
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protection from neurodegeneration in the 6-hydroxydopamine (6-OHDA) model of Parkinson's with novel 1-hydroxypyridin-2-one metal chelators†
David G.
Workman
a,
Andrew
Tsatsanis
b,
Frank W.
Lewis
a,
John P.
Boyle
c,
Maryam
Mousadoust
a,
Nishani T.
Hettiarachchi
c,
Michael
Hunter
a,
Chris S.
Peers
c,
David
Tétard
*a and
James A.
Duce
*bd
aDepartment of Applied Sciences, Faculty of Health and Life Sciences, Northumbria University, Newcastle upon Tyne, Tyne and Wear NE1 8ST, UK. E-mail: david.tetard@northumbria.ac.uk; Fax: +44 (0)0191 227 3519; Tel: +44 (0)191 227 4934
bSchool of Biomedical Sciences, The Faculty of Biological Sciences, University of Leeds, Leeds, West Yorkshire LS2 9JT, UK. E-mail: j.a.duce@leeds.ac.uk; Tel: +44 (0)113 343 3075
cLICAMM, Faculty of Medicine and Health, University of Leeds, Leeds, West Yorkshire, UK. E-mail: c.s.peers@leeds.ac.uk; Fax: +44 (0)113 343 4803; Tel: +44 (0)113 343 4174
dOxidation Biology Unit, The Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, Victoria, Australia
First published on 10th March 2015
Abstract
Brain iron accumulation has been associated with inciting the generation of oxidative stress in a host of chronic neurological diseases, including Parkinson's disease. Using the catecholaminergic neurotoxin 6-hydroxydopamine to lesion cellular dopaminergic pathways as a model of Parkinson's disease in culture, a selection of 1-hydroxypyridin-2-one (1,2-HOPO) metal chelators were synthesized and their neuroprotective properties were compared to the 3-hydroxypyridin-4-one; deferiprone (3,4-HOPO; DFP). Protection against 6-OHDA and iron insult by the novel compounds 6 and 9 was comparable to DFP. Iron associated changes by 6-OHDA imply that the neuroprotective capacity of these compounds are due to chelation of the neuronal labile iron pool and the requirement of the iron binding moiety of compound 6 for efficacy supported this hypothesis. In conclusion, two novel 1,2-HOPO's and DFP have comparable neuroprotection against Parkinsonian-associated neurotoxins and supports the continued development of hydroxypyridinone compounds as a non-toxic therapeutic agent in the treatment of neurodegenerative disease.
Introduction
There is longstanding evidence of a selective iron increase in the substantia nigra pars compacta (SNc) of patients with Parkinson's disease (PD)1,2 as well as a rapid accumulation of iron in Parkinsonian models that use neurotoxins such 6-hydroxydopamine (6-OHDA),3 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP),4,5 rotenone6 or paraquat.5 These models are also capable of disrupting PD relevant catecholaminergic systems and cause oxidative toxicity that lead to the death of the dopaminergic neurons.7 The significance of this enrichment of iron with PD has even led to the suggestion of its use as a biomarker for early diagnosis of PD.8Iron plays critical roles in a number of key pathways in the cell, including electron transport and the metabolism of various neurotransmitters including dopamine (DA), noradrenaline and gamma-aminobutyric acid as well as in dopamine 2 receptor (D2R) function.9 The requirement for iron in DA metabolism is mainly through its production; tyrosine hydroxylase (TH) is a non-heme iron enzyme that uses molecular oxygen to hydroxylate tyrosine to form L-dihydroxyphenylalanine (L-DOPA).10 The co-localisation of unregulated iron and a potent reductant such as DA in circumstances where regulation of iron is perturbed, such as occurs in the SNc in PD, is also considered to increase the likelihood of unregulated and potentially toxic interactions such as protein and lipid oxidation as well as damage to DNA.11 Such an example is iron induced α-synuclein (α-syn) aggregation12,13 and the presence of the redox-active form of this metal with α-syn in Lewy bodies.14 In the healthy brain these interactions are minimised as the intracellular and extracellular levels of iron are tightly regulated.8,15
For some years, due to the primary role excess localised iron has in accelerating dopaminergic neuron death, iron chelating agents have been considered as a potential therapeutic avenue for PD.16,17 Metal chelating compounds, such as desferrioxamine (DFO) and more recently deferiprone (DFP) have been entered into clinical use to treat peripheral iron toxicity, such as acute iron poisoning and the iron overload conditions of thalassemia major and sickle cell anemia.18–20 However, chelating agents suitable for the treatment of iron accumulating neurodegenerative disorders such as PD must fulfil a careful mediation of additional requirements. In particular, their lipophilic ability to permeate the blood-brain barrier (BBB) must not be too great as to cause its rapid extraction by the liver and their iron affinity must be high enough to selectively bind iron in excess but not so high as to inhibit essential iron-containing enzymes that regulate neurotransmission, such as TH. While existing chelating agents such as DFO have been used as a potential PD therapeutic, its high iron affinity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β110 = 30.5),21 hydrophilicity and molecular size, render it not suitable for efficient clinical use. Indeed, DFO administration in models of neurodegenerative disease have indicated that large doses are required for minimal benefit when peripherally administered, and any benefits reported have mostly arisen from direct administration to the brain.22,23
β110 = 30.5),21 hydrophilicity and molecular size, render it not suitable for efficient clinical use. Indeed, DFO administration in models of neurodegenerative disease have indicated that large doses are required for minimal benefit when peripherally administered, and any benefits reported have mostly arisen from direct administration to the brain.22,23
Currently, hydroxyquinolines and hydroxypyridinones are the only two major classes of molecules that contain a sufficient bidentate metal binding moiety and fit the requirements described above. The 8-hydroxyquinoline chemical class of compounds (e.g. clioquinol and PBT2) are effective in vitro at preventing toxicity caused by the pathological aggregation of misfolded proteins including α-syn24 and are thought to restore brain biometals to their correct anatomical compartments by performing as transition metal ionophores.25,26 However the mixed N- and O-donor metal binding site of hydroxyquinoline based compounds are not conducive to the stabilization of hard Lewis acids such as ferric iron (Fe3+), thus explaining clioquinol's very low affinity for Fe3+.27 Instead, the relatively hard bidentate O-donor ligand common with 3-hydroxypyridin-4-ones (e.g. DFP) and several natural siderophores,28 are expected to be better at forming a neutral iron complex and accordingly have also demonstrated to be neuroprotective against iron-related toxicity29 and provided a promising outcome on a recent clinical trial in PD.30
Despite the promising therapeutic value of hydroxypyridinones in neurodegenerative disease, little investigation in models of neurotoxicity has been performed on analogues other than DFP and its derivatives. In this study, a selection of novel 1-hydroxypyridin-2-one compounds were synthesized and examined for neuroprotection against the 6-OHDA neurotoxicity model of PD. The critical involvement of iron chelation in protection from 6-OHDA was illustrated through the neuroprotective inefficiency of additional synthesized analogues of the most promising candidate compound that do not have the ability to bind metal. Further, comparisons between DFP and the novel compounds were carried out to investigate protective capacity through attenuated oxidative stress and mobilized cellular iron.
Results
Synthesis
The known 1-hydroxypyridin-2-one carboxylic acid 2 was synthesized by a slight modification to the literature method of Xu et al. (Scheme 1).31 Treatment of 6-hydroxypicolinic acid 1 with peracetic acid in acetic acid (rather than in the mixture of trifluoroacetic acid–trifluoroacetic anhydride–acetic acid previously used) generated 2 with an improved yield of 77%.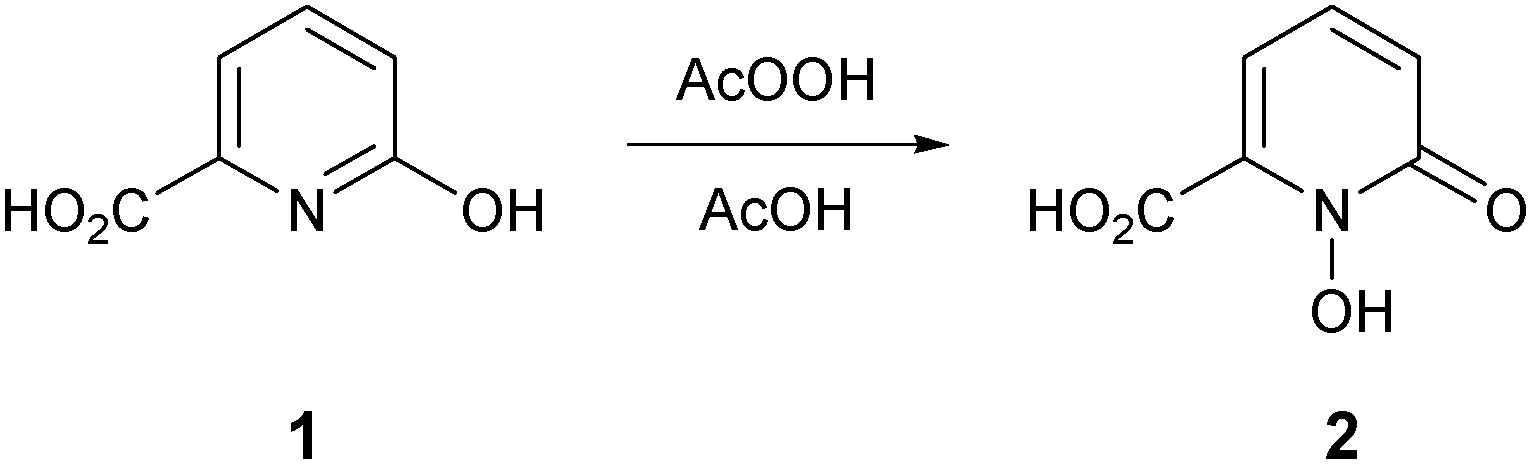 | ||
| Scheme 1 Synthesis of HOPO acid 2. | ||
The novel 1-hydroxypyridin-2-one 6 was synthesized as shown in Scheme 2. Treatment of acid 2 with thionyl chloride in methanol afforded the known methyl ester 332 in 96% yield. To protect the chelating N-hydroxyl functionality, we chose the allyl protecting group rather than the benzyl protecting group, owing to the wide variety of methods for the removal of the former and also to mitigate the risk of cleavage of the benzylic C–O bond in compound 6 under standard hydrogenation conditions. O-Allylation of ester 3 with K2CO3/allyl bromide generated 4 in 94% yield. Reduction of the ester group of 4 was accomplished with sodium borohydride in methanol–tetrahydrofuran (THF) at reflux and subsequent O-allyl deprotection of alcohol 5 (Pd/C and trifluoroacetic acid)33 gave the desired 1-hydroxypyridin-2-one 6 in 60% yield (Scheme 2).
