
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-cadherin-mediated cell adhesion is regulated by extracellular Zn2+
E.
Heiliger
a,
A.
Osmanagic
a,
H.
Haase
bc,
N.
Golenhofen
d,
A. M.
Grabrucker
de,
A.
Weth
f and
W.
Baumgartner
*af
aDepartment of Cellular Neurobionics, RWTH-Aachen University, Helmertweg 3, 52074 Aachen, Germany
bInstitute of Immunology, RWTH-Aachen University, Pauwelstr. 30, 52074 Aachen, Germany
cDepartment of Food Chemistry and Toxicology, Berlin Institute of Technology, Gustav-Meyer-Allee 25, D-13355 Berlin, Germany
dInstitute of Anatomy and Cell Biology, Ulm University, Albert-Einstein-Allee 11, Ulm, Germany
eWG Molecular Analysis of Synaptopathies, Neurology Dept., Neurocenter of Ulm University, Albert-Einstein-Allee 11, Ulm, Germany
fInstitute of Biomedical Mechatronics, Johannes Kepler University of Linz, Altenbergerstr. 69, Linz, Austria
First published on 5th January 2015
Abstract
Synapses in the central nervous system (CNS) are highly dynamic structures that undergo reorganisation in response to synaptic activity. Dysfunctional structural synaptic plasticity is associated with impaired brain function and several neurological disorders. As response to synaptic activity, dendritic spines of excitatory synapses were reported to undergo alterations in their molecular structure and morphology leading to increased postsynaptic density size and spine volume. For these structural changes a transient activity-dependent weakening of synaptic adhesion will be necessary. Here, we report that zinc can modulate N-cadherin-mediated adhesion. Quantification of binding activity was performed using laser tweezer technique. Our results show that increased levels of zinc abolished N-cadherin binding without altering the number of N-cadherin molecules expressed at the cell surface. Furthermore, zinc directly interacted with N-cadherin and the regulatory role was found to take place under physiological zinc concentrations within minutes. Given that zinc is released at zincergic synapses in the CNS, our findings may contribute to mechanistic insights in the interplay between zinc signalling, activation of glutamate receptors and downstream pathways, and the coordination of pre- and postsynaptic changes via trans-synaptic cell adhesion complexes, all finally contributing to synaptic plasticity.
Introduction
Zinc is the most prevalent trace metal in the brain and plays an important role in processes such as synaptic plasticity, the underlying mechanism of learning and memory. While most of the total brain zinc exists in a protein-bound state (>80%), “free” zinc as Zn2+-ions can be found predominantly within synaptic vesicles,1,2 where it is released with glutamate from glutamatergic (zincergic) presynaptic terminals.3,4 In particular, zinc-containing neurons are found predominantly in limbic and cerebrocortical regions,5 where the release of zinc upon synaptic activity is followed by an increase in Zn2+ concentration in the synaptic cleft as well as the postsynaptic intracellular compartment. Here zinc might enter mainly through NMDA and AMPA receptors, and calcium-channels6 or is released within the postsynapse from internal zinc stores and metallothioneins (MTs).7–9Within the synaptic cleft, zinc is known to bind to various neuronal ion channels and receptors, such as GPR39,10 TrkB receptors,11 and NMDA and kainate receptors.12–14 To that end, free zinc levels are tightly regulated, for example by zinc transporters such as Zrt-Irt-like proteins (ZIP) and the zinc transporter (ZnT) family. Intriguingly, the zinc exporter ZnT-1 was found enriched at postsynapses15 hinting at an additional zinc extrusion mechanism directly localized to synapses. Disruption of zinc signaling is involved in several neurodegenerative and neuropsychiatric disorders such as Alzheimer's disease (AD), Autism Spectrum Disorder (ASD), and Schizophrenia (SCZ).16 Moreover, zinc deficient rodent animal models display impairments in memory and learning.17–19 Thus, it is of major interest to investigate the molecular mechanisms by which zinc might regulate morphological and physiological changes at synapses underlying synaptic plasticity.
There is increasing evidence that cell adhesion molecules are involved in modulation of synaptic structure and function.20–22 Trans-synaptic cell adhesion is mediated by cadherin complexes that modify adhesive contacts, activate signal transduction pathways, and regulate synapse formation and plasticity.23 Inhibition of N-cadherin function, the most widespread cadherin of the nervous system, has profound effects on activity-dependent spine enlargement and suppresses long term potentiation (LTP).24–26 Transient weakening of adhesion contacts with subsequent formation of new contacts may be one prerequisite for structural reorganization of synapses. However, how morphological changes and synaptic function are coordinately regulated has not yet been identified. In the past we have proposed a model that activity-dependent local changes of ion concentrations within the synaptic cleft during synaptic activity may initiate such a weakening of adhesion contacts. We could show that N-cadherin binding is regulated by extracellular Ca2+ ions as well as protons.27–29 Thus, modulation of adhesive activity of N-cadherin by local changes of ion concentrations may be one mechanism of how N-cadherin mediates synaptic remodeling.
In this study we followed the hypothesis that transient local changes of the free zinc concentration in the synaptic cleft occurring during synaptic activity might affect N-cadherin binding activity and thereby modulates synaptic morphology and function. We found a direct regulatory interaction of zinc and N-cadherin. Thus, the interplay between zinc and N-cadherin might provide a molecular basis for the structural alterations accompanying plasticity of zincergic excitatory synapses.
Results
Zinc dependency of N-cadherin binding
To investigate if N-cadherin binding is modulated by Zn2+ we studied the trans-interaction of recombinant N-cadherins with endogenous N-cadherins expressed on the surface of PC-12 cells. A fusion protein of the extracellular domain of N-cadherin and the Fc-portion of human IgG (N-cad-Fc) was generated to yield functional cadherin dimers as described previously.28,29 N-cad-Fc-coated microbeads were allowed to settle 15 min on the cell surface of PC-12 cells leading to adherens-like junctions between microbeads and cells as shown by the recruitment of characteristic proteins of adherens junctions to bead-cell-contacts.27,29,30 Subsequently the laser tweezer technique was applied to quantify the amount of beads binding to cells in dependence of the zinc concentration in the culture medium. This experimental model of measuring cadherin-binding activity at bead-cell-contacts is a very useful system since it allows exact definition of one side of the adhesion contact (i.e. the coated beads) and allows on the other hand measuring cadherin binding in a cellular context.This technique was applied to N-cad-Fc-coated beads on the cell surface of PC-12 cells using culture medium with various concentrations of zinc. The free zinc concentration of the medium was measured by spectroscopic analysis using the zinc-dependent fluorophore FluoZin-3. Table 1 shows the results of these measurements of the culture media after addition of various amounts of ZnCl2 as well as the values after chelation of zinc (TPEN) and at saturated conditions. The control culture medium (DMEM plus 5% FCS, no addition of zinc) contained about 259 pM of free Zn2+. By addition of 2 μmol l−1 zinc chloride the free Zn2+ concentration was actually increased to 385 pM. Most of the added zinc ions were obviously buffered by zinc-binding molecules of the culture medium.
