DOI:
10.1039/C4MT00304G
(Critical Review)
Metallomics, 2015,
7, 730-742
The role of vanadium in biology
Received
24th November 2014
, Accepted 9th January 2015
First published on 13th January 2015
Abstract
Vanadium is special in at least two respects: on the one hand, the tetrahedral anion vanadate(V) is similar to the phosphate anion; vanadate can thus interact with various physiological substrates that are otherwise functionalized by phosphate. On the other hand, the transition metal vanadium can easily expand its sphere beyond tetrahedral coordination, and switch between the oxidation states +V, +IV and +III in a physiological environment. The similarity between vanadate and phosphate may account for the antidiabetic potential of vanadium compounds with carrier ligands such as maltolate and picolinate, and also for vanadium's mediation in cardiovascular and neuronal defects. Other potential medicinal applications of more complex vanadium coordination compounds, for example in the treatment of parasitic tropical diseases, may also be rooted in the specific properties of the ligand sphere. The ease of the change in the oxidation state of vanadium is employed by prokarya (bacteria and cyanobacteria) as well as by eukarya (algae and fungi) in respiratory and enzymatic functions. Macroalgae (seaweeds), fungi, lichens and Streptomyces bacteria have available haloperoxidases, and hence enzymes that enable the 2-electron oxidation of halide X− with peroxide, catalyzed by a Lewis-acidic VV center. The X+ species thus formed can be employed to oxidatively halogenate organic substrates, a fact with implications also for the chemical processes in the atmosphere. Vanadium-dependent nitrogenases in bacteria (Azotobacter) and cyanobacteria (Anabaena) convert N2 + H+ to NH4+ + H2, but are also receptive for alternative substrates such as CO and C2H2. Among the enigmas to be solved with respect to the utilization of vanadium in nature is the accumulation of VIII by some sea squirts and fan worms, as well as the purport of the nonoxido VIV compound amavadin in the fly agaric.

Dieter Rehder
| Dieter Rehder studied chemistry and astronomy at the University of Hamburg, Germany. After receiving his PhD in chemistry, he lectured at the College for Tobacco Technology and Bio-Engineering in Hamburg (until 1973), and at the College of Arts Science & Technology in Kingston/Jamaica (1973–1975). After returning, he became habilitated and Full Professor (1984) in Hamburg. His main fields of attention are Organometallic Chemistry, NMR Spectroscopy (metal nuclei), Bioinorganic Chemistry (vanadium), and Exoplanets/Interstellar Chemistry. He received the Vanadis Award for outstanding research in vanadium chemistry in 2006. In 2008/2009, he was guest lecturer at the University of Lund (Sweden). |
1. General and background
Earth's crust, plus the water reservoirs and the atmosphere, contain an average of 135 ppm vanadium (exceeding the vanadium concentration in the Universe by a factor of ca. 135). This makes vanadium the 21st most abundant element in the outer regions of our planet. Generally, vanadium is rather dissipated, i.e. vanadium-based minerals are comparatively rare. A “famous” representative is vanadinite, a lead orthovanadate of the formula PbCl2·3Pb3(VO4)2 that is closely linked to the discovery of vanadium by Andrés Manuel del Río y Fernandez in Mexico in 1801.1 Enrichment of vanadium has been observed in soils and rocks in volcanic areas, and in crude oil, oil-shales, asphalts, peat, and bitumen. In crude oil, vanadium contents – with vanadium present in the form of VO2+-porphyrins – can go up to 0.12%. This accumulation of vanadium goes back to the extraction of VO2+ (“vanadyl”) from shale that is being pervaded by kerosene. In coal bottom ash, total vanadium concentrations can go up to 0.7 g per kg of dry weight.2 The occurrence of “fossil” vanadium poses potential environmental and health problems, in as far as burning coal and oil produces vanadium oxides that become absorbed to dust particles. As detailed below, vanadium oxides can cause health hazards; furthermore, vanadium oxides are powerful catalysts in the oxidation of, for example, SO2 to SO3 (and hence sulfuric acid).
Seawater contains vanadium in the form of ion pairs Na+H2VO4− at a concentration typically between 30 and 35 nM, making vanadium the second to the most abundant transition metal in the oceans, overtopped only by molybdenum in the form of molybdate MoO42− at concentrations of 100 nM. This comparatively high abundance of vanadate(V) in seawater does have consequences with respect to utilization of vanadium by macro-algae, and thus indirectly also for a role of vanadium in the global ozone balance – as will be detailed later in this review. Other marine organisms, namely sea squirts (ascidians) and some fan worms, also make recourse to vanadate, although without any apparent biological benefit.
The average vanadium concentration in fresh water, ground water and potable water is 10 nM, with peak concentrations in volcanic areas going up to 2.5 μM. The average vanadium content in edibles, where vanadium is mainly present in the form of the vanadyl species, amounts to 5–30 μg kg−1. Under ambient conditions, the daily oral intake of vanadium via potable water and food varies between 10 μg and 2 mg. This is clearly beyond the no-effect level of 10 mg vanadium per day and per kg body mass.
After an oral uptake of vanadium compounds, speciation occurs by the saliva, in the stomach and in the intestinal tract. The main part of vanadium is thus converted to sparingly soluble VO(OH)2, most of which is excreted via the faeces, minimizing or even excluding adverse effects that otherwise might be caused by unphysiologically high vanadium levels. Vanadate(V) is more easily resorbed than the soluble vanadyl species, and this can principally cause health problems, for example in households with lead water pipe systems, where drinking water enriched with phosphate can contain appreciable amounts of vanadate: phosphate remobilizes vanadate from otherwise insoluble lead vanadate deposited in the wall of the pipe system.3 As noted above, an additional source for vanadium intake are aerosols in the breathing air. In urban areas, vanadium contents in the breathing air can go up to 103 ng m−3 of vanadium, and hence two to three orders of magnitude more than in rural areas. Incomplete combustion of fossil fuels is a major source of vanadium oxides absorbed to particulate matter in the air. In the alveoli of the lung, vanadium oxides VOx can be converted (oxidatively) to vanadate and thus become resorbed. In addition, direct pulmonary problems can result from high aerial loads of VOx,4a a main issue for labourers exposed to excessive aerial VOx concentrations at the working place. The maximum allowable concentration (MAC) of V2O5 at the workplace has been set to 0.05 mg m−3. For a recent review of the direct and indirect toxicity of V2O5, see Fortoul et al.4b
Once in the bloodstream, vanadium – in the form of vanadate and vanadyl – binds to serum proteins, in particular to transferrin,5a an issue that originally was attended to by Chasteen.5b,c Vanadium is then distributed to the tissues of the inner compartment (heart, liver, and kidney) and the outer compartment (brain, muscle, adipose tissues). All in all, vanadium contents in the blood are reduced to about 30% within one day.6 Bone, however, and to some extent also the kidneys, can hold back vanadate/vanadyl. In the apatite of the bones, vanadate can substitute for phosphate; here, the half-life of vanadium amounts to about 5 days.7 Elimination of resorbed vanadium occurs via the urinary tract; in the kidneys; VO2+ can be retained intermittently, for example by coordination to dangling NH2 groups of proteins.
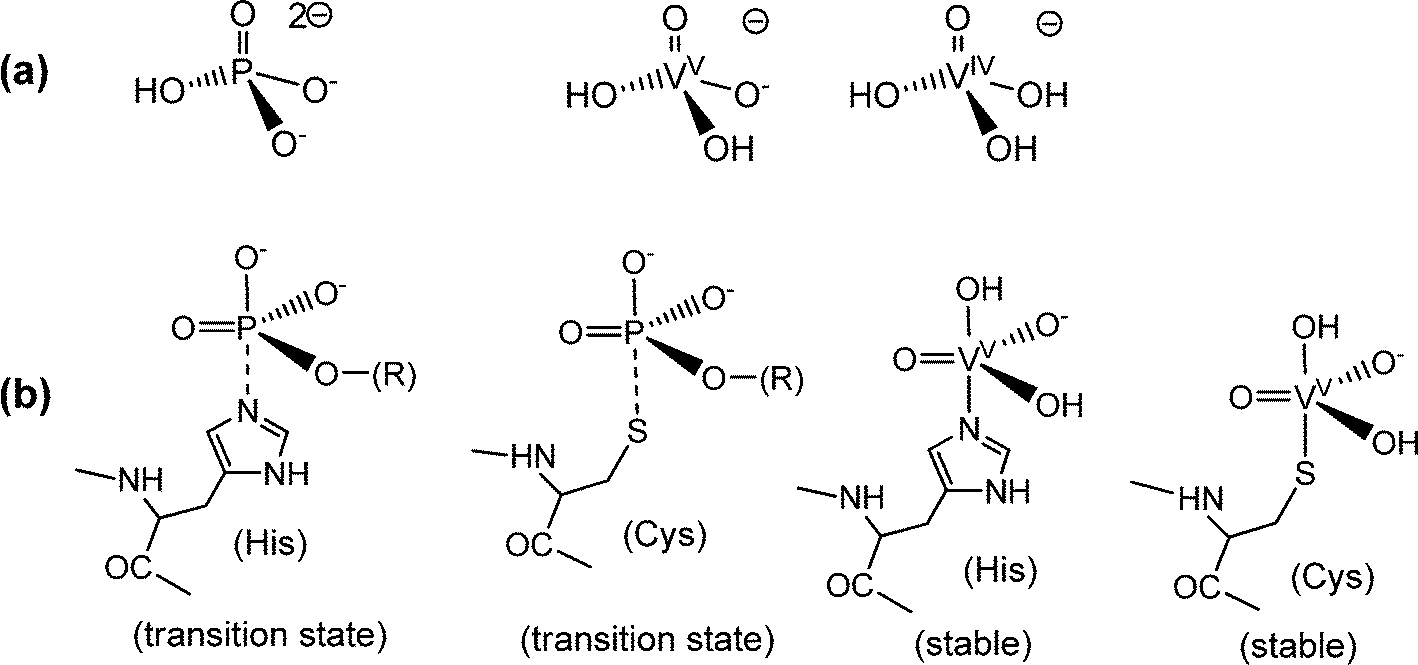
The ability of vanadate to substitute for phosphate in apatite points to a pronounced similarity between vanadate and phosphate (Fig. 1a), and hence towards a possible general role of vanadate in physiological processes involving phosphate. The tetrahedral anions vanadate and phosphate are in fact structural analogs of almost the same size, with a volume of the circumscribing spheres of 125 and 102 Å3, respectively.8 Vanadate can thus easily substitute for phosphate in enzymes such as phosphatases and kinases. But this fact also implies distinct differences between vanadate and phosphate: vanadium in vanadate easily attains the stable coordination number five – commonly in a somewhat distorted trigonal-bipyramidal coordination environment (Fig. 1b) – while penta-coordination is just a transition state in the case of phosphate. Consequently, the replacement of phosphate for vanadate results in the inhibition of the enzyme, as initially documented by Chasteen5b and established for the inhibition of a Na,K-ATPase by Cantley almost four decades ago.9 With an association constant of 2.4 × 108 M−1, vanadate forms a stable five-coordinate complex with the ATPase, involving bidentate binding of an aspartate carboxylate. An additional feature that distinguishes phosphate and vanadate at physiologically relevant concentrations is the protonation state at about neutral pH: at pH 7, vanadate is almost exclusively present in its diprotonated form, eqn (1a), while phosphate exists as a mix of mono- and dihydrogenphosphate, eqn (1b). Further, in contrast to phosphate, vanadate(V) is susceptible to reduction to oxidovanadium(IV), eqn (2), and vanadium(III).
| | | H2VO4− ⇆ HVO42− + H+ pKa = 8.2 | (1a) |
| | | H2PO4− ⇆ HPO42− + H+ pKa = 6.7 | (1b) |
| | | H2VO4− + 4H+ + e− ⇆ VO2+ + 3H2O EpH=7 = −0.34 V | (2) |
The most common oxidation states of vanadium in biological systems are the +V (d0) and +IV (d1) states, but the +III (d2) is also principally available and has been shown to be realized in the final storage form of vanadium in ascidians (Section 2). In vanadium-dependent nitrogenase (Section 4.1), supposedly the +II state also comes in; this oxidation state of vanadium is otherwise unstable at physiological and environmental conditions. In close relationship to the ease of redox interconversion between vanadium(V) and -(IV), vanadium can play a crucial role in balancing the level of reactive oxygen species. As examples, the generation of the hydroxo radical as initiated by VO2+, and the elimination of the superoxide by vanadate, are depicted in eqn (3).
| | | VO2+ + H2O2 + 3H2O → H2VO4− + OH− + ˙OH + 4H+ | (3a) |
| | | H2VO4− + 4H+ + ˙O2− → VO2+ + O2 + 3H2O | (3b) |
2. Unspecific accumulation of vanadium compounds in living organisms
Three groups of organisms have so far been identified that accumulate vanadium without any apparent benefit, viz. (i) several Amanita mushrooms such as the fly agaric, (ii) marine polychaeta fan worms, and (iii) ascidians. In ascidians, specialized blood cells termed vanadocytes take up vanadium; the highest vanadium contents have been found in Ascidia gemmata, with vanadium concentrations of up to 350 mM – hence enrichment from seawater by a factor of 107! The role of vanadium is obscure; since vanadium is toxic at higher concentrations, the accumulation in ascidians (and some fan worms) might thus hint towards being poisonous for potential predators. In any case, vanadium does not take over a role in oxygen transfer, as originally suggested, and was coined by the term hemovanadin, by Henze, who was the first to provide evidence for the accumulation of vanadium in the blood of ascidians more than a century ago,13a an area that was later pioneered by Kustin13b,c and, more recently, by Michibata et al. (vide infra).