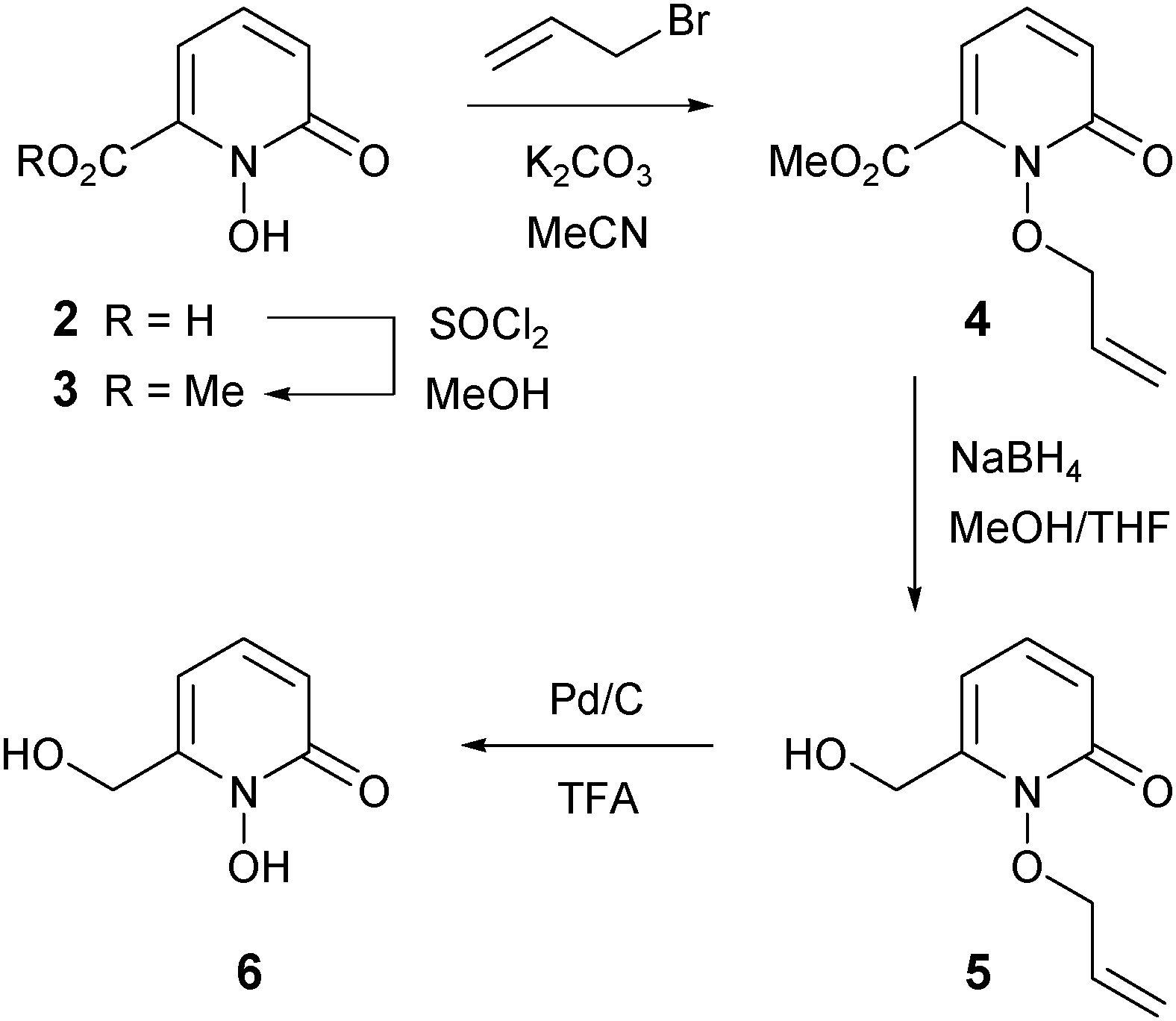 | ||
| Scheme 2 Synthesis of HOPO 6. | ||
In order to determine if the location of the carboxylic acid group in compound 2 had any significant effect on neuroprotection and iron chelation, we synthesized the novel regioisomeric compound 9 (Scheme 3). Initially, we attempted to synthesize 9 by direct oxidation of 2-hydroxynicotinic acid. This oxidation reaction failed using several different oxidants and only starting materials were recovered, presumably because 2-hydroxynicotinic acid exists predominantly as the less reactive keto tautomer. To overcome this problem, we instead synthesized 9 from 2-chloronicotinic acid 7 in a manner analogous to that previously used to synthesize 2 from 6-bromopicolinic acid.34 Oxidation of 7 proceeded smoothly to afford the N-oxide intermediate 8 in 42% yield, and hydrolysis of 8 using potassium hydroxide in water generated the novel 1-hydroxypyridin-2-one 9 in 92% yield (Scheme 3).
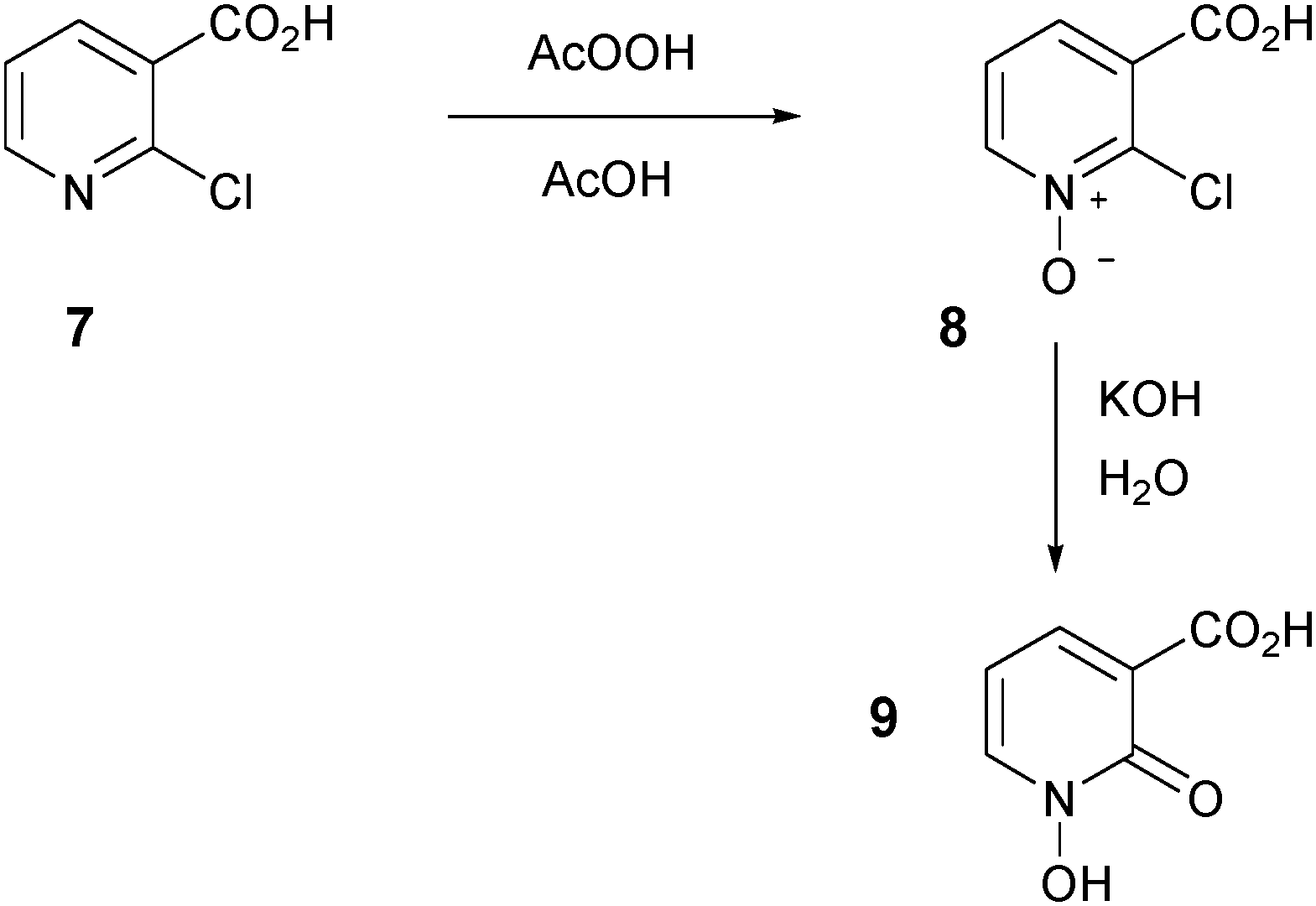 | ||
| Scheme 3 Synthesis of HOPO acid 9. | ||
Confirmation of the structure of acid 9 was unambiguously obtained using single crystal X-ray crystallography. Fig. 1 indicates there is hydrogen bonding between the hydrogens of both the carboxylic acid group (O(1)) and the hydroxamic acid group (O(4)) and the carbonyl oxygen at O(3), with hydrogen bond lengths of 1.75 Å and 2.10 Å. Consequently, the structure of 9 is near planar with torsion angles of O(1)–C(1)–C(2)–C(3) of 178.8°, O(1)–C(1)–C(2)–C(6) of 0.1°, O(2)–C(1)–C(2)–C(3) of −0.1°, and O(4)–N–C(6)–O(3) of 0.5°, respectively. Full details of data collection and structural refinement are shown in the ESI.†
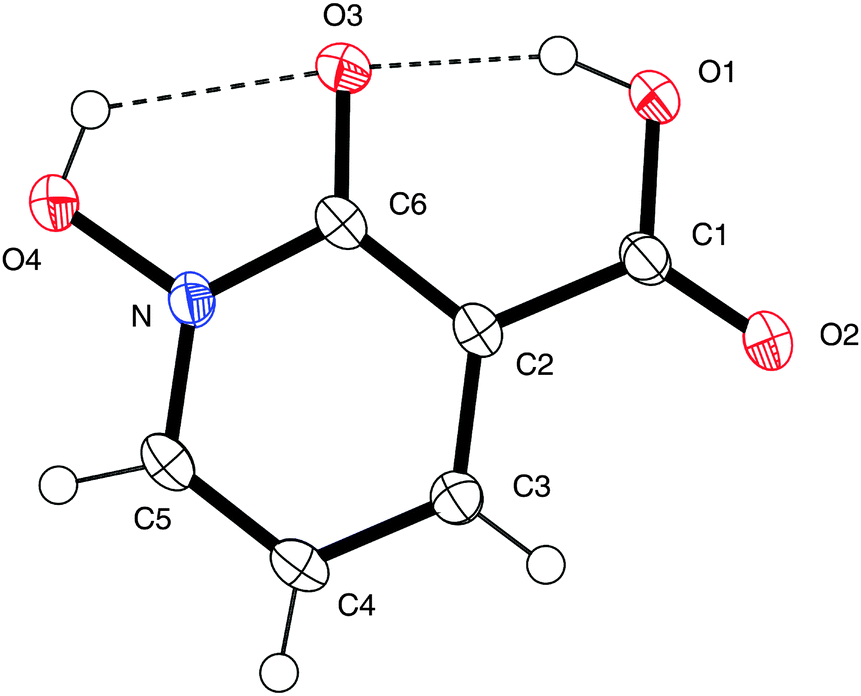 | ||
| Fig. 1 X-ray crystal structure of acid 9. Thermal ellipsoids are shown at 50% probability. One of the four asymmetric units is shown for clarity. Hydrogen bonds are shown by dotted lines. CCDC 983801. | ||
We also synthesized structural analogues of compound 6 that lack the cyclic hydroxamic acid metal chelating functionality. N-oxide 11 and pyridone 12 were synthesized and screened to provide information on whether the drug efficacy of 6 was due to its metal chelating ability. The known N-oxide 11 was prepared by oxidation of 2-(hydroxymethyl)pyridine 10 with m-CPBA in chloroform (Scheme 4).35 The known pyridone 12 was synthesized by the reduction of acid 1 with borane in THF (Scheme 5).36
 | ||
| Scheme 4 Synthesis of N-oxide 11. | ||
 | ||
| Scheme 5 Synthesis of pyridone 12. | ||
6-OHDA neuroprotection by novel 1,2-HOPO derivatives