| Added substance | Fluorescence (arbitrary units) | Free Zn2+ (pM) |
|---|---|---|
| TPEN | 601 | 0 |
| 0 μM Zn2+ | 1556 ± 109 | 259 |
| 0.5 μM Zn2+ | 1798 ± 130 | 290.5 |
| 1 μM Zn2+ | 1862 ± 58 | 322 |
| 1.5 μM Zn2+ | 2117 ± 65 | 353.5 |
| 2 μM Zn2+ | 2002 ± 145 | 385 |
| 250 μM Zn2+ | 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 787 787 |
— |
The percentage of N-cad-Fc-coated beads tightly bound to the cell surface (resisting displacement by the laser tweezer) was counted at different zinc-concentrations and the results are depicted in Fig. 1. Under control conditions, i.e. using cell culture medium without additional zinc, 43 ± 3.2% of N-cad-Fc-coated beads adhered to PC-12 cells. Specificity of bead binding was controlled by the use of Fc-coated beads as well as by measuring binding activity at Ca2+-free conditions, i.e. after addition of 5 mM EGTA. Under these conditions 15 ± 3.5% and 17 ± 4.5% beads were bound, respectively, reflecting the amount of unspecific bead binding (Fig. 2). Unspecific bead binding (Fc-coated beads) was not affected by addition of zinc (Fig. 1). If the zinc-concentration in the medium was increased cadherin-mediated bead binding dropped to basal levels. At a free Zn2+-concentration of 385 pM only 16 ± 2.5% of N-cad-Fc-coated beads were bound which represents unspecific bead binding. Half-maximal binding activity (KD) was observed at roughly 310 pM free Zn2+. This value was estimated at the concentration where the linear interpolation-curve of the data crosses the 50% difference of minimal and maximal binding activity.
 | ||
| Fig. 1 Zinc dependence of N-cadherin mediated adhesion. The percentage of N-cad-Fc coated microbeads adhering to the surface of PC-12 cells was measured at various extracellular Zn2+ concentrations using the laser tweezer technique. The free Zn2+ concentration is given on the x-axis as well as the amount of Zn2+ added to the culture medium to achieve these free Zn2+ levels. N-cadherin-mediated bead adhesion decreased with increasing Zn2+ concentration (KD ≈ 310 pM). Fc-coated beads showed only low binding which was independent of Zn2+ and resembled unspecific binding activity. Data are presented as means ± standard deviation. All data points represent at least three individual experiments with probing of 100 beads. | ||
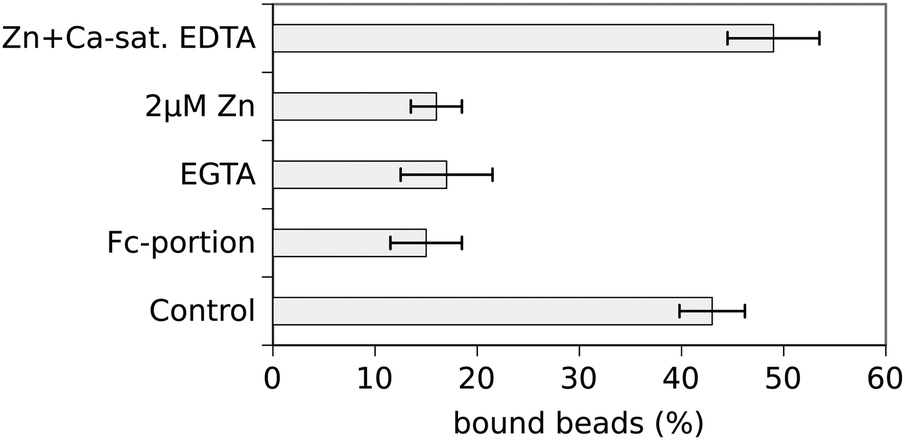 | ||
| Fig. 2 Control experiments demonstrating specificity of bead binding. Unspecific bead binding amounted to ∼15% as assessed by the use of Fc-coated beads or N-cad-Fc-coated beads in the absence of Ca2+ (EGTA). N-cad-Fc-coated beads bound to 43% at control conditions (see also Fig. 1) whereas it dropped to basal levels after addition of 2 μM total Zn2+ (corresponding to 385 pM free Zn2+). Chelation of extracellular Zn2+ by addition of calcium-saturated EDTA restored bead binding confirming the Zn2+-dependency of N-cadherin mediated adhesion. | ||
Further control experiments were performed to guarantee specificity of bead binding and to rule out any unspecific effects due to changes of extracellular zinc. As already described above unspecific bead binding was determined by the use of Fc-coated beads and Ca2+-free conditions. To demonstrate that the observed effect of inhibition of cadherin-mediated bead binding after addition of 2 μM Zn2+ is really mediated by zinc, Zn2+ was specifically chelated after 15 min of incubation by addition of 10 μM Ca2+-saturated EDTA for another 15 min. EDTA has a higher affinity for zinc than for calcium resulting in chelating of zinc without lowering the free calcium concentration. Under this condition bead binding was restored and even increased to 49 ± 4.5% showing that the observed effect was really mediated by extracellular Zn2+ (Fig. 2). It has to be emphasised that EDTA cannot enter the cell and thus effects on the cytosolic zinc-concentration can be ruled out. Removal of Zn2+ from a cell by depletion of extracellular Zn2+ would take place on a totally different time scale. Thus the extracellular Zn2+ is responsible for the observed effects.