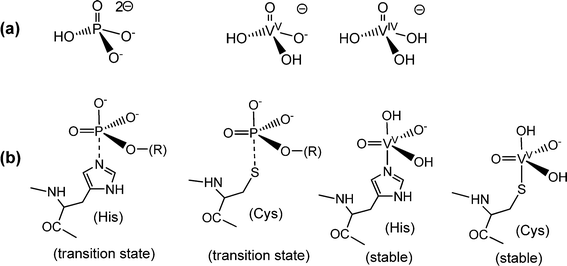 |
| | Fig. 1 Analogies and differences between phosphate and vanadates. (a) The predominant protonation states of phosphate and of the vanadates(V) and -(IV) at neutral pH. Due to the low solubility of VO(OH)2, dissolved vanadate(IV) H3VO4− (the coordination sphere of VIV can be expanded by aqua ligands) is restricted to the nanomolar concentration range. (b) Penta-coordinate species evolving from the interaction between phosphate (and phosphate esters) or vanadate with peptide–protein residues: phosphate forms labile transition states only, symbolized here by a dashed P⋯N bond to histidine or a P⋯S bond to cysteinate (serinate – not shown – is a third alternative), while vanadate ascertains stable complexes. Examples are the binding of vanadate to a histidine residue in vanadate-dependent haloperoxidases (Section 4.2) and in rat prostate acid phosphatase,10a and the coordination of vanadate to a cysteinate residue in phosphotyrosyl phosphatase.10b This coordination mode has also been invoked for the inhibitory effect of vanadate towards intracellular protein tyrosine phosphatase in the context of the insulin enhancing properties of vanadate (Section 5).11 The hydroxide in the apical position can be replaced by, for example, tyrosinate. For an overview of structural details, see Crans et al.12 | |
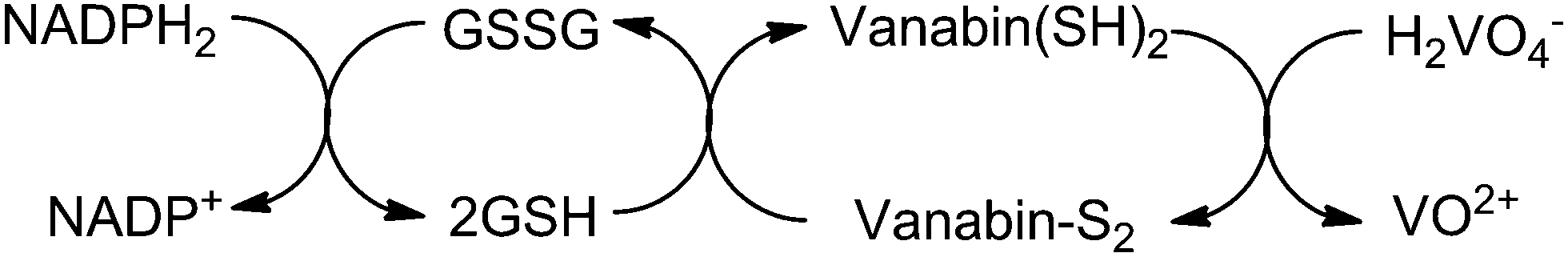
Vanadium enters the ascidians, assisted by Na+-dependent phosphate transporters, as vanadate(V) HnVO4(3−n)− (n = 2, 1), which is then reduced to V(IV) (VO2+) and further to V(III), the storage form in the vanadocytes, essentially [V(H2O)5HSO4]2+/[V(H2O)6]3+. Several key proteins are related to the accumulation and reduction of vanadium, among these so-called vanabins. Vanabins are low molecular mass proteins rich in cysteinyl residues. A well investigated representative is vanabin2, isolated from Ascidia sydneiensis samea. Vanabin 2 is a 13.2 kDa protein consisting of 120 amino acids,14 18 of which are cysteines that form nine disulfide bonds. Vanabin2 acts as a reductase for vanadate (Scheme 1) and intermittent store for vanadyl VO2+. The reductase activity has been traced back to cysteine residues,15 the binding of VO2+ is associated with dangling amino groups provided by lysine and arginine residues. The final reduction step (VO2+ → V3+) so far remains obscure.
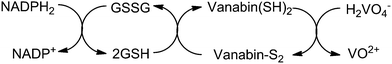 |
| | Scheme 1 The reduction of vanadate(V) as catalyzed by vanabin2: in a first step, the reduced from of nicotine-adenine dinucleotide, NADPH2, reduces glutathione disulfide GSSG to glutathione GSH. In a second step, the disulfide form of vanabin2 is reduced to the thiol form which, in the final step, reduces vanadate to vanadyl VO2+. Based on ref. 15. | |
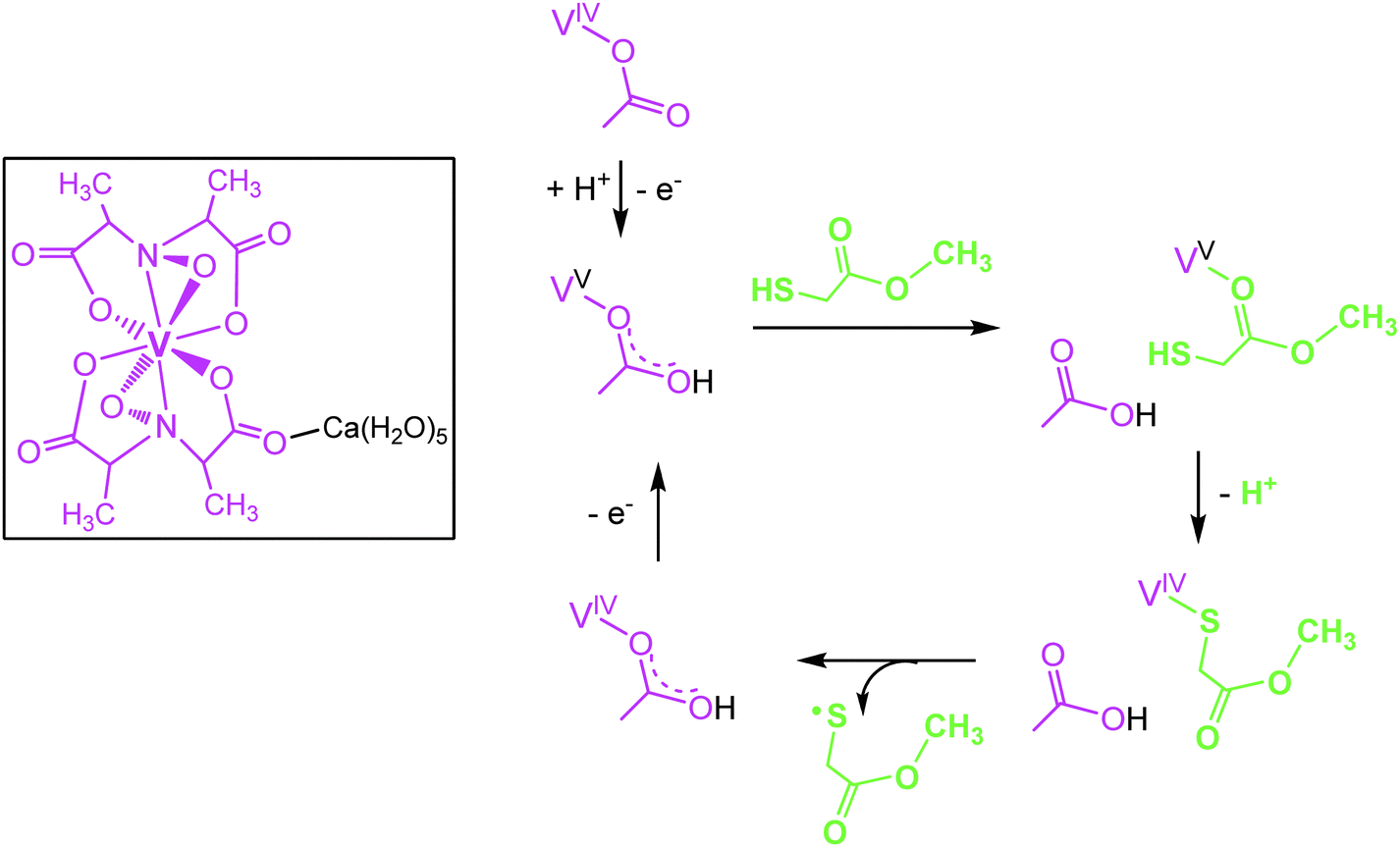
Several representatives of the genus Amanita contain a non-oxido vanadium(IV) complex Ca(H2O)5[Δ-V(S,S-hidpa)2] termed amavadin (Fig. 2), in which vanadium is octa-coordinated to two hidpa3− ligands, where hidpa3− is short for N-hydroxyimino-2,2′diisopropionate(3−). The bulb and the lamella of the mushroom are particularly rich in vanadium; in A. muscaria, the fly agaric (also known as toadstool), vanadium levels can go up to 1 g amavadin per kg of dry mass, hence an enrichment by a factor of 103 to 104 with respect to the average vanadium concentrations in soil. The biological function of amavadin is elusive; it has been proposed that amavadin is an evolutionary overcome relic of an oxidase or peroxidase. The amavadin anion in its oxidized (vanadium(V)) form, [Δ-V(S,S-hidpa)2]−, can in fact act as a peroxidase and, in the absence of a substrate, as a catalase.16 The catalase activity is depicted in eqn (4), the peroxidase activity in the presence of thiols, resulting in the formation of disulfides, in eqn (5). In eqn (4) and (5), the reduced and oxidized forms of amavadin are symbolized by {VIV} and {VV}. Mechanistically, the one-electron oxidation of thiols, such as mercapto-methylacetate, likely proceeds via a protonated VV and a VIV–thiol intermediate17 as exemplified in Fig. 2. Interestingly, the protonated VV form of amavadin, H+[V(hidpa)2]−, also exerts bromoperoxidase activity,18 an ability otherwise restricted to naturally occurring vanadate-dependent bromoperoxidases; Section 4.2.
| | | 2{VV} + 2H2O2 → 2{VIV} + 1½O2 + H2O + 2H+ | (4) |
| | | {VV} + RSH → {VIV} + ½R-S-S-R + H+ | (5) |
3. Vanadium in bacterial respiration
A whole array of bacteria can employ vanadium in varying biological functions.19a Well documented examples are the bacterial vanadium-dependent nitrogenases and haloperoxidases (both enzymes will be dealt with in detail in Section 4), and bacteria that resort to vanadate(V) as an electron acceptor in respiration. Vanadium has also been reported to optionally replace molybdenum in the periplasmatic nitrate reductase of Pseudomonas isachenkovii:19b nitrate reductases catalyze the two-electron reduction of nitrate to nitrite.
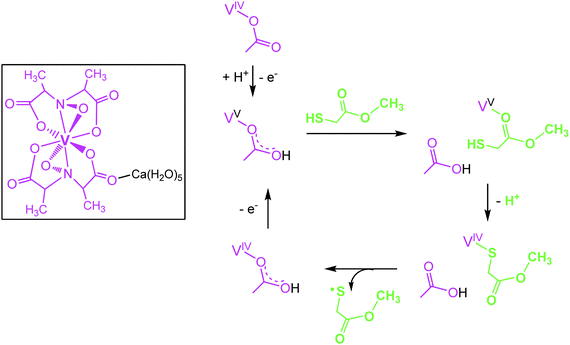 |
| | Fig. 2 The vanadium compound amavadin (framed) from the fly agaric (Amanita muscaria), and intermediate steps in the oxidation of mercapto-methylacetate (green), catalyzed by amavadin (purple) as suggested by DFT calculations (from ref. 17; modified). | |
The facultative anaerobic chemolitotrophic bacterium P. isachenkovii reduces vanadate, primarily to VO2+ in the form of sparingly soluble VO(OH)2, resorting to, inter alia, H2 or CO as electron donors in vanadate reduction, eqn (6a) and (6b).20 Other bacterial strains can equally use vanadate as an electron acceptor in respiratory and/or dissimilatory reduction. Respiratory reduction refers to a process coupled to proton translocation and thus ATP generation and growth, while dissimilatory reduction occurs without a proton motive force. Specific strains of Shewanella oneidensis, a Gram-negative soil bacterium, utilizes lactate and formate as electron sources in respiratory reduction,21eqn (7), while Vibrio parahaemolyticus, thriving in brackish salt water and causing gastrointestinal dysfunctions when ingested, promotes dissimilatory vanadate reduction by making recourse to glycerol or formate.22 The strict anaerobe Geobacter metallireducens effectively employs metal ions and metalates with the metal in a high oxidation state (including vanadate,23eqn (8)) as electron acceptors in the oxidation of organics such as fatty acids and alcohols, and thus contributes to the detoxification of ground water with high metal ion loads. Finally, P. isachenkovii, as well as vanadate-respiring anaerobic bacteria isolated from deep sea hydrothermal vents,24 have been shown to partially reduce vanadate(V) to vanadium(III), eqn (9).