Initial selection studies with the novel 1,2-HOPO compounds 2, 6 and 9 as well as the commercially available 3,4-HOPO; DFP were carried out to determine cell viability at the highest dose introduced to the cells. At 100 μM no significant change in cell viability was identified upon introduction of compounds alone to the media for 24 hours, as measured by MTT assay (Fig. 2A). These compounds were then evaluated for neuroprotective capability in the 6-OHDA cellular model of PD. Variable efficiency in neuroprotection was observed between compounds, whereby compounds 6 and 9 displayed similar defence against 6-OHDA neurotoxicity as DFP and compound 2 was only able to restore 20% of cell viability by 100 μM. (Fig. 2B–E). Comparing compound 2 with 9 suggests that the location of the carboxylic acid has an effect on neuroprotection against 6-OHDA.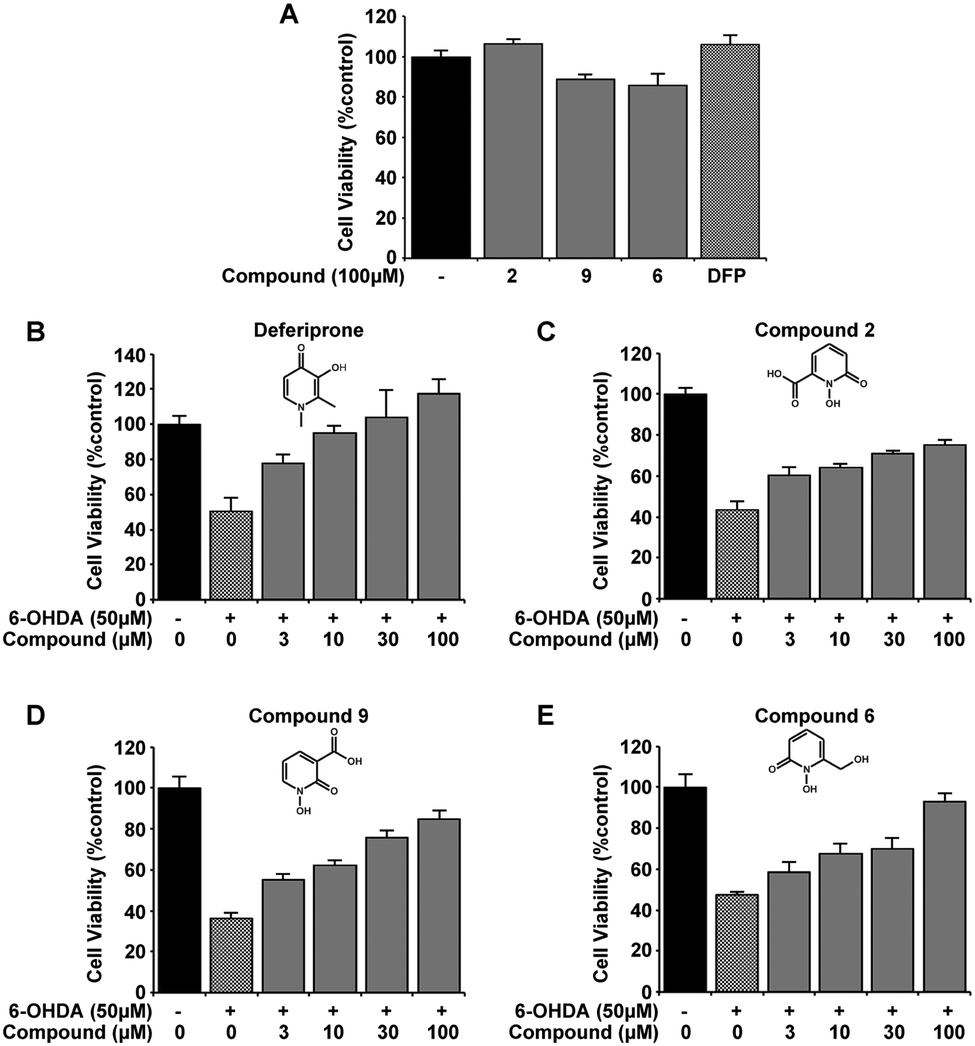 | ||
| Fig. 2 Novel 1,2-HOPO compounds have neuroprotection comparable to DFP against 6-OHDA excitotoxicity in the SH-SY5Y neuroblastoma cell line. Neither the 3 novel 1,2-HOPO compounds 2, 6 and 9, nor DFP (100 μM) affected cell viability after 24 h when extracellularly added (A). Incubation with 6-OHDA (50 μM) for 24 h decreases cell viability by ∼60% as measured by MTT assay. However, co-incubation with compounds DFP, 2, 6, and 9 at varying concentrations (3, 10, 30 and 100 μM) was dose-dependently able to protect against 6-OHDA induced cell death. At 100 μM of each compound, cell viability compared to non-treated cells for DFP = 117 ± 8.3%, compound 2 = 75 ± 2%, compound 9 = 84 ± 4.2% and compound 6 = 93 ± 4% (B–E). Experiments were carried out 3 times with each experiment in triplicate. Data is mean ± standard error. The presence of compounds at all concentrations with 6-OHDA indicated a protection in cell viability at p > 0.001 against 6-OHDA alone. | ||
From the novel 1,2-HOPO compounds 2, 6 and 9, compound 6 was selected as the most efficient at protection against 6-OHDA (at 100 μM mean cell viability was 75%, 84% and 93% respectively). To evaluate if the iron binding hydroxamic acid moiety of compound 6 was necessary for neuroprotection, the non-metal binding analogues 5, 11 and 12 were tested in the same 6-OHDA cell viability assay. Whilst the minimal modifications to their molecular structure did not impair cell viability (Fig. 3A), all analogue compounds were unable to protect against 6-OHDA neurotoxicity (Fig. 3B–E), demonstrating that the hydroxamic acid metal binding functionality in 6 was indeed essential for neuroprotection.