Cellular uptake of Zn2+ is negligible
In order to see whether significant amounts of Zn2+ were taken up by the PC12-cells under the conditions investigated, cells were loaded with the membrane permeable Zn2+-indicator FluoZin-3 AM prior to exposure to extracellular Zn2+. The uptake was quantified by measuring the corresponding fluorescence over time. The results are depicted in Fig. 3.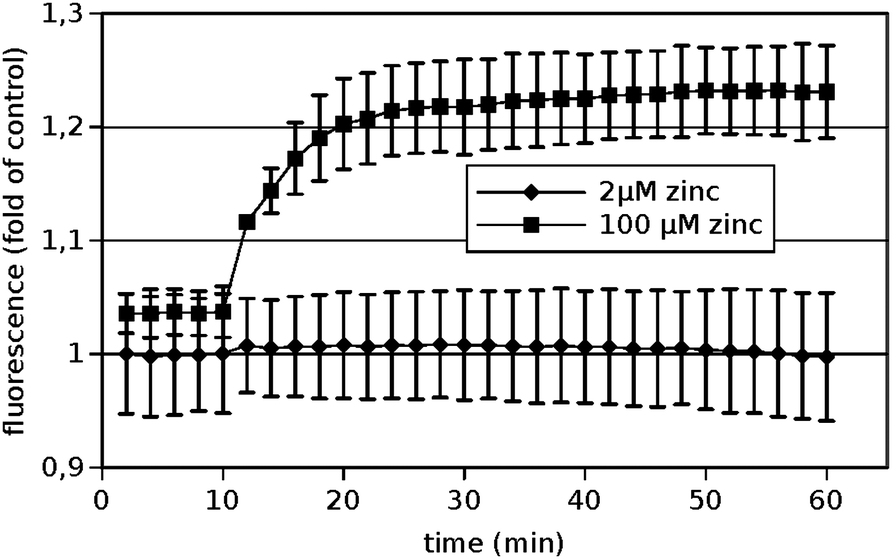 | ||
| Fig. 3 Uptake of Zn2+ into PC12 cells. PC-12 cells were loaded with the Zn2+-selective fluorescent probe FluoZin-3. After 10 minutes recording of the baseline, the indicated final concentrations of Zn2+ were added. Data are shown as means from n = 3 independent experiments. | ||
Clearly no significant amounts of zinc were taken up within one hour under the conditions used for the cadherin binding assays, i.e. 2 μM total added zinc (corresponding to 385 pM free Zn2+). If an unphysiological high Zn2+-concentration of 100 μM was added, a significant increase could be observed demonstrating that the method is in principle applicable. Thus the described effects of Zn2+ on N-cadherin mediated adhesion were mediated by the extracellular Zn2+ and not by elevated intracellular zinc levels.
Cellular growth and viability are not changed by Zn2+
To test if cellular growth was impaired by the addition of Zn2+ in the range used, PC-12 cells were grown in normal culture medium supplemented with increasing concentrations of Zn2+ in the range from 0 to 2 μM (added total concentration) for 24 h and 72 h, respectively. The total number of cells was determined by flow cytometry and the amount of viable cells was determined by using a MTT assay. At concentrations up to 2 μM of added Zn2+ no significant alteration of the number of total or viable cells could be observed (data not shown).Zinc does not change N-cadherin expression
Next we checked whether the amount of N-cadherin expressed on the cell surface was altered by the addition of Zn2+. For that purpose cells were treated with trypsin in the absence of Ca2+, resulting in proteolysis of the cadherin extracellular domain if cadherins were inserted in the plasma membrane. Fig. 4A shows a western blot of cell lysates of PC-12 cells incubated in culture medium without or with additional zinc (2 μM) and after treatment with trypsin or without.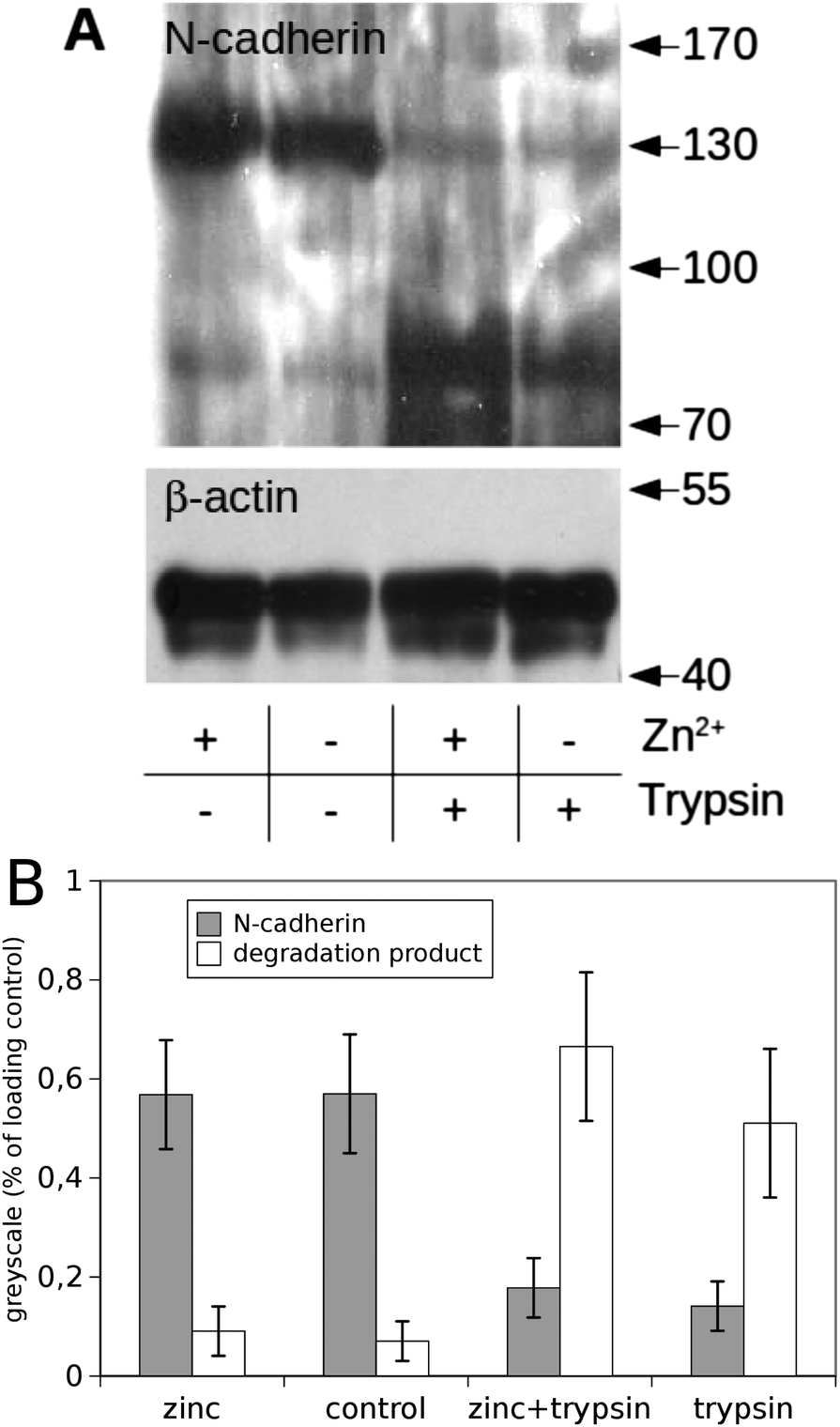 | ||
| Fig. 4 N-cadherin levels in PC12-cells with and without zinc treatment. PC-12 cells were exposed to culture media with (+) or without (−) additional Zn2+ and treated subsequently with trypsin resulting in proteolysis of the extracellular domain of N-cadherin if cadherins are inserted in the plasma membrane. A representative western blot (A) shows that trypsin treatment resulted in a nearly complete proteolysis of N-cadherin independent of Zn2+ indicating nearly complete surface expression of N-cadherin. Equal amounts of protein were loaded onto each lane; β-actin served as loading control. The greyscale analysis of three independent blots is shown in (B). | ||
Without trypsin treatment an immunoreactive band for N-cadherin could be seen at the expected height of ∼130 kDa independent of the Zn2+ concentration. A greyscale analysis of three blots is shown in Fig. 4B. This analysis indicates that the total amount of N-cadherin was not affected by zinc. Trypsin treatment led to nearly complete proteolysis of cadherins at both conditions (with/without addition of Zn2+). The band at 130 kDa disappeared and a degradation product at about 80 kDa became dominant. A western blot for actin served as loading control. Thus, the amount of N-cadherin localised in the plasma membrane was not altered by elevated zinc-concentrations within the investigated time window of up to 30 minutes.