| | | H2VO4− + ½H2 + H+ → VO(OH)2 + H2O | (6a) |
| | | 2H2VO4− + CO + 2H+ → 2VO(OH)2 + CO2 + H2O | (6b) |
| | | 2H2VO4− + HCO2− + 3H+ → 2VO(OH)2 + CO2 + 2H2O | (7) |
| | | 4H2VO4− + CH3CO2− + 5H+ → 4VO(OH)2 + 2CO2 + 4H2O | (8) |
| | | H2VO4− + HCO2− + 2H+ → V(OH)3 + CO2 + H2O | (9) |
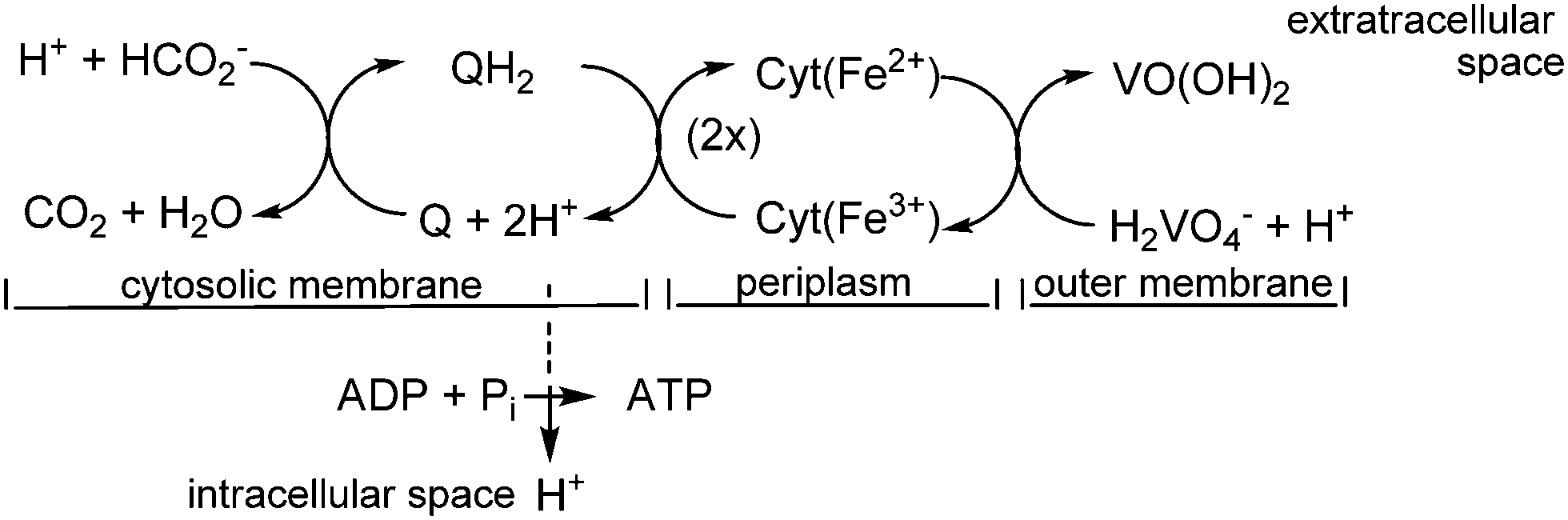
In the case of S. oneidensis, a terminal vanadate reductase associated with the outer membrane of the organism catalyzes the reduction of vanadate to vanadyl. VO(OH)2 becomes deposited mainly in the periplasm and at the outer membrane. The electrons for the reduction of H2VO4− to VO2+ are commonly delivered through the oxidation – in the cytosolic membrane – of lactate to pyruvate, or of formate to CO2. The electrons are then shuttled to the outer membrane by cytochrome c type haem proteins across the periplasmatic space, and finally to membrane-associated vanadate. Scheme 2 provides a simplified picture of this situation.
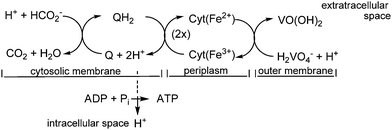 |
| | Scheme 2 A simplified view of the electron transfer across the cellular membrane, starting with the oxidation of formate (cytosolic membrane), and terminating with the reduction of vanadate at the outer membrane. Q/QH2 is menaquinone/-hydroquinone. The electron transport from the inner (the cytosolic) to the outer membrane is accomplished via a cascade of cytochrome c type haem proteins. Vanadium ends up in a mineralized form based on VIVO(OH)2. The H+ transport into the intracellular space is coupled to the formation of energy-rich adenosine triphosphate (ATP) from ADP and inorganic phosphate HPO42− (Pi). | |
In addition to bacteria, mesophilic and thermophilic methanogenic archaea such as Methanothermobacter thermautotrophicus can also reduce VV to VIV, diverting the electron transfer in such a way that methanogenesis is inhibited.25 The reduction of vanadate by prokarya, resulting in the formation of vanadyl (and, in part, also of VIII) has likely contributed – and still contributes – to the mineralization of soluble vanadate. Minerals such as häggite VIIIO(OH)·VIVO(OH)2, simplotite CaVIV4O9 and sherwoodite Ca9Al2VIV4VV24O80·xH2O are examples.
4. Nitrogenases and haloperoxidases
To date, the only indubitably established naturally occurring vanadium-dependent enzymes are either haloperoxidases or nitrogenases. Vanadium nitrogenases VNases can be present, along with and iron-only nitrogenases and the (phylogenetically younger and far more prevalent) molybdenum nitrogenases, in bacterial strains belonging to the genus Azotobacter and in cyanobacteria of the genera Anabaena and Nostoc. The vanadium-based Azotobacter nitrogenase is more effective than its molybdenum analogue at low temperatures, and is predominantly expressed when Mo is limited. Vanadate-dependent haloperoxidases VHPOs have been found in marine macroalgae, in terrestrial fungi, lichens, Streptomyces bacteria and in cyanobacteria, in part along with heme-dependent peroxidases.26 VHPOs are rather widespread and are directly involved in the utilization of halides in aquatic (essentially marine) environments either for synthesizing halogenated organics or, in the case of chloroperoxidases, either in the organism's defense against bacterial and viral affliction, or to get access to a host. Indirectly, VHPOs are also involved in atmospheric issues, the removal of ozone in particular (Section 4.3).
4.1 Vanadium nitrogenases
Proteobacteria such as the soil bacteria Azotobacter vinelandii and A. chroococcum are equipped with VNase, as are the filamentous cyanobacteria Anabaena nostoc,27A. azotica, and A. variabilis. More recently, a VNase has also been detected in lichens of the genus Peltigera,28 thriving in high latitude ecosystems. These lichens contain cyanobacterial symbionts belonging to the genus Nostoc of the family Nostocaceae, related to Anabaena, another genus that is a member of this specific family of cyanobacteria. VNases catalyze the reduction of N2 to NH4+, thus bio-mimicking the Haber–Bosch synthesis. In the natural system, the reduction of N2 to NH4+ (and commonly some hydrazine) is coupled to the reduction of H+ to H2;29a the overall ATP-driven reaction is represented by eqn (10). Several substrates other than N2 (and H+) can also undergo nitrogenase-catalyzed reduction; examples are CO and acetylene,29beqn (11) and (12), and HCN.29c Acetylene is reduced to ethane, eqn (11), CO to ethene, ethane, propene and propane, eqn (12). The reduction of CO is reminiscent of the Fischer–Tropsch process. The reductive conversion of HCN, formulated non-stoichiometrically in eqn (13), affords several products, among these ammonia, methyleneimine, methylamine and formaldehyde.| | | N2 + 14H+ + 12e− → 2NH4+ + 3H2 | (10) |
| | HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH + 4H+ + 4e− → C2H6 CH + 4H+ + 4e− → C2H6 | (11) |
| | | CO + nH+ + ne− → → C2H6, C2H4, C3H6, C3H8 | (12) |
| | HCN + nH+ + ne− → → NH3, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH, CH3NH2, HCHO NH, CH3NH2, HCHO | (13) |
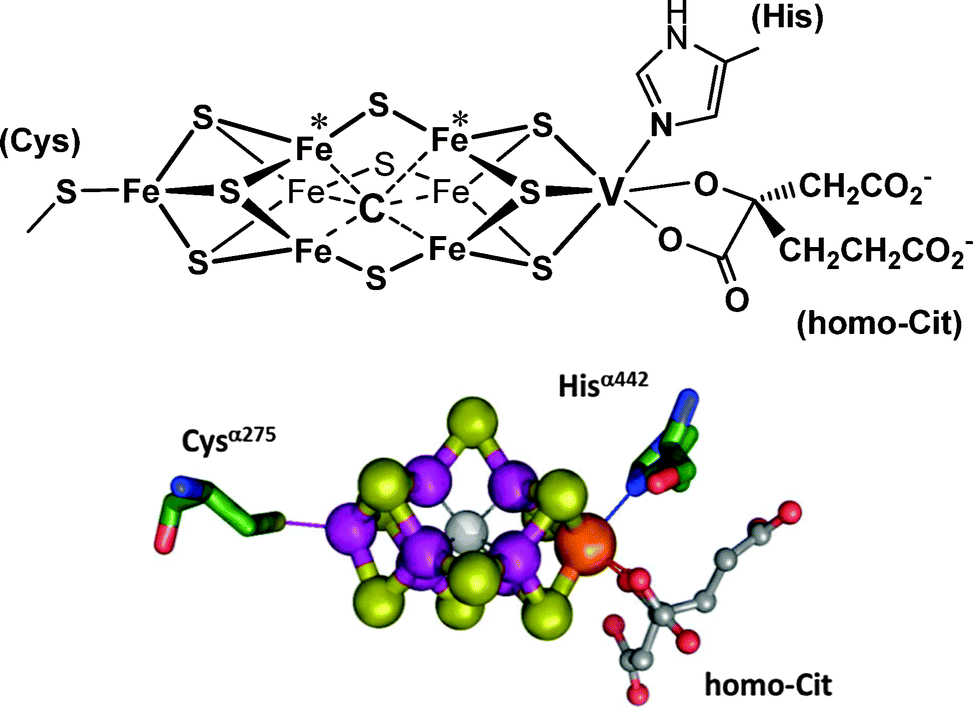
The VFe-protein and the MoFe proteins are biochemically similar, although they differ somewhat in their substructures, viz. α2β2 for the MoFe protein and α2β2δ2 for the VFe-protein. Once isolated from the bacteria, VNases are sufficiently less robust than their molybdenum analogues, and so far direct structural information (i.e. information obtained from X-ray structure analyses) is not available. However, indirect information from various sources (such as extended X-ray absorption, electron paramagnetic resonance EPR, Mößbauer, and magnetic circular dichroism spectroscopies) indicates a buildup similar to that of the Mo-nitrogenases.30 Correspondingly, the central unit of the vanadium–iron protein (Fig. 3) – the so-called M clusters or FeVCo, where the direct reduction of N2 to NH4+ occurs – is a cage system formed by seven iron ions plus one vanadium ion. The metal centres are bridged by nine S2−. Six of the iron centres of the Fe7 cage are additionally linked to an interstitial light atom that, in analogy to the Mo-nitrogenase, supposedly is carbon in the form of carbide μ6-C4−.31 The cluster is connected to the protein matrix via a cysteinate (coordinated to one of the iron centres), and the Nε of a histidine residue, coordinated to vanadium. A homocitrate completes the coordination sphere of vanadium. The EPR spectra of the FeVCo adopt a pattern that is characteristic of a spin S = 3/2 state for the vanadium centre, hence suggesting high-spin V+II.30b
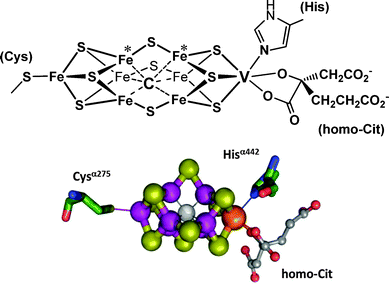 |
| | Fig. 3 The structure of the M cluster {VFe7(μ6-C)(μ2-S)3(μ3-S)6} of vanadium nitrogenase. Schematic view, ball-and-stick representation (adapted with permission from ref. 30b; © J. Am. Chem. Soc.). Potential sites for (side-on) binding of N2 are the iron centres labeled with an asterisk.32 Electrons for the N2 reduction are delivered via an adjacent [4Fe–4S] ferredoxin-type cluster. The complete system is encoded by the vnfHDK genes. | |
4.2 Vanadate-dependent haloperoxidases
Along with co-factor free and a haem-based haloperoxidases, vanadate-dependent haloperoxidases VHPOs represent a third powerful regime for the oxidation, by hydrogen peroxide, of halides. VHPOs are differentiated with respect to their primary halide substrate(s): iodoperoxidases VIPOs oxidize iodide only, bromoperoxidases VBrPOs oxidize iodide and bromide (and also, to some extent, chloride), and chloroperoxidases VClPOs have a sufficiently high oxidation potential to oxidatively attack chloride along with iodide and bromide. In contrast (and somewhat surprisingly), a vanadate-substituted acid phosphatase isolated from the embryonic axes of the kidney bean Phaseolus vulgaris exhibits chloroperoxidases activity, but no bromo- and iodoperoxidases activity.33
Common substrates for marine VHPOs are the halides I−, Br− and Cl−; these ions are present in seawater at average concentrations of 0.47 μM (iodide), 0.82 mM (bromide) and 0.55 M (chloride). Other substrates, the pseudohalides cyanide and thiocyanate in particular, are also oxidized, as are organic compounds such as sulfides R2S. The latter are oxidized to sulfoxides; in the case of prochiral sulfides (RSR′) as substrate, the oxidation occurs enantioselectively. This is noteworthy since the vanadate-based reaction centre itself (vide infra) is non-chiral, suggesting influencing factors that go back to the active site protein pocket. The oxidant is hydrogen peroxide H2O2, the concentration of which in seawater can go up to 0.25 μM, but is otherwise quite variable. All of these reactions are two-electron oxidations. Eqn (14) to (17) exemplify a selection of the oxidation reactions; X− represents a halide. In reactions (14) to (16), protons are consumed; the oxidation of sulfides, eqn (17), affords just catalytic amounts of H+. “HOX” in eqn (14) actually is an equilibrium mixture of HOX, X2 and X3−. In the absence of a substrate, singlet oxygen 1O2 is released.