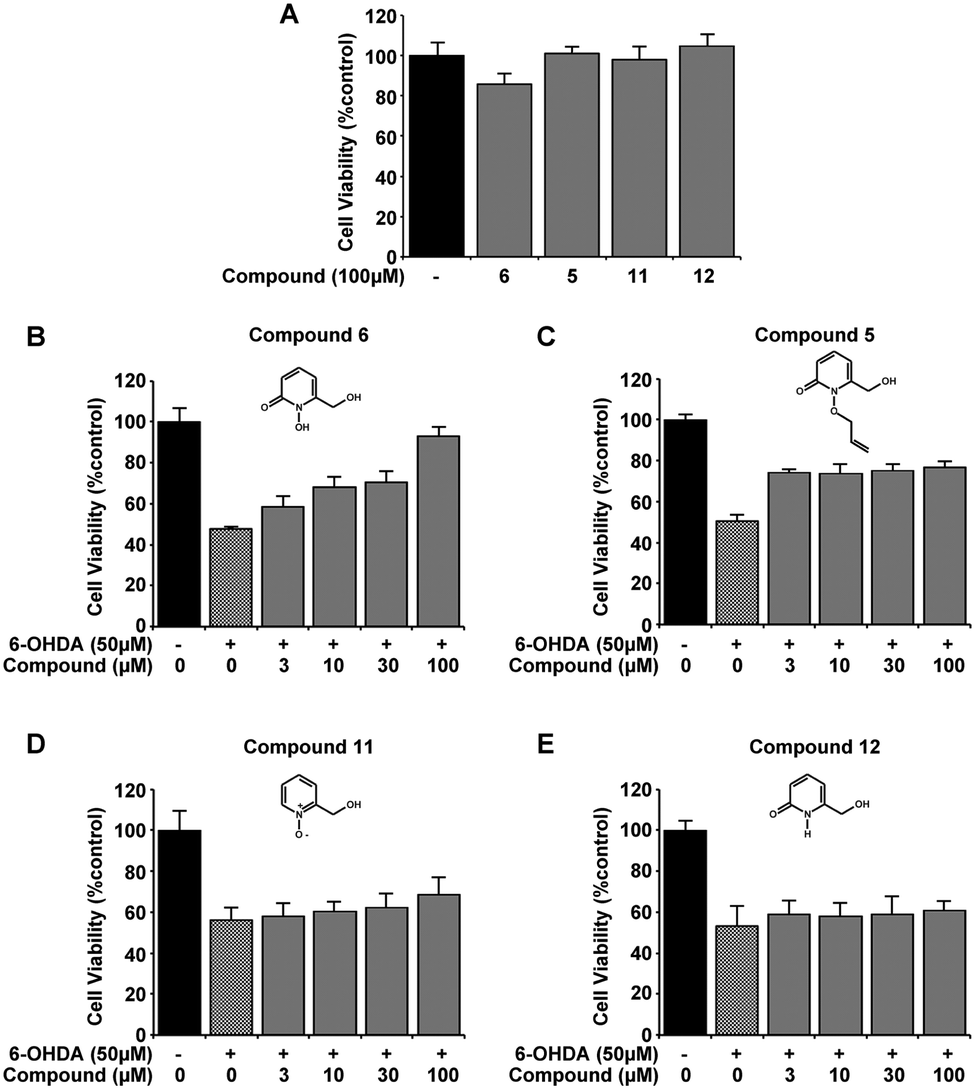 | ||
| Fig. 3 The metal binding moiety of compound 6 is required to protect 6-OHDA excitotoxicity in the SH-SY5Y neuroblastoma cell line. None of the 3 derivatives of compound 6 that lack the cyclic hydroxamic acid moiety required for metal binding was found to be detrimental to cell viability at 100 μM, as measured by MTT assay (A). Despite the structural similarity in all compounds, compound 6 was able to restore cell viability with 50 μM 6-OHDA (B) whereas compounds 5, 11 and 12 were incapable of protecting against 6-OHDA induced cell toxicity (C–E). Experiments were carried out 3 times with each experiment in triplicate. Data is mean ± standard error. Significance in 6-OHDA protection was only reached with compound 6 (p > 0.001). | ||
The neuronal labile iron pool is reduced by compound 6 and DFP
The same novel 1,2-HOPO compounds were also investigated for their capacity to reduce the cytoplasmic labile iron pool (LIP) in SH-SY5Y neuroblastoma cultures. DFP has previously been shown to efficiently cross the plasma membrane and protect against iron induced oxidative stress by reducing the LIP.29,37 We therefore compared compounds 2, 6 and 9 to DFP's ability to chelate LIP using the calcein-AM assay (Fig. 4A). In correlation with 6-OHDA protection, compounds 6 and 9 were similar to DFP in reducing the LIP of iron loaded cells, whereas compound 2 had little effect. As predicted, ablation of the iron binding moiety in compound 6 (as in compounds 5, 11 and 12) also negated LIP chelation by the compound (Fig. 4B). The indirect measurement of LIP through iron response protein expression also illustrated that ferritin (FT) and transferrin (TfR) expression was kept at a level comparable to cell without prior iron loading when compounds 6 and DFP were added for 12 hours following iron loading. Incubation with compound 12 however showed no significant effect compared to cells with iron loading alone (Fig. 4C–E).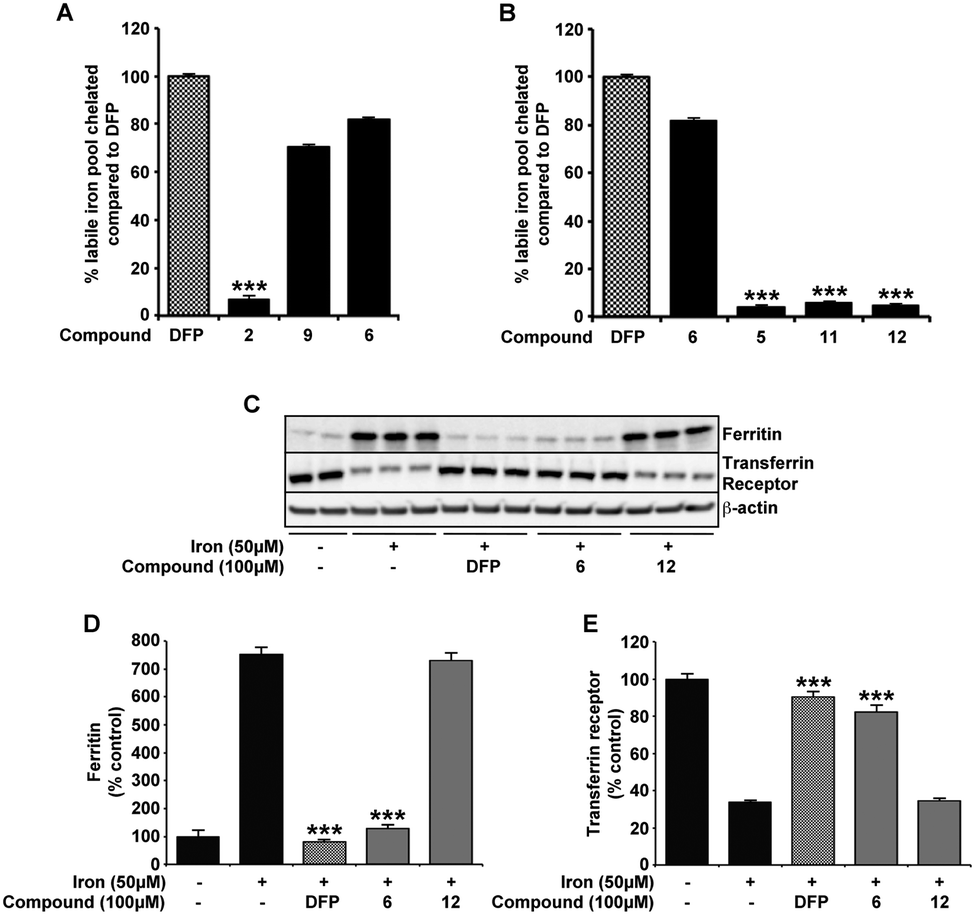 | ||
| Fig. 4 Compounds 6, 9 and DFP have comparable affinity for intracellular labile pool of iron. In SH-SY5Y cultures preincubated with iron (FAC; 50 μM), the acute response of each compound (100 μM) on the cytoplasmic labile iron pool (LIP) was evaluated by calcein-AM assay and iron response protein expression. Using the capacity of iron to quench calcein fluorescence, compounds 6 and 9 have comparable efficiency as DFP at entering the cell and binding the cytoplasmic pool of iron (A). In contrast, compound 2 and the derivatives of 6 without the metal binding moiety (compounds 5, 11 and 12) were unable to reduce LIP quenched calcein fluorescence (A and B). Accordingly, LIP changes modified the expression of the iron response proteins FT and TfR (C–E). After loading of cells with iron for 24 h, the addition of DFP and compound 6 incubation in media for a further 12 h altered FT and TfR to levels observed in cells without iron treatment. In contrast, 12 added after iron had no effect on iron response protein expression. Data in A and B are means ± S.E. of 3 experiments, performed in duplicate, *** p < 0.001 compared to DFP using two-tailed t tests. Data in D and E are means ± S.E. of 3 experiments, *** p < 0.001 compared to cells incubated with iron only using two-tailed t tests. | ||
Alterations in the neuronal labile iron pool by 6-OHDA are negated with compound 6 and DFP
Here, we confirm previous studies that strongly suggest an iron involvement in 6-OHDA neurotoxicity,3,38,39 whereby an elevation in neuronal LIP was evident through changes in iron response protein expression upon 6-OHDA (50 μM) incubation (Fig. 5). Protection against 6-OHDA by co-incubation of DFP and compound 6 (Fig. 2) correlate with both compounds also reducing the neuronal cytoplasmic LIP, as illustrated by the expression of the iron response proteins FT and TfR matching that observed in the untreated samples. Support for the necessity of the metal binding moiety of compound 6 in protecting against 6-OHDA-induced changes to LIP was again provided by the inability of compound 12 to alter the iron response protein levels (Fig. 5).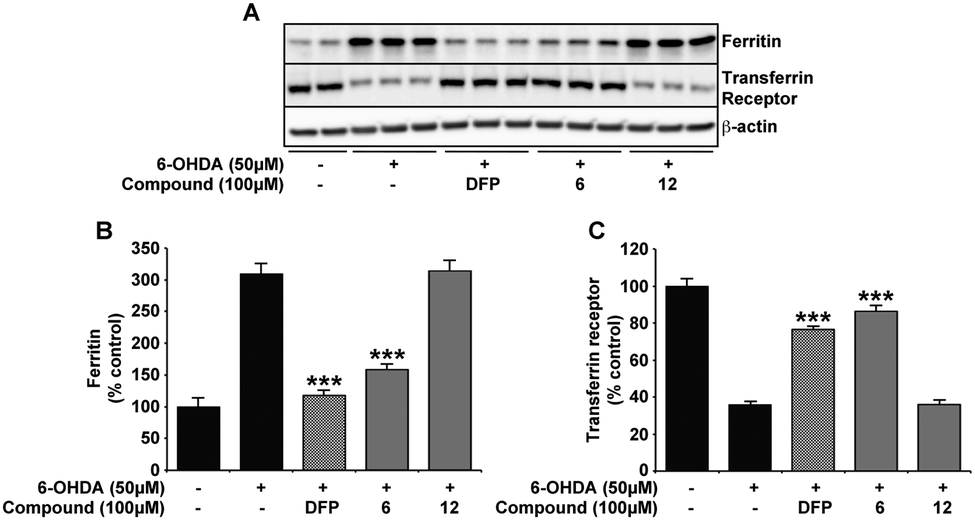 | ||
| Fig. 5 DFP and compound 6 negate labile iron pool elevation associated with 6-OHDA in the SH-SY5Y neuroblastoma cell line. Consistent with previous evidence of dysregulated iron homoeostasis upon 6-OHDA insult,3,38,39 FT is elevated and TfR decreased in cells incubated for 24 h with 6-OHDA (50 μM). Co-incubation with either DFP or compound 6 at 100 μM attenuated this response to 6-OHDA, with FT and TfR expression analogous to SH-SY5Y without treatment. However, compound 12 was unable to alter iron response protein expression in the presence of 6-OHDA. Data in B and C are means ± S.E. of 3 experiments, *** p < 0.001 compared to 6-OHDA only using two-tailed t tests. | ||
Neuroprotection against oxidative insult by compound 6 and DFP
Against both iron and 6-OHDA oxidative insult, confirmation of normal cellular function in the presence of compound 6 and DFP was confirmed using cell viability markers that are known to be altered in the SNc of PD patients and a range of Parkinsonian models.29,40 Tyrosine hydroxylase (TH) is the rate limiting enzyme in the synthesis of dopamine that requires iron and is highly sensitive to ROS whereas synaptophysin (Syn) is a nerve terminal marker that indicates damage to synapses. The presence of either compound 6 or DFP protected against the evident reduction in both TH and Syn upon iron (50 μM) or 6-OHDA (50 μM) incubation (Fig. 6). As with all other data provided, compound 12 had no significant protective effect on either insult.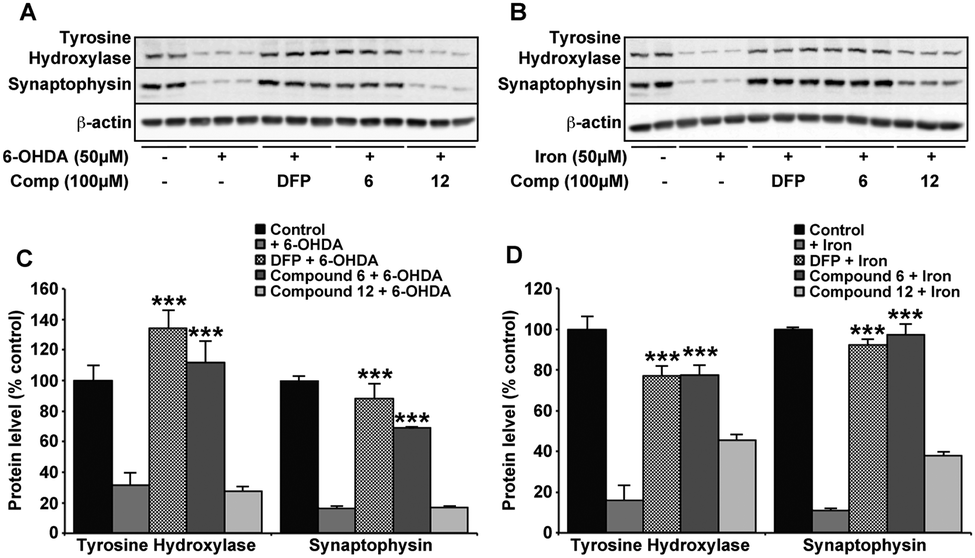 | ||
| Fig. 6 Markers associated with PD-related neurotoxicity are unchanged with DFP and compound 6 after 6-OHDA. In the same conditions for 6-OHDA (50 μM) (A and C) and iron (50 μM) (B and D) induced neurotoxicity, the known markers of neuronal function; tyrosine hydroxylase (TH) and synaptophysin (Syn), are both reduced. The presence of DFP or compound 6 (100 μM) with either insult prevented the loss in expression of either marker, whereas compound 12 had no effect. Data are means ± S.E. of 2 experiments, *** p < 0.001 compared to 6-OHDA (C) or iron (D) alone using two-tailed t tests. | ||
Discussion
A number of iron chelators including DFP have been successfully employed in preclinical studies of PD using a range of models including iron, 6-OHDA and MPTP.38 While protective against most causes of dopaminergic neurodegeneration, to our knowledge there has only been one study on DFP neuroprotection against 6-OHDA.23 This reported the ability of DFP at 10 mg kg−1 twice daily for 5 days, to attenuate the loss of TH positive neurons in the SNc and dopamine levels in the striatum. Here, we provide further evidence that 6-OHDA neurotoxicity is facilitated by neuronal iron dyshomeostasis and, by supporting the neuroprotective capacity of DFP in this model, reinforce a mechanism of protection as being via iron-associated oxidative stress. In turn, disrupted neuronal iron homeostasis has similar downstream consequences to those identified in PD. These include disrupted expression and activity of proteins that require iron for their structure and function (as seen with TH) as well as synaptic transmission (as seen with synaptophysin) and suggests that iron dyshomeostasis may be an early event in the progression of the disease.As well as providing mechanistic support for DFP protection in the 6-OHDA model, we investigated whether other hydroxypyridones were protective to the same insult. Accumulative evidence supports the use of 1,2-HOPO metal chelators in a number of applications, such as actinide sequestering,41 magnetic resonance imaging,42 treatment of iron overload43 and lanthanide luminescence.44 However, to date 1,2-HOPOs have not been therapeutically investigated in a model of neurodegeneration.