Binding of Zn2+ to N-cadherin
To investigate if reduction of N-cadherin binding activity by Zn2+ is mediated by a direct binding of Zn2+ to N-cadherin molecules or by any indirect effects the amount of zinc bound to N-cad-Fc was measured by atomic absorption spectrometry (AAS). 100 μg of N-cad-Fc were incubated with or without 10 μM of zinc in 100 μl HBSS. Then the proteins were washed three times with zinc-free buffer by using Centricon centrifugation filters. After that the samples were subjected to AAS to determine the total amount of zinc contained. The concentration of zinc in the N-cad-Fc sample preincubated with zinc was 4.32 μM in comparison to 0.56 μM in the sample without prior zinc incubation. To rule out zinc binding to the Fc-portion of the fusion protein control experiments with Fc instead of N-cad-Fc were performed. No significant changes of the zinc content of the Fc-portion depending on prior zinc incubation could be detected. Thus, zinc can directly bind to the extracellular domain of N-cadherin. Assuming a molecular weight of N-cad-Fc of 130 kD, one finds that about one zinc-ion per cadherin molecule is bound.In an alternative approach direct Zn2+-binding was proven using a FluoZin-3 based fluorescence spectroscopy approach which confirmed the interaction and led to a stoichiometric ratio of about 2 Zn2+-ions per cadherin molecule. While the direct binding of Zn2+ to the extracellular domain of N-cadherin could be shown by these approaches beyond doubt, only a lower limit of the stoichiometry can be given. This is due to the fact that the experimental procedure, especially the washing steps which take quite some time, allow the dissociation of formerly bound ions.
Interference of Zn2+ and Ca2+-binding to N-cadherin
Clearly Zn2+ directly binds to N-cadherin and thereby alters cadherin trans-interaction. How this functions on the molecular level has to be determined. As cadherins are calcium dependent adhesion proteins, it is tempting to assume that Zn2+-binding could interfere with the Ca2+-binding, which is known to be vital for cadherin function. To test for this hypothesis, affinity chromatography was performed on a N-cadherin-functionalised column and a BSA-functionalised column for control. The columns were initially equilibrated with Ca2+ in the presence and absence of Zn2+ and then eluated with Ca2+-free buffer. Then the Ca2+-concentration in the different eluation-fractions was determined photometrically. A typical result is shown in Fig. 5. What can clearly be seen is that from the N-cadherin column Ca2+ is eluated rather slowly. Up to 1.5 minutes after the bulk of Ca2+ from the exclusion volume of the column has passed, free Ca2+ ions are still detectable, representing the release of the bound Ca2+ from the immobilised N-cadherin molecules. This is in contrast to the BSA-functionalised column where Ca2+ drops down simply exponentially, i.e. a straight line can be observed in the logarithmic representation. After 160 s of eluation time no Ca2+ could be detected in the eluate from the BSA-column with our detection method. In the presence of Zn2+ however, there is no such difference between BSA and N-cadherin functionalised columns. This indicates that Zn2+ binding to N-cadherin somehow impaired the Ca2+-binding very fast as the presence of Zn2+ in the Ca2+-containing equilibration buffer instantaneously prevents Ca2+-binding. When instead of Zn2+, Cd2+ was used, no change in Ca2+-eluation could be observed (data not shown) indicating a Zn2+-specific effect.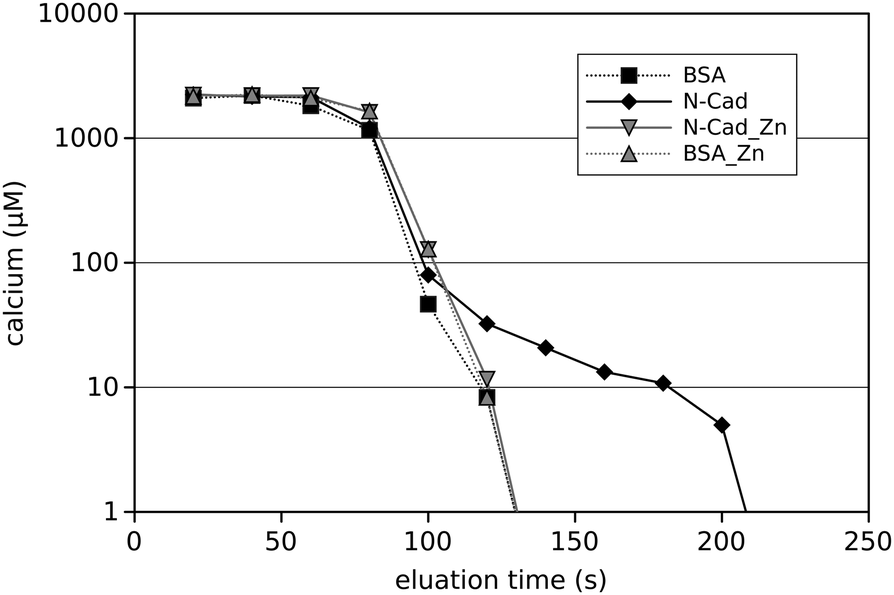 | ||
| Fig. 5 Ca2+-eluation from protein columns in the presence and absence of Zn2+. Prior to eluation with a Ca2+-free buffer the columns packed with either N-cadherin (solid lines) or BSA (dashed lines) were equilibrated with Ca2+ in the presence (grey lines) or absence (black lines) of Zn2+. The free Ca2+-concentration was determined by measuring the transmission at 600 nm of an Eriochrome Black T-indicator solution. | ||
Discussion