| | | X− + H2O2 + H+ → HOX + H2O | (14) |
| | | CN− + H2O2 + H+ → HOCN + H2O | (15) |
| | | SCN− + RH + H2O2 + H+ → SCN−R + 2H2O | (16) |
| | | RR′S + H2O2 → RR′SO + H2O | (17) |
The oxidation of halide exemplified by eqn (14) is a generalization in as far as free hypohalous acid HOX (or hypohalite XO−) does not necessarily emerge. Rather, a substrate can be halogenated directly, presumably via the intermediate formation of an “X+” species (such as Br2 or Br3− in the case of X− = bromide).34 For X = Cl, however, hypochlorous acid HOCl is commonly directly employed. VClPOs are predominantly found in Streptomyces bacteria,35 where they serve as chlorinating agents for complex organics, and in terrestrial fungi. Fungi such as Curvularia inaequalis36 can use HOCl to oxidatively degrade the lignocellulose in the cell wall of their “host”, thus allowing access of the fungal hypha to the intracellular space of the host.37 The C. inaequalis VClPO has also been shown to possess antimicrobial activity, for example against the intestinal bacterium Enterococcus faecalis.38a Furthermore, an alkolophilic mutant of the VClPO has a broad antimicrobial activity against Gram-negative and Gram-positive bacteria, and also exhibits virucidal activity.38b
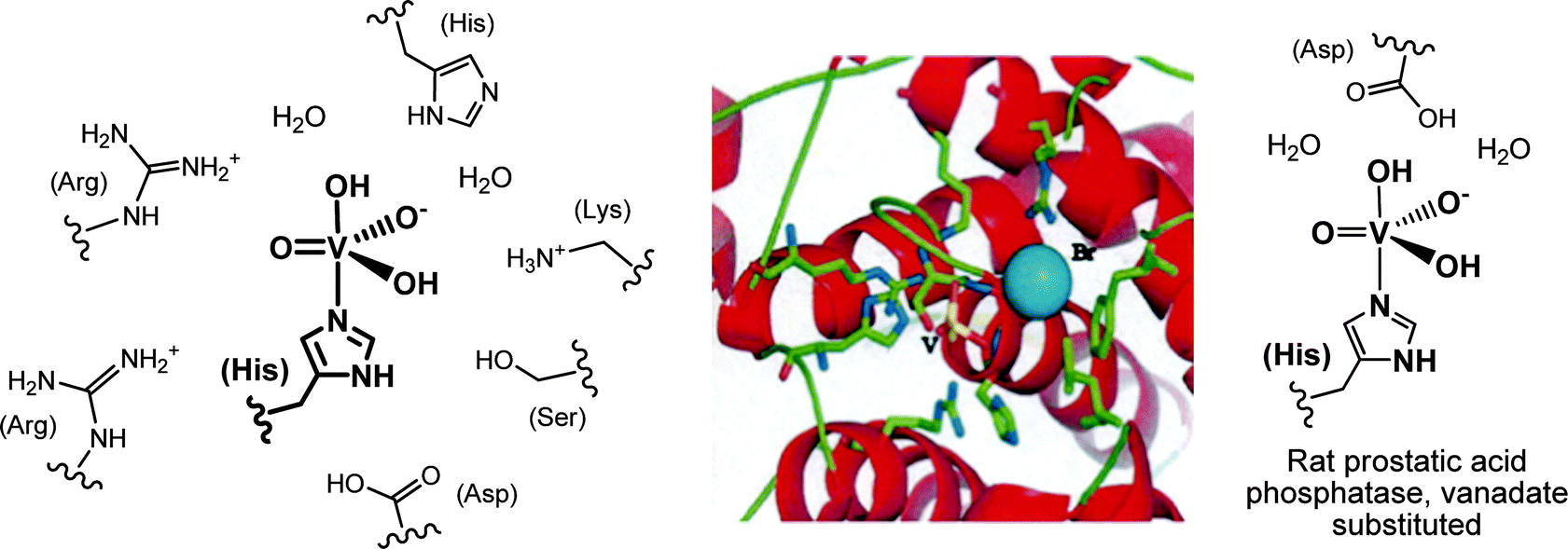
In the VHPOs, vanadate H2VO4− is linked to the Nε of a histidine in the enzyme's active centre, and is additionally stabilized through various hydrogen bonding interactions to neighbouring amino acid residues, as sketched in Fig. 4 for the VBrPO from the marine green macroalga Ascophyllum nodosum (known as knotted wrack or knotted kelp) and the red seaweed Corallina pilulifera. The vanadium(V) centre is in a trigonal-bipyramidal coordination environment, with histidine in one of the axial positions. A. nodosum contains two homologous bromoperoxidases, VBrPO(AnI) and VBrPO(AnII), both of which have been thoroughly characterized: VBrPO(AnI) is a homodimer of 557 amino acids per subunit,39 and VBrPO(AnII) is a hexamer with 641 amino acids in each subunit.40 The sequence homology for the two homologues is 41%. The coordination environment of vanadium in the VClPOs41 is identical to that of the VBrPOs. Interestingly, the buildup of the active centre in VHPOs is very much the same as in rat prostatic acid phosphatase,10 when phosphate is replaced by vanadate (Fig. 4, right).
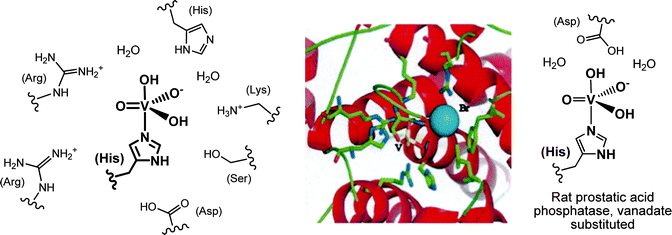 |
| | Fig. 4 Left: the vanadate(V) centre of the vanadate-dependent bromoperoxidase from A. nodosum. Most of the close-by amino acid residues in (hydrogen-bonding) contact with the active center, plus two water molecules, are shown. Centre: the environment of vanadate (yellow), and the location of bromide (blue) in the C. pilulifera peroxidase.42 Bromide is positioned within hydrogen bonding distance (ca. 3.0 Å) between vanadate and an arginine residue. Reproduced with permission, © Elsevier. Vanadate, deeply buried in the protein, is accessible via a positively charged channel. Right: the vanadate centre in the vanadate variant of rat prostatic acid phosphatase. | |
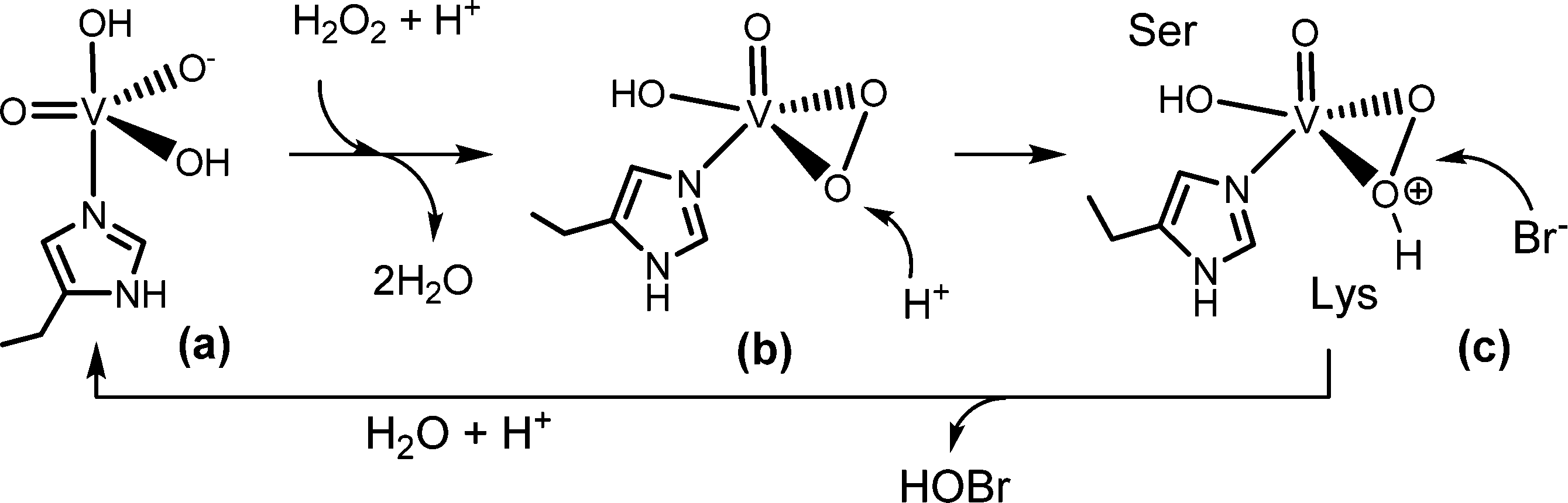
The substrate halide does not bind directly to vanadium. Rather, it interacts peripherally with the active centre, as shown in Fig. 4. During turnover, peroxido and hydroperoxido intermediates are involved; here, vanadium is in an environment in-between a trigonal-bipyramidal and tetragonal-pyramidal arrangement,43Scheme 3. The hydroperoxido intermediate is attacked by the halide, generating hypohalous acid and thus restoring the starting situation; for the net reaction see eqn (14). The oxidation state of vanadium(+V) does not change during turn over; the catalytic VV centre thus functions as a Lewis acid rather than a redox catalyst.
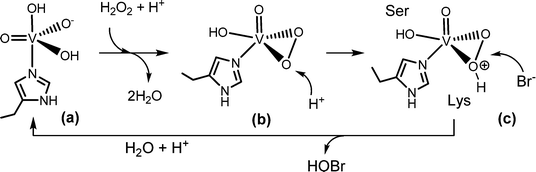 |
| | Scheme 3 Proposed mechanism for the 2-electron oxidation of bromide to a Br+ species such as hypobromous acid HOBr.43b Serine44 and lysine (and/or arginine) play an intrinsic role in activating the (hydro)peroxido intermediate and as a director for bromide. | |
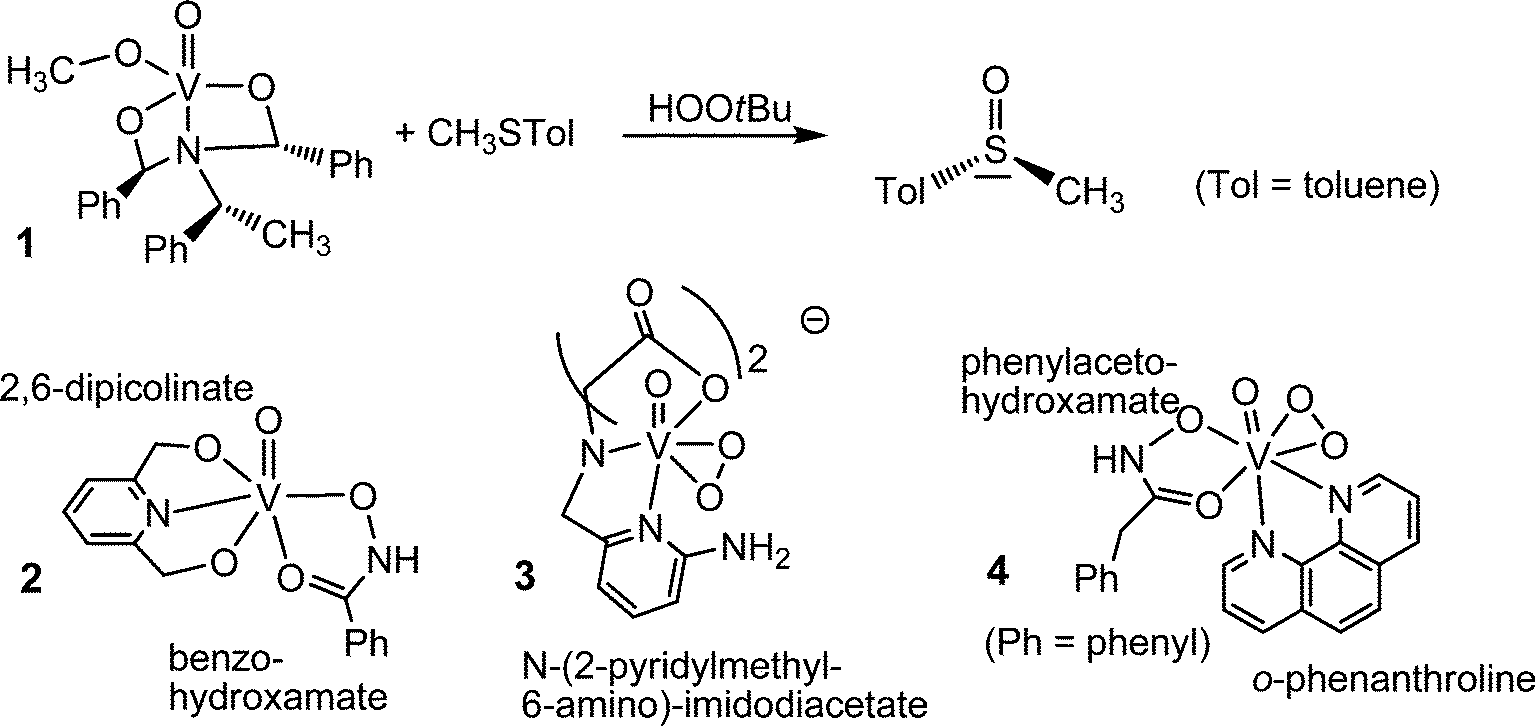
To speak in a general way, oxidovanadium(V) compounds are inherently active in the catalysis of oxidation reactions. The peroxidase activity of the VHPOs in particular has thus inspired various groups to “copy” the naturally occurring enzyme by devising molecular models in which vanadium is in a similar arrangement as in the VHPOs. Even simpler vanadium compounds, such as nanoparticulate vanadium pentoxide n-V2O5 has equally been shown to be an efficient oxidant: n-V2O5 counteracts biofouling, for example of submerged ships’ hulls.45 Molecular “models” for the active centre of VHPOs can thus greatly deviate from the actual vanadium environment in VHPOs. A selection of models is collated in Fig. 5. The oxidovanadium(V) complex 1 in Fig. 5 is a comparatively close model of the active centre of the peroxidases in as far as vanadium is in an approximately trigonal bipyramidal environment constituted of one nitrogen donor opposite of the oxido ligand, and three oxygen donors in the equatorial plane. The complex catalyzes the enantioselective oxidation of prochiral sulfides to chiral sulfoxides.46 Complex 2 is another example where an NO4 coordination sphere is realized; 2 catalyzes peroxidative brominations.47 The complexes 348 and 449 exemplify crystallographically characterized peroxido intermediates.