The parent 1,2-HOPO; 1-hydroxypyridin-2-one and the dimethylamide derivative of 1,2-HOPO 2 have pKa values of 5.845–49 and 5.1734 respectively. This compares with a pKa of 9.9 for DFP,46 suggesting a lower membrane penetration for 1,2-HOPOs such as compound 6 and 9 compared to 3,4-HOPOs such as DFP if only considering pKa. However, intriguingly DFO, a compound known for its lack of membrane permeability, has pKa values similar to DFP (dependent on hydroxamic acid groups these range between 9.70 and 8.30).50 We therefore suggest partition coefficients (logP) should also be considered in predicting membrane permeability. As one of the properties used in Lipinski's ‘rule of 5’ to predict oral activity of potential drug molecules, it has also been useful in predicting membrane permeability.51 Using Hyperchem software, the calculated logP for DFP is −0.81 (compared to the measured logP of −0.7752) whereas the calculated value for compound 6 is −0.54 and that of compound 9 is −0.51. Whilst care must be taken in the interpretation of these calculated values, the slightly higher logP values for 6 and 9 suggest analogous or slightly better membrane permeability to DFP and strengthens the ability of all 3 compounds to cross the blood brain barrier as well as to penetrate cellular plasma membranes.
Despite the affinity of 3,4-HOPOs for Fe3+ (pFe3+ of DFP = 20.5)45–47 being greater than the predicted affinity for compounds 6 and 9 (based on the measured pFe3+ of 16 for the parent 1,2-HOPO46), rescue of neuronal iron toxicity in our conditions was comparative between DFP and the novel 1,2-HOPO compounds 6 and 9. 6-OHDA neuroprotection and normalization of proteins associated with neurotoxic damage were also similar between these compounds. Information attained from the 1,2-HOPO compounds that had less protective properties suggest that the location of the carboxylic acid group on the 1,2-HOPO (compound 2vs.9) and the presence of the metal binding hydroxamic acid moiety (compounds 6vs.5, 11 and 12) are important for the defence of iron-associated 6-OHDA induced neurotoxicity.
While the use of DFP and other strong iron chelators in neurodegenerative disease has largely evolved from their success in treating peripheral iron overload in disorders such as thalassemia, it is increasingly evident that strongly chelating compounds may be less efficient in comparatively milder iron dyshomeostatic disorders. Largely due to the size and iron affinity of DFP (pFe3+ = 20.5), its increased ability to gain access to the active sites of metalloenzymes such as tyrosine hydroxylase52 and redistribute iron from areas in which they are required may causes potential hazards to normal neuronal function. It has thus become more apparent that with iron homeostasis being so precisely balanced within the brain, iron chelators with a weaker affinity for iron (pFe3+ < 20) may be more viable therapeutic candidates.52 These weaker chelating compounds are still required to redistribute iron from areas of high lability to protect against ROS production (e.g.: hydroxyl radical formation through the Fenton reaction), but have an iron affinity weak enough not to extract this metal from essential metalloenzymes. Indeed, these compounds would ideally bind iron in areas where the labile neuronal iron pool is pathologically high, redistribute it to regions of requirement and relinquish the bound iron to higher metal affinity apo-enzymes such as transferrin (pFe3+ = 20.3)19 for normal function.
Conclusions
In summary, two new iron bidentate chelators belonging to the family of 1,2-HOPO metal chelators show DFP comparative potential as a non-toxic therapeutic in the Parkinsonian models of iron and 6-OHDA. Future directions require studies in animal models and verification of the blood brain barrier permeability, but the continual development of these novel compounds can assist in the therapeutic impact already achieved in the removal of excess iron in neurodegenerative diseases. However, it is important to note that despite the proven efficacy in PD clinical trials with DFP,30 patients suffering from a complex neurodegenerative disease such as PD are unlikely to have a substantial response to a drug with a single target mechanism of action. This suggests that these diseases are best treated either by multiple drugs, each acting on different disease pathways, or by a single drug designed to act on multiple pathological pathways. Therapeutic compounds that counteract more than one disease pathology are increasingly being considered as more promising therapies for PD and related dementia's as they are considered to have a reduced chance of detrimental side effects.53,54 Modification of the 1,2-HOPO's bidentate structure to attach additional disease modifying moieties may provide a future novel multifunctional therapeutic compound with strong iron ionophoric capabilities. This work is currently underway in our laboratories.Experimental
General procedures
All solvents and reagents were purchased from Sigma-Aldrich, Acros Organics or Alfa-Aesar and used without further purification unless otherwise specified. Reactions were monitored by TLC using silica gel with UV254 fluorescent indicator. Uncorrected melting points were measured in open capillary tubes using a DigiMelt MPA161 SRS instrument. Elemental microanalyses were carried out at Newcastle University. IR spectra were recorded on a Perkin-Elmer Paragon 1000 FT-IR Spectrometer. NMR spectra were recorded on either a JEOL JNM-EX270FT Delta spectrometer (270.17 MHz for 1H NMR, 67.93 MHz for 13C NMR) or on a JEOL ECS400FT Delta spectrometer (399.78 MHz for 1H NMR, 100.53 MHz for 13C NMR). Chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane as internal standard. Coupling constants (J) are measured in hertz. Multiplets are reported as follows: b = broad, s = singlet, d = doublet, dd = double doublet, t = triplet, q = quartet, m = multiplet, app d = apparent doublet, app t = apparent triplet. Low resolution mass spectra were obtained in methanol solutions on a Thermo Finnigan LCQ Advantage MS detector using electrospray ionisation (ESI). High resolution mass spectra were obtained on a Finnigan MAT900XLT high-resolution double focussing MS spectrometer using nano-electrospray ionisation (NESI) at the EPSRC National Mass Spectrometry Service (University of Swansea). Column chromatography was conducted using 0.060–0.20 mm silica gel (70–230 mesh), and automated flash column chromatography was performed using a Biotage Isolera One ISO-1SV instrument. The partition coefficients (logP) for compounds 6, 9 and DFP were calculated using Hyperchem software (available at: http://www.hyper.com/).
1-Hydroxy-6-oxo-1,6-dihydropyridine-2-carboxylic acid 231
To a suspension of 6-hydroxypicolinic acid 1 (26.20 g, 188 mmol) in glacial acetic acid (160 mL) was carefully added peroxyacetic acid (36–40%, 80 mL). The temperature was carefully raised to 80 °C and stirring was continued for 12 h. The flask was allowed to cool to room temperature and the resulting solid precipitate was collected by filtration and washed with diethyl ether, affording the title compound 2 as a cream solid (18.17 g, 77%). Mp 223–226 °C (from AcOH, Lit.31 216 °C). νmax(neat)/cm−1 3114 (O–H), 1611 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1505 (CO), 1198. δH(399.8 MHz, DMSO-d6) 6.65 (1H, dd, J 7.3, 1.8, 3-H), 6.73 (1H, dd, J 9.2, 1.8, 5-H), 7.46 (1H, dd, J 8.7, 6.9, 4-H). δC(100.5 MHz, DMSO-d6) 106.8 (ArC), 120.8 (ArC), 137.3 (ArC), 139.5 (quat), 157.7 (quat), 162.4 (quat).
O), 1505 (CO), 1198. δH(399.8 MHz, DMSO-d6) 6.65 (1H, dd, J 7.3, 1.8, 3-H), 6.73 (1H, dd, J 9.2, 1.8, 5-H), 7.46 (1H, dd, J 8.7, 6.9, 4-H). δC(100.5 MHz, DMSO-d6) 106.8 (ArC), 120.8 (ArC), 137.3 (ArC), 139.5 (quat), 157.7 (quat), 162.4 (quat).
Methyl 1-hydroxy-6-oxo-1,6-dihydropyridine-2-carboxylate 332
To a suspension of acid 2 (15.73 g, 101 mmol) in methanol (200 mL) at 0 °C was added thionyl chloride (31.00 g, 426 mmol) dropwise. The mixture was heated under reflux for 4 h. The solution was then allowed to cool to room temperature and the solvent was removed in vacuo to afford the title compound 3 as a cream solid (16.47 g, 96%). Mp 106–108 °C (from MeOH, Lit.31 90–92 °C). νmax(neat)/cm−1 3115 (O–H), 1732 (CO), 1505 (CO), 1204. δH(399.8 MHz, DMSO-d6) 3.87 (3H, s, CO2CH3), 6.53 (1H, dd, J 6.9, 1.4, 3-H), 6.69 (1H, dd, J 9.2, 1.4, 5-H), 7.45 (1H, dd, J 9.2, 6.9, 4-H). δC(100.5 MHz, DMSO-d6) 53.8 (CO2CH3), 105.8 (ArC), 122.6 (ArC), 137.8 (ArC), 138.8 (quat), 158.1 (quat), 161.4 (quat).
Methyl 6-oxo-1-(allyloxy)-1,6-dihydropyridine-2-carboxylate 455
To a solution of compound 3 (16.47 g, 97 mmol) in acetonitrile (200 mL) was added potassium carbonate (32.11 g, 232 mmol), followed by allyl bromide (28.10 g, 232 mmol). The flask was heated under reflux for 4 h before the reaction mixture was filtered and the solvent removed under high vacuum. The residue was dissolved in toluene (100 mL) and the solvent was evaporated in vacuo to afford the title compound 4 as a white crystalline solid (19.12 g, 94%). Mp 65–67 °C (from toluene). Found C, 57.06; H, 5.38; N, 6.67%; C10H11NO4 requires C, 57.41; H, 5.30; N, 6.70%. νmax(neat)/cm−1 3462, 3078, 2953, 1735 (CO), 1661 (CO), 1586 (CC), 1445, 1275, 1209, 1136. δH(399.8 MHz, DMSO-d6) 3.94 (3H, s, CO2CH3), 4.90 (2H, d, J 6.9, CH2CHCH2), 5.42 (2H, m, CH2CHCH2), 6.08 (1H, m, CH2CHCH2), 6.53 (1H, dd, J 6.9, 1.8, 3-H), 6.80 (1H, dd, J 9.2, 1.8, 5-H), 7.31 (1H, dd, J 9.6, 6.9, 4-H). δC(100.5 MHz, DMSO-d6) 53.3 (CH2CHCH2), 78.1 (CO2CH3), 107.9 (CH2CHCH2), 121.9 (CH2CHCH2), 126.0 (ArC), 130.6 (ArC), 137.2 (ArC), 138.7 (quat), 158.8 (quat), 160.6 (quat). m/z (NESI) 210.0759 ([M + H]+); C10H12NO4 requires 210.0766.
6-(Hydroxymethyl)-1-(allyloxy)pyridin-2(1H)-one 5
To a suspension of compound 4 (19.12 g, 92 mmol) in THF (200 mL) was added solid sodium borohydride (25.07 g, 663 mmol) in small portions. The solution was heated under reflux for 15 min. Methanol (14 mL) was then added dropwise at reflux over 2 h. The solution was then cooled to 0 °C, quenched by careful addition of saturated aqueous ammonium chloride (25 mL) and stirring was continued for 15 min. The solvents were removed in vacuo and the residue was extracted with dichloromethane (3 × 25 mL). The combined organic extracts were dried and evaporated to afford the title compound 5 as an off-white solid (10.83 g, 65%). Mp 101–104 °C (from DCM). Found C, 59.23; H, 6.23; N, 7.59%; C9H11NO3 requires C, 59.66; H, 6.12; N, 7.73%. νmax(neat)/cm−1 3198 (O–H), 2896, 2842, 1650 (CO), 1560 (CC), 1441, 1154, 1093. δH(399.8 MHz, CDCl3, Me4Si) 2.74 (1H, t, J 6.4, CH2OH), 4.68 (2H, d, J 6.4, CH2OH), 4.84 (2H, d, J 6.4, CH2CHCH2), 5.43 (2H, m, CH2CHCH2), 6.06 (1H, m, CH2CHCH2), 6.24 (1H, d, J 6.9, 3-H), 6.50 (1H, dd, J 9.2, 1.4, 5-H), 7.30 (1H, dd, J 9.2, 6.9 4-H). δC(100.5 MHz, CDCl3, Me4Si) 59.4 (CH2CHCH2), 103.7 (CH2CHCH2), 120.4 (CH2CHCH2), 122.6 (ArC), 130.3 (ArC), 138.7 (ArC), 148.9 (quat), 159.7 (quat). m/z (NESI) 182.0811 ([M + H]+); C9H12NO3 requires 182.0817.
1-Hydroxy-6-(hydroxymethyl)pyridin-2(1H)-one 6
To a solution of compound 5 (0.21 g, 1.06 mmol) in 20% water in dioxane (10 mL) was added trifluoroacetic acid (0.11 mL). The solution was heated under reflux for 1 hour. Palladium on activated carbon (10%, Pd/C, 0.02 g) was then added and the mixture was heated under reflux for a further 20 h. The mixture was filtered and the filtrate was evaporated in vacuo to afford the title compound 6 as a brown solid (0.08 g, 60%). Mp 65–66 °C (from H2O/dioxane). Found C, 49.79; H, 4.64; N, 9.21%; C6H7NO3 requires C, 51.06; H, 5.06; N, 9.92%. νmax(neat)/cm−1 3248 (O–H), 3082, 2914, 1637 (CO), 1540, 1178, 1094. δH(399.8 MHz, DMSO-d6) 3.33 (1H, br s, OH), 4.47 (2H, s, CH2OH), 5.55 (1H, br s, OH), 6.23 (1H, dd, J 7.1, 1.8, 3-H), 6.36 (1H, dd, J 8.9, 1.8, 5-H), 7.34 (1H, dd, J 8.9, 7.1, 4-H). δC(100.5 MHz, DMSO-d6) 57.7 (CH2OH), 101.3 (ArC), 115.9 (ArC), 137.4 (ArC), 148.5 (quat), 158.4 (quat). m/z (LRMS, ESI) 142.00 ([M + H]+); C6H8NO3 requires 142.05. 140.00 (M−); C6H6NO3 requires 140.03.
2-Chloropyridine-3-carboxylic acid N-oxide 8
To a solution of 2-chloronicotinic acid 7 (12.00 g, 76 mmol) in glacial acetic acid (90 mL) was added peroxyacetic acid (36–40%, 35 mL). The solution was carefully raised to 80 °C and stirring was continued for 10 h. The solution was cooled to room temperature and the resulting precipitate was filtered and washed with diethyl ether. The solid was allowed to dry in air to afford the title compound 8 as a white powder (5.56 g, 42%). Mp 209–210 °C (from AcOH, Lit.56 195 °C). δH(399.8 MHz, DMSO-d6) 7.49 (1H, dd, J 7.8, 6.9, 5-H), 7.69 (1H, dd, J 7.8, 1.4, 4-H), 8.58 (1H, dd, J 6.4, 1.4, 6-H). δC(100.5 MHz, DMSO-d6) 124.6 (ArC), 126.4 (ArC), 132.3 (ArC), 140.0 (quat), 142.3 (quat), 164.9 (quat). m/z (LRMS, ESI) 173.94 ([M + H]+). m/z (NESI) 173.9954 ([M + H]+); C6H5NO3Cl requires 173.9958.1-Hydroxy-2-oxo-1,2-dihydropyridine-3-carboxylic acid 9
A solution of compound 8 (7.85 g, 45 mmol) in aqueous potassium hydroxide (10%, 125 mL) was stirred at 70 °C for 3 days. The solution was then cooled to 0 °C and concentrated hydrochloric acid was added to pH 1. The precipitated solid was collected by filtration and washed with water. The crude solid was then recrystallised from methanol to afford the title compound 9 as white needles (6.51 g, 92%). Mp 211–215 °C (from MeOH). δH(399.8 MHz, DMSO-d6) 6.65 (1H, t, J 6.9, 5-H), 8.27 (1H, dd, J 7.3, 1.8, 6-H), 8.45 (1H, dd, J 6.9, 1.8, 4-H). δC(100.5 MHz, DMSO-d6) 107.5 (ArC), 117.6 (ArC), 142.3 (ArC), 142.8 (quat), 160.7 (quat), 165.0 (quat). m/z (LRMS, ESI) 154.06 (M−). m/z (NESI) 154.0136 (M−); C6H4NO4 requires 154.0140.2-(Hydroxymethyl)pyridine N-oxide 1135
To a solution of m-CPBA (14.67 g, 50 wt% in H2O, 42.5 mmol) in chloroform (30 mL) was slowly added a solution of 2-(hydroxymethyl)pyridine 10 (3.85 g, 35.25 mmol) in chloroform (15 mL). The solution was heated at 65 °C for 20 h. The solution was then allowed to cool to room temperature and was dried and evaporated. The resulting residue was triturated with diethyl ether (150 mL) and the suspension was heated under reflux for 30 min. The insoluble solid was filtered and washed with diethyl ether (20 mL) and allowed to dry in air to afford the title compound 11 as a white solid (2.77 g, 63%). δH(399.8 MHz, CDCl3, Me4Si) 4.79 (2H, s, CH2OH), 7.23–7.38 (3H, m, 3-H, 4-H and 5-H), 8.22 (1H, d, J 6.4, 6-H). δC(100.5 MHz, CDCl3, Me4Si) 61.4 (CH2OH), 124.7 (ArC), 124.8 (ArC), 127.1 (ArC), 139.5 (ArC), 150.0 (quat).6-(Hydroxymethyl)pyridin-2(1H)-one 1236
To a suspension of acid 1 (1.00 g, 7.19 mmol) in dry THF (35 mL) was added a solution of borane in THF (35.9 mL, 1 M, 5 eq.) dropwise. After the addition was complete, the flask was heated to 65 °C for 24 h. The solution was then cooled to 0 °C and methanol (30 mL) was added. The flask was heated at 65° overnight and the solvent was evaporated. Methanol (50 mL) was added and the solvent was again evaporated. The resulting solid was triturated with acetone (30 mL), and then filtered and washed with methanol (5 mL) and acetone (40 mL) to afford the title compound 12 as a white solid (0.47 g, 52%). δH(399.8 MHz, DMSO-d6) 4.22 (2H, d, J 5.9, CH2OH), 5.36 (1H, t, J 5.9, CH2OH), 6.09–6.13 (2H, m, 3-H and 5-H), 7.34 (1H, dd, J 9.1, 6.8, 4-H). δC(100.5 MHz, DMSO-d6) 60.0 (CH2OH), 102.1 (ArC), 118.0 (ArC), 141.4 (ArC), 150.4 (quat), 163.2 (quat).Crystal structure determination
Crystals of acid 9 suitable for X-ray analysis were obtained by slow evaporation of a chloroform solution of 9. 3871 independent reflection data were collected at 150 K with an Agilent Technologies ‘XCALIBUR Atlas Gemini ultra’ CCD diffractometer using MoKα radiation (λ = 0.71073 Å). The crystal was mounted on a mounting loop in an inert perfluoropolyether oil (Fomblin) and cooled to 150 K using a cryostream. The structure was solved using direct methods with the SHELXS program. The structure was refined by least squares methods on F2 using the SHELXS program. Crystal data have been deposited at the Cambridge Crystallographic Data Centre as CCDC 983801. Data collection and structural refinement details are shown in the ESI.†Analysis of compound toxicity and neuroprotection against 6-OHDA insult on SH-SY5Y neuroblastoma cells
SH-SY5Y neuroblastoma cells were plated at 20000 cells per well in a 96 well microplate and left to adhere to well surface overnight in media (50% advanced minimum essential medium (MEM), 50% Ham's F12 medium, 1% L-glutamine) with 2% fetal bovine serum (FBS). When testing for 6-hydroxydopamine hydrobromide (6-OHDA) neurotoxicity, a stock concentration of 10 mM was prepared just before use and stored in the dark at 4 °C until added to cells. Media was then replaced with 100 μL per well of serum free media containing varying concentrations of the novel compounds ranging from 0 to 100 μM and 50 μM 6-OHDA as necessary. After a further 24 h incubation at 37 °C in a 5% CO2 environment, cell viability was measured using the tetrazolium dye 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). For this colorimetric assay, 11 μL of MTT (5 mg mL−1) was added to the 100 μL media in the well and incubated for 3 h at 37 °C. After this time an equal volume of solubilizing solution (24 mL isopropyl alcohol, 1 mL HCl) was added to each well and thoroughly mixed to lyse the cells. Absorption was measured at 570 nm using a Tecan Sunrise spectrophotometer (Model: Sunrise-Basic Tecan). Cell viability was calculated as a percentage compared to the untreated control.
Measurement of the neuronal cytoplasmic labile iron pool using calcein-AM
The use of calcein-AM to measure neuronal cytoplasmic labile iron pool was adapted from a previously reported procedure.57 In brief, SH-SY5Y cells plated at 20000 cells per well in a black 96-well microplate were treated with ferric ammonium citrate (FAC; 50 μM) for 6 h in serum-free media. Cells were washed twice with phosphate buffered saline (PBS) after which calcein-AM (60 nM) was added. Fluorescence at an excitation of 485 nm and emission of 535 nm measurements were started immediately and taken every minute using a Biotek Fluorescence microplate reader for 10 min or until a consistent minimum reading had been reached. In triplicate, the compounds of interest (100 μM) were rapidly added and fluorescence measurements started again immediately. Readings were continued for a further 5 min or until a plateau had been reached. The percentage labile iron pool chelated was calculated as the ΔF (the difference in Fluorescence from before and after compound addition) compared to DFP as a known lipid permeable chelator.37
Analysis of iron and 6-OHDA changes to iron response proteins
For investigating compound effect on iron response protein expression after iron loading, SH-SY5Y's prepared 6-well plates, were incubated in serum-free media with FAC (50 μM) for 24 h. After iron loading, iron containing media was replaced with fresh serum-free media alone or in the presence of the compound of interest (100 μM) and incubated for a further 12 h. To evaluate iron response protein expression to each compound on 6-OHDA, a similar procedure to the MTT assay was followed. Therefore, SH-SY5Y's prepared in 6-well plates, were incubated in serum-free media with 6-OHDA (50 μM) with or without the compound (100 μM) for 24 h. After each experimental condition, cells were washed twice with cold PBS, before collection and homogenizing in RIPA buffer (150 mM NaCl, 1% (v/v) Nonidet P-40, 1% (w/v) sodium deoxycholate, 0.1% (v/v) SDS, 25 mM Tris-HCl, pH 7.6) with EDTA-free protease inhibitor cocktail (Complete; Roche). Lysates were clarified by centrifugation at 14000 × g for 15 min.
Western blot analysis
As determined by BCA assay, 10 μg protein from each experimental condition was separated either on 10% PAGE (Tris-Glycine, BioRad) for 22C11 and TfR or 4–20% PAGE (Tris-Glycine, BioRad) for FT, TH and Syn. Resolved proteins transferred to polyvinylidene difluoride membranes (Hybond-P, Pierce) were probed with mouse anti-APP (1:1000, 22C11, in house), mouse anti-TfR (1:2000, H68.4, Invitrogen), rabbit anti-Ferritin (1:1000, Cell Signaling Technology), mouse anti-TH (1:1000, AB152, Millipore) or mouse anti-Syn (1:1000, D35E4, Cell Signaling Technology) and the appropriate secondary antibodies. The load control was mouse anti-β-actin (1:5000, AC15, Sigma). Proteins of interest were visualized with ECL (Pierce) and a LAS-3000 Imaging Suite, and analyzed using Multi Gauge (Fuji). Densitometry using Image J (NIH) was performed in triplicate on 3 separate experiments. All quantitation was standardized against β-actin levels.
Acknowledgements
We are grateful to Prof. Colin Masters for providing access to the 22C11 antibody, and to Dr Ross Harrington (Newcastle University) for obtaining the X-ray crystal structure of acid 9. Work carried out in Leeds was supported by Parkinson's UK, Alzheimer's Research UK and the European Research Council. We thank the Healthcare Infection Society and Northumbria University for funding two PhD studentships (D. G. W. and M. H.). We also thank Northumbria University for the award of an Anniversary Research Fellowship to F. W. L.Notes and references
- D. Berg, M. Gerlach, M. B. Youdim, K. L. Double, L. Zecca, P. Riederer and G. Becker, J. Neurochem., 2001, 79, 225–236 CrossRef CAS PubMed.
- D. T. Dexter, A. Carayon, F. Javoy-Agid, Y. Agid, F. R. Wells, S. E. Daniel, A. J. Lees, P. Jenner and C. D. Marsden, Brain, 1991, 114, 1953–1975 CrossRef PubMed.
- H. Y. Zhang, N. D. Wang, N. Song, H. M. Xu, L. M. Shi, H. Jiang and J. X. Xie, BioMetals, 2013, 26, 705–714 CrossRef CAS PubMed.
- B. A. Faucheux, M. E. Martin, C. Beaumont, S. Hunot, J. J. Hauw, Y. Agid and E. C. Hirsch, J. Neurochem., 2002, 83, 320–330 CrossRef CAS PubMed.
- C. B. Carroll, M. L. Zeissler, N. Chadborn, K. Gibson, G. Williams, J. P. Zajicek, K. E. Morrison and C. O. Hanemann, Neurochem. Int., 2011, 59, 73–80 CrossRef CAS PubMed.
- P. G. Mastroberardino, E. K. Hoffman, M. P. Horowitz, R. Betarbet, G. Taylor, D. Cheng, H. M. Na, C. A. Gutekunst, M. Gearing, J. Q. Trojanowski, M. Anderson, C. T. Chu, J. Peng and J. T. Greenamyre, Neurobiol. Dis., 2009, 34, 417–431 CrossRef CAS PubMed.
- J. R. Cannon and J. T. Greenamyre, Prog. Brain Res., 2010, 184, 17–33 CAS.
- D. Berg, Neurochem. Res., 2007, 32, 1646–1654 CrossRef CAS PubMed.
- T. J. Sobotka, P. Whittaker, J. M. Sobotka, R. E. Brodie, D. Y. Quander, M. Robl, M. Bryant and C. N. Barton, Physiol. Behav., 1996, 59, 213–219 CrossRef CAS PubMed.
- K. E. Goodwill, C. Sabatier and R. C. Stevens, Biochemistry, 1998, 37, 13437–13445 CrossRef CAS PubMed.
- Z. I. Alam, A. Jenner, S. E. Daniel, A. J. Lees, N. Cairns, C. D. Marsden, P. Jenner and B. Halliwell, J. Neurochem., 1997, 69, 1196–1203 CrossRef CAS PubMed.
- M. Hashimoto, L. J. Hsu, Y. Xia, A. Takeda, A. Sisk, M. Sundsmo and E. Masliah, NeuroReport, 1999, 10, 717–721 CrossRef CAS PubMed.
- N. Ostrerova-Golts, L. Petrucelli, J. Hardy, J. M. Lee, M. Farer and B. Wolozin, J. Neurosci., 2000, 20, 6048–6054 CAS.
- R. J. Castellani, S. L. Siedlak, G. Perry and M. A. Smith, Acta Neuropathol., 2000, 100, 111–114 CrossRef CAS PubMed.
- T. Moos, T. Rosengren Nielsen, T. Skjorringe and E. H. Morgan, J. Neurochem., 2007, 103, 1730–1740 CrossRef CAS PubMed.
- S. Bolognin, D. Drago, L. Messori and P. Zatta, Med. Res. Rev., 2009, 29, 547–570 CrossRef CAS PubMed.
- R. C. Hider, S. Roy, Y. M. Ma, X. L. Kong and J. Preston, Metallomics, 2011, 3, 239–249 RSC.
- D. S. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2005, 57, 547–583 CrossRef CAS PubMed.
- M. Amélia Santos, S. M. Marques and S. Chaves, Coord. Chem. Rev., 2012, 256, 240–259 CrossRef.
- Z. D. Liu and R. C. Hider, Coord. Chem. Rev., 2002, 232, 151–171 CrossRef CAS.
- S. Konetschny-Rapp, G. Jung, K. N. Raymond, J. Meiwes and H. Zaehner, J. Am. Chem. Soc., 1992, 114, 2224–2230 CrossRef CAS.
- D. Ben-Shachar, G. Eshel, P. Riederer and M. B. Youdim, Ann. Neurol., 1992, 32(suppl), S105–S110 CrossRef CAS PubMed.
- D. T. Dexter, S. A. Statton, C. Whitmore, W. Freinbichler, P. Weinberger, K. F. Tipton, L. Della Corte, R. J. Ward and R. R. Crichton, J. Neural Transm., 2011, 118, 223–231 CrossRef CAS PubMed.
- D. F. Tardiff, M. L. Tucci, K. A. Caldwell, G. A. Caldwell and S. Lindquist, J. Biol. Chem., 2012, 287, 4107–4120 CrossRef CAS PubMed.
- P. A. Adlard, L. Bica, A. R. White, M. Nurjono, G. Filiz, P. J. Crouch, P. S. Donnelly, R. Cappai, D. I. Finkelstein and A. I. Bush, PLoS One, 2011, 6, e17669 CAS.
- P. J. Crouch, M. S. Savva, L. W. Hung, P. S. Donnelly, A. I. Mot, S. J. Parker, M. A. Greenough, I. Volitakis, P. A. Adlard, R. A. Cherny, C. L. Masters, A. I. Bush, K. J. Barnham and A. R. White, J. Neurochem., 2011, 119, 220–230 CrossRef CAS PubMed.
- S. Bandyopadhyay, X. Huang, H. Cho, N. H. Greig, M. B. Youdim and J. T. Rogers, J. Neural Transm., Suppl., 2006, 237–247 CAS.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637–657 RSC.
- A. Gaeta, F. Molina-Holgado, X. L. Kong, S. Salvage, S. Fakih, P. T. Francis, R. J. Williams and R. C. Hider, Bioorg. Med. Chem., 2011, 19, 1285–1297 CrossRef CAS PubMed.
- D. Devos, C. Moreau, J. C. Devedjian, J. Kluza, M. Petrault, C. Laloux, A. Jonneaux, G. Ryckewaert, G. Garcon, N. Rouaix, A. Duhamel, P. Jissendi, K. Dujardin, F. Auger, L. Ravasi, L. Hopes, G. Grolez, W. Firdaus, B. Sablonniere, I. Strubi-Vuillaume, N. Zahr, A. Destee, J. C. Corvol, D. Poltl, M. Leist, C. Rose, L. Defebvre, P. Marchetti, Z. I. Cabantchik and R. Bordet, Antioxid. Redox Signaling, 2014, 21, 195–210 CrossRef CAS PubMed.
- J. Xu, P. W. Durbin, B. Kullgren, S. N. Ebbe, L. C. Uhlir and K. N. Raymond, J. Med. Chem., 2002, 45, 3963–3971 CrossRef CAS PubMed.
- R. Burgada, T. Bailly, J. P. Noël, J. M. Gomis, A. Valleix, E. Ansoborlo, M. H. Hengé-Napoli, F. Paquet and P. Gourmelon, J. Labelled Compd. Radiopharm., 2001, 44, 13–19 CrossRef CAS.
- D. Liu, R. Chen, L. Hong and M. J. Sofia, Tetrahedron Lett., 1998, 39, 4951–4954 CrossRef CAS.
- R. C. Scarrow, P. E. Riley, K. Abu-Dari, D. L. White and K. N. Raymond, Inorg. Chem., 1985, 24, 954–967 CrossRef CAS.
- A. Landa, A. Minkkila, G. Blay and K. A. Jorgensen, Chem. – Eur. J., 2006, 12, 3472–3483 CrossRef CAS PubMed.
- L. M. Garrick, D. M. Green, D. B. Hauze, K. L. Kees, J. T. Lundquist IV, C. W. Mann, J. F. Mehlmann, J. C. Pelletier, J. F. Rogers Jr and J. E. Wrobel, US2006/0019965A1, 2006.
- H. Glickstein, R. B. El, G. Link, W. Breuer, A. M. Konijn, C. Hershko, H. Nick and Z. I. Cabantchik, Blood, 2006, 108, 3195–3203 CrossRef CAS PubMed.
- O. Weinreb, S. Mandel, M. B. Youdim and T. Amit, Free Radical Biol. Med., 2013, 62, 52–64 CrossRef CAS PubMed.
- H. Jiang, N. Song, H. Xu, S. Zhang, J. Wang and J. Xie, Cell Res., 2010, 20, 345–356 CrossRef CAS PubMed.
- S. Wakita, Y. Izumi, T. Matsuo, T. Kume, Y. Takada-Takatori, H. Sawada and A. Akaike, J. Neurosci. Methods, 2010, 192, 83–89 CrossRef CAS PubMed.
- A. E. Gorden, J. Xu, K. N. Raymond and P. Durbin, Chem. Rev., 2003, 103, 4207–4282 CrossRef CAS PubMed.
- J. Xu, D. G. Churchill, M. Botta and K. N. Raymond, Inorg. Chem., 2004, 43, 5492–5494 CrossRef CAS PubMed.
- I. Turcot, A. Stintzi, J. Xu and K. N. Raymond, JBIC, J. Biol. Inorg. Chem., 2000, 5, 634–641 CrossRef CAS PubMed.
- E. G. Moore, J. Xu, C. J. Jocher, E. J. Werner and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 10648–10649 CrossRef CAS PubMed.
- R. C. Hider and Z. D. Liu, Curr. Med. Chem., 2003, 10, 1051–1064 CrossRef CAS PubMed.
- T. Zhou, Y. Ma, X. Kong and R. C. Hider, Dalton Trans., 2012, 41, 6371–6389 RSC.
- Z. D. Liu and R. C. Hider, Med. Res. Rev., 2002, 22, 26–64 CrossRef CAS PubMed.
- Y. J. Li and A. E. Martell, Inorg. Chim. Acta, 1993, 214, 103–111 CrossRef CAS.
- R. C. Scarrow, P. E. Riley, K. Abu-Dari, D. L. White and K. N. Raymond, Inorg. Chem., 1985, 24, 954–967 CrossRef CAS.
- D. W. Whisenhunt Jr., M. P. Neu, Z. Hou, J. Xu, D. C. Hoffman and K. N. Raymond, Inorg. Chem., 1996, 35, 4128–4136 CrossRef.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- Z. D. Liu, M. Lockwood, S. Rose, A. E. Theobald and R. C. Hider, Biochem. Pharmacol., 2001, 61, 285–290 CrossRef CAS PubMed.
- M. B. Youdim, Exp. Neurobiol., 2010, 19, 1–14 CrossRef PubMed.
- W. J. Geldenhuys, M. B. Youdim, R. T. Carroll and C. J. Van der Schyf, Prog. Neurobiol., 2011, 94, 347–359 CrossRef CAS PubMed.
- M. Nechab, D. N. Kumar, C. Philouze, C. Einhorn and J. Einhorn, Angew. Chem., Int. Ed., 2007, 46, 3080–3083 CrossRef CAS PubMed.
- A. Puszko and Z. Talik, Pol. J. Chem., 1992, 66, 1427–1430 CAS.
- O. Kakhlon and Z. I. Cabantchik, Free Radical Biol. Med., 2002, 33, 1037–1046 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data collection and structural refinement details for acid 9. CCDC 983801. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4mt00326h |
| This journal is © The Royal Society of Chemistry 2015 |