Cadherins are localised at synaptic sites bordering the active zone.31–33 As mentioned above cadherins have various functions at synapses, such as in synaptogenesis and in synaptic plasticity at mature synapses.34 They affect synaptic structure and function, for example formation of LTP. Several mechanisms how cadherins regulate synaptic plasticity are possible, which are not necessarily mutually exclusive. Trans-synaptic interactions might activate intracellular signalling pathways thereby influencing synaptic strength. Furthermore, LTP of synapses is accompanied by an increase in synaptically localised cadherin such as N-cadherin.24 Despite the fact that cadherins are Ca2+-dependent adhesion molecules and Ca2+-binding to the extracellular domain is necessary for their proper function, little attention has been drawn to the possibility that binding of Ca2+ or other ions to the extracellular domain might be one important mechanism to regulate the strength of adhesion. Interestingly, adhesive activity of N-cadherin is regulated between 0.2 and 1.3 mM Ca2+ (KD = 0.7 mM).27 In addition, N-cadherin binding has been shown to be regulated by the extracellular concentration of protons.29 This is of special interest because transient changes of ion concentrations (such as Ca2+ and H+) in the synaptic cleft will occur during high synaptic activity and thus, regulation of N-cadherin binding under such conditions might be one mechanism by which cell adhesion is regulated and synaptic plasticity is influenced.Here we report Zn2+ binding to N-cadherin and regulation of N-cadherin binding activity by extracellular Zn2+. Zinc was already shown to play an important role in synaptic transmission and serves as an endogenous neuromodulator. Moreover, zinc is an important factor regulating dynamic processes mediating the stability of the post synaptic density (PSD).8,35 Zinc is selectively stored in, and co-released with neurotransmitters from glutamatergic presynaptic vesicles.6 Thereby, vesicular zinc transiently elevates local concentrations in the synaptic cleft. However, the exact amount of such release is not known and controversially discussed. Total Zn2+ in human cerebrospinal fluid is 150 nM,36 and a basal concentration of 38.6 nM extracellular Zn2+ has been measured by in vivo microdialysis in the hippocampi of rats.37 As a result of Zn2+-buffering by proteins and low molecular weight ligands, the concentration of free Zn2+ in biological fluids is usually several orders of magnitude lower than the total amount. Resting free extracellular Zn2+ is close to the detection limit of most experimental techniques for its quantitation, and is probably in the picomolar concentration range, maybe even as low as 1 pM.3 However, others reported free baseline Zn2+-concentrations in the nanomolar range.37 Upon synaptic transmission, Zn2+ is transiently released, and the concentration of extracellular free Zn2+ is elevated to nanomolar or even micromolar values.38–40 Thus, the exact values of the free Zn2+-concentrations in the brain are not known and might vary within the synaptic cleft. But, baseline concentrations within the picomolar range and nanomolar to micromolar concentrations after synaptic activity seem to be most realistic. In this paper, we could show that N-cadherin responds to zinc in a low concentration, displaying high affinity. Half maximal binding was already reached at a concentration of about 310 pM free zinc. This argues for a basal binding under physiological conditions with few free zinc ions present and decreased binding upon synaptic activity with high amounts of locally released free zinc. Non-physiological conditions such as zinc deficiency might stabilise N-cadherin further, impairing structural changes necessary for the implementation of synaptic plasticity. In this context it is interesting to mention that reduction of zinc attenuates hippocampal LTP induction and cognitive memory.41
Moreover, we could show that zinc directly binds to the extracellular domain of N-cadherin. A sequence analysis of N-cadherin using Metaldetector v2.0 software42 revealed no typical zinc-binding motifs. However, in this approach only the primary sequence was analysed. There might be intermolecular zinc-binding sites (within the cadherin-dimer) or binding sites formed by different subdomains of the protein. Thus, a detailed investigation of the 3D-structure would be necessary to identify the exact location of the interaction site. A hint comes from the experiments showing that Zn2+ can prevent Ca2+-binding to N-cadherin, while Cd2+ or Mg2+ can not. If this is due to a competition of Zn2+ and Ca2+ for the same binding site or if Zn2+ binds to another site leading to conformational or electrostatic effects rendering Ca2+-binding impossible can not be concluded from the results available so far. This has to be determined in future studies. However, Zn2+ is released from synapses during neuronal activity physiologically and elevated Zn2+-levels in the physiological range dramatically modulate N-cadherin binding.