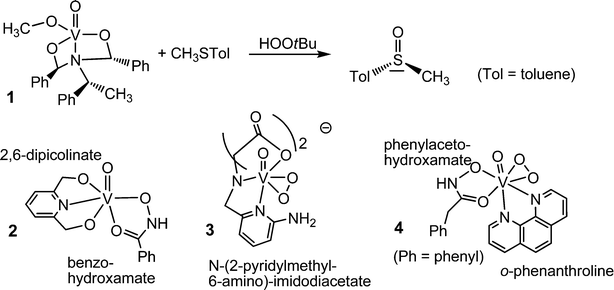 |
| | Fig. 5 A selection of vanadium complexes that model the active centre of vanadate-dependent haloperoxidases (1 and 2), and the intermediate peroxido state (3 and 4). | |
4.3 Impact on atmospheric chemistry
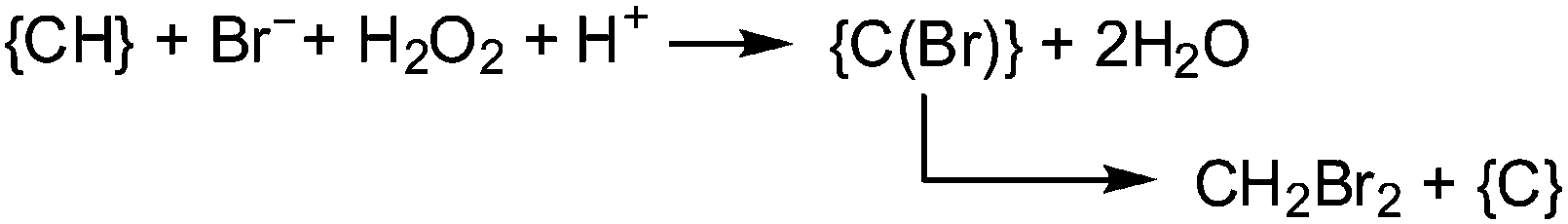
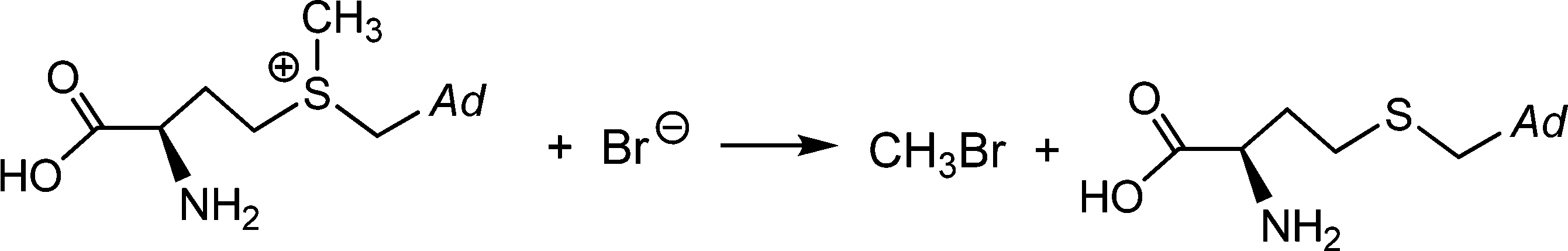
Brown macroalgae such as A. nodosum and Laminaria digitata, as well as the red seaweed Delisia pulchra efficiently brominate a broad variety of organic substrates; these products of halogenation include halomethanes. Monosubstituted halomethanes are preferentially generated in the frame of a nucleophilic attack of halide to the CH3S+ site of S-adenosylmethionine, Scheme 4,50 and hence without participation of VHPOs, while the synthesis of di- and tri-substituted halomethanes (including hetero-substituted ones such as CHBr2I and CHBr2Cl) is catalyzed by VHPOs with recourse to reactive dissolved organic matter {CH}51 and (hydrogen)peroxide. {CH} is available externally from environmental sources or produced as a by-product in photosynthesis. For a net reaction course yielding dibromomethane see eqn (18).52 It should be noted that, along with the marine production of halogenated methanes, terrestrial contributions are involved, in particular soils in tropical areas.| | 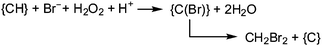 | (18) |
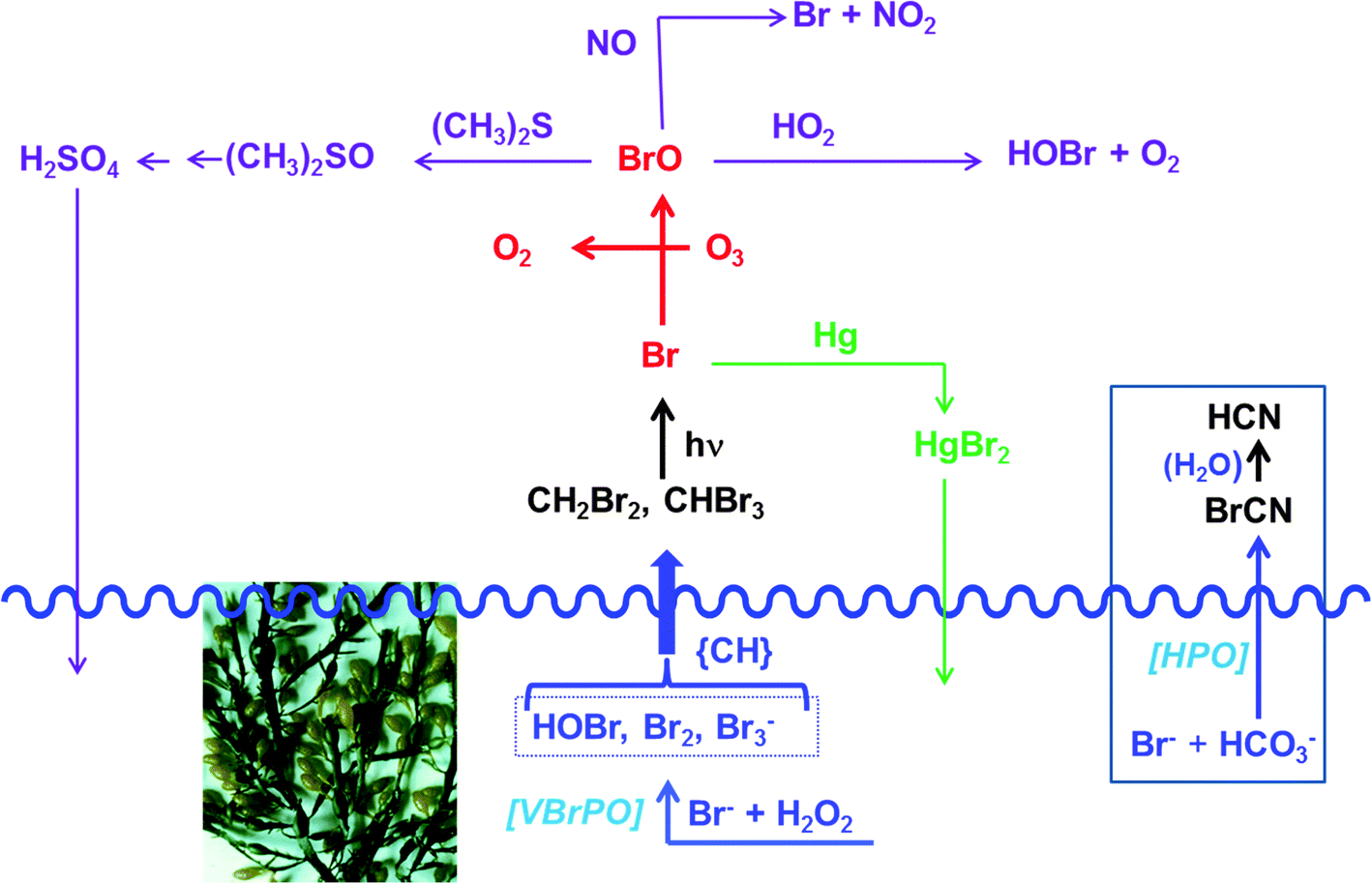
In Fig. 6, several of the atmospheric processes involving halomethanes are exemplified for the bromomethanes CH4−xBrx (x = 2, 3) as primary products introduced into the troposphere; for similar scenarios with iodomethanes CH4−xIx see, for example, ref. 53. The overall coastal flux of CH4−xBrx has been estimated to be 2.5 × 109 moles per year.54a Accordingly, light-induced photolysis in the troposphere generates Br radicals that catalyze the depletion of ozone (red in Fig. 6), forming, along with O2, bromine monoxide BrO. About 20–30% of the atmospheric ozone depletion is attributed to BrO. Br is then partially regenerated from BrO via 2BrO → 2Br + O2 or BrO + O3 → Br + 2O2 (not shown in Fig. 6). BrO can then react with atmospheric constituents such as hydroperoxide radicals HO2, dimethyl sulfide, and nitrous oxide.54a These reaction paths are sketched in mauve in Fig. 6. The hypobromous acid recovered in the reaction between BrO and HO2 can additionally be involved in a symproportionation with bromide and thus recover bromine Br2, eqn (19). Photolytic splitting by light then regenerates bromine radicals. This reaction sequence is occasionally referred to as “bromine explosion”.54b
| | | HOBr + Br− + H+ → Br2 + H2O | (19) |
The reaction with dimethylsulfide is of some interest in as far as (CH3)2S is a main constituent in sulfur cycling between the hydrosphere and the atmosphere. In the marine environment, (CH3)2S is released as a metabolite of dimethylsulfoniopropionate,57 which in turn is produced in a reaction cascade from sulfate via hydrogensulfide and methionine. The atmospheric re-oxidation of (CH3)2S to dimethylsulfoxide and further to sulfuric acid/sulfate helps to replenish the marine sulfate contents. Another interesting branching out is the oxidation of atmospheric elemental mercury. Mercury is released into the atmosphere in the frame of volcanism, anthropogenic activity and, from seawater and soils, after methylation of inorganic mercuric compounds (Hg2+ → CH3HgI → CH3HgCl → (CH3)2Hg) and subsequent volatilization and photolysis of the dimethylmercury ((CH3)2Hg → Hg + C2H6). As shown in green in Fig. 6, bromine radicals can reoxidize Hg0 to Hg2+, which then becomes redeposited.58
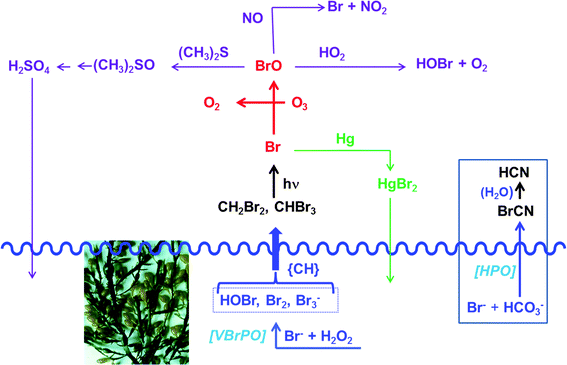 |
| | Fig. 6 Selected atmospheric products/processes, exemplified here for bromomethanes, the formation of which is initiated by apoplastic bromoperoxidase VBrPO such as present in the brown macroalga A. nodosum (shown). For the release of the comparable iodomethanes by the brown alga Laminaria digitata see ref. 55. {CH} symbolizes an organic substrate (dissolved organic matter), serving as a precursor for di- and tribrominated methanes. For additional details see the text. The waved blue line indicates the sea level. Included in this figure (see the box to the right) is the formation of highly toxic cyanogen bromide BrCN from bromide and hydrogencarbonate by the benthic diatom Nitzschia cf pellucida. This diatom employs a yet to be identified haloperoxidase HPO.56 | |
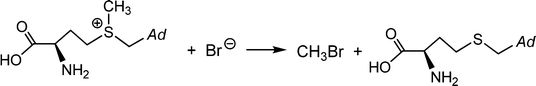 |
| | Scheme 4 The formation of monobromomethane from bromide and S-adenosylmethionine; Ad = adenosine. | |
5. Vanadium's potential role in medicinal applications
In the introductory Section 1, the similarity between vanadate and phosphate has been pointed out (see e.g.Fig. 1), enabling vanadate to substitute for phosphate in phosphate-dependent physiological processes, such as those depending on, or regulated by, phosphatases, kinases, phosphomutases and -diesterases, ATPases and ribonucleases. The pharmacological activity of vanadate in the amelioration of diabetic symptoms is closely related to this specific interchange. The first clinical application of vanadate, in the form of aqueous solutions, in fact goes back to the end of the 19th century.59 In addition to vanadium's anti-diabetic (insulin-enhancing) effect, vanadium compounds have been shown to have pharmacological activity in the treatment of parasitic diseases, malign tumors, bacterial and viral infections. A plethora of vanadium-based medications has been investigated in this respect, a selection of which can be found in recent reviews.8,11 In most cases, oxidovanadium(IV) and -(V) coordination compounds with organic ligand systems coordinating through O-, N- and S-functions have been looked at, but a few traditional as well as more recent examples for the cancerostatic activity of organovanadium compounds, containing the π bonding cyclopentadienyl system, have also appeared (see below).
