
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vesicular disruption of lysosomal targeting organometallic polyarginine bioconjugates†
Annika
Gross‡
*a,
Hamed
Alborzinia
b,
Stefania
Piantavigna
c,
Lisandra L.
Martin
c,
Stefan
Wölfl
b and
Nils
Metzler-Nolte
*a
aDepartment of Chemistry and Biochemistry, University of Bochum, Universitätsstrasse 150, D-44801 Bochum, Germany. E-mail: Nils.Metzler-Nolte@ruhr-uni-bochum.de; Annika.Gross@ruhr-uni-bochum.de; Fax: +49 (0)234-32 14378; Tel: +49 (0)234 32 28152
bInstitute of Pharmacy and Molecular Biotechnology, Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 364, D-69120 Heidelberg, Germany
cSchool of Chemistry, Monash University, Clayton, Victoria, Australia
First published on 22nd January 2015
Abstract
Compounds which are able to destabilize the lysosomal membrane have been proposed as interesting candidates for targeted anticancer drugs due to the pronounced lysosomal changes in cancer cells. For this purpose, metallocene derivatives of a cell penetrating polyarginine peptide M–(Arg)9(Phe)2Lys–NH2 (where M = ferrocene carboxylate or ruthenocene carboxylate) were designed and their biological activities were investigated in detail. The ferrocenoyl- and ruthenocenoyl polyarginine bioconjugates were synthesized via Fmoc solid-phase peptide synthesis (SPPS) protocols on a microwave-assisted synthesizer. After HPLC purification >98% purity was observed for all conjugates. Their interaction with supported biomimetic membranes was investigated on a quartz crystal microbalance (QCM) and revealed a very strong binding of the metallocene peptides and their metal-free congeners to an artificial eukaryotic membrane model (DMPC–cholesterol). To demonstrate their antiproliferative utility as cytotoxic compounds for a targeted anticancer drug, cell viability (by the crystal violet assay), apoptosis (flow cytometry, Ann V/PI staining), induction of reactive oxygen species (ROS, by flow cytometry with dihydroethidium staining), and changes in cancer cell metabolism, e.g. respiration and glycolysis, were studied. Our results reveal only a weak toxicity for the metal-free polyarginine peptide, which could be significantly enhanced (to ca. 50 μM against HeLa cells in the best case) by coupling ferrocene or ruthenocene carboxylates to the N-terminus of the peptide. The investigation of the cellular uptake and intracellular localization by fluorescence microscopy revealed an enhanced vesicular disruption by the metallocene bioconjugate compared to the metal-free derivative which could be triggered by light and chemicals. Further studies of apoptosis, respiration, glycolysis and ROS formation reveal the superior characteristics of the metallocene compounds. While most cells remain viable even at 300 μM of the metal free bioconjugate 1, most cells are dead or in late stages of apoptosis at 200 μM of the ruthenocene derivative 3, and at 100 μM of the most active ferrocene derivative 2, however, all show very little sign of necrosis. Also, the metal free compound 1 does not induce ROS formation but both metallocene–polyarginine bioconjugates are clearly associated with enhanced intracellular ROS levels, with levels for the redox-active ferrocene derivative being two times higher than for the structurally very similar but redox-silent ruthenocene derivative. We propose that such metallocene–polyarginine peptides induce lysosomal membrane permeabilization and thereby could be developed towards targeted anticancer drugs.
Introduction
Lysosomes are important intracellular vesicles responsible for degradation of cellular components. They maintain a lower pH than the rest of the cell, and contain numerous hydrolytic enzymes such as hydrolases, peptidases, phosphatases, and cathepsins. For a long time, lysosomal membrane permeabilization (LMP) was only known to release hydrolases in an unspecific manner, resulting in uncontrolled necrosis. More recently, it was established that this is valid only in the case of massive lysosomal leakage, whereas minor leakage seems to be an important part of the apoptotic pathway.1 Therefore, targeting of lysosomes was proposed as an interesting, novel pathway for cancer therapy. Controlled LMP is capable to either initiate early events of cell death by releasing cathepsins, or to amplify later events in the apoptotic pathway by modulation of various apoptotic stimuli, including death receptor activation.2 Also, reactive oxygen species (ROS) play an important role since they lead to membrane destabilization via peroxidation of membrane lipids. Alternatively, modification in the intracellular redox balance may start a redox-dependent signaling cascade, again resulting in LMP.2 Iron complexes or iron-containing proteins increase lysosomal vulnerability,3–7 and furthermore, changes in the membrane structure may lead to a revised fluidity, which can result in lysosomal destabilization.8–12 In terms of therapeutic potential, lysosomotropic detergents – i.e. basic compounds that accumulate in acidic lysosomes and disrupt them from within – were considered as putative anticancer drugs.1 Cancer cells and particularly multidrug-resistant cancer cells may be targeted by this method,1 due to their strong lysosomal changes which are associated with invasive growth and angiogenesis in cancer cells.13,14 However, these changes of the lysosomes for example in expression and activity of lysosomal cysteine cathepsins correlate with the metastatic capacity and aggressiveness of tumors and should sensitize cells to the lysosomal cell death pathway even when apoptosis is inhibited.15For our studies we assumed that basic polyarginine peptides, also known as cell-penetrating peptides (CPP), might be capable of targeting lysosomes and inducing LMP.16–18 CPPs are artificially designed peptides or variegated sequences based on natural peptides or proteins to guide a wide variety of cargoes into living cells.19–21 Polyarginine peptides can enter cells in a receptor-independent way, gaining access into nearly all types of cells and tissues. This propensity has made them optimal tools to deliver molecular cargoes into cells. The mechanism is not known in detail,22,23 but one possible mode of action is the interaction of the cationic peptide with the cell surface glycosaminoglycans, followed by an energy dependent cellular uptake mechanism, probably via endocytosis. In live cell experiments endocytotic vesicles are visible. To escape the vesicles, heparan sulfate proteoglycans have to be degraded by heparanases to free the peptide.24 The basic nature of polyarginines makes them promising candidates for LMP. This study was designed as a first step to test such assumptions, with the purpose of further developing such peptides and their derivatives as anticancer drugs in the future.
Since iron compounds were able to enhance vesicular disruption3–7 we were interested in the effect of metallocenes, especially ferrocene, linked to the basic peptide polyarginine for the purpose of lysosomal disruption towards anticancer therapy. Recently, organometallic compounds have attracted great attention and their applications in medicinal chemistry have been reviewed.25–27 Ferrocene is the by far most important and most investigated metallocene in medicinal chemistry.28–30 It is very stable towards air and water, which makes it an ideal workhorse within a biological context. Furthermore, it has interesting characteristics due to its redox properties like its ability to undergo Fenton chemistry, which makes intracellular ROS production possible. Ruthenocene is very similar to ferrocene regarding its structure, lipophilicity and size, but it is more stable and not redox active, hence unable to induce Fenton chemistry. There is a wide variety of ferrocene conjugates known, but much less attention has been directed to the investigation of ruthenocene bioconjugates. In a pioneering study, our group recently presented the synthesis of a ruthenocene PNA oligomer and peptide conjugates.31–35
For the investigation of metallocene–polyarginine bioconjugates as candidates for LMP, we functionalize the peptide with ferrocene (2, 5, Fig. 1) or ruthenocene (3, 6). Furthermore, the compounds were labeled with the fluorophore FITC (5, 6) to investigate their intracellular biological properties in terms of cytotoxicity and lysosomal localization. We have chosen a polyarginine peptide based on an optimized sequence by Iversen et al. which contains two phenylalanines in addition to the arginine core sequence for an improved interaction with the cell membrane.36 As a bonus, these two phenylalanine residues facilitate detection during HPLC purification due to their absorption in the UV.
Experimental section
General experimental conditions
All reagents and HPLC-grade solvents were purchased from Acros (Geel, Belgium), Aldrich/Sigma/Fluka (Deisenhofen, Germany), E. Merck (Darmstadt, Germany), Novabiochem (Laufelfingen, Switzerland) and IRIS Biotech (Marktredwitz, Germany) and were used without further purification. The Rink Amide linker (IRIS Biotech) and only L-amino acids were used throughout the synthesis. FITC was ordered from Fluka Chemicals, DMF from Roth. All materials for cell culture were purchased from Invitrogen Corporation (Karlsruhe, Germany) unless otherwise specified. HPLC fractions of all products were frozen in liquid nitrogen and lyophilized using a Christ Alpha 1-4 LD plus freeze dryer. All aqueous solutions were prepared from Millipore® water and filtered with a 0.22 μm syringe filter before use.Instrumentation and analytical measurements
A Liberty Microwave Peptide Synthesizer from CEM was used for peptide synthesis. MALDI-TOF mass spectra were recorded on ‘Daltonics® Autoflex’ or Ultraflex III instruments (Bruker, Bremerhaven, Germany) in linear mode with positive polarity using sinapinic acid as the matrix. ESI mass spectra were recorded on an Esquire 6000 instrument (Bruker, Bremerhaven, Germany). Nuclear magnetic resonance spectra were recorded on a Bruker DRX 600 MHz spectrometer (Bruker, Karlsruhe, Germany). 1H and 13C chemical shifts are given in ppm and were referenced with the residual solvent resonances relative to tetramethylsilane (TMS). Absorption was measured with a Tecan Microplate Platereader Saphire2 (Tecan, Crailsheim, Germany). Uptake studies were implemented on the Olympus IX51 fluorescence microscope equipped with an Olympus XC10 camera and localization studies were performed on a Leica DMIRE2 confocal microscope containing a Leica TCS SP2 and Leica CTRMIC.HPLC analysis and purification
HPLC analysis and purifications were carried out using C18 analytical (Varian Dynamax, 4.6 mm × 250 mm) and C18 semipreparative (Varian Dynamax, 21.4 mm × 250 mm) columns on a customised Varian Prostar instrument. Linear gradient, 5–95% MeCN in 18 min, eluents: H2O and MeCN both containing 0.1% (v/v) TFA. Analytical (flow rate: 1.0 mL min−1) and preparative (flow rate: 4.0 mL min−1) runs were performed with a linear gradient of A (95% millipore® water, 5% MeCN, 0.1% TFA (v/v/v)) and B (5% millipore® water, 95% MeCN, 0.1% TFA (v/v/v)). Analytical runs: t = 0 min: 0% B; t = 18 min: 100% B; t = 26 min: 0% B; t = 30 min: 0% B. Preparative runs: t = 0 min: 0% B; t = 45 min: 50% B; t = 55 min: 100% B; t = 65 min: 0% B. All samples were filtered before injection using a 0.22 μm syringe filter. Spectra were recorded at 254 nm and ambient temperature, retention times (tR/[min]) were noted in each case.Solid-phase peptide synthesis
The resin bound Fmoc-polyarginine was synthesized on an automated peptide synthesizer using standard protocols (amino acid coupling: TBTU in DMF (0.5 M), HOBt in DMF (0.5 M), DIPEA in NMP (2 M) and amino acids in DMF (0.2 M); arginine coupling: 25 min, 75 °C, 0 W followed by 5 min, 75 °C, 25 W; standard amino acid coupling: 5 min, 75 °C, 24 W. For deprotection: 20% piperidine in DMF: initial deprotection: 0.5 min, 75 °C, 30 W followed by deprotection: 3 min, 75 °C, 50 W). Afterwards, aliquots of 100 mg peptide containing resin was transferred into a filter-containing syringe for further derivatization. For the synthesis of all compounds, the N-terminal Fmoc group was deprotected and either acetylated (1, 4) or coupled with ferrocenecarboxylic acid (2, 5) or ruthenocenecarboxylic acid (3, 6). After each step the resin was washed 5 times using 2 mL DMF.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:4:6 equiv.) in DMF for 1 h or 3 h for the coupling of ferrocene- or ruthenocenecarboxylic acid, respectively.
:2.5:2.5) or TFA/phenol/TIS (2 mL, 85:10:5) for 6 h at room temperature. The resin was filtered and washed with 0.5 mL TFA. Addition of cold diethyl ether yielded a precipitate, which was washed repeatedly with diethyl ether. The crude product was dissolved in MeCN–water, filtered, lyophilized and afterwards purified and analyzed with RP-HPLC and finally characterized with MALDI-TOF mass spectrometry.
:4:4:6 equiv.) in DMF for 1 h or 3 h for the coupling of ferrocene- or ruthenocenecarboxylic acid, respectively.
:2.5:2.5) or TFA/phenol/TIS (2 mL, 85:10:5) for 6 h at room temperature. The resin was filtered and washed with 0.5 mL TFA. Addition of cold diethyl ether yielded a precipitate, which was washed repeatedly with diethyl ether. The crude product was dissolved in MeCN–water, filtered, lyophilized and afterwards purified and analyzed with RP-HPLC and finally characterized with MALDI-TOF mass spectrometry.
1: white solid, C80H143N41O13 (1887.26 g mol−1): MS (MALDI-TOF): m/z 1888.0 [M + H]+, 1910.0 [M + Na]+, HPLC: tR = 10.5 min. 1H NMR (90% H2O: 10% D2O, 600.13 MHz): δ = 8.49–8.42 (m, 6H, HNH,Arg), 8.37–8.33 (m, 4H, HNH,Arg), 8.33 (m, 1H, HNH,Phe1), 8.23 (m, 1H, HNH,Lys), 8.21 (m, 1H, HNH,Phe2), 7.57 (br, 3H, Hζ,Lys), 7.39 (m, 2H, Hphenyl,PheA), 7.38 (m, 2H, Hphenyl,PheB), 7.35 (m, 2H, 1Hphenyl,PheA, 1Hphenyl,PheB), 7.28 (m, 2H, Hphenyl,PheA), 7.27 (m, 2H, Hphenyl,PheB), 6.71 (br, 36H, Hη,Arg), 4.65 (m, 1H, Hα,Phe1), 4.63 (m, 1H, Hα,Phe2), 4.39–4.28 (m, 9H, Hα,Arg), 4.20 (m, 1H, Hα,Lys), 3.29–3.14 (m, 18H, Hδ,Arg), 3.11 (m, 1H, Hβ2,Phe1), 3.10 (m, 1H, Hβ2,Phe2), 3.06 (m, 1H, Hε2,Lys), 3.04 (m, 1H, Hε3,Lys), 3.03 (m, 1H, Hβ3,Phe1), 3.01 (m, 1H, Hβ3,Phe2), 2.10 (s, 3H, HAcetyl), 1.90–1.84 (m, 9H, Hβ2,Arg), 1.89–1.45 (m, 18H, Hγ,Arg), 1.83–1.75 (m, 9H, Hβ3,Arg), 1.82 (m, 1H, Hβ2,Lys), 1.74 (m, 1H, Hγ2,Lys), 1.72 (m, 1H, Hβ3,Lys), 1.71 (m, 1H, Hγ3,Lys), 1.43 (m, 1H, Hγ2,Lys), 1.40 (m, 1H, Hγ3,Lys). 13C NMR (90% H2O: 10% D2O, 150.92 MHz): δ = 182.6, 175.9 (CON), 174.6 (CON), 174.3 (CON), 173.6 (CON), 173.5 (CON), 173.4 (CON), 173.3 (CON), 173.1 (CON), 172.7 (CON), 172.2 (CON), 172.1 (CON), 136.2, 136.1 (CγPhe1,Phe2), 129.3, 129.2, 128.8, 128.7 (Cδ,Phe1, Cδ,Phe2, Cε,Phe1, Cε,Phe2), 127.2, 127.1 (Cζ,Phe1, Cζ,Phe2), 54.8 (Cα,Phe1, Cα,Phe2), 54.6–53.1 (Cα,Arg), 53.5 (Cα,Lys), 40.9–40.7 (Cδ,Arg), 39.5 (Cε,Lys), 37.5 (Cβ,PheA, Cβ,PheB), 30.5 (Cβ,Lys), 28.5–27.6 (Cβ,Arg), 26.6 (Cδ,Lys), 24.9–24.1 (Cγ,Arg), 22.1 (Cγ,Lys).
2: orange solid, C89H149FeN41O13 (2056.16 g mol−1): MS (ESI, pos.): m/z 1029.4 [M + 2H]2+, 686.8 [M + 3H]3+, 515.6 [M + 4H]4+, 412.9 [M + 5H]5+, 344.4 [M + 6H]6+, HPLC: tR = 11.4 min. 1H NMR (DMSO-d6, 600.13 MHz): δ = 8.32 (d, J = 7.1 Hz, 1H, HNH,Phe1), 8.07 (m, 7H, 1HNH,Lys, 6HNH,Arg), 8.02 (m, 2H, 1HNH,Arg, 1HNH,Phe2), 7.97 (d, J = 7.5 Hz, 1H, HNH,Arg), 7.92 (d, J = 7.7 Hz, 1H, HNH,Arg), 7.80–7.60 (m, 12H, 9Hε,Arg, 3Hζ,Lys), 7.60–6.80 (m, 46H, 36Hη,Arg, 10Hphenyl,Phe1,Phe2), 4.89 (m, 1H, HCp2), 4.87 (m, 1H, HCp5), 4.57 (m, 2H, 1Hα,Phe1, 1Hα,Phe2), 4.42 (m, 2H, HCp3,4), 4.41–4.17 (m, 9H, Hα, Arg), 4.23 (s, 5H, HCp′), 4.20 (m, 1H, Hα,Lys), 3.22–3.00 (m, 18H, Hδ,Arg), 3.08 (m, 1H, Hβ2,Phe1), 3.03 (m, 1H, Hβ2,Phe2), 2.88 (m, 1H, Hβ3,Phe1), 2.79 (m, 2H, Hε,Lys), 2.77 (m, 1H, Hβ3,Phe2), 1.84–1.62 (m, 9H, Hβ2,Arg), 1.71 (m, 1H, Hβ2,Lys), 1.62–1.40 (m, 9H, Hβ3,Arg), 1.57 (m, 2H, Hδ,Lys), 1.56 (m, 1H, Hβ3,Lys), 1.33 (m, 2H, Hγ,Lys). 13C NMR (DMSO-d6, 150.92 MHz): δ = 174.1 (CON), 173.0 (CON), 172.3 (CON), 172.0 (CON), 171.8 (CON), 171.6 (CON), 171.0 (CON), 170.7 (CON), 130.1, 130.0, 129.0, 128.8 (Cδ,Phe1, Cδ,Phe2, Cζ,Phe1, Cζ,Phe2), 127.2, 121.4 (Cε,Phe1, Cε,Phe2), 70.8 (CCp3,4), 69.9 (CCp′), 69.8 (CCp2,5), 54.4 (Cα,Phe1,Phe2), 53.6–52.7 (Cα,Arg), 52.9 (Cα,Lys), 41.1 (Cδ,Arg), 39.3 (Cε,Lys), 38.3 (Cβ,Phe2), 38.1 (Cβ,Phe1), 32.2 (Cβ,Lys), 29.8 (Cβ,Arg), 27.4 (Cδ,Lys), 25.6 (Cγ,Arg), 22.8 (Cγ,Lys).
3: white solid, C89H149N41O13Ru (2102.0 g mol−1): MS (MALDI-TOF, Sinap.): m/z 2103.1 [M + H]+, HPLC: tR = 12.8 min. 1H NMR (DMSO-d6, 400.13 MHz): δ = 8.27 (d, J = 7.5 Hz, 1H, HNH,Phe1), 8.16–8.11 (m, 2H, HNH,Arg), 8.06 (d, J = 7.8 Hz, 1H, HNH,Lys), 8.00–7.90 (m, 3H, HNH,Arg), 7.98 (d, J = 7.1 Hz, 1H, HNH,Phe2), 7.88–7.72 (m, 4H, HNH,Arg), 7.85–7.72 (m, 12H, 9Hε,Arg, 3Hζ,Lys), 7.55–7.02 (m, 46H, 36HηArg, 4Hδ,Phe, 4Hε,Phe, 2Hζ,Phe), 5.23 (m, 1H, HCp2), 5.20 (m, 1H, HCp5), 4.70 (m, 2H, HCp3,4), 4.58 (s, 5H, HCp′), 4.52 (m, 1H, Hα,Phe1), 4.50 (m, 1H, Hα,Phe2), 4.33–4.14 (m, 9H, Hα, Arg), 4.16 (m, 1H, Hα,Lys), 3.15–2.69 (m, 18H, Hδ,Arg), 3.04 (m, 1H, Hβ2,Phe1), 2.98 (m, 1H, Hβ2,Phe2), 2.84 (m, 1H, Hβ3,Phe1), 2.75 (m, 2H, Hε,Lys), 2.72 (m, 1H, Hβ3,Phe2), 1.69–1.58 (m, 18H, Hβ,Arg), 1.67 (m, 2H, Hβ,Lys), 1.57–1.33 (m, 18H, Hγ,Arg), 1.52 (m, 2H, Hδ,Lys), 1.29 (m, 2H, Hγ,Lys). 13C NMR (DMSO-d6, 100.61 MHz): δ = 173.3 (CON), 171.8 (CON), 171.7 (CON), 171.5 (CON), 171.4 (CON), 171.3 (CON), 171.1 (CON), 170.9 (CON), 170.7 (CON), 168.5 (CON), 137.6, 137.5 (Cγ,Phe1, Cγ,Phe2), 129.3, 128.8, 128.1, 128.0, 123.1, 120.6 (Cδ,Phe1, Cδ,Phe2, Cε,Phe1, Cε,Phe2, Cζ,Phe1, Cζ,Phe2), 72.1 (CCp3,4), 71.6 (CCp′), 70.5 (CCp5), 70.3 (CCp2), 54.1, 53.6 (Cα,Phe1, Cα,Phe2), 52.6–51.9 (Cα,Arg), 52.1 (Cα,Lys), 41.738.7 (Cδ,Arg), 38.4 (Cε,Lys), 37.6 (Cβ,Phe2), 37.5 (Cβ,Phe1), 31.4 (Cβ,Lys), 31.3 (Cδ,Lys), 29.4–28.3 (Cβ,Arg), 25.3, 25.0–24.8 (Cγ,Arg), 22.0 (Cγ,Lys).
4: yellow solid, C101H154N42O18S (2275.21 g mol−1): MS (MALDI-TOF, Sinap.): m/z 2282.7 [M]+; MS (ESI, pos.): m/z 1138.8 [M + 2H]2+, 944.4 [M–FITC + 3H]2+, 759.6 [M + 3H]3+, 629.9 [M − FITC + 4H]3+, HPLC: tR = 13.1 min.
5: yellow solid, C110H160FeN42O18S (2445.20 g mol−1): MS (MALDI-TOF, Sinap.): m/z 2447.1 [M + H]+, MS (ESI, pos.): m/z 1223.8 [M + 2H]2+, 1029.4 [M − FITC + 3H]2+, 816.8 [M + 4H]3+, 686.6 [M − FITC + 3H]3+, HPLC: tR = 14.8 min.
6: yellow solid, C110H160N42O18RuS (2491.17 g mol−1): MS (ESI, pos.): m/z 831.5 [M + 3H]3+, 701.7 [M − FITC + 4H]3+, 390.0 [FITC + H]+. HPLC: tR = 14.9 min.
Membrane studies
A Quartz crystal microbalance (QCM) was used to assess the interaction between polyarginine peptides and a supported biomimetic membrane (SBM). This approach has been extensively explained in previous works.37,38 Briefly, QCM experiments were performed using the E4 system with flow cells (Q-Sense, Västra Frölunda, Sweden). The QCM instrument measures the relative changes to the resonance frequency (Δf) of the sensor over the course of the experiment. The variation in the resonance frequency is proportional to the change of the mass (Δm) coupled to the sensor surface by the Sauerbrey equation:37| Δm = −C(Δfn/n) |
:3 v/v) or DMPC/DMPG (4:1 v/v), suspended in a 20 mM PBS buffer with 100 mM of NaCl, into the QCM chamber. The deposition occurred thus in situ, onto a gold sensor, which has been previously modified with a layer of 3-mercaptopropionic acid (MPA) or with 6-mercaptohexanoic acid (MHA). Once a stable bilayer (membrane) was formed, various concentrations of peptide, dissolved in PBS, were introduced. The interaction of the peptide and subsequent effects were recorded in real time at the various harmonics (3rd, 5th, 7th and 9th), simultaneously. However, only the 7th is represented in this study, since the other harmonics did not show any substantial difference. The last step of the experiment, after the incubation time for the peptide–membrane system, was a PBS flush with the purpose of removing any peptide molecule weakly bound to the membrane. All experiments were conducted at a temperature of 19.10 ± 0.05 °C.
Cell culture
Cellular uptake
The uptake was investigated by fluorescence microscopy using conjugates labeled with the fluorophore FITC (compounds 4, 5 and 6). 8 × 104 cells were seeded in flat bottom 24 well plates and incubated for 24 h at 37 °C and 5% (HepG2) or 10% CO2 (IMIM-PC2, PT45). Due to an enhanced solubility of the compounds, 0.5% DMSO (maximal concentration in the cell medium) was added. The compounds were dissolved in a DMSO–PBS solution, added to the cell medium and incubated with the cells for 14 h using five different concentrations (1, 5, 10, 25, 50 μM). After completion of the incubation, the cell medium was removed and the cells were washed five times with PBS. The PBS was replaced with cell culture medium and the fluorescence of the cells was measured on a fluorescence microscope Olympus IX51 (magnification 100, 200 or 400 times). To assess the effect of the cellular uptake qualitatively the amount of fluorescent cells and the intensity of the fluorescence were compared visually.Colocalization
Colocalization studies were performed using confocal microscopy. For colocalization, commercially available dyes were used according to the specifications of the manufacturer for live cell experiments. To visualize lysosomes LysoTracker Red® was used, for endosomes the FM 4-64® dye was employed.2.5 × 104 cells were seeded in manually poly-L-lysine coated ibidi μ-slides (8 wells) for 14 h. The cells were incubated with 10 μM of 5 for 2 h. 30 min before the end of the incubation time LysoTracker Red® (75 nM) or FM 4-64® (10 μM) were added. After completion of the incubation, the culture medium was removed and the cells were washed 5 times using PBS and replaced with RPMI 1640 medium without phenol red and supplements. The colocalization was measured on a Leica confocal microscope. Measurement parameters used for colocalization studies of 5 are as followed: excitation wavelength of 488 nm and emission range between 500–525 nm, for LysoTraker Red® excitation wavelength of 543 nm and emission range of 590–620 nm, for FM 4-64® excitation wavelength of 514 nm and emission range of 700–800 nm. Pictures were recorded using the sequential imaging mode. To obtain one image, an average of at least three recorded images and an average of two recorded lines were used.
Cytotoxicity experiments
To determine the activity of 2 and 3 in comparison to 1, two antiproliferative assays, resazurin and crystal violet were performed (see ESI† for details). 6000 cells per well of HeLa, PT45 and HepG2 cells were incubated in 96-well plates at 37 °C and 10% CO2 for 24 h. To determine the initial cell viability and biomass, a t0-plate with HeLa, HepG2 and PT45 was additionally plated and after 24 h incubation the resazurin assay was performed. Subsequently the cells were fixed with 0.2% glutardialdehyde solution, followed by the crystal violet assay. In the remaining plates the medium was replaced with medium containing 1, 2 or 3 with 0.5% DMSO added to improve the solubility of the compound. Concentrations between 1–1000 μM were used and later adapted regarding their antiproliferative activity. Each concentration was tested six times in parallel, as positive control, cisplatin was used and as negative control, medium containing 0.5% DMSO. To determine the effect of metallocenes, ferrocene- and ruthenocenecarboxylic acid were tested. The incubation time of the compounds was 48 h. Then, the compound-containing medium was removed, the cells were washed with PBS and the resazurin assay and afterwards the crystal violet assay (see ESI†) was performed. The mean absorption of the initial cell plate (t0-plate) for each cell line was subtracted from the absorption of each experiment and control. For both assays the negative control (non-treated) was set to 100%.Apoptosis
ROS measurements
After the treatment of HeLa cells with indicated concentrations of the compound, the cells were collected, washed and re-suspended (2.5 × 105 cells per 0.5 mL) in FACS buffer [D-PBS (Gibco) with 1% BSA (PAA)]. 1.25 μl of 5 mM dihydroethidium (D1168, Molecular Probes, Invitrogen) solution was added to each sample followed by 15 minutes incubation at RT in the dark. Signal intensity in which represent the intracellular ROS level was analyzed using a FACS® Calibur (Becton Dickinson) and CellQuest Pro (BD) analysis software. Excitation and emission settings were 488 nm and 564–606 nm (FL2 channel).Cell cycle studies
After 24 h treatment of 5 × 105 HeLa cells with indicated concentrations of substances, cells were harvested and fixed with 70% ethanol. Fixed cells were kept at −20 °C for at least one day and then washed with PBS and stained with propidium iodine for 30 min in the dark. Samples were then analyzed by FACS® Calibur to determine the DNA content and cell cycle analysis.BIONAS real-time cell metabolism assay
Approximately 2 × 105 HeLa cells were seeded in 450 μl medium in each biosensor chips (SC1000) 24 h prior insertion of the chips into the Bionas 2500 analyzer (Bionas, Rostock, Germany). The cell number used results in approximately 80% cell confluence on the chip surface after 24 h. During the analysis of cell metabolism, cells were fed with DMEM running medium (PAN Cat. Nr. P03-0010) without sodium bicarbonate, and only buffered with 1 mM Hepes, supplemented 0.1% FCS. A Bionas 2500 analyzing system was used to continuously record two important physiological cellular parameters over time: oxygen consumption, change in medium pH.39 The metabolic sensor chips (SC1000) include ion-sensitive field effect transistors (ISFETs) to record pH changes which are linked with glycolytic activity of the cells, and Clark-type electrodes to monitor oxygen consumption representing mitochondrial activity. To measure the activity of the compounds we included the three following steps: (i) 6 h equilibration with running medium (RM). (ii) drug incubation with substances freshly dissolved in medium at the indicated concentrations for treatment periods of up to 24 h, and (iii) a drug-free step in which cells are again fed with running medium only. At the end of each experiment, the cells were killed by addition of 0.2% Triton X-100 to get a basic signal without living cells on the sensor surface as a negative control.Results
Synthesis
The peptides and peptide conjugates used in this study are shown in Fig. 1. For our purpose, we adapted the polyarginine sequence designed by Iversen et al. with minor changes.36 Instead of including a cysteine as part of the original sequence, we integrated a lysine to accommodate the fluorophore FITC. | ||
| Fig. 1 Conjugates 1–6. | ||
The synthesis route of the bioconjugates reported in this article is shown in Scheme 1. The polyarginine peptide was prepared by solid-phase peptide synthesis (SPPS) using the Fmoc technique. The complete peptide sequence ((Arg)9–(Phe)2–Lys) was assembled on an automated, microwave assisted synthesizer. All subsequent steps were performed manually. After deprotection of the terminal Fmoc group the peptide was either acetylated (1, 4), or ferrocenecarboxylic acid (2, 5) and ruthenocenecarboxylic acid (3, 6) were coupled to the N-terminus of the sequence, respectively. In compounds 1, 2 and 3 the fluorophore is absent (see Fig. 1), with the sequence containing an unmodified lysine. The synthesis of compounds 4, 5 and 6 required an orthogonally cleavable protecting group to bind FITC after deprotection. To this end, we have chosen the rather acid labile Mtt group. The fluorophore labeling was achieved by removal of the Mtt group by 1% TFA, followed by FITC coupling to the side chain of the unprotected lysine.
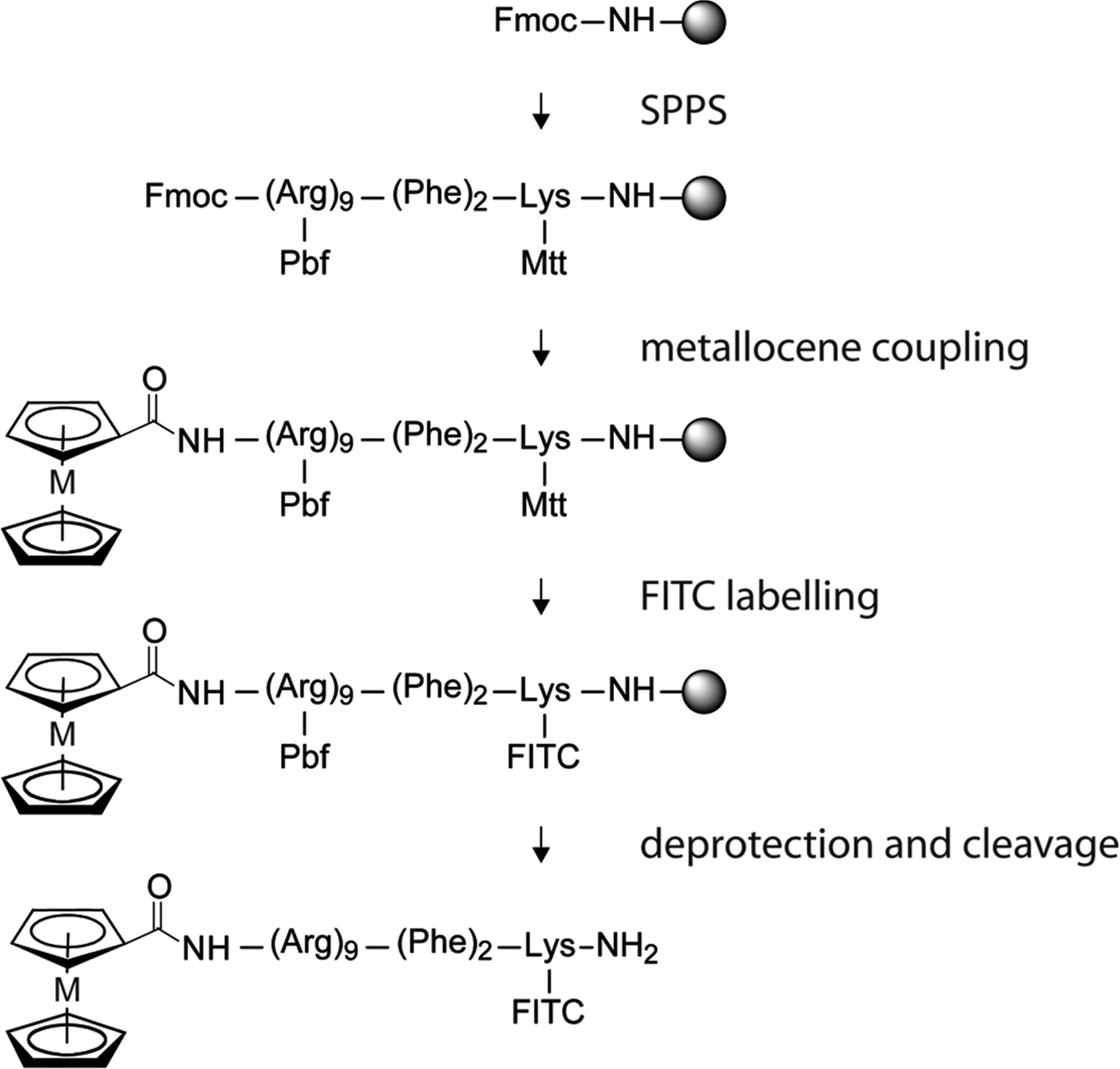 | ||
| Scheme 1 Schematic preparation of FITC-labeled metallocene polyarginine conjugates; M = Fe (5), Ru (6). | ||
The conjugates were successfully cleaved from the resin after 6 hours by 85–95% TFA, which leaves the metallocene units untouched. All compounds were obtained successfully in a good yield and purity. The crude product of 1 and 3 were obtained with a purity of >95%.
The conjugates were purified by reverse-phase HPLC on a C18 column and found to be >98% pure by subsequent analytical HPLC of all metallocene-containing fractions. The identity of the purified products was verified by mass spectrometry (ESI and MALDI-TOF). Furthermore, compounds 1, 2 and 3 were additionally characterized by 1D and 2D NMR spectroscopy.
HPLC chromatograms of the purified compounds reveal a short retention time of the bioconjugate due to the hydrophilicity of the peptide moiety. Coupling of ferrocene or ruthenocene to polyarginine results in a higher lipophilicity which was further increased by the FITC label (5, 6). Ferrocene and ruthenocene conjugation show a longer but comparable retention time, due to the comparable lipophilicity of these metallocenes (see Experimental section and Fig. S1, ESI†).
Biological studies
The biological properties were examined to evaluate the potential of metallocene polyarginine bioconjugates towards lysosomal destabilization and possible antiproliferative activity. Therefore, we studied their cytotoxicity, cellular uptake, localization and vesicular/membrane destabilization on live cells and on artificial membranes. To gain further insights into the mode of action, induction of apoptosis and necrosis, ROS formation, and their influence on the fluidity of a eukaryotic-mimic membrane were studied. Furthermore, the impact on cell cycle, cellular respiration and glycolysis were also investigated.:1 v/v) (eukaryotic mimic).38,40
 | ||
| Fig. 2 Plots of frequency change vs. time (Δf − t plots) obtained for peptides 1 (A) and 3 (B) interacting with a DMPC–cholesterol membrane. The concentrations used are 1, 5 and 10 μM (from the lightest to the darkest trace). The 7th harmonic only is shown. The vertical dash line indicates the time when the peptide flow stopped (II), while I. corresponds to the beginning of the peptide injection into the QCM chamber. | ||
The QCM traces reveal, firstly, that peptides bind very strongly in a trans-membrane manner with no indication of membrane removal (see Fig. S2, ESI†). Furthermore, the change in frequency is the same for both peptides (1, 3) (Δf = −8 Hz for 5 μM). To notice also that this frequency change occurs for all the three concentrations tested, with a difference of ±0.3 Hz. Indeed, the peptides at these three concentrations nearly saturate the membrane, since the traces start to flatten when the peptide flow terminates (II). Similar behaviour was observed also for the peptide 2, which showed a similar uptake (see Fig. S3, ESI†). However, these peptides acted as membranolytic peptides towards negatively charged membranes (DMPC/DMPG, 4:1 v/v), that mimic a bacterial cell membrane.38,41 Indeed, these peptides inserted into the membrane although this binding is immediately followed by removal of material (mass), presumably lipid-rich molecules, which corresponds to an increase in frequency (see Fig. S4, ESI†).
Fluorophore labeled bioconjugates 4, 5 and 6 were tested on three cell lines, HepG2, IMIM-PC2 and PT45, revealing a good uptake in all three cell lines. The HepG2 cell line was previously found to give reliable and reproducible results in cell uptake studies of metallocene–peptide conjugates.28,42 The other two cell lines are derived from pancreatic cancers, which are more difficult to treat clinically and show a reduced uptake in general.
Incubation of cells with a high concentration of 50 μM reveals a good cellular uptake into the tested cell lines (Fig. 3A). Interestingly, here the breakage of the vesicular structure was observed during exposure to the excitation light of the microscope at 488 nm (Fig. 3B).
 | ||
| Fig. 3 Cellular uptake of 5 (50 μM) in PT45 cells after 14 h incubation. (A) fluorescence image, t = 0; (B) fluorescence image, t = 3 min; (C) phase contrast. FITC-filter, 200× magnification. | ||
Fig. S5 (see ESI†) presents the results of the cellular uptake studies of PT45 at 10 μM, bearing in mind, that the uptake into the other two cell lines HepG2 and IMIM-PC2 is comparable (data not shown). Cellular uptake studies of the polyarginine bioconjugates 4, 5 and 6 revealed a good uptake in PT45 cells (Fig. S5, ESI†) appearing in a vesicular perinuclear localization. By visual inspection, an incubation of 10 μM of 4 results in a cellular uptake in around 50% of the cells. The amount of fluorescent cells increases with higher peptide concentration. Photo-induced breakage of the vesicular structure was observed for 4 by the microscope's excitation light after a relatively long exposure time of approximately 5 min (Fig. S5B, ESI†), which could be impelled by a higher compound concentration, revealing a concentration dependent light-induced vesicular breakage for the fluorophore labeled polyarginine peptide.
Uptake studies of the metallocene peptides 5 and 6 show a stronger destabilization of the vesicles and an enhanced breakage (see Fig. 3 and Fig. S5D–I, ESI†) in comparison to 4, which is in agreement with the observed higher uptake for both compounds. Furthermore, with a higher peptide concentration the exposure time could be dramatically reduced. Therefore, as seen in Fig. 3A the breakage of the vesicular structure starts immediately.
Incubation of the cells with a 10 μM concentration of the metallocene bioconjugates 5 and 6 exhibit a significant uptake as seen by an intense fluorescence and photo induced breakage of vesicles in nearly all plated PT45 cells (Fig. S5D–I, ESI†). They show a vesicular, perinuclear localisation comparable to 4, but additionally exhibit a low cytosolic localization, indicating a light independent leakage of compound out of the vesicles into the cytosol. However, metallocene peptides 5 and 6 reveal an enhance uptake compared to 4. This is presented by an increased content of fluorescent cells exhibiting a higher intensity of the fluorescence and a quicker and more extensive light induced breakage of the vesicular structure, with a homogeneous distribution in the cytosol and a higher intensity in the nucleus. The efficiency of the cellular uptake increases in the order acetyl-polyarginine (4) < ruthenocenoyl-polyarginine (6) < ferrocenoyl-polyarginine (5).
Colocalization of 5 results in a detained uptake of FM 4-64® (Fig. 4A–C). Cells containing FM 4-64® after the incubation reveal the release of 5 into the cell body indicating the chemically induced disruption of the vesicles. Breakage of vesicles due to incubation with FM4-46® was not seen by us with previous tested bioconjugates and shows the sensitivity of the cells towards the ferrocene containing polyarginine (2, 5), not be seen with other ferrocene conjugates.35
 | ||
| Fig. 4 Colocalization study in HepG2 cells; (A) 5 (10 μM), (B) FM 4-64® (10 μM), (C) overlay of A and B. (D) 5 (10 μM), (E) LysoTracker Red® (75 nM), (F) overlay of D and E. | ||
Partial colocalization was observed with LysoTracker Red® (Fig. 4D–F). No light induced disruption was visible by confocal microscopy. We observed leakage of vesicles in the fluorescence microscopy after 14 h incubation, however, not by using confocal microscopy after 2 h incubation. This might originate from the different incubation times and light intensity/energy. In conclusion we can observe enhanced leakage for metallocene conjugates, as well as enhanced photoinduced breakage of vesicles by coupling of ferrocene and ruthenocene and enhanced chemical induced breakage for ferrocene polyarginine.
| Compound | HeLa [μM] | PT45 [μM] | HepG2 [μM] |
|---|---|---|---|
| 1 | 179 ± 36 | 246 ± 39 | 291 ± 38 |
| 2 | 48 ± 9 | 92 ± 14 | 69 ± 7 |
| 3 | 70 ± 9 | 124 ± 22 | 102 ± 12 |
| FcC(O)OH, RcC(O)OH | >1000 | >1000 | >1000 |
| Cisplatin | 1.3 ± 0.2 | 0.9 ± 0.2 | 2.4 ± 0.4 |
This antiproliferative effect was increased by functionalization of the peptide with the metallocenes ruthenocene or ferrocene (3, 2) by 2–4-fold. Interestingly, the enhanced toxicity is dependent on the metallocene coupled to the peptide. Both metallocenes do enhance the bioconjugates' lipophilicity and therefore expected unspecific uptake in a comparable way, as shown by HPLC retention times as well as previous logP experiments on metallocene peptide bioconjugates.35 Nevertheless, both bioconjugates show a distinct difference (around 1.5-fold) in their toxicity. This result either indicates a higher cellular uptake of the toxic polyarginine peptide sequence by coupling to ferrocene- over ruthenocene or an additional cytotoxic effect originating from the ferrocene moiety such as enhanced ROS production for example due to the ability of iron(II) center in ferrocene to undergo Fenton chemistry directly as shown by Osella and coworkers.49
HeLa cells were treated for 48 h with indicated concentrations of compounds 1, 2 and 3, and flow cytometry analysis upon Annexin V/propidium iodide (AnnV/PI) staining was performed. AnnV staining indicates apoptototic cells, whereas propidium iodide stains only dead cells/necrotic cells. Single staining of AnnV reveals early apoptosis, combined AnnV–PI coloring shows late apoptosis whereas PI staining alone reveals necrosis due to complete cell membrane destruction. As shown in Fig. 5, none of the tested compounds induce necrosis. Depending on their antiproliferative activity the compounds promote early and late apoptosis. For the metal free bioconjugate 1, most cells are still viable even at a fairly high concentration of 300 μM, whereas upon treatment with 200 μM of the ruthenocene derivative 3 most cells are dead or are in the stage of late apoptosis. The iron containing compound 2, the most active compound of this series shows mainly late apoptosis already at a concentration of 100 μM also without inducing necrosis. Even at those high concentrations there is no sign of necrosis induction (see also Fig. S6, ESI†).
 | ||
| Fig. 5 Apoptosis induction of 1, 2 and 3 in HeLa cells at the indicated concentrations after 48 h. | ||
The intracellular ROS level of 1, 2 and 3 were quantified upon treatment of HeLa cells (Fig. 6). Cells were treated with indicated concentrations of the compound and the intracellular ROS levels were measured after 48 h using flow cytometry upon dihydroethidium staining.
 | ||
| Fig. 6 Intracellular ROS level of HeLa cells treated with 1, 2 and 3 at indicated concentrations for 48 h. | ||
The metal free reference compound 1 does not induce ROS formation compared to the non treated cells (NT). However, the metallocene–polyarginine bioconjugates (2, 3) clearly reveal an enhanced intracellular ROS induction. As expected the iron containing bioconjugate 2 shows the highest ROS formation of the three tested compounds. Whether this is the consequence of the higher toxicity and therefore only a secondary effect of a higher apoptosis induction or whether this result is primarily due to the redox chemistry of the iron of the ferrocene head group itself is not clear and needs further investigation. For compound 3, no redox activity is expected of the Ru(II) under the intracellular condition present. Therefore, here the increase in ROS seems to be the result of the induced apoptosis. Compound 2 exhibits a two times higher ROS induction than 3 which is in agreement with the antiproliferative activity of the compounds and therefore, seems to be a secondary effect of the apoptosis. This assumption is consistent with the rather high concentration needed to induce ROS formation in relation to the IC50 values.
 | ||
| Fig. 7 (a) Standard acidification and (b) standard respiration rate of 1, 2 and 3 (100 μM) in HeLa cells. | ||
The result reveals a negative influence of 1, 2 and 3 (at concentrations of 100 μM) towards the respiration and acidification resulting from an almost immediate inhibition of glycolysis and respiration. However, the way the respiration as well as glycolysis have been effected shows a general toxicity rather than a direct block of the energy production. Moreover the ferrocene compound (2) and to a lesser degree also the ruthenocene derivative (3) reveal an enhanced distortion of the energy production compared to the metal free compound 1. Interestingly, after removal of the metal free compound 1, the cells show a rapid recovery especially in the acidification rate and a slower but constant recovery of the respiration rate. However, compound 3 reveals only a weak recovery of the acidification whereas 2 shows no recovery of the cells at all after compound removal.
Discussion
An important matter of discussion is the origin of the antiproliferative activity in poly-Arg peptides. A review of the literature reveals that the toxicity of polyarginine itself is discussed controversially.46–48 Previously, Miklan et al. reported a ferrocenoyl-polyarginine compound with a slightly modified polyarginine sequence than the here presented ferrocenoyl-polyarginine 2.48 This already published ferrocene conjugate has shown a comparable toxicity to 2, but the origin of its activity was assumed to be due to the ferrocene functionalization. In their study the unmodified peptide revealed no cytotoxic effect up to a concentration of 10 mM. However, in our study also the polyarginine sequence alone reveals a weak but distinct activity.For our system, we propose that the bioconjugate's toxicity is not based on the ferrocene or ruthenocene moieties, which is in agreement with the lack of an antiproliferative effect of our previously studied ruthenocenoyl and monosubstituted ferrocenoyl peptides and PNA bioconjugates.32,33,35
More likely, the cytotoxicity is the result of a higher uptake of the weakly active polyarginine peptide when coupled to a metallocene. Both metallocenes are not charged to contribute to the electrostatic binding mode of polyarginine, but both metallocenes do enhance the bioconjugates' lipophilicity in a comparable way, as shown by their HPLC retention times. Nevertheless, they show a distinct difference (around 1.5-fold) in their toxicity. This either indicates a superior uptake of ferrocene over ruthenocene or an additional cytotoxic effect of ferrocene, for example enhanced ROS production due to its ability to undergo Fenton chemistry. In our studies we observed an increased uptake for both metallocene conjugates (ruthenocene < ferrocene) in comparison to the acetylated peptide as evaluated by fluorescence microscopy (Fig. S5, ESI†). This result is supported by previous uptake studies, where we could show that ferrocene-derivatized PNA reveals an enhanced uptake (by a factor of ca. 4) over its ruthenocene analogue in HT-29 cells, quantified by an AAS study.32 Therefore, the higher cytotoxic activity is probably an indicator for metallocene-enhanced uptake rather than for metal-based toxicity.
If we assume that the toxicity of the peptide is dependent only on the intracellular compound concentration with no additional cytotoxic effect, we can postulate from the IC50 values a semi-quantitative value of 2- to 3-fold enhanced uptake of 3 by the ruthenocene headgroup compared to 1. For ferrocene, we can expect an iron based enhancement of the toxicity due to its redox properties and therefore, an enhanced ROS production followed by enhanced lysosomal leakage. Here, the comparison of uptake and cytotoxicity might be more intricate.
Conclusion
In this study, a series of metallocene polyarginine bioconjugates was synthesized by Fmoc-SPPS to investigate their potential to disrupt lysosomal membranes and to act as LMP inducing compounds. LMP inducing compounds have been proposed as interesting compounds for tumor targeting by releasing cathepsins and induce a number of apoptotic stimuli. Metallocene bioconjugates as well as their fluorophore-labelled derivatives containing the ferrocene or ruthenocene headgroup were successfully synthesized and compared to the acetylated, metal free peptide regarding their potential to induce LMP. In addition to ferrocene–peptide conjugates, this paper also reports the synthesis of the first conjugates of ruthenocene with cell penetrating peptide derivatives.To investigate their biological behaviour the compounds were tested on an artificial mammalian membrane, but no membranolytic activity could be observed. This finding supports a lysosomal disruption based on the basic guanidine residues of the polyarginine peptide due to protonation inside the more acidic lysosomes. However, increased vulnerability of vesicles could be shown by fluorescence microscopy by photo- as well as chemical-induced leakage. This increased vulnerability was found to be concentration dependent (between 10–50 μM). That the compounds actually reach lysosomes could be shown by fluorescence microscopy co-localization studies of the ferrocene peptide 5 which revealed a vesicular distribution and at least a partial localization in lysosomes. By visual inspection the best cellular uptake of all tested compounds was observed for the ferrocene conjugate over ruthenocene and the metal free derivatives. Furthermore, this differential uptake, in the order ferrocenoyl- > ruthenocenoyl- > acetyl-polyarginine, also correlates with the toxicity of the compounds. As shown by the crystal violet antiproliferative assay, ferrocene increases the antiproliferative activity of polyarginines around 3–4-fold and ruthenocene by 2–3-fold, depending on the cell line, and compared to the acetylated, but metal-free compound 1. After finding an increased vulnerability of vesicles which correlates with toxicity we were interested to get more detailed insights into the biological effects of these compounds, notably in induction of apoptosis and/or necrosis since both have been reported to be effects of LMP. In our assays no increased necrosis compared to the non-treated cells was observed, while especially 2 and 3 show strong induction of apoptosis. ROS induction and cell cycle arrest revealed only minor changes, which might be a secondary effect of apoptosis induction. The biological activity of metallocene–polyarginine conjugates is also reflected in other experiments such as respiration and acidification rate. Here, the metal-free compound (1) shows minor activity compared to the metal containing conjugates with a rapid recovery after compound removal, whereas 2 and 3 reveal higher activity, with no or only weak recovery.
All taken together, the metallocene–polyarginine bioconjugates 2 and 3 clearly enhance the properties of the metal-free compound 1 regarding their antiproliferative and lysosomal destabilizing properties and therefore, we propose these compounds as interesting candidates for the development of lysosomal targeting anticancer drugs.
Acknowledgements
This research was supported by the German Research Foundation (DFG) through the Research Unit “Biological Function of Organometallic compounds” (FOR 630, http://www.rub.de/for630) and the Cluster of Excellence RESOLV (EXC 1069). A.G. thanks Joachim Lügger for synthesizing ruthenocene carboxylic acid, the RUB Research School for further financial support, and Prof. Hahn for permission to use cell culture facilities in their lab.References
- L. Groth-Pedersen and M. Jaattela, Combating apoptosis and multidrug resistant cancers by targeting lysosomes, Cancer Lett., 2013, 332, 265–274 CrossRef CAS PubMed.
- A.-C. Johansson, H. Appelqvist, C. Nilsson, K. Kagedal, K. Roberg and K. Oellinger, Regulation of apoptosis-associated lysosomal membrane permeabilization, Apoptosis, 2010, 15, 527–540 CrossRef CAS PubMed.
- B. Garner, W. Li, K. Roberg and U. T. Brunk, On the cytoprotective role of ferritin in macrophages and its ability to enhance lysosomal stability, Free Radical Res., 1997, 27, 487–500 CrossRef CAS PubMed.
- H. L. Persson, K. J. Nilsson and U. T. Brunk, Novel cellular defenses against iron and oxidation: Ferritin and autophagocytosis preserve lysosomal stability in airway epithelium, Redox Rep., 2001, 6, 57–63 CrossRef CAS PubMed.
- H. L. Persson, T. Kurz, J. W. Eaton and U. T. Brunk, Radiation-induced cell death: Importance of lysosomal destabilization, Biochem. J., 2005, 389, 877–884 CrossRef CAS PubMed.
- H. L. Persson, Z. Yu, O. Tirosh, J. W. Eaton and U. T. Brunk, Prevention of oxidant-induced cell death by lysosomotropic iron chelators, Free Radical Biol. Med., 2003, 34, 1295–1305 CrossRef CAS PubMed.
- Z. Yu, H. L. Persson, J. W. Eaton and U. T. Brunk, Intralysosomal iron: A major determinant of oxidant-induced cell death, Free Radical Biol. Med., 2003, 34, 1243–1252 CrossRef CAS PubMed.
- K. Kagedal, M. Zhao, I. Svensson and U. T. Brunk, Sphingosine-induced apoptosis is dependent on lysosomal proteases, Biochem. J., 2001, 359, 335–343 CrossRef CAS PubMed.
- H.-F. Zhao, X. Wang and G.-J. Zhang, Lysosome destabilization by cytosolic extracts, putative involvement of Ca2+/phospholipase C, FEBS Lett., 2005, 579, 1551–1556 CrossRef CAS PubMed.
- G. Zhang, Y.-P. Yi and G.-J. Zhang, Effects of arachidonic acid on the lysosomal ion permeability and osmotic stability, J. Bioenerg. Biomembr., 2006, 38, 75–82 CrossRef CAS PubMed.
- J.-S. Hu, Y.-B. Li, J.-W. Wang, L. Sun and G.-J. Zhang, Mechanism of lysophosphatidylcholine-induced lysosome destabilization, J. Membr. Biol., 2007, 215, 27–35 CrossRef CAS PubMed.
- Y. P. Yi, X. Wang, G. Zhang, T. S. Fu and G. J. Zhang, Phosphatidic acid osmotically destabilizes lysosomes through increased permeability to K+ and H+, Gen. Physiol. Biophys., 2006, 25, 149–160 CAS.
- J. A. Joyce, A. Baruch, K. Chehade, N. Meyer-Morse, E. Giraudo, F.-Y. Tsai, D. C. Greenbaum, J. H. Hager, M. Bogyo and D. Hanahan, Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis, Cancer Cell, 2004, 5, 443–453 CrossRef CAS PubMed.
- T. Kirkegaard and M. Jaeaettelae, Lysosomal involvement in cell death and cancer, Biochim. Biophys. Acta, Mol. Cell Res., 2009, 1793, 746–754 CrossRef CAS PubMed.
- N. Fehrenbacher, L. Bastholm, T. Kirkegaard-Sorensen, B. Rafn, T. Bottzauw, C. Nielsen, E. Weber, S. Shirasawa, T. Kallunki and M. Jaeaettelae, Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2, Cancer Res., 2008, 68, 6623–6633 CrossRef CAS PubMed.
- E. Vives, P. Brodin and B. Lebleu, A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef CAS PubMed.
- P. M. Fischer, E. Krausz and D. P. Lane, Cellular delivery of impermeable effector molecules in the form of conjugates with peptides capable of mediating membrane translocation, Bioconjugate Chem., 2001, 12, 825–841 CrossRef CAS PubMed.
- S. Futaki, T. Suzuki, W. Ohashi, T. Yagami, S. Tanaka, K. Ueda and Y. Sugiura, Arginine-rich peptides: An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery, J. Biol. Chem., 2001, 276, 5836–5840 CrossRef CAS PubMed.
- A. Berchanski and A. Lapidot, Bacterial RNAse P RNA is a drug target for aminoglycoside-arginine conjugates, Bioconjugate Chem., 2008, 19, 1896–1906 CrossRef CAS PubMed.
- S. R. Schwarze, A. Ho, A. Vocero-Akbani and S. F. Dowdy, In vivo protein transduction: Delivery of a biologically active protein into the mouse, Science, 1999, 285, 1569–1572 CrossRef CAS PubMed.
- J. B. Rothbard, E. Kreider, K. Pattabiraman, E. T. Pelkey, C. L. VanDeusen, L. Wright, B. L. Wylie and P. A. Wender, Arginine-rich molecular transporters for drugs: The role of backbone and side chain variations on cellular uptake, Cell-Penetrating Pept., 2002, 141–160 CAS.
- J. P. Richard, K. Melikov, E. Vives, C. Ramos, B. Verbeure, M. J. Gait, L. V. Chernomordik and B. Lebleu, Cell-penetrating peptides, J. Biol. Chem., 2003, 278, 585–590 CrossRef CAS PubMed.
- S. M. Fuchs and R. T. Raines, Polyarginine as a multifunctional fusion tag, Protein Sci., 2005, 14, 1538–1544 CrossRef CAS PubMed.
- S. M. Fuchs and R. T. Raines, Pathway for polyarginine entry into mammalian cells, Biochemistry, 2004, 43, 2438–2444 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, Organometallic anticancer compounds, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- G. Gasser and N. Metzler-Nolte, The potential of organometallic complexes in medicinal chemistry, Curr. Opin. Chem. Biol., 2012, 16, 84–91 CrossRef CAS PubMed.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Challenges and opportunities in the development of organometallic anticancer drugs, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- F. Noor, A. Wuestholz, R. Kinscherf and N. Metzler-Nolte, A cobaltocenium-peptide bioconjugate shows enhanced cellular uptake and directed nuclear delivery, Angew. Chem., Int. Ed., 2005, 44, 2429–2432 CrossRef CAS PubMed.
- E. Hillard, A. Vessieres, L. Thouin, G. Jaouen and C. Amatore, Ferrocene-mediated proton-coupled electron transfer in a series of ferrocifen-type breast-cancer drug candidates, Angew. Chem., Int. Ed., 2006, 45, 285–290 CrossRef CAS PubMed.
- C. Biot, F. Nosten, L. Fraisse, D. Ter-Minassian, J. Khalife and D. Dive, The antimalarial ferroquine: From bench to clinic, Parasite, 2011, 18, 207–214 CrossRef CAS PubMed.
- A. Gross and N. Metzler-Nolte, Synthesis and characterization of a ruthenocenoyl bioconjugate with the cyclic octapeptide octreotate, J. Organomet. Chem., 2009, 694, 1185–1188 CrossRef CAS.
- A. Gross, N. Huesken, J. Schur, L. Raszeja, I. Ott and N. Metzler-Nolte, A ruthenocene-PNA bioconjugate - synthesis, characterization, cytotoxicity and AAS-detected cellular uptake, Bioconjugate Chem., 2012, 23, 1764–1774 CrossRef CAS PubMed.
- A. Gross, M. Neukamm and N. Metzler-Nolte, Synthesis and cytotoxicity of a bimetallic ruthenocene dicobalt-hexacarbonyl alkyne peptide bioconjugate, Dalton Trans., 2011, 40, 1382–1386 RSC.
- M. Patra and N. Metzler-Nolte, Azidomethyl-ruthenocene: Facile synthesis of a useful metallocene derivative and its application in the ‘click’ labelling of biomolecules, Chem. Commun., 2011, 47, 11444–11446 RSC.
- A. Gross, D. Habig and N. Metzler-Nolte, Synthesis and structure-activity relationship study of organometallic bioconjugates of the cyclic octapeptide octreotate, ChemBioChem, 2013, 14, 2472–2479 CrossRef CAS PubMed.
- H. M. Moulton, M. H. Nelson, S. A. Hatlevig, M. T. Reddy and P. L. Iversen, Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides, Bioconjugate Chem., 2004, 15, 290–299 CrossRef CAS PubMed.
- G. A. McCubbin, S. Praporski, S. Piantavigna, D. Knappe, R. Hoffmann, J. H. Bowie, F. Separovic and L. L. Martin, QCM-D fingerprinting of membrane-active peptides, Eur. Biophys. J., 2011, 40, 437–446 CrossRef CAS PubMed.
- S. Piantavigna, G. A. McCubbin, S. Boehnke, B. Graham, L. Spiccia and L. L. Martin, A mechanistic investigation of cell-penetrating Tat peptides with supported lipid membranes, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 1811–1817 CrossRef CAS PubMed.
- H. Alborzinia, S. Can, P. Holenya, C. Scholl, E. Lederer, I. Kitanovic and S. Woelfl, Real-time monitoring of cisplatin-induced cell death, PLoS One, 2011, 6, e19714 CAS.
- S.-R. Ji, Y. Wu and S.-F. Sui, Cholesterol is an important factor affecting the membrane insertion of β-amyloid peptide (Aβ1-40), which may potentially inhibit the fibril formation, J. Biol. Chem., 2002, 277, 6273–6279 CrossRef CAS PubMed.
- P. Czihal, D. Knappe, S. Fritsche, M. Zahn, N. Berthold, S. Piantavigna, U. Mueller, S. Van Dorpe, N. Herth, A. Binas, G. Koehler, B. De Spiegeleer, L. L. Martin, O. Nolte, N. Straeter, G. Alber and R. Hoffmann, Api88 is a novel antibacterial designer peptide to treat systemic infections with multidrug-resistant gram-negative pathogens, ACS Chem. Biol., 2012, 7, 1281–1291 CrossRef CAS PubMed.
- F. Noor, R. Kinscherf, G. A. Bonaterra, S. Walczak, S. Woelfl and N. Metzler-Nolte, Enhanced cellular uptake and cytotoxicity studies of organometallic bioconjugates of the NLS peptide in Hep G2 cells, ChemBioChem, 2009, 10, 493–502 CrossRef CAS PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Investigation of the alamar blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef.
- R. J. Gillies, N. Didier and M. Denton, Determination of cell number in monolayer cultures, Anal. Biochem., 1986, 159, 109–113 CrossRef CAS PubMed.
- Y.-I. Yang, D.-W. Jung, D.-G. Bai, G.-S. Yoo and J.-K. Choi, Counterion-dye staining method for DNA in agarose gels using crystal violet and methyl orange, Electrophoresis, 2001, 22, 855–859 CrossRef CAS PubMed.
- S. W. Jones, R. Christison, K. Bundell, C. J. Voyce, S. M. V. Brockbank, P. Newham and M. A. Lindsay, Characterization of cell-penetrating peptide-mediated peptide delivery, Br. J. Pharmacol., 2005, 145, 1093–1102 CrossRef CAS PubMed.
- K. H. Ko, C. J. Lee, C. Y. Shin, M. Jo and K. C. Kim, Inhibition of mucin release from airway goblet cells by polycationic peptides, Am. J. Physiol.: Lung Cell. Mol. Physiol., 1999, 277, 811–815 Search PubMed.
- Z. Miklan, R. Szabo, V. Zsoldos-Mady, J. Remenyi, Z. Banoczi and F. Hudecz, New ferrocene containing peptide conjugates: Synthesis and effect on human leukemia (HL-60) cells, Biopolymers, 2007, 88, 108–114 CrossRef CAS PubMed.
- D. Osella, M. Ferrali, P. Zanello, F. Laschi, M. Fontani, C. Nervi and G. Cavigiolio, On the mechanism of the antitumor activity of ferrocenium derivatives, Inorg. Chim. Acta, 2000, 306, 42–48 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: HPLC chromatograms of chosen compounds (1, 2, 3 and 5) Fig. S1, membrane studies Fig. S2–S4, cellular uptake data of 4, 5 and 6 in PT45 cells Fig. S5, resazurin assay of 1, 2 and 3 Table S1, apoptosis study S6, cell cycle experiment S7 and abbreviations. See DOI: 10.1039/c4mt00255e |
| ‡ Current address: Institute of Pharmacy, Technische Universität Braunschweig, Beethovenstr. 55, D-38106 Braunschweig, Germany. |
| This journal is © The Royal Society of Chemistry 2015 |