Experimental
Cell culture and recombinant N-cadherin-Fc
PC12 rat pheochromocytoma cells (ECACC 88022401), endogenously expressing N-cadherin, were cultured in DMEM containing 5% FCS and 10% horse serum (Biochrom, Berlin, Germany), 2 mM glutamine, 50 U ml−1 penicillin-G and 50 μg streptomycin at 37 °C in a humidified atmosphere (5% CO2). N-cadherin-Fc fusion protein (mouse N-cadherin extracellular domain fused to Fc-portion of human IgG1, called N-cad-Fc in the following) was generated and purified as described previously.29 Briefly, Chinese hamster ovary cells were stable transfected, and the secreted N-cadherin-Fc was purified from the culture supernatants by affinity chromatography using protein A agarose (Merck, Darmstadt, Germany).Coating of polystyrene beads
N-cad-Fc was coupled to protein A-coated superparamagnetic polystyrene microbeads (Dynabeads, diameter 2.8 μm, Dynal, Oslo) as described.27,29 Briefly, after vortexing 10 μl bead solution were washed 3× in 100 μl of buffer A (100 mM Na-phosphate, pH 8.1) by sedimenting the beads via a magnet and reuptake in buffer A. The washed beads were suspended in 100 μl buffer A containing 10 μg of cadherin-Fc protein and allowed to react for 30 min at room temperature under permanent slow overhead rotation. After washing 3× in 100 μl buffer B (200 mM triethanolamine, pH 9.0) beads were incubated for 45 min in 100 μl buffer B containing 0.54 mg dimethyl pimelimidate·2HCl (DMP, Pierce, Rockford, USA) at RT. Free DMP was blocked by two washes for 30 min at 37 °C in 100 μl 100 mM Tris pH 8.0. Finally the beads were washed 3× in Hanks Balanced Salt Solution (HBSS, Gibco, Karlsruhe). Beads were stored for up to 7 days in HBSS containing 0.1% BSA and 0.02% sodium azide at 4 °C at slow overhead rotation to avoid sedimentation.Laser tweezer experiments
A home-built laser tweezer setup was used as described before.27 The beam of a Nd:YAG laser (1064 nm) was expanded to fill the back aperture of a high NA-objective (Plan Neofluar, 100 × 1.3 oil, Zeiss), coupled through the epiillumination port of an Axiovert 40 inverted microscope (Zeiss, Oberkochen, Germany) and reflected to the objective by a dicroic mirror (FT510, Zeiss).Zn2+ was adjusted in the decomplemented DMEM containing 5% FCS by addition of increasing volumes of an isotonic, i.e. 0.3 M ZnCl2 solution that 0–2 μmol l−1 of Zn2+ were added in total. Decomplementation of the serum was carried out at 56 °C for 60 min. Protein coated polystyrene microbeads were allowed to interact with the cell surface of monolayers for 15 min. Then in each experiment 100 beads were probed during the following 5–10 min by the laser tweezer. At least three experiments with different batches of cells and N-cad-Fc-coated beads were performed for each experimental condition investigated.
Specificity of cadherin–Fc binding was investigated by Ca2+-depletion of the culture medium (addition of 5 mM of EGTA). Furthermore, control beads were coated with human Fc fragment only and these beads were applied to the cells and probed by the laser tweezer.
Free Zn2+ measurements in cell culture medium
Free Zn2+ was measured using FluoZin-3.43,44 Different amounts of ZnCl2 were added to DMEM + 5% FCS covering the range used for the laser tweezer experiments. The medium was incubated for 30 min at 37 °C, corresponding to the incubation time in the laser tweezer experiment and then subjected to a fluoroscopic determination of the free Zn2+-concentration. FluoZin-3 (tetrapotassium salt, Invitrogen, Germany) was added to a final concentration of 1 μM and incubated for 15 min. 100 μl of each sample were transferred into 96-well plates. The resulting fluorescence F was recorded on a Tecan Ultra 384 fluorescence well plate reader (Tecan, Crailsheim, Germany) at room temperature. Free zinc concentrations were calculated using the formula by Grynkiewicz et al.:45with a dissociation constant for the Zn2+–FluoZin-3 complex of KD = 8.9 nM. For the determination of the background fluorescence and the maximal fluorescence Fmin and Fmax, the medium was incubated with 10 μM of the Zn2+-chelator N,N,N′,N′-tetrakis (TPEN) or ZnSO4 (250 μM), respectively. All measurements for Zn2+-determination were carried out using three independent samples (n = 3).
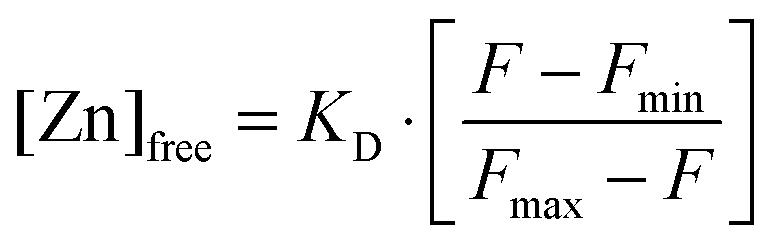
Trypsin treatment of cultured cells
PC-12 cells grown in Petri dishes were incubated for 15 min in DMEM + 5% FCS with 2 μmol l−1 added ZnCl2. After that cell layers were either scratched from the petri dish with a rubber policeman in Laemmli sample buffer or treated with 0.125% trypsin and 2 mM EDTA in PBS for 7 min at 37°. After addition of 200 μl FCS to stop trypsin activity cells were washed three times in PBS and dissolved in Laemmli sample buffer. These samples were subjected to SDS-PAGE and western blotting.Electrophoresis and western blotting
Cell lysates were dissolved in Laemmli sample buffer and protein content was determined by amido black staining.46,47 Immunoblots were performed with equal amount of protein per lane after separation with SDS-PAGE (7.5% polyacrylamide). Membranes were blocked with 5% low-fat milk for 1 h at room temperature in phosphate-buffered saline containing 0.05% (v/v) Tween 20 and incubated with the respective primary antibody overnight at 4 °C. The antibody ab18203 against N-cadherin (Abcam, Cambridge, UK) was used at a dilution of 1:1500 and the β-actin antibody CLONE AC-15 (Sigma, St. Louis, MS) at a dilution of 1:5000. As secondary antibodies horseradish peroxidase (HRP)-labeled goat anti-rabbit and goat anti-mouse IgG (Jackson Immunoresearch, West Grove, PA) both diluted 1:3000 were used. Bound antibodies were detected with the enhanced chemiluminescence technique (Amersham). Grey-scale analysis was performed using on the blots which were scanned using 8-bit grey-scale digitalization by employing the image software Gimp 2.8.