This section will be constrained to a brief treatise of a few selected examples, thus providing an overview of the inherent capability of vanadium compounds in coping with diseases. It should be noted, however, that so far vanadium compounds have not yet been introduced into actual medicinal applications. It should further be pointed out that vanadium compounds undergo at least partial biotransformation in the blood and other body fluids, i.e. the pharmaceutically active species commonly is not the same as the applied compound.60
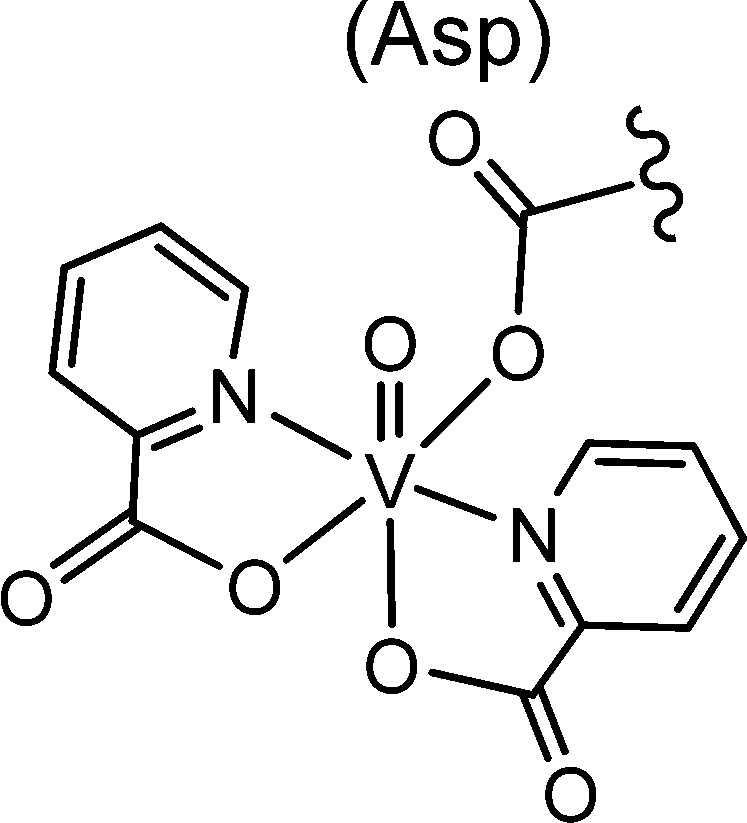
The most important transporter for H2VO4−, VO2+ (and vanadium compounds with an accessible coordination site) in blood is serum transferrin Tf. VO2+ also binds to immunoglobulin and serum albumin, though less efficiently than to Tf. In addition, red blood cells contribute in the uptake, transport and subsequent distribution, provided that VO2+/VO2+ is coordinated to an appropriate carrier, i.e. a ligand system that is able to transcend cellular membranes.61 Inside the erythrocytes, VO2+ is reduced to VO2+ which is then partially released from the genuine “drug” and binds to, inter alia, haemoglobin Hb. However, Hb can also coordinate to the intact complex, likely via a histidine residue; an example is VO(maltol)Hb. Other proteins can likewise stabilize intact vanadium complexes, as recently demonstrated for the coordination of the VO(picolinate)2 moiety to a carboxylate oxygen of a side chain aspartate of a lysozyme, Fig. 7.62a Lysozymes are glycoside hydrolases. Picolinatovanadium complexes have otherwise intensively been studied for their insulin-enhancing properties in animal and laboratory models.62b,c
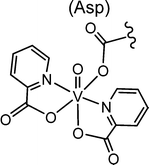 |
| | Fig. 7 Coordination of the VIVO(pic)2 unit (pic = picolinate(1−)) to the aspartate residue Asp52 of hen egg white lyosyme.62b,c | |
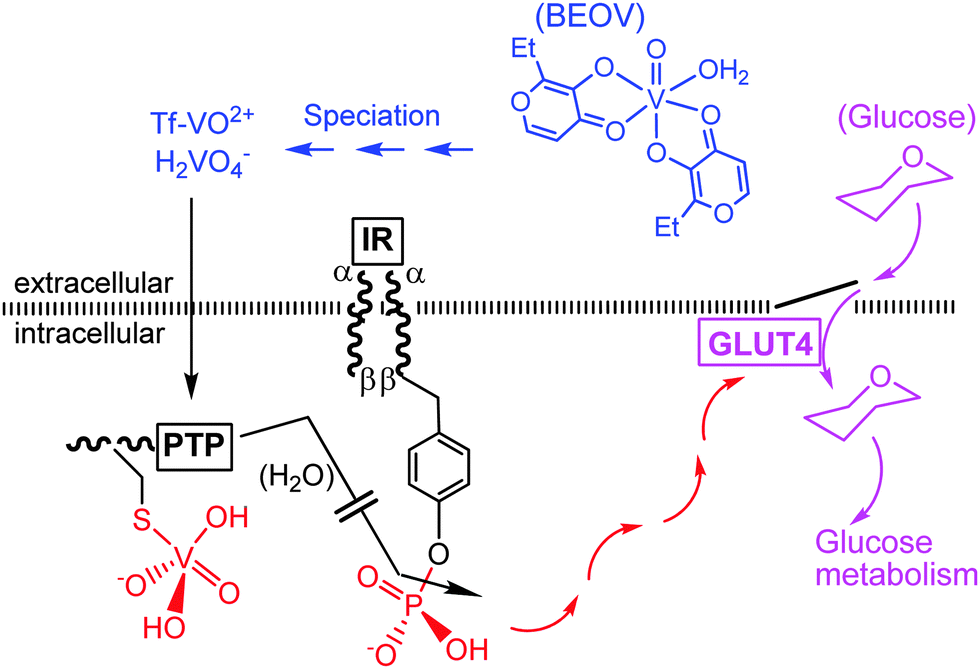
First clinical trials with simple inorganic vanadium compounds in diabetic individuals have been performed in the last decade of the bygone century.63,64 An advanced clinical study – clinical tests phase IIa – has been carried out more recently with the insulin-enhancing VO2+ maltolato complex BEOV = VO(ethylmaltol)2(H2O).7 A possible and simplified mode of action is illustrated in Fig. 8. Accordingly, BEOV undergoes (partial) speciation in blood serum. The speciation includes removal of the maltolato ligand, coordination of the VO2+ moiety to Tf, and/or oxidation to vanadate. Both the Tf complex and vanadate can enter the intracellular space via endocytosis and through phosphate channels, respectively. The insulin receptor IR is a trans-membrane receptor having at its disposal tyrosine residues linked to the intracellular β subunits. Docking of insulin to the extracellular α subunit promotes phosphorylation of the tyrosines. In the absence of insulin (type I diabetes) or in the case of insufficient insulin response of the receptor (common type II diabetes), a protein tyrosine phosphatase PTP counteracts the phosphorylation of IRβ and thus the signaling path (red arrows) responsible for the cellular uptake of glucose (mauve arrows) by the glucose transporter GLUT4. This is the point where vanadate comes in: vanadate strongly coordinates to a cysteine residue of the PTP, thus preventing dephosphorylation of the IRβ subunits and restoring the signaling path.
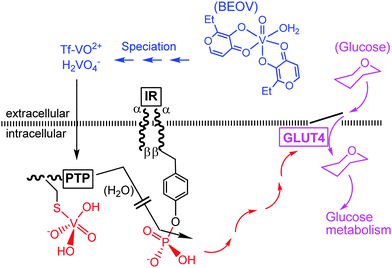 |
| | Fig. 8 A simplified illustration of the action of vanadate as an insulin-mimetic/enhancing agent (red and mauve traces). BEOV is bis(ethylmaltolato)oxidovanadium(IV); Tf = transferrin, IR = insulin receptor, PTP = protein tyrosine phosphatase. GLUT4 is a glucose transporter. For additional details, see the text. | |
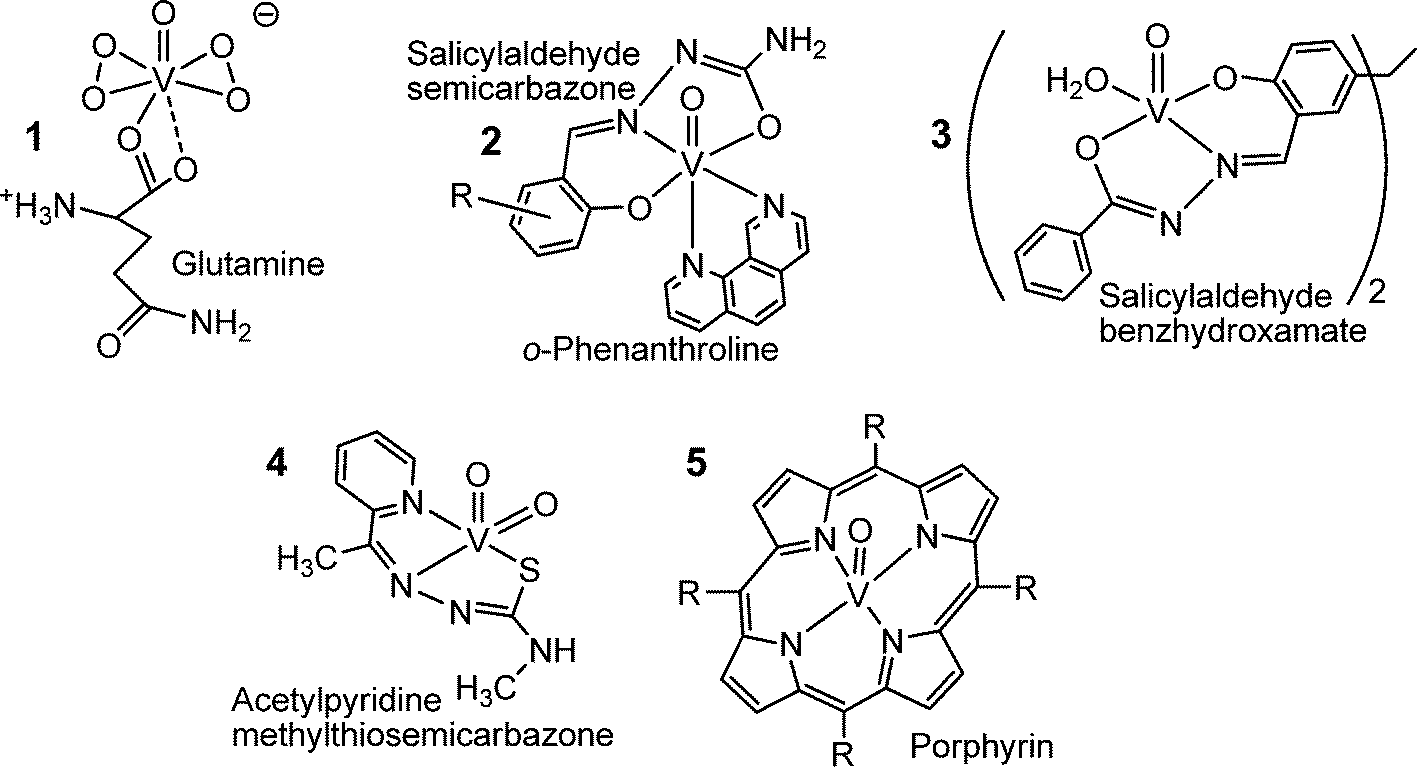
Fig. 9 provides a compilation of examples of an increasing number of vanadium coordination compounds that display in vitro and/or in vivo activity against parasitic tropical diseases (1, 2 and 3), bacterial (4) and viral (5) infections. The bis(peroxido)vanadium complex 1 is effective against the Leishmania flagellates, hence the protozoan parasites responsible for leishmaniasis,65 a wide-spread disease that is transmitted by sandflies predominantly in tropical and subtropical areas. People infected by the Leishmania parasite suffer from cutaneous and visceral infections. The potentiality of compound 1 can likely be attributed to the formation of radicals such as superoxide and nitrous oxide. Compound 2 is a potential therapeutic tool to fight Chagas disease,66 also known as American tryposomiasis, and sleeping disease. Swollen lymph nodes as well as cardiac and digestive disorders are typical symptoms for Chagas disease. The disease is triggered by Trypanosomas cruzi, a protozoa transmitted by the kissing bug (Triatoma infestans).
