 10.1039/C4MT00301B.
- K. H. Scheller, T. H. J. Abel, P. E. Polanyi, P. K. Wenk, B. E. Fischer and H. Sigel, Metal Ion/Buffer Interactions, Eur. J. Biochem., 1980, 107, 455–466 CrossRef CAS PubMed.
- D. Slomczynski, J. P. Nakas and S. W. Tanenbaum, Production and Characterization of Laccase from Botrytis cinerea 61-34, Appl. Environ. Microbiol., 1995, 61, 907–912 CAS.
- P. Atkins and J. d. Paula, Physical Chemistry, Freeman, New York, 7th edn, 2002, p. 172 Search PubMed.
- C. Kim, W. W. Lorenz, J. T. Hoopes and J. F. Dean, Oxidation of phenolate siderophores by the multicopper oxidase encoded by the Escherichia coli yacK gene, J. Bacteriol., 2001, 183, 4866–4875 CrossRef CAS PubMed.
- S. A. Roberts, G. F. Wildner, G. Grass, A. Weichsel, A. Ambrus, C. Rensing and W. R. Montfort, A Labile Regulatory Copper Ion Lies Near the T1 Copper Site in the Multicopper Oxidase CueO, J. Biol. Chem., 2003, 278, 31958–31963 CrossRef CAS PubMed.
- Notably, no copper was detected in sites Cu6 and Cu7 in a Cu(II) X-ray crystal structure, but the experiment was conducted in solution of pH 4.6 (ref. 12) and such acidic pH is expected to disfavour even weak Cu(II) binding.
- D. L. Huffman, J. Huyett, F. W. Outten, P. E. Doan, L. A. Finney, B. M. Hoffman and T. V. O'Halloran, Spectroscopy of Cu(II)-PcoC and the multicopper oxidase function of PcoA, two essential components of Escherichia coli pco copper resistance operon, Biochemistry, 2002, 41, 10046–10055 CrossRef CAS PubMed.
- K. Y. Djoko, Z. Xiao and A. G. Wedd, Copper Resistance in E. coli: The Multicopper Oxidase PcoA Catalyzes Oxidation of Copper(I) in Cu(I)Cu(II)-PcoC, ChemBioChem, 2008, 9, 1579–1582 CrossRef CAS PubMed.
- S. Puig, J. Lee, M. Lau and D. J. Thiele, Biochemical and genetic analyses of yeast and human high affinity copper transporters suggest a conserved mechanism for copper uptake, J. Biol. Chem., 2002, 277, 26021–26030 CrossRef CAS PubMed.
- K. L. Haas, A. B. Putterman, D. R. White, D. J. Thiele and K. J. Franz, Model Peptides Provide New Insights into the Role of Histidine Residues as Potential Ligands in Human Cellular Copper Acquisition via Ctr1, J. Am. Chem. Soc., 2011, 133, 4427–4437 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1 and S2; Fig. S1–S5. See DOI: 10.1039/c5mt00001g |
| This journal is © The Royal Society of Chemistry 2015 |