Measurement of intracellular free zinc
Cells were loaded with 1 μM FluoZin-3 AM (Invitrogen, Karlsruhe, Germany) in loading buffer (25 mM HEPES, pH 7.35, 120 mM NaCl, 5.4 mM KCl, 5 mM glucose, 1.3 mM CaCl2, 1 mM MgCl2, 1 mM NaH2PO4, 0.3% bovine serum albumin) for 30 min at 37 °C. Subsequently, cells were washed with culture medium and transferred into a 96 well plate at a density of 1 × 106 cells per ml. The resulting fluorescence was recorded on a Tecan Ultra 384 fluorescence well plate reader (Tecan, Crailsheim, Germany) using an excitation wavelength of 485 nm and measuring the emission at 535 nm.MTT assay
Cells were seeded onto 96 well plates and incubated with different Zn2+-concentrations. Subsequently, cells were treated for three hours with 0.01% (w/v) MTT (methylthiazolyldiphenyl-tetrazolium bromide, Sigma) in normal culture medium, lysed in isopropanol and the absorption determined on a Tecan Sunrise well plate reader (Crailsheim, Germany) at 570 nm, using a reference wavelength of 700 nm.Determination of direct Zn2+–cadherin-binding using atomic absorption spectrometry (AAS)
N-cad-Fc or pure Fc-fragments at a concentration of 1 mg ml−1 in HBSS were incubated for 15 min with 10 μM ZnCl2. Then the proteins were washed 5 times with HBSS using Centricon centrifugation filters. One centrifugation step for removal of excess liquid took approximately 3–5 min. Then these samples were subjected to AAS. As negative control the proteins were washed the same way but the incubation step with ZnCl2 was omitted. All reagents for atomic absorption were of appropriate quality for trace element analysis (TraceSelect, Fluka, Germany). Samples were diluted 1:10 in ultrapure water containing 0.2% (v/v) HNO3 and were analysed by flame AAS on a Perkin Elmer AAnalyst 800 instrument and quantified with a standard curve between 0 and 1 mg l−1 zinc in 0.2% (v/v) HNO3. Acetylene flow was set to 2.0 l min−1 and oxidant (air) to 17.0 l min−1, measuring the absorption at 213.9 nm (slit 0.7 nm).
Confirmation of direct Zn2+–cadherin-binding using FluoZin-3A
As alternative approach to proof that Zn2+ directly binds to N-cad-Fc 100 μl samples of HBSS containing 1 μM ZnCl2 were incubated with increasing amounts of N-cad-Fc from 0 to 1 μM. Then 1 μM of FluoZin-3A was added and the free Zn2+-concentration was determined as described above.Ca2+-binding to N-cadherin in the presence and absence of Zn2+
CNBr-activated sepharose (100 mg; Sigma, Taufkirchen, Germany) was allowed to swell for 45 min at 4 °C in 1 mM HCl (10 ml). The swollen sepharose (∼1 ml) was transferred to a column (diameter 5 mm) and washed with 100 ml of 1 mM HCl, followed by 3 ml of distilled water. The column was equilibrated with 1 ml of coupling buffer (100 mM NaHCO3, 500 mM NaCl, pH 8.4) and then loaded with 1 ml of coupling buffer containing a mixture of 0.8 mg ml−1 N-cadherin-Fc and 0.8 mg ml−1 bovine serum albumin (BSA) and allowed to react for 2 h at RT under slow overhead rotation. Afterwards the column was washed once with 3 ml coupling buffer, followed by a wash with 300 ml blocking buffer (200 mM glycine, pH 8.0) and then subjected to incubation for 3 h at RT in blocking buffer. After three washes with 3 ml acetate buffer (100 mM acetate, 500 mM NaCl, pH 4.5), the column was washed and equilibrated with 10 ml of HBSS containing 2 mM Ca2+. For control experiments a second column was packed identically to the above described but only BSA was coupled to the sepharose.For determination of Ca2+-binding the N-cadherin- as well as the BSA-functionalised column were equilibrated with 5 ml of a Ca2+-containing HEPES-buffer (5 mM HEPES pH 7.4, 150 mM NaCl, 2 mM CaCl2, ±1 μM ZnCl2). Then the column was eluated with the HEPES-buffer lacking Ca2+ and Zn2+ with a flow velocity of about 1.1 ml min−1. Fractions of 348 μl (5 drops) were collected. The Ca2+-concentration in the fractions was determined photometrically using the Ca2+-sensitive chromophore Eriochrome Black-T. For this 500 μM Eriochrome Black-T were dissolved in 100 mM Tris (pH 11) and 100 μl of the fractions were added to 500 μl of the chromophore solution. If the chromophore was saturated, the measurement was repeated but only 10 μl of the fraction were added to the indicator solution. The Ca2+-concentration was determined photometrically by measuring the transmission at 600 nm. Calibration was done using samples of defined Ca2+-concentration.
Conclusions
Here, we show that zinc binds to the extracellular domain of N-cadherin and modulates N-cadherin-mediated adhesion. Increased levels of zinc abolished N-cadherin binding (KD ≈ 310 pM). Our results imply a model, where synaptic activity leads to an increase in the extracellular zinc concentration within the synaptic cleft which leads, among other effects, to reduced N-cadherin adhesion, thereby opening a short time window for structural changes that might accompany processes such as synaptic plasticity and LTP.Acknowledgements
Prof. Lothar Rink is acknowledged for helpful discussions. The work was financially supported by the Deutsche Forschungsgemeinschaft BA 2272/10-1 and the framework of the COMET K2 Austrian Center of Competence in Mechatronics (ACCM/LCM).Notes and references
- T. B. Cole, H. J. Wenzel, K. E. Kafer, P. A. Schwartzkroin and R. D. Palmiter, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1716–1721 CrossRef CAS.
- R. D. Palmiter, T. B. Cole, C. J. Quaife and S. D. Findley, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14934–14939 CrossRef CAS.
- C. J. Frederickson and A. I. Bush, BioMetals, 2001, 14, 353–366 CrossRef CAS.
- C. J. Frederickson and D. W. Moncrieff, Biol. Signals, 1994, 3, 127–139 CrossRef CAS PubMed.
- C. J. Frederickson and G. Danscher, Prog. Brain Res., 1990, 83, 71–84 CAS.
- C. J. Frederickson, J. Y. Koh and A. I. Bush, Nat. Rev. Neurosci., 2005, 6, 449–462 CrossRef CAS PubMed.
- A. M. Grabrucker, Dev. Neurobiol., 2014, 74, 136–146 CrossRef CAS PubMed.
- S. Grabrucker, L. Jannetti, M. Eckert, S. Gaub, R. Chhabra, S. Pfaender, K. Mangus, P. P. Reddy, V. Rankovic, M. J. Schmeisser, M. R. Kreutz, G. Ehret, T. M. Boeckers and A. M. Grabrucker, Brain, 2014, 137, 137–152 CrossRef PubMed.
- J. Y. Lee, J. H. Kim, R. D. Palmiter and J. Y. Koh, Exp. Neurol., 2003, 184, 337–347 CrossRef CAS.
- E. Chorin, O. Vinograd, I. Fleidervish, D. Gilad, S. Herrmann, I. Sekler, E. Aizenman and M. Hershfinkel, J. Neurosci., 2011, 31, 12916–12926 CrossRef CAS PubMed.
- Y. Z. Huang, E. Pan, Z. Q. Xiong and J. O. McNamara, Neuron, 2008, 57, 546–558 CrossRef CAS PubMed.
- D. D. Mott, M. Benveniste and R. J. Dingledine, J. Neurosci., 2008, 28, 1659–1671 CrossRef CAS PubMed.
- J. Veran, J. Kumar, P. S. Pinheiro, A. Athane, M. L. Mayer, D. Perrais and C. Mulle, Neuron, 2012, 76, 565–578 CrossRef CAS PubMed.
- A. M. Vergnano, N. Rebola, L. P. Savtchenko, P. S. Pinheiro, M. Casado, B. L. Kieffer, D. A. Rusakov, C. Mulle and P. Paoletti, Neuron, 2014, 82, 1101–1114 CrossRef CAS PubMed.
- C. Sindreu, A. Bayes, X. Altafaj and J. Perez-Clausell, Mol. Brain, 2014, 7, 16 CrossRef PubMed.
- S. Pfaender and A. M. Grabrucker, Metallomics, 2014, 6, 960–977 RSC.
- D. F. Caldwell, D. Oberleas, J. J. Clancy and A. S. Prasad, Proc. Soc. Exp. Biol. Med., 1970, 133, 1417–1421 CrossRef CAS.
- K. A. Keller, A. Grider and J. A. Coffield, Physiol. Behav., 2001, 72, 339–348 CrossRef CAS.
- G. Martel, C. Hevi, N. Kane-Goldsmith and G. P. Shumyatsky, Behav. Brain Res., 2011, 223, 233–238 CrossRef CAS PubMed.
- D. L. Benson, L. M. Schnapp, L. Shapiro and G. W. Huntley, Trends Cell Biol., 2000, 10, 473–482 CrossRef CAS.
- G. W. Huntley, O. Gil and O. Bozdagi, Neuroscientist, 2002, 8, 221–233 CrossRef CAS PubMed.
- S. Murase, E. Mosser and E. M. Schuman, Neuron, 2002, 35, 91–105 CrossRef CAS.
- J. Arikkath and L. F. Reichardt, Trends Neurosci., 2008, 31, 487–494 CrossRef CAS PubMed.
- O. Bozdagi, W. Shan, H. Tanaka, D. L. Benson and G. W. Huntley, Neuron, 2000, 28, 245–259 CrossRef CAS.
- O. Bozdagi, X. B. Wang, J. S. Nikitczuk, T. R. Anderson, E. B. Bloss, G. L. Radice, Q. Zhou, D. L. Benson and G. W. Huntley, J. Neurosci., 2010, 30, 9984–9989 CrossRef CAS PubMed.
- L. Tang, C. P. Hung and E. M. Schuman, Neuron, 1998, 20, 1165–1175 CrossRef CAS.
- W. Baumgartner, N. Golenhofen, N. Grundhofer, J. Wiegand and D. Drenckhahn, J. Neurosci., 2003, 23, 11008–11014 CAS.
- W. Baumgartner, A. Osmanagic, M. Gebhard, S. Kraemer and N. Golenhofen, Synapse, 2013, 67, 705–715 CrossRef CAS PubMed.
- W. M. Heupel, W. Baumgartner, B. Laymann, D. Drenckhahn and N. Golenhofen, Mol. Cell. Neurosci., 2008, 37, 548–558 CrossRef CAS PubMed.
- W. Baumgartner, G. J. Schutz, J. Wiegand, N. Golenhofen and D. Drenckhahn, J. Cell Sci., 2003, 116, 1001–1011 CrossRef CAS.
- B. Bartelt-Kirbach, K. Langer-Fischer and N. Golenhofen, Cell Commun. Adhes., 2010, 17, 75–82 CrossRef CAS PubMed.
- A. M. Fannon and D. R. Colman, Neuron, 1996, 17, 423–434 CrossRef CAS.
- N. Uchida, Y. Honjo, K. R. Johnson, M. J. Wheelock and M. Takeichi, J. Cell Biol., 1996, 135, 767–779 CrossRef CAS.
- M. B. Dalva, A. C. McClelland and M. S. Kayser, Nat. Rev. Neurosci., 2007, 8, 206–220 CrossRef CAS PubMed.
- H. H. Jan, I. T. Chen, Y. Y. Tsai and Y. C. Chang, J. Neurochem., 2002, 83, 525–534 CrossRef CAS.
- C. O. Hershey, L. A. Hershey, A. Varnes, S. D. Vibhakar, P. Lavin and W. H. Strain, Neurology, 1983, 33, 1350–1353 CrossRef CAS.
- C. J. Frederickson, L. J. Giblin, A. Krezel, D. J. McAdoo, R. N. Mueller, Y. Zeng, R. V. Balaji, R. Masalha, R. B. Thompson, C. A. Fierke, J. M. Sarvey, M. de Valdenebro, D. S. Prough and M. H. Zornow, Exp. Neurol., 2006, 198, 285–293 CrossRef CAS PubMed.
- S. Y. Assaf and S. H. Chung, Nature, 1984, 308, 734–736 CrossRef CAS.
- G. A. Howell, M. G. Welch and C. J. Frederickson, Nature, 1984, 308, 736–738 CrossRef CAS.
- A. R. Kay, J. Neurosci., 2003, 23, 6847–6855 CAS.
- A. Takeda, S. Takada, M. Ando, K. Itagaki, H. Tamano, M. Suzuki, H. Iwaki and N. Oku, Neuroscience, 2010, 171, 443–450 CrossRef CAS PubMed.
- A. Passerini, M. Lippi and P. Frasconi, Nucleic Acids Res., 2011, 39, W288–W292 CrossRef CAS PubMed.
- H. Haase, S. Hebel, G. Engelhardt and L. Rink, Anal. Biochem., 2006, 352, 222–230 CrossRef CAS PubMed.
- L. M. Plum, A. Brieger, G. Engelhardt, S. Hebel, A. Nessel, M. Arlt, J. Kaltenberg, U. Schwaneberg, M. Huber, L. Rink and H. Haase, Metallomics, 2014, 6, 1277–1287 RSC.
- G. Grynkiewicz, M. Poenie and R. Y. Tsien, J. Biol. Chem., 1985, 260, 3440–3450 CAS.
- A. Dieckmann-Schuppert and H. J. Schnittler, Cell Tissue Res., 1997, 288, 119–126 CrossRef CAS.
- W. Heinzel, A. Vogt, E. Kallee and W. Faller, J. Lab. Clin. Med., 1965, 66, 334–340 CAS.
| This journal is © The Royal Society of Chemistry 2015 |