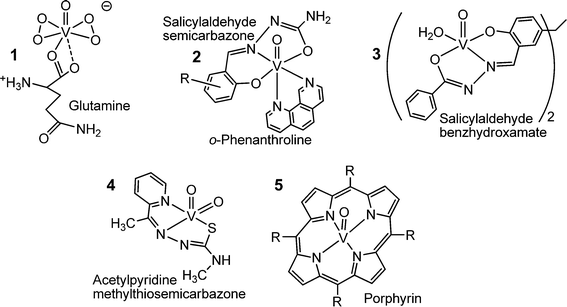 |
| | Fig. 9 Oxidovanadium complexes that are active against parasites causing tropical diseases such as leishmaniasis (1), Chagas disease and sleeping sickness (trypanosomiasis) (2), amoebiasis (3), or fight bacterial infections (such as caused by Mycobacterium tuberculosis, compound 4) and viral infections such as HIV (5). | |
Gastrointestinal infections and, in more serious cases, also liver abcesses go along with amoebiasis, brought about by the amoeba Entamoeba histolytica. The hydrazine complex 3 is more effective against the parasite than metronidazole, a common medication against amoebiasis.67 Complex 4 in Fig. 9, with a thiosemicarbazone ligand linked to the dioxidovanadium(V) fragment features anti-tuberculosis activity,68 and several oxidovanadium(IV) porphyrin complexes, 5 in Fig. 9,69 have been shown to efficiently inhibit the human immunodeficiency virus (HIV) that causes AIDS. Antibacterial and antiviral activities have also been reported for polyoxidovanadates such as [V15O36(CO3)]7−.70
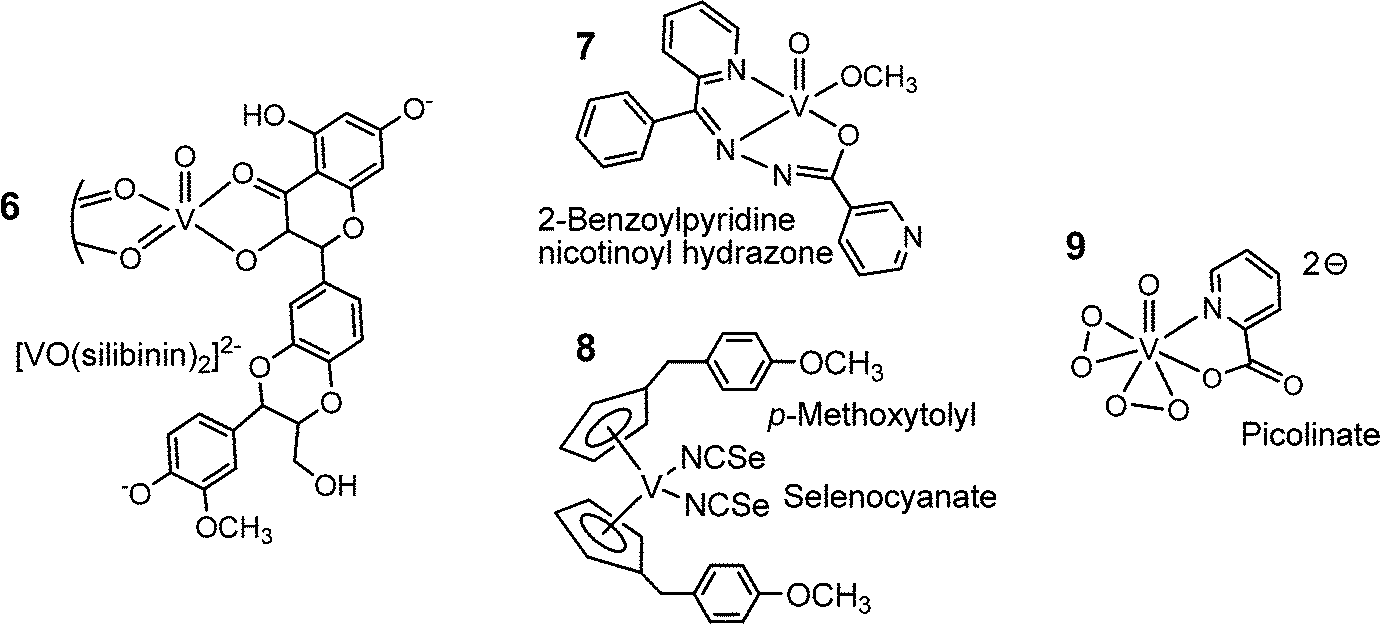
Several of the compounds mentioned above, in particular the porphyrin complex 5 and the polyoxidovanadates, are also active in cancer treatment. More specifically, the complexes 6, 7 and 8 in Fig. 10 exhibit anti-cancer activity. The flavonoid ligand silibinin in 6 has been isolated from extracts of the milk thistle (Silybum marianum); the corresponding vanadium complex inhibits the viability of human osteosarcoma cells.71 The nicotinoylhydrazone complex 7 shows anti-cancer activity against cervical cancer,72 and the vanadocene derivative 8 is cytotoxic against renal cancer cells.73 The cytotoxicity of compound 8 carries on a long-standing tradition in cancer research with titanocenes and vanadocenes, originally going back to Köpf and Köpf-Meier.74 Finally, vanadium complexes have been proven to have neuroprotective and cardio-protective effects. Examples are the bis(peroxido)-picolinato complex 9 in Fig. 10, and bis(maltolato)-oxidovanadium (Fig. 8). The maltolato complex attenuates myocardial reperfusion,75i.e. blood flow is restored to tissues that have had their blood supply cut off, and complex 9 promotes neuroprotection, for example in the case of cervical spinal cord injury (spinal trauma).76
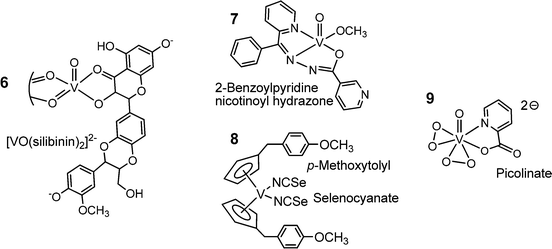 |
| | Fig. 10 A selection of vanadium compounds that exhibit an anti-cancer potential (6, 7, 8) or attenuate myocardial reperfusion (9). The structure of 6 has been deduced from EPR details provided in ref. 77. | |
6. Concluding remarks
Along with the transition elements Mo, W, Mn, Fe, Co, Ni, Cu and Zn, vanadium is an essential bioelement, but in contrast to most of these elements (namely Mo, Mn, Fe, Co, Cu and Zn), which are generally essential for all life forms, functional vanadium compounds have so far been detected only in the form of vanadium nitrogenases and vanadate-dependent haloperoxidases in a comparatively restricted number of organisms. Further, some bacteria may employ vanadium in nitrate reductases, and vanadate is used by various anaerobic prokarya in respiration, commonly along with other transition metal compounds containing the metal in a high oxidation state. A few groups of organisms – sea squirts, fan worms, and Amanita mushrooms – accumulate vanadium without an apparent benefit or, to formulate this issue more cautiously, so far without an apparent reason why these organisms recur to vanadium in its biologically rather unusual forms VIII (sea squirts and fan worms) and non-oxido VIV (amavadin).
As far as the well-established vanadium-dependent nitrogenases and peroxidases are concerned, their impact on nature is noteworthy: the vanadium nitrogenases of soil bacteria such as Azotobacter and planctic cyanobacteria (Anabaena) contribute substantially to nitrogen fixations, and hence to the supply of ammonium for the global plant growth, while vanadate-dependent haloperoxidases in macroalgae are co-responsible for the supply of methylhalides to the atmosphere and thus the regulation of ozone levels. In addition, chloroperoxidases have a noteworthy potential in the defense against microbial spoliation.
The omnipresence of vanadium in our food, in drinking water and the surroundings on the one hand, and the striking similarity of vanadate and phosphate on the other hand (the differences between the transition metal vanadium and the main group element phosphorus notwithstanding), suggest that vanadium also attains a general role in life, in as far as vanadate can interfere with phosphate in a variety of metabolic processes, reflected for example by the dramatically increasing number of protein structures in which phosphate is replaced by vanadate,78 and by the fact that vanadate – and simple vanadium compounds as likely precursors for vanadate – can ameliorate the outcome of diabetes mellitus.7,79 The phosphate-vanadate antagonism, as well as the specific properties of vanadium coordination compounds implemented by the ligand system, have increasingly motivated working groups to explore and to fathom the potentiality of vanadium complexes in combatting parasitic (tropical) diseases, bacterial and viral infections, but also in the treatment of various bodily dysfunctions, such as uncontrolled cell growth (cancer), cardiovascular and neuronal problems.
To date, little resilience detail is known about the perspective essentiality of vanadium and its handling in higher organisms, a fact that should encourage enhanced research activities directed towards, for example, vanadium's role in the treatment of the diseases and bodily dysfunctions sketched above. Similar considerations apply to the mechanisms of action of unphysiologically high exposure to vanadium and its concomitant (potential) toxicity.4b
References
- D. Rehder, Dalton Trans., 2013, 42, 11749–11761 RSC.
- F. Aydin, A. Saydut, B. Gunduz, I. Aydin and C. Hamanci, Clean: Soil, Air, Water, 2012, 40, 444–448 CrossRef PubMed.
- T. L. Gerke, K. G. Scheckel and M. R Schock, Environ. Sci. Technol., 2009, 43, 4412–4418 CrossRef.
-
(a) J. O. Olopade and J. R. Condor, Curr. Top. Toxicol., 2011, 7, 33–39 Search PubMed;
(b) T. I. Fortoul, V. Rodriguez-Lara, A. Gonzáles-Villalva, M. Rojas-Lemus, G. Cano-Gutiérrez, M. Ustarroz-Cano, L. Colín-Barenque, P. Bizarro-Nevares, I. García-Pealez, L. F. Montaño, R. S. Jimenez-Martinez, N. Lopez-Valdez, M. L. Ruiz-Guerrero, N. A. Meléndez-García, F. A. García-Ibarra, V. Martínez-Baez, D. Zapata Alfaro, A. Muñiz-Rivera-Cambas, L. S. López-Zepeda, E. M. Quezada-Maldonado and S. Cervantes-Yépez, Inorg. Chim. Acta, 2014, 420, 8–15 CrossRef PubMed.
-
(a) A. Gorzsás, I. Andersson and L. Pettersson, Eur. J. Inorg. Chem., 2006, 3559–3565 CrossRef PubMed;
(b)
N. D. Chasteen, in Metal Ions in Biological Systems, ed. H. Sigel and A. Sigel, Marcel Dekker Inc., New York, 1995, ch. 7, vol. 31 Search PubMed;
(c) N. D. Chasteen, R. J. DeKoch, B. L. Rogers and M. W. Hanna, J. Am. Chem. Soc., 1973, 95, 1301–1309 CrossRef.
- G. Heinemann, B. Fichti and W. Vogt, Clin. Pharmacol., 2003, 55, 241–245 CrossRef.
- K. H. Thomson, J. Lichter, C. LeBel, M. C. Scaife, J. H. McNeill and C. Orvig, J. Inorg. Biochem., 2009, 103, 554–558 CrossRef PubMed.
- D. Rehder, Met. Ions Life Sci., 2013, 13, 139–169 Search PubMed.
- L. C. Cantley Jr., L. Josephson, R. Warner, M. Yanagisawa, C. Lechene and G. Guidotti, J. Biol. Chem., 1977, 252, 7421–7423 Search PubMed.
-
(a) Y. Lindquist, G. Schneider and P. Vihko, Eur. J. Biochem., 1994, 221, 139–142 CrossRef PubMed;
(b) M. Zhang, M. Zhou, R. L. van Etten and C. V. Stauffacher, Biochemistry, 1997, 36, 15–23 CrossRef PubMed.
- D. Rehder, Future Med. Chem., 2012, 4, 1823–1837 CrossRef PubMed.
- D. C. Crans, M. L. Tarlton and C. C. McLauchlan, Eur. J. Inorg. Chem., 2014, 4450–4458 CrossRef PubMed.
-
(a) M. Henze, Z. Physiol. Chem., 1917, 72, 494–501 CrossRef PubMed;
(b) A. L. Dingley, K. Kustin, I. G. Macara and G. C. McLeod, Biochim. Biophys. Acta, 1981, 649, 493–502 CrossRef;
(c)
K. Kustin and W. E. Robinson, in Metal Ions in Biological Systems, ed. H. Sigel and A. Sigel, Marcel Dekker Inc., New York, 1995, ch. 15, vol. 31 Search PubMed.
- T. Hamada, M. Asanuma, T. Ueki, F. Hayashi, N. Kobayashi, S. Yokoyama, H. Michibata and H. Hirota, J. Am. Chem. Soc., 2005, 127, 4216–4222 CrossRef PubMed.
- S. Yamamoto, K. Matsuo, H. Michibata and T. Ueki, Inorg. Chim. Acta, 2014, 420, 47–52 CrossRef PubMed.
- J. A. L. da Silva, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2013, 257, 2388–2400 CrossRef PubMed.
- L. Bertini, V. Barbieri, P. Fantucci, L. De Goia and G. Zampella, Dalton Trans., 2011, 40, 7704–7712 RSC.
- G. Zampella, L. Bertini and L. De Goia, Chem. Commun., 2014, 50, 304–307 RSC.
-
(a) D. Rehder, Org. Biomol. Chem., 2008, 6, 957–964 RSC;
(b) A. N. Antipov, N. N. Lyalikova, T. V. Khijniak and N. P. L’vov, FEBS Lett., 1998, 441, 257–260 CrossRef.
- N. A. Yukova, N. D. Saveljeva and N. N. Lyalikova, Mikrobiologiya, 1993, 62, 597–603 Search PubMed.
- W. Carpentier, L. De Smet, J. Van Beeumen and A. Brigé, J. Bacteriol., 2005, 187, 3293–3301 CrossRef PubMed.
-
W. Carpentier, PhD thesis, University of Gent, 2005.
- I. Ortiz-Bernad, R. T. Anderson, H. A. Vrionis and D. R. Loveley, Appl. Environ. Microbiol., 2004, 70, 3091–3095 CrossRef CAS.
- J. T. Csotonyi, E. Stackebrandt and Y. Yorkov, Appl. Environ. Microbiol., 2006, 72, 4950–4956 CrossRef PubMed.
- J. Zhang, H. Dong, L. Zhao, R. McCarrick and A. Agrawal, Chem. Geol., 2014, 370, 29–39 CrossRef PubMed.
- M. Bernroitner, M. Zamocky, P. G. Furtmüller, G. A. Pescheck and C. Obinger, J. Exp. Bot., 2009, 60, 423–440 CrossRef PubMed.
- G. Boison, C. Steingen, L. J. Stal and H. Bothe, Arch. Microbiol., 2006, 186, 367–376 CrossRef PubMed.
-
(a) B. P. Hodkinson, J. L. Allen, L. L. Forrest, B. Goffinet, E. Sérusiaux, Ó. S. Andrésson, V. Miao, J.-P. Bellenger and F. Lutzoni, Eur. J. Phycol., 2014, 49, 11–19 CrossRef;
(b) R. Darnajoux, J. Constantin, J. Miadlikowska, F. Lutzoni and J.-P. Bellenger, New Phytol., 2014, 202, 765–771 CrossRef PubMed.
-
(a) C. C. Lee, Y. Hu and M. W. Ribbe, Angew. Chem., Int. Ed., 2010, 50, 5545–5547 CrossRef PubMed;
(b) Y. Hu, C. C. Lee and M. W. Ribbe, Science, 2011, 333, 753–755 CrossRef PubMed;
(c) K. Fisher, M. J. Dilworth and W. E. Newton, Biochemistry, 2006, 45, 4190–4198 CrossRef PubMed.
-
(a) R. R. Eady, Coord. Chem. Rev., 2003, 237, 23–30 CrossRef;
(b) A. W. Fay, M. A. Blank, C. C. Lee, Y. Hu, K. O. Hodgson, B. Hedman and M. W. Ribbe, J. Am. Chem. Soc., 2010, 132, 12612–12618 CrossRef PubMed.
- J. A. Wiig, C. C. Lee, Y. Hu and M. W. Ribbe, J. Am. Chem. Soc., 2013, 135, 4982–4983 CrossRef PubMed.
- I. Dance, Dalton Trans., 2011, 40, 5516–5527 RSC.
- T. Yoneyama, M. Shiozawa, M. Nakamura, T. Suzuki, Y. Sagane, Y. Katoh, T. Watanabe and T. Ohyama, J. Biol. Chem., 2004, 36, 37477–37484 CrossRef PubMed.
- J. N. Carter-Franklin and A. Butler, J. Am. Chem. Soc., 2004, 126, 15060–15066 CrossRef PubMed.
- L. Kaysser, P. Bernhardt, S.-J. Nam, S. Loesgen, J. G. Ruby, P. Skewes-Cox, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2012, 134, 11988–11991 CrossRef CAS PubMed.
- A. Messerschmidt and R. Wever, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 392–396 CrossRef.
- J. M. Winter and B. S. Moore, J. Biol. Chem., 2009, 284, 18577–18581 CrossRef PubMed.
-
(a) I. F. Persoon, M. A. Hoogenkamp, A. Bury, P. R. Wesselink, A. F. Hartog, R. Wever and W. Crielaard, J. Endod., 2012, 38, 72–74 CrossRef PubMed;
(b) R. Renirie, A. Dewilde, C. Pierlot, R. Wever, D. Hober and J.-M. Aubry, J. Appl. Microbiol., 2008, 105, 364–370 CrossRef PubMed.
- M. Weyand, H.-J. Hecht, M. Kiess, M. F. Liaud, H. Vilter and D. Schomburg, J. Mol. Biol., 1999, 293, 595–611 CrossRef CAS PubMed.
-
(a) D. Wischang, M. Radlow, H. Schulz, H. Vilter, L. Viehweger, M. O. Allmeyer, C. Kegler, J. Herrmann, R. Müller, F. Gaillard, L. Delage, C. Leblanc and J. Hartung, Bioorg. Chem., 2012, 44, 25–34 CrossRef CAS PubMed;
(b) D. Wischang, M. Radlow and J. Hartung, Dalton Trans., 2013, 42, 11926–11940 RSC.
- A. Messerschmidt and R. Wever, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 392–396 CrossRef CAS.
- J. Littlechild, E. Garcia Rodriguez and M. Isupov, J. Inorg. Biochem., 2009, 103, 617–621 CrossRef CAS PubMed.
-
(a) A. Messerschmidt, L. Prade and R. Wever, Biol. Chem., 1997, 378, 309–315 CrossRef CAS PubMed;
(b) G. Zampella, P. Fantucci, V. L. Pecoraro and L. De Goia, J. Am. Chem. Soc., 2005, 127, 953–960 CrossRef CAS PubMed.
- V. Kraehmer and D. Rehder, Dalton Trans., 2012, 41, 5225–5234 RSC.
- F. Natalio, R. André, A. F. Hartog, B. Stoll, K. P. Jochum, R. Wever and W. Tremel, Nat. Nanotechnol., 2012, 7, 530–535 CrossRef CAS PubMed.
- G. Santoni, G. Licini and D. Rehder, Chem. – Eur. J., 2003, 9, 4700–4708 CrossRef CAS PubMed.
- S. Patra, S. Chatterjee, T. K. Si and K. K. Mukherjea, Dalton Trans., 2013, 42, 13425–13435 RSC.
- C. Kimblin, X. Bu and A. Butler, Inorg. Chem., 2002, 41, 161–163 CrossRef CAS PubMed.
- T. K. Si, S. S. Paul, M. G. B. Drew and K. K. Mukherjea, Dalton Trans., 2012, 41, 5805–5815 RSC.
- S. La Barre, P. Potin, C. Leblanc and L. Delage, Mar. Drugs, 2010, 8, 988–1010 CrossRef CAS PubMed.
- C. Yu Lin and S. L. Manley, Limnol. Oceanogr., 2012, 57, 1857–1866 CrossRef.
- R. Wever and M. A. van der Horst, Dalton Trans., 2013, 42, 11778–11786 RSC.
- G. McFiggans, C. S. E. Bale, S. M. Ball, J. M. Beames, W. J. Bloss, L. J. Carpenter, J. Dorsey, R. Dunk, M. J. Flynn, K. L. Fumeaux, M. W. Gallagher, D. E. Heard, A. M. Hollingsworth, K. Hornsby, T. Ingham, C. E. Jones, R. L. Jones, L. J. Kramer, J. M. Langridge, C. Leblanc, J.-P. LeCrane, J. D. Lee, R. J. Leigh, I. Longley, A. S. Mahajan, P. S. Monks, H. Oetjen, A. J. Orr-Ewing, J. M. C. Plane, P. Potin, A. J. L. Shillings, F. Thomas, R. von Glasow, R. Wada, L. K. Whalley and J. D. Whitehead, Atmos. Chem. Phys., 2010, 10, 2975–2999 CrossRef CAS.
-
(a) L. J. Carpenter, C. E. Jones, R. M. Dunk, K. E. Horsby and J. Woeltje, Atmos. Chem. Phys., 2009, 9, 1805–1816 CrossRef CAS;
(b) A. Saiz-Lopez and R. von Glasow, Chem. Soc. Rev., 2012, 41, 6448–6472 RSC.
-
(a) F. C. Küpper, L. J. Carpenter, G. B. McFiggans, C. J. Palmer, T. J. Waite, E.-M. Boneberg, S. Woitsch, M. Weiller, R. Abela, D. Grolimund, P. Potin, A. Butler, G. W. Luther III, P. M. H. Kroneck, W. Meyer-Klaucke and M. C. Feiters, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6954–6958 CrossRef PubMed;
(b) C. Paul and G. Pohnert, Nat. Prod. Rep., 2011, 28, 186–195 RSC.
-
(a) B. Vanelslander, C. Paul, J. Grueneberg, E. K. Prince, J. Gillard, K. Sabbe, G. Pohnert and W. Vyverman, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2412–2417 CrossRef CAS PubMed;
(b) M. Syrpas, E. Ruysbergh, L. Blommaert, B. Vanelslander, K. Sabbe, W. Vyverman, N. De Kimpe and S. Mangelinckx, Mar. Drugs, 2014, 12, 352–367 CrossRef CAS PubMed.
- M. Vila-Costa, R. Simó, H. Harada, J. M. Gasol, D. Slezak and R. P. Kiene, Science, 2006, 314, 652–654 CrossRef PubMed.
- C. W. Moore, D. Obrist, A. Steffen, R. M. Staebler, T. A. Douglas, A. Richter and S. V. Nghiem, Nature, 2014, 506, 81–84 CrossRef CAS PubMed.
- B. Lyonnet, X. Martz and E. Martin, La Presse Medicale, 1899, 32, 191–192 Search PubMed.
- Y. Yoshikawa, H. Sakurai, D. C. Crans, G. Micera and E. Garribba, Dalton Trans., 2014, 43, 6965–6972 RSC.
- D. Sanna, M. Serra, G. Micera and E. Garribba, Inorg. Chem., 2014, 53, 1449–1464 CrossRef CAS PubMed.
-
(a) M. F. A. Santos, I. Correira, A. R. Oliveira, E. Garribba, J. Costa Pessoa and T. Santos-Silva, Eur. J. Inorg. Chem., 2014, 3293–3297 CrossRef CAS PubMed;
(b) G. R. Willsky, L.-H. Chi, M. Godzala III, P. J. Kostyniak, J. J. Smee, A. M. Trujillo, J. A. Alfano, W. Ding, Z. Hu and C. C. Crans, Coord. Chem. Rev., 2011, 255, 2258–2269 CrossRef CAS PubMed;
(c) A. D. Sostarecz, E. Gaidamauskas, S. Distin, S. J. Bonetti, N. E. Levinger and D. C. Crans, Chem. – Eur. J., 2014, 20, 5149–5159 CrossRef CAS PubMed.
-
(a) A. B. Goldfine, D. C. Simonson, F. Folli, M.-E. Patti and C. R. Kahn, J. Clin. Endocrinol. Metab., 1995, 80, 3311–3320 CAS;
(b) N. Cohen, M. Halberstam, P. Shlimovich, C. J. Chang, H. Shamoon and L. Rossetti, J. Clin. Invest., 1995, 95, 2501–2509 CrossRef CAS PubMed.
- G. R. Willsky, A. B. Goldfine, P. J. Kostyniak, J. H. McNeill, L. Q. Yang, H. R. Khan and D. C. Crans, J. Inorg. Biochem., 2001, 85, 33–42 CrossRef CAS.
- A. Kumar Haldar, S. Banerjee, K. Naskar, D. Kalita, N. S. Islam and S. Roy, Exp. Parasitol., 2009, 122, 145–154 CrossRef PubMed.
- M. Fernández, L. Becco, I. Correia, J. Benitez, O. E. Piro, G. A. Echeverria, A. Madeiros, M. Comini, M. L. Lavaggi, M. Gonzáles, H. Cerecetto, V. Moreno, J. Costa Pessoa, B. Garat and D. Gambino, J. Inorg. Biochem., 2013, 127, 150–160 CrossRef PubMed.
- M. R. Maurya, A. Alam Khan, A. Azam, S. Ranjan, N. Mondal, A. Kumar, F. Avecilla and J. Costa Pessoa, Dalton Trans., 2010, 39, 1345–1360 RSC.
- P. I. da S. Maia, F. R. Pavan, C. Q. F. Leite, S. S. Lemos, C. F. de Sousa, A. A. Batista, O. R. Nascimento, J. Ellena, E. E. Castellano, E. Niquet and V. M. Deflon, Polyhedron, 2009, 28, 398–406 CrossRef PubMed.
- R. W.-Y. Sun, D.-L. Ma, E. L.-M. Wong and C.-M. Che, Dalton Trans., 2007, 4884–4892 Search PubMed.
- T. Yamase, J. Mater. Chem., 2005, 15, 4773–4782 RSC.
- I. E. Leon, V. Porro, A. L. Di Virgilio, L. G. Naso, P. A. M. Williams, M. Bollati-Fogolín and S. B. Etcheverry, JBIC, J. Biol. Inorg. Chem., 2014, 19, 59–74 CrossRef CAS PubMed.
- R. S. Nair, M. Kuriakose, V. Somasundaram, V. Shenoi, M. R. Prathapachandra Kurup and P. Srinivas, Life Sci., 2014, 116, 90–97 CrossRef CAS PubMed.
- I. Fichtner, J. Claffey, A. Deally, B. Gleeson, M. Hogan, M. Rivera Marcelova, H. Müller-Bunz, H. Weber and M. Tacke, J. Organomet. Chem., 2010, 695, 1175–1181 CrossRef CAS PubMed.
- P. Köpf-Maier and H. Köpf, Drugs Future, 1986, 11, 297–319 Search PubMed.
- D. A. Liem, C. C. Gho, B. C. Gho, S. Kazim, O. C. Manintveld, P. D. Verdouw and D. J. Duncker, J. Pharmacol. Exp. Ther., 2004, 309, 1256–1262 CrossRef CAS PubMed.
- C. L. Walker, M. J. Walker, N.-K. Liu, E. C. Risberg, X. Gao, J. Chen and X.-M. Xu, PLoS One, 2012, 7, e3002 Search PubMed.
- L. G. Naso, E. G. Ferrer, N. Butenko, I. Cavaco, L. Lezama, T. Rojo, S. B. Etcheverry and P. A. M. Williams, JBIC, J. Biol. Inorg. Chem., 2011, 16, 653–668 CrossRef CAS PubMed.
- J. Costa Pessoa, E. Garribba, M. F. A. Santos and T. Santos-Silva, Vanadium in proteins: a structural overview, Coord. Chem. Rev., 2015 Search PubMed , in press.
- J. A. Meyer and D. M. Spence, Metallomics, 2009, 1, 32–41 RSC.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence