
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-responsive regulation of two novel Helicobacter pylori NikR-targeted genes†
M. D.
Jones
a,
I.
Ademi
a,
X.
Yin
a,
Y.
Gong
b and
D. B.
Zamble
*a
aDepartment of Chemistry, University of Toronto, 80 St. George St., Toronto, ON, M5S 3H6, Canada. E-mail: dzamble@chem.utoronto.ca
bDepartment of Cell and Systems Biology, the Centre for the Analysis of Genome Evolution and Function, University of Toronto, Canada
First published on 2nd December 2014
Abstract
Nickel is an essential transition metal for the survival of Helicobacter pylori in the acidic human stomach. The nickel-responsive transcriptional regulator HpNikR is important for maintaining healthy cytosolic nickel concentrations through the regulation of multiple genes, but its complete regulon and role in nickel homeostasis are not well understood. To investigate potential gene targets of HpNikR, ChIP sequencing was performed using H. pylori grown at neutral pH in nickel-supplemented media and this experiment identified HPG27_866 (frpB2) and HPG27_1499 (ceuE). These two genes are annotated to encode a putative iron transporter and a nickel-binding, periplasmic component of an ABC transporter, respectively. In vitro DNA-binding assays revealed that HpNikR binds both gene promoter sequences in a nickel-responsive manner with affinities on the order of ∼10−7 M. The recognition sites of HpNikR were identified and loosely correlate with the HpNikR pseudo-consensus sequence (TATTATT-N11-AATAATA). Quantitative PCR experiments revealed that HPG27_866 and HPG27_1499 are transcriptionally repressed following growth of H. pylori G27 in nickel-supplemented media, and that this response is dependent on HpNikR. In contrast, iron supplementation results in activation of HPG27_1499, but no impact on the expression of HPG27_866 was observed. Metal analysis of the Δ866 strain revealed that HPG27_866 has an impact on nickel accumulation. These studies demonstrate that HPG27_866 and HPG27_1499 are both direct targets of HpNikR and that HPG27_866 influences nickel uptake in H. pylori.
Introduction
The transition metal nickel plays an important role in the survival of many organisms because it serves as a catalytic co-factor for a variety of enzymes.1–3 A case in point is H. pylori, the persistent human pathogen that is estimated to infect 50% of the global population.4 This microorganism is the causal agent of chronic gastritis, peptic ulcer disease, and gastric mucosa associated lymphoid tissues (MALT) lymphoma, and is the most important risk factor for gastric cancer.5,6H. pylori are able to survive and thrive under the acidic conditions of the stomach lining, where they encounter pH fluctuations that range from 6 to as low as 1.7–9 A primary component of H. pylori acid defence is the nickel metalloenzyme urease, which generates ammonia and bicarbonate from urea to buffer the cytoplasm and periplasm.5,10–12 Nickel is also a component of [NiFe]-hydrogenase, an enzyme needed for efficient colonization because it allows H. pylori to use the hydrogen gas produced by other host flora as an energy source.13 Although nickel is a nutritional requirement in this organism and a substantial amount of the metal metabolism of H. pylori is devoted to the production of these two obligate nickel enzymes,14 the metal can be toxic to H. pylori if it accumulates inside the cell at high concentrations.15 Intracellular nickel distribution is tightly controlled in H. pylori through the efforts of metalloproteins that function in uptake, efflux, storage, and metalloenzyme assembly.1,14 Regulation of these metal homeostasis systems is primarily at the transcriptional level, controlled by a nickel-responsive transcription factor called HpNikR.16,17HpNikR is a pleiotropic regulator, acting as either a repressor or an activator of a variety of genes.16,18,19 Ni(II)-HpNikR regulates transcription upon binding to the promoters of genes that encode factors involved in nickel import (nixA, fecA, frpB4, exbB), nickel storage (hpn), enzymatic activity (ureAB), autoregulation (nikR), as well as genes involved in iron homeostasis (fur, pfr) and possibly other cellular processes.18,20–23HpNikR is also involved in the global response of H. pylori to acidic conditions,24,25 and there is evidence that the activity of HpNikR is directly modulated by changes in pH.21 The mechanism by which nickel or protons activate DNA binding by HpNikR remains unresolved.20,21,24,26,27 Furthermore, a strong consensus DNA-binding sequence for HpNikR has not been identified. Several iterations of alignments of the DNA recognition sites on multiple HpNikR target promoters highlighted an A/T-rich pseudo-palindromic recognition sequence,28,29 with varying degrees of conservation observed. Furthermore, less sequence conservation correlates with weaker binding in vitro,20 suggesting a mechanism of hierarchical regulation. However, several HpNikR-controlled genes have been identified that lack any sequence homology with the proposed consensus,22 consistent with a more complicated strategy for DNA recognition by HpNikR.30
Understanding the complete physiological function of HpNikR requires that all of the HpNikR targets be identified. To this end, we performed a chromatin immunoprecipitation experiment followed by sequencing (ChIP-seq), which revealed the promoters of HPG27_866 and HPG27_1499. HPG27_866 encodes FrpB2, a putative iron-responsive outer membrane protein,31–33 whereas HPG27_1499 encodes the H. pylori homolog of CeuE, the periplasmic binding protein (PBP) component of an ABC transporter that has been associated with iron networks and more recently with nickel uptake.33–35 Neither of these genes were a part of the defined HpNikR regulon, but given the association of both gene products with metal homeostasis, we examined whether they were under the control of HpNikR. To validate these genes as HpNikR targets, DNA binding to the promoter sequences by HpNikR was investigated in vitro. Purified HpNikR binds to both DNA sequences in a nickel-dependent manner, and the recognition sites were mapped out. Analysis of transcription in H. pylori revealed that both genes are repressed upon supplementation of the growth media with nickel and that this response is dependent on HpNikR. In contrast to a previous report,32 iron-responsive regulation of HPG27_866 was not observed in this study, but upregulation of HPG27_1499 was detected. Furthermore, deletion of HPG27_866 revealed that the gene product has an impact on nickel uptake. These results provide insight into the role of HpNikR as a global regulator of multiple genes in H. pylori and the HpNikR mechanism of action in nickel homeostasis.
Experimental
Bacterial strains and vector construction
The ΔnikR H. pylori G27 strain was generously donated by Dr Peter Chivers (School of Biological and Biomedical Sciences, Durham University, UK).36 The pET24bhpnikr vector was generated previously.37 The promoters of 866 and 1499 were PCR amplified from the G27 genome by using the p866F/p866R and p1499F/p1499R primers, respectively (see Table S1 (ESI†) for primer sequences) and inserted into a pBS-SK(+) vector following digestion with the EagI and SalI restriction enzymes.Protein expression and purification
The pET24bhpnikr plasmid bearing the HpNikR gene was transformed into BL21(DE3) Star E. coli cells and expressed and purified as previously described.37 Briefly, the bacteria were grown in LB media containing 50 μg ml−1 kanamycin to an optical density of 0.8–0.9 at 600 nm (OD600) and protein expression was induced with 0.3 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 15 °C for 4 hours. The cells were then lysed via sonication in protein buffer (20 mM Tris, pH 7.6 and 100 mM NaCl) and centrifuged at 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 1 hour at 4 °C. HpNikR was purified from the supernatant by using Ni-NTA resin followed by dialysis in protein buffer containing 10 mM EDTA and 2 mM DTT. The dialyzed protein was further purified by anion exchange chromatography on a UnoQ column (Bio-Rad) run with a NaCl gradient in 20 mM Tris, pH 7.6, and the protein eluted at approximately 200 mM NaCl. Fractions containing protein were identified via 12% SDS-PAGE, concentrated, and stored at 4 °C. HpNikR concentration was determined using electronic absorption spectroscopy with a predicted extinction coefficient of 8480 M−1 cm−1 at 280 nm and the oxidation state of the protein was detected by performing a 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) assay as previously described.37 Only protein that was >90% reduced was used.
000g for 1 hour at 4 °C. HpNikR was purified from the supernatant by using Ni-NTA resin followed by dialysis in protein buffer containing 10 mM EDTA and 2 mM DTT. The dialyzed protein was further purified by anion exchange chromatography on a UnoQ column (Bio-Rad) run with a NaCl gradient in 20 mM Tris, pH 7.6, and the protein eluted at approximately 200 mM NaCl. Fractions containing protein were identified via 12% SDS-PAGE, concentrated, and stored at 4 °C. HpNikR concentration was determined using electronic absorption spectroscopy with a predicted extinction coefficient of 8480 M−1 cm−1 at 280 nm and the oxidation state of the protein was detected by performing a 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) assay as previously described.37 Only protein that was >90% reduced was used.
Electrophoretic mobility shift assays
The 194 bp DNA probes containing the sequences from the 866 and 1499 promoters were PCR amplified from their respective pBS plasmids by using the p866F/p866R and p1499F/p1499R primers, respectively (Table S1, ESI†). The DNA radiolabeling and mobility shift assay experiments were performed as described previously.37 The samples were resolved on 6% native polyacrylamide gels prepared with Tris-borate (TB) containing either 800 μM NiSO4, 3 mM MgSO4, or 1 mM EDTA in TB running buffer supplemented with the same amount of metal or chelator. The gels were dried and exposed overnight to a phosphor screen, scanned with Pharos FX Plus Molecular Imager (BioRad) and analysed using Quantity One software. The fraction of DNA bound was determined by measuring the intensity of the bound DNA divided by the total intensity of the overall lane. Data were fitted to a Hill equation: r = [HpNikR]n/((Kdapp)n + [HpNikR]n) where r is the fraction of DNA bound to protein and Kdapp is the protein concentration required for 50% DNA binding.
DNase I footprinting assays
DNA probes of the 866 and 1499 promoters were radiolabeled as described above. To achieve single-strand labelling, the 5′ ends of the labelled oligonucleotides were cut with SalI, generating 185 bp and 179 bp probes, respectively. To remove the 5′ fragment, the DNA probes were resolved on a 7% native polyacrylamide gel, and the DNA was eluted from the gel piece by electro-dialysis at 100 V for 2 hours and then precipitated with ethanol at −20 °C. The amount of labelled probe was quantified by liquid scintillation counting on a Tri-Carb Scintillation Counter (Packard). To localize the footprint and confirm that the correct DNA sequence was obtained, the Maxam–Gilbert reaction for guanine bases was performed as described.38,39Increasing concentrations of holo-HpNikR were incubated for 1 hour at room temperature with the radiolabeled promoters in DNase binding buffer (100 mM KCl, 20 mM Tris-HCl, pH 7.6, 1 mM MgCl2, 1 mM CaCl2 and 0.5% (v/v) glycerol and 0.01% IGEPAL). Following incubation, 1 μL of 0.4 U of an RNase-free DNase I enzyme (Invitrogen) was added to each 20 μL sample and quenched after 4 min at room temperature by the addition of 80 μL DNase stop buffer (20 mM Tris, pH 8, 40 mM EDTA, 1% (w/v) SDS, 0.1 μg μL−1 herring sperm DNA). The proteins were extracted from the samples by using a 24:25:1 phenol:chloroform:isoamyl alcohol mixture and the DNA was precipitated with ethanol at −20 °C overnight. The samples were then centrifuged, and the DNA was resuspended in 4 μL formamide loading dye and denatured at 90 °C for 3 min before loading onto a pre-run 8% denaturing polyacrylamide gel. The samples were then resolved at 1700 V for 2 hours in TBE buffer (89 mM Tris-HCl, pH 7.5, 87 mM borate and 20 mM EDTA). The gel was vacuum-dried and exposed overnight to a phosphor screen, scanned with a Pharos FX Plus Molecular Imager (Bio-Rad) and analysed using Quantity One software. The degree of protection was determined by quantifying the intensity of 2 bands within the recognition sequence relative to a reference band. Half maximal binding was calculated by fitting the data to the Hill equation with a variable Hill coefficient as described for the electrophoretic mobility shift assays.
H. pylori growth conditions and nickel exposure
Wild-type H. pylori G27 and its isogenic ΔnikR strain were plated on Columbia agar plates containing 5% horse blood (Thermo Scientific) under a microaerobic environment (5% O2, 10% CO2 and 85% N2) at 37 °C for two days, then replated on new Columbia blood agar plates and grown for a further 22 hours. The colonies were then cultured in 30 mL of Brucella broth with 10% fetal bovine serum (Gibco BRL) and 25 μg mL−1 Helicobacter Selective Supplement (Oxoid) while shaking at 100 rpm at 37 °C under the microaerobic environment for 10–15 hours up to an OD600 of 0.5–0.7. For overnight exposure to nickel, the medium was supplemented with 10 μM NiCl2. Samples for qPCR from the overnight exposure were collected by pelleting an aliquot of the cells such that the final number of bacteria per sample was ∼4 × 107. For transient nickel exposure, H. pylori grown overnight without added nickel were transferred to 5 mL of fresh Brucella broth at pH 7.0 to a final OD600 of 0.2, supplemented with 50 mM urea and with or without 100 μM NiCl2. The cells were incubated for 15 minutes at 37 °C in a microaerobic environment and then pelleted at 4000 rpm at 4 °C for 10 minutes. The supernatant was removed using a vacuum pump and the pellets were either immediately stored at −80 °C or lysed for RNA isolation. After the exposure but prior to centrifugation, 10 μL aliquots of the cultures were collected and serially diluted ten-fold in Brucella broth three times. An aliquot of 5 μM from each dilution was spotted on a Columbia blood agar plate and incubated for 3 days to measure cell viability.Chromatin immunoprecipitation
The Helicobacter pylori strain G27 was grown as a liquid culture at pH 7.0 supplemented with 20 μM NiCl2 as described above. The ChIP experiment was carried out using the One-Day Chromatin Immunoprecipitation Kit (Millipore), with a bacterial sample size of 2 × 108 cells and five thirty-second pulses in a bath sonicator to lyse the cells and shear the DNA to an average fragment size of 200–500 bp. The remainder of the protocol was as prescribed by the kit. Rabbit polyclonal anti-HpNikR antibody (raised against purified HpNikR, Division of Comparative Medicine, University of Toronto) was purified from serum by Western blotting using purified HpNikR protein as previously described.40 The recovered genomic DNA was sequenced by Illumina sequencing at ACTG Corporation (Toronto, ON). The reads for ChIP and control DNA isolated with IgG were 35284339 and 39211483, respectively. The reads were aligned to the H. pylori G27 genomic DNA sequence using the Novoalign short sequence alignment software (novocraft.com). The location information of uniquely mapped reads was converted to BED (Browser Extensible Data) format and used as input for the peak finding software SISSRs (Site Identification from Short Sequence Reads).41 SISSRs was run for binding site identification with the parameters D = 0.001, 0.01, 0.1; e = 1, 5, 10; p = 0.1, 0.01, 0.001 and w = 2, 4, 8, 16. A php script was used to merge the identified sequences. These hits were matched with promoters for various genes in H. pylori G27 and used to generate a list of potential regulation targets of HpNikR.
H. pylori knockout construction
To generate a knockout of the HPG27_866 gene in H. pylori G27, a DNA construct containing the aphA-3 kanamycin resistance cassette from Campylobacter coli flanked by two homological regions corresponding to the 500 bases upstream and downstream of the HPG27_866 gene was synthesized (BioBasic Canada, Inc.). The construct was transformed into wild-type H. pylori G27 via natural transformation, as previously described.42 Briefly, a lawn of H. pylori was collected from a Columbia blood agar plate grown for two days, resuspended in Brucella broth, spotted onto another blood agar plate and incubated under microaerophilic conditions at 37 °C for 4 hours. At that point, 2 μg of the knockout construct DNA was applied to the spot, given time to dry and then further incubated for 12 hours. The spot was then resuspended in Brucella broth and plated onto blood agar plates containing 50 μg mL−1 kanamycin. Colonies of transformants were collected after 3 days. Successful incorporation of the resistance cassette was checked by amplifying the genomic DNA of the transformants around the HPG27_866 gene by PCR using the primers sKO866_F and sKO866_R (Table S1, ESI†). The upstream and downstream junctions of the recombination region were also amplified using the primers sKO866up_F, sKO866up_R, sKO866dwn_F and sKO866dwn_R (Table S1, ESI†) and the products were sequenced to ensure that the genes flanking the aphA-3 cassette were not disrupted.RNA isolation and reverse transcription
H. pylori G27 RNA was isolated by resuspending and lysing the bacterial pellet for 5 min in 1 mL of the TRIzol reagent (Invitrogen), according to product protocols. The aqueous layer extracted from the TRIzol was washed twice with chloroform and the RNA precipitated with isopropanol for at least 2 hours at −20 °C. The RNA was then pelleted and washed with 70% ethanol. The resulting RNA pellet was air-dried, resuspended in 30 μL DEPC-treated water and treated with 178 U of RNase-free DNase I (Invitrogen) to remove trace genomic DNA. The crude RNA was then further cleaned using Aurum Total RNA Mini Kit (Bio-Rad). Purified RNA was quantified using an ND-1000 Nanodrop spectrophotometer and analysed using 1.5% agarose gel electrophoresis to monitor RNA integrity. Only RNA with an A260/A280 ratio greater than 1.8 was used for reverse transcription. RNA from each condition was then diluted to 50 ng μL−1 and converted to cDNA using the iScript cDNA Synthesis Kit (Bio-Rad) according to product protocol with 500 ng of RNA as the template. The remaining RNA was stored at −80 °C and the cDNA was stored at −20 °C.Quantitative PCR
All quantitative PCR was performed by using a CFX96 Real-Time System (Bio-Rad) with SsoFast Master Mix (Bio-Rad). Samples were prepared according to the product protocols, with 400 nM combined primer concentration per well. The q16Sf/r, qgyrf/r, qUreAf/r, q866f/r and q1499f/r primers were used to monitor the expression of 16S rRNA, gyr, ureA, 866 and 1499 transcripts, respectively (Table S1, ESI†). The optimal primer annealing temperature of 55 °C was determined through temperature gradient qPCR and all primers used gave efficiency values between 90 and 110%. All experiments were performed with 45 cycles of a two-step program (5 seconds at 95 °C, 10 seconds at 55 °C) in technical triplicate for at least three biological replicates. A melting curve was also measured at the end of the reaction to ensure the specificity of each primer. The relative expression of the target genes was normalized to the 16S rRNA and gyr levels for each condition and then relative to expression in wild type H. pylori in the absence of nickel. The mRNA expression levels of rpoA were also examined as a possible reference gene, but its expression was not sufficiently constant under the investigated conditions to be included in these studies (data not shown). Compared to alternate housekeeping genes, 16S and gyr expression produced the smallest combined coefficient of variation (<0.25) and M value (<0.5) as calculated by geNorm (BioRad) (data not shown). No-template controls (NTC) and no-reverse transcriptase controls (NRT) were also run for each primer pair to confirm the lack of primer–dimer formation/DNA contamination and genomic DNA contamination, respectively.Inductively coupled plasma mass spectrometry
All ICP-MS experiments were performed on a Sciex ELAN 6100 ICP Mass Spectrometer (Perkin Elmer). Overnight cell samples grown in media with and without 10 μM supplemented nickel (as described above) were collected by spinning down an aliquot of cells such that the final OD600 = 0.4 in 1 mL. Two samples of each strain/condition were collected as technical replicates per biological replicate. The cells were washed with 1 mL PBSE (PBS, pH 7.4, 1 mM EDTA) twice, pelleted and frozen at −20 °C until analysis. Each sample was digested in 550 μL 35% trace-metal-free HNO3 (BDH Chemicals) and 50 μL of each neat sample was removed and diluted 1/10 in 35% HNO3. An In-112 standard (5 ppb in 35% HNO3) was run before each set of samples to correct for signal fluctuations in the instrument. The Ni-60 signals were measured from the neat samples and Mg-24 signals from the 1/10 dilutions, because the Mg-24 signal saturated the detector in the neat samples. The raw counts for each isotope were collected as an average of six readings in five sweeps. The Mg-24 signal was used as an internal control to correct the Ni-60 signals for the loss of cells during washing and pipetting.Results
Identifying and analyzing new HpNikR targets
HpNikR is pleiotropic transcription factor that controls a wide repertoire of promoters, and the extent of its activity is not clear. To explore the regulon of HpNikR and discover new targets, chromatin immunoprecipitation sequencing (ChIP-seq) was performed, a method that has been widely used to define the regulatory network of many transcription factors.43,44 A pilot experiment using H. pylori grown at neutral pH in media supplemented with 20 μM nickel revealed known HpNikR targets including nikR22,45,46 and fecA3.22,45–47 Several well-characterized targets such as ureA22,23,27,45,46 were not identified under these experimental conditions, but the reason for this deficiency is not clear. A quantitative Western blot experiment using cell lysates from wild-type H. pylori revealed that the HpNikR protein is present in the bacteria at an intracellular concentration of roughly 10 μM (data not shown), so the absence of any confirmed target is not a result of insufficient HpNikR expression.Several novel promoters were also identified as putative targets of HpNikR (Table S2, ESI†). Two genes identified in this initial experiment are HPG27_1499 and HPG27_866, referred to as 1499 and 866 in this report, which respectively encode CeuE, the periplasmic protein component of a putative transporter of the ABC superfamily33,34 and FrpB2, an iron-responsive outer membrane protein.31–33 Both of these genes were identified as possible targets of HpNikR in a previous microarray study of the SS1 strain of H. pylori,45 but direct regulation of these genes by HpNikR has not been demonstrated. Given this precedence, the possibility of nickel-responsive regulation of these genes by HpNikR was explored.
Nickel-activated HpNikR directly binds to the target promoters
To validate the results of the ChIP-seq experiment, we examined whether HpNikR can bind directly to the two promoter sequences by using electrophoretic mobility shift assays (EMSAs). In the presence of 800 μM NiSO4, HpNikR binds to the 1499 sequence at half-maximal saturation with a protein concentration of 140 nM (Fig. 1, Table 1). Under the same conditions, HpNikR also binds to the 866 promoter with an affinity of approximately 60 nM (Fig. 1, Table 1). In both cases, no visible shift was observed when the experiment was run with 75 μM NiSO4 (data not shown). DNA binding by holo-HpNikR (loaded with 1:1 nickel) was also observed when 3 mM MgSO4 was included in the gel and running buffer (Fig. 2). The calculated Ni(II)-HpNikR affinities for the 1499 and 866 sequences that were measured in excess Mg(II) were similar to those measured in excess Ni(II) and revealed that approximately 100 nM protein was required for half-maximal binding on both promoters (Table 1). These results are consistent with previous observations that an excess of divalent cations are required to detect clear DNA binding by nickel-loaded HpNikR.26,37,48
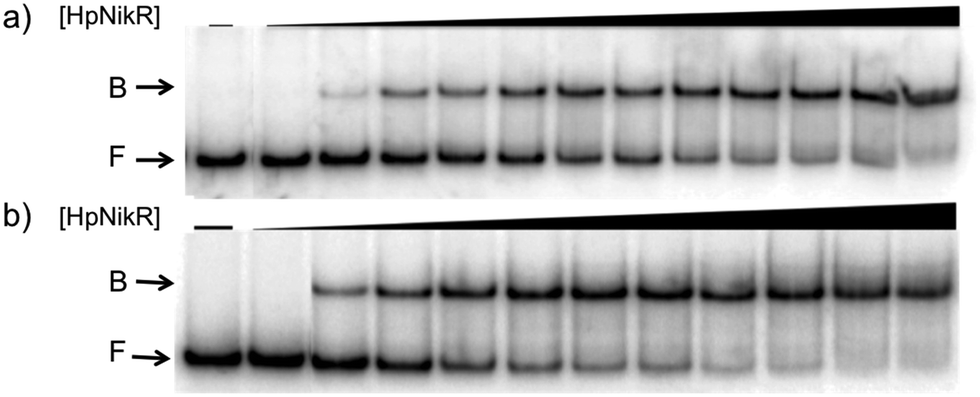 | ||
| Fig. 1 DNA binding of HpNikR in excess nickel. Oligonucleotides containing the sequence from the HPG27_1499 promoter (a) or the HPG27_866 promoter (b) were incubated with holo-HpNikR (1–600 nM or 0.1 nM to 1 μM, respectively). The reactions were analysed on 6% native polyacrylamide gels with 800 μM NiSO4 in the gel and running buffer. F indicates free DNA and B indicates protein-bound DNA. The data were analysed as described in the Experimental section and the average protein concentrations required for half-maximal saturation are listed in Table 1. | ||
| EMSA | DNase footprint | |||
|---|---|---|---|---|
| Holo-HpNikR | Holo-HpNikR | Excess Ni(II) | ||
| 800 μM NiSO4 | 3 mM MgSO4 | 1:1 Ni(II) |
35 μM NiCl2 | |
| N/D – Not determined.a Values are the average protein concentrations required for half-maximal saturation and standard deviation from at least three experiments. | ||||
| 1499 | 140 ± 30 | 130 ± 80 | 230 ± 80 | 330 ± 40 |
| Hill coefficient | 1.3 ± 0.3 | 1.9 ± 0.8 | 2.1 ± 0.3 | 2.02 ± 0.04 |
| 866 | 60 ± 50 | 22 ± 5 | 210 ± 70 | N/D |
| Hill coefficient | 1.4 ± 0.2 | 1.9 ± 0.4 | 1.7 ± 0.4 | N/D |
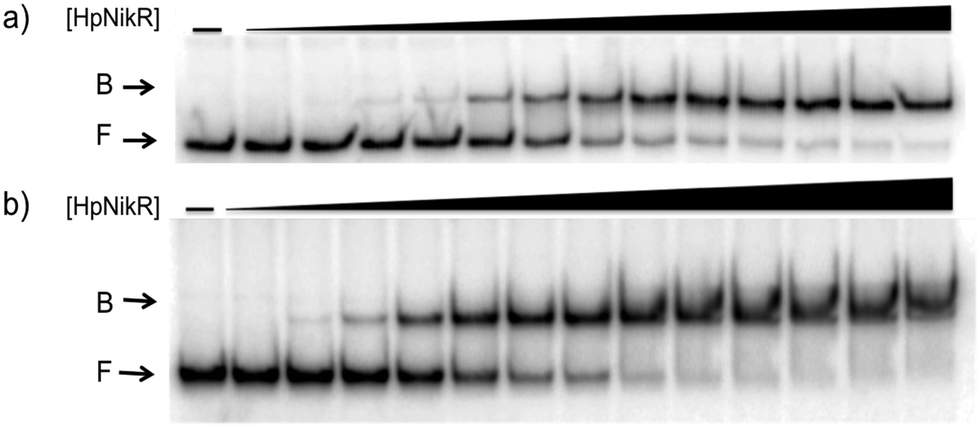 | ||
| Fig. 2 Magnesium as a requirement of holo-HpNikR for DNA binding. Holo-HpNikR (0.1–600 nM) was incubated with either the 1499 promoter sequence (a) or the 866 promoter sequence (b) and analysed on a 6% native polyacrylamide gel with 3 mM MgSO4 in the gel and running buffer. F indicates free DNA and B indicates protein-bound DNA. The data were analysed as described in the Experimental section and the average protein concentrations required for half-maximal saturation are listed in Table 1. | ||
In contrast, apo-HpNikR did not interact with either promoter in the presence of 1 mM EDTA, which was added to the gel and buffer to ensure that HpNikR was metal-free (data not shown). Mobility shift assays performed with apo-HpNikR in the presence of 3 mM MgSO4 in the gel and running buffer revealed weak but detectable binding to the 866 and 1499 promoters (Fig. S1, ESI†). Apo-HpNikR binding in the presence of excess Mg(II) was only detected with >50 μM protein, a concentration several orders of magnitude higher than that required for DNA binding by holo-HpNikR.
Localizing the HpNikR binding site
To locate the binding sites of HpNikR on the 1499 and 866 promoters, DNase I footprinting assays with holo-HpNikR were performed. These assays revealed a 37 bp protected region on the 1499 sequence (Fig. 3) and titration of Ni(II)-HpNikR into the reactions resulted in half-maximal DNA binding with 230 nM protein, comparable to the affinity measured by EMSA. Similarly, a 37 bp footprint was detected on the 866 promoter with a calculated affinity of 210 nM (Fig. 4). Both recognition sequences are AT-rich and loosely follow the pseudo-palindromic consensus sequence (TATTATT-N11-AATAATA) proposed by Delany et al. (Fig. 5),28 as well as the modified version suggested by Stoof et al. (TRWYA-N15-TRWYA).29 Furthermore, the recognition sequence on the 866 promoter matches the region predicted by bioinformatic analysis of the frpB2 promoter in H. pylori 26695, albeit with some minor changes in the flanking and bridging sequences.29 Finally, the same footprint region and binding constant were observed when HpNikR was incubated with the 1499 promoter in the presence of excess nickel (Fig. S2, ESI†), which suggests that excess nickel is not required for tight binding of the promoter in this footprinting experiment. Altogether, these results demonstrate that the promoters of 866 and 1499 are directly bound by HpNikR in a nickel-dependent manner.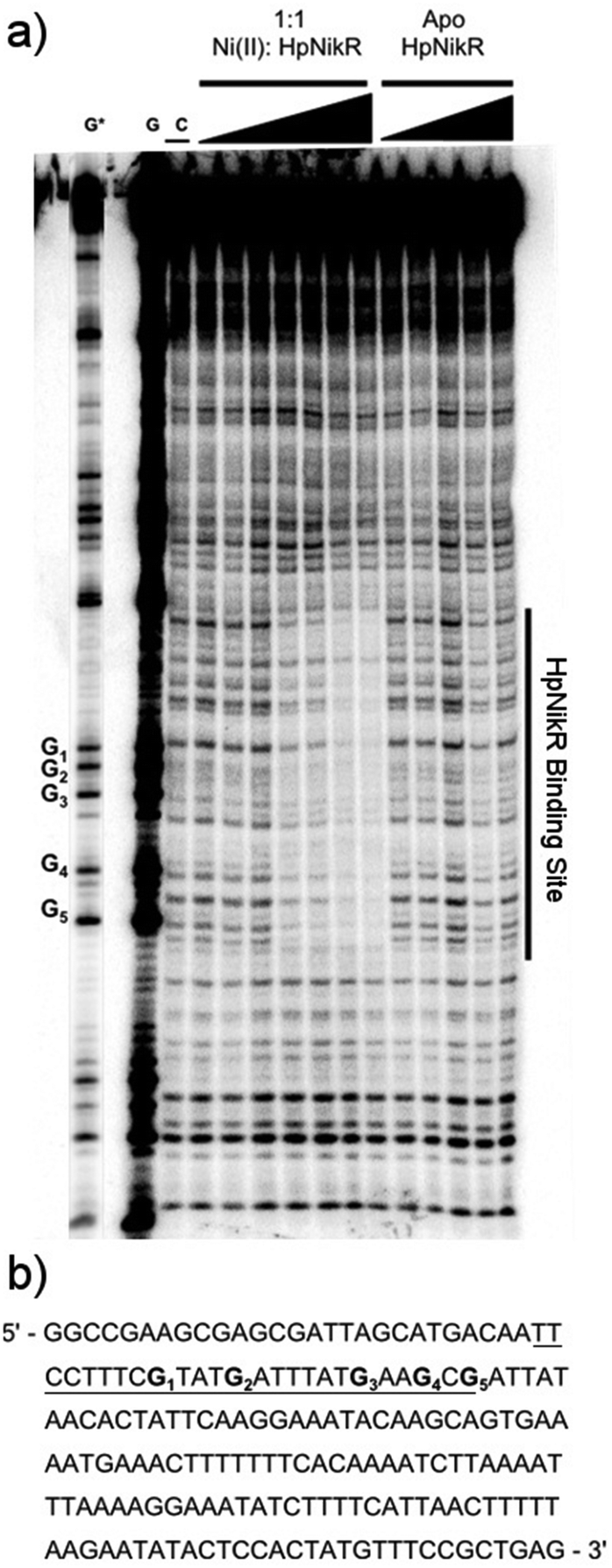 | ||
| Fig. 3 DNA binding by HpNikR on the 1499 promoter sequence. (a) Apo-HpNikR (50 nM–5 μM) and holo-HpNikR (50 nM–5 μM) were incubated with 32P-radiolabeled 179 bp DNA probe containing the HPG27_1499 promoter sequence before cleavage with DNase I. The reactions were resolved on an 8% denaturing polyacrylamide gel and the area of protection is indicated. Key: G and G*, Maxam–Gilbert G reaction with longer and shorter reaction times, respectively; C, control DNase I reaction in the absence HpNikR. The apparent DNA-binding affinity, determined to be 230 ± 80 nM with a Hill coefficient of 2.1 ± 0.3, is an average from four independent experiments. (b) DNA sequence of the 1499 promoter. The binding region is underlined and the guanines within the footprint are bolded and noted on the footprint gel. Note that the 1499 probe used is the reverse complement of the genomic promoter sequence, thus the start codon is upstream of the indicated binding site. | ||
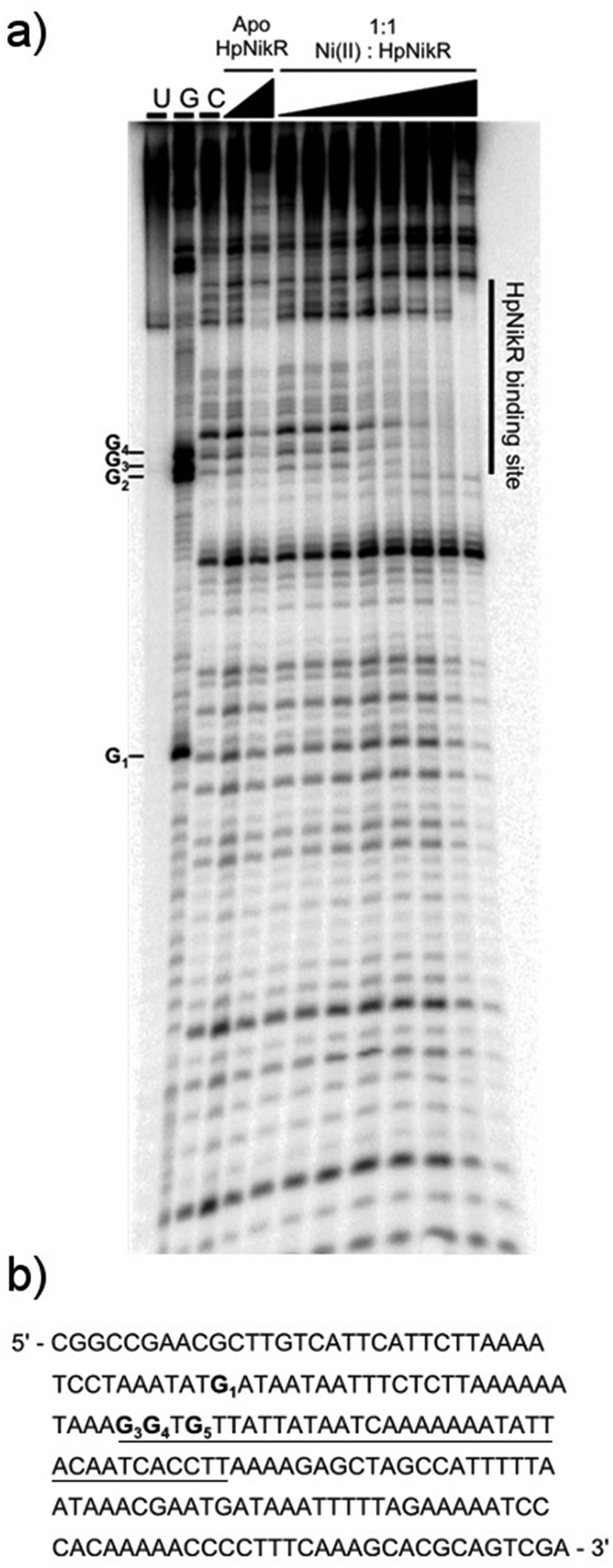 | ||
| Fig. 4 DNA binding by HpNikR on the 866 promoter. (a) Apo-HpNikR (100 nM, 10 μM) and holo-HpNikR (100 nM–10 μM) were incubated with 32P-radiolabeled 185 bp DNA probe containing the HPG27_866 promoter sequence prior to the addition of DNase I. The reactions were resolved on an 8% denaturing polyacrylamide gel and the area of protection is indicated. Key: U, undigested 190 bp probe; G, Maxam–Gilbert G reaction; C, control DNase I reaction of 185 bp probe with no HpNikR. The apparent DNA-binding affinity was determined to be 210 ± 70 nM with a Hill coefficient of 1.7 ± 0.4, calculated as an average from three independent experiments. (b) DNA sequence of the 866 promoter region. The binding region is underlined and the guanines within the footprint are bolded and noted on the footprint gel. Note that the 1499 probe used is the reverse complement of the genomic promoter sequence, thus the start codon is upstream of the indicated binding site. | ||
 | ||
| Fig. 5 Alignment of the HpNikR binding sites from select promoters. The two parts of the consensus sequence are bolded, in red, and numbered according to Evans et al. for the aligned sequences.62 Consensus 1 and Consensus 2 represent the DNA recognition sites proposed by Delany et al.28 and Stoof et al.,29 respectively. N represents any nucleotide and W represents either an A or T nucleotide. Clustal Omega was used to align the promoters and generate the image. | ||
In vivo regulation of 1499 and 866 by HpNikR
To determine the influence of nickel on the transcription of 1499 and 866 and the role of HpNikR in this response, quantitative PCR (qPCR) was performed. Wild-type H. pylori G27 and an isogenic ΔnikR strain were grown in the absence or presence of nickel. Two different conditions were applied, either an overnight incubation in media supplemented with 10 μM nickel, or a shorter 15 min exposure to higher nickel concentrations (100 μM). As a positive control for our in vivo analysis, ureA gene transcription was also examined. The ureA gene encodes the large subunit of the H. pylori urease enzyme, which is an abundant nickel metalloenzyme in H. pylori that is crucial for acid resistance and is a critical virulence factor for H. pylori infection.11,49–51 Upregulation of ureA upon nickel exposure has been observed previously, attributed to the activity of HpNikR, and recently it was discovered that ureA is among the first genes activated by HpNikR when H. pylori are exposed to increased nickel concentrations.22,23,27,52In this study, the qPCR data reveal a statistically significant upregulation of ureA upon exposure of wild-type H. pylori to 10 μM nickel (Fig. 6), in agreement with previous studies.22,27,52 No significant difference in ureA transcription was detected in the ΔnikR strain upon nickel supplementation, consistent with the role of HpNikR as the primary nickel-responsive regulator in the cell. In contrast to the impact on ureA, the transcription levels of 866 and 1499 were significantly repressed when the wild-type bacteria were grown in nickel-supplemented media (Fig. 6), whereas no change in expression was observed for either gene when nickel was added to the ΔnikR knockout strain. These results suggest that 866 and 1499 are directly regulated by nickel-activated HpNikR, consistent with the in vitro DNA-binding data.
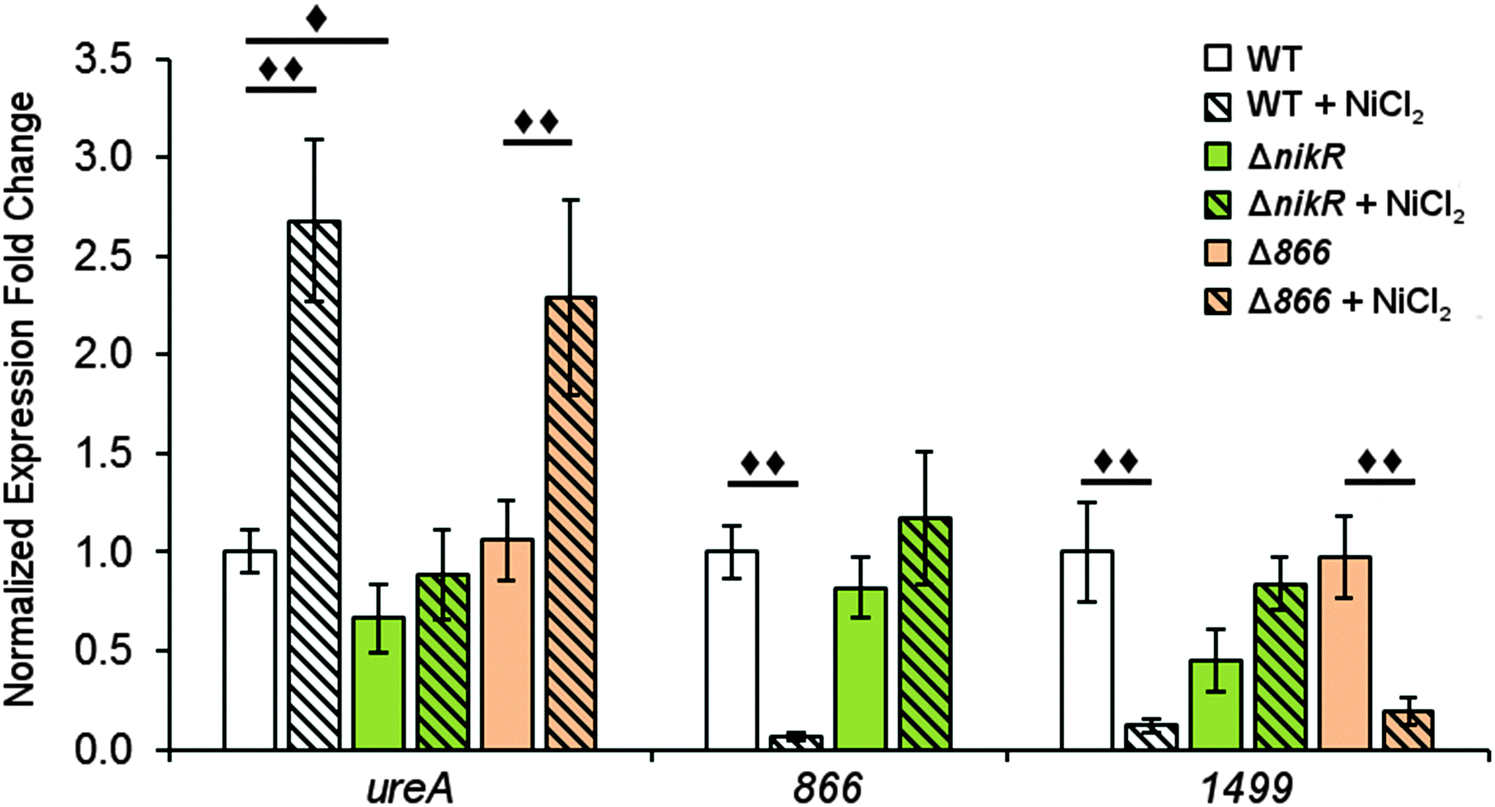 | ||
| Fig. 6 Expression of H. pylori G27 genes after overnight nickel exposure. Quantitative PCR was used to analyse H. pylori G27 (wild-type, ΔnikR or Δ866) grown overnight to midlog phase in the presence or absence of a 10 μM NiCl2 media supplement. Expression of the ureA, 866 and 1499 genes were normalized to 16S and gyr expression levels and relative to wild-type expression of the gene in the absence of supplemental nickel. Statistically significant differences are represented by diamonds (◆ = P < 0.05, ◆◆ = P < 0.01). All data represent the average values from at least three biological replicates with three technical replicates each and error bars represent one standard error of the mean. | ||
Similar trends were observed when the bacteria were grown with higher levels of nickel but for a shorter time (Fig. S3, ESI†). Nickel supplementation results in activation of ureA gene transcription and repression of the 866 and 1499 genes in the wild-type strain. A small but significant reduction in ureA expression was observed in ΔnikR strain under these growth conditions, and while this latter observation could suggest a nickel-responsive component in ureA regulation that is not directly controlled by HpNikR, as previously suggested,22 the exact physiological basis behind this effect is not known. In addition, under the short exposure conditions, 1499 transcription was upregulated in the ΔnikR strain versus the wild-type strain when grown in unsupplemented media. One possible explanation is that apo-HpNikR is regulating 1499 in vivo, but this model is not consistent with the extremely weak DNA binding observed in vitro with apo-HpNikR on the 1499 promoter. Instead, it is more likely that trace levels of holo-HpNikR cause some repression, which is alleviated in the knock-out strain, or that the 50 mM urea added to the cultures for the short exposures affected relative transcription levels, as urea was previously observed to affect global transcription levels in H. pylori.53
There is evidence for overlap between the HpNikR and HpFur regulons.18,28 Furthermore, both 866 and 1499 are regulated in an iron-responsive fashion in some strains,32,33,35 but not in others,54–56 so iron-mediated regulation was also investigated in this study (Fig. 7). No significant change was observed in ureA transcription when either the wild-type or ΔnikR G27 bacteria were exposed to iron-supplemented media, in agreement with previous reports.52,55 Similarly, the expression of 866 was unaffected by iron. In contrast, the expression of 1499 was observed to increase significantly in wild-type H. pylori when the bacteria were exposed to FeCl3. This effect was not observed with the ΔnikR mutant, suggesting that the iron-dependent component of 1499 regulation may be mediated through HpNikR.
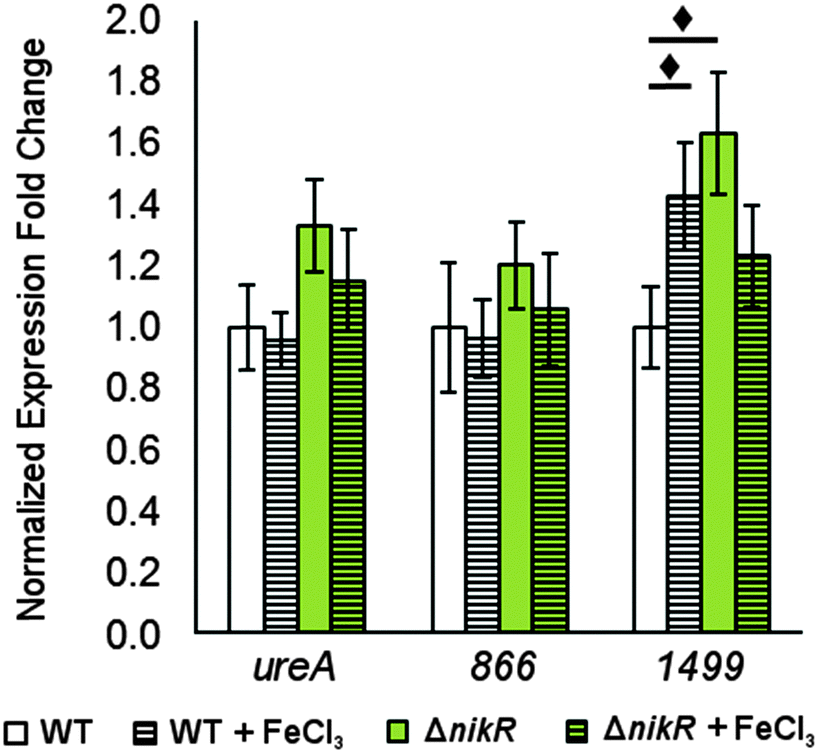 | ||
| Fig. 7 Changes in gene expression in response to transient iron exposure. Quantitative PCR for H. pylori G27 and its isogenic ΔnikR mutant grown to mid-log phase and exposed to 100 μM FeCl3 for 30 minutes. Fold expression is relative to 16S and gyr expression under the same conditions and normalized to WT without supplemented metal. Statistically significant differences between expression levels (P < 0.05) are marked with a diamond. The data represent the average values from at least three independent biological and technical replicates, and the error bars indicate one standard deviation. | ||
Metal uptake through HPG27_866
Given the nickel-responsive regulation of 866, we examined the effects of an 866 knockout on gene transcription and nickel uptake by H. pylori G27. The Δ866 strain did not exhibit any significant changes in the growth rate versus the wild type, with or without nickel supplementation of the media (data not shown), suggesting that 866 is not critical for H. pylori growth under standard laboratory conditions. Furthermore, the baseline and nickel-dependent regulation of ureA and 1499 transcription were not significantly different in the Δ866 strain versus the wild-type bacteria (Fig. 6), suggesting that any impact of 866 is insufficient to influence the activity of HpNikR.To directly observe changes in metal uptake as a result of 866 gene expression, total nickel levels in the wild-type, ΔnikR and Δ866 H. pylori were compared by using inductively-coupled plasma mass spectrometry (ICP-MS, Fig. 8).15,33,57 Under standard growth conditions, nickel accumulation in the ΔnikR strain increased 1.4-fold relative to the wild-type, consistent with previous experiments with an H. hepaticus ΔnikR strain.58HpNikR operates in a negative feedback loop to downregulate nickel import into the cell by repressing the transcription of several nickel uptake genes in a nickel-dependent manner.18,19 Therefore, the lack of this HpNikR-mediated nickel homeostasis could account for the relative increase in nickel in the ΔnikR strain. In contrast, total nickel was significantly lower in the Δ866 strain versus the wild type (Fig. 8), demonstrating that this transporter does influence nickel accumulation. Furthermore, the amounts of 59Co, 63Cu and 64Zn were not significantly different between the three strains (data not shown). The levels of 56Fe could not be accurately measured because of interference from atmospheric argon.59 Nickel accumulation was also examined in bacteria grown in nickel-supplemented media. Under these conditions, the total nickel content increased markedly in all three strains, and again there was a relative increase in the ΔnikR strain compared with the wild-type bacteria, but no significant difference was detected in the Δ866 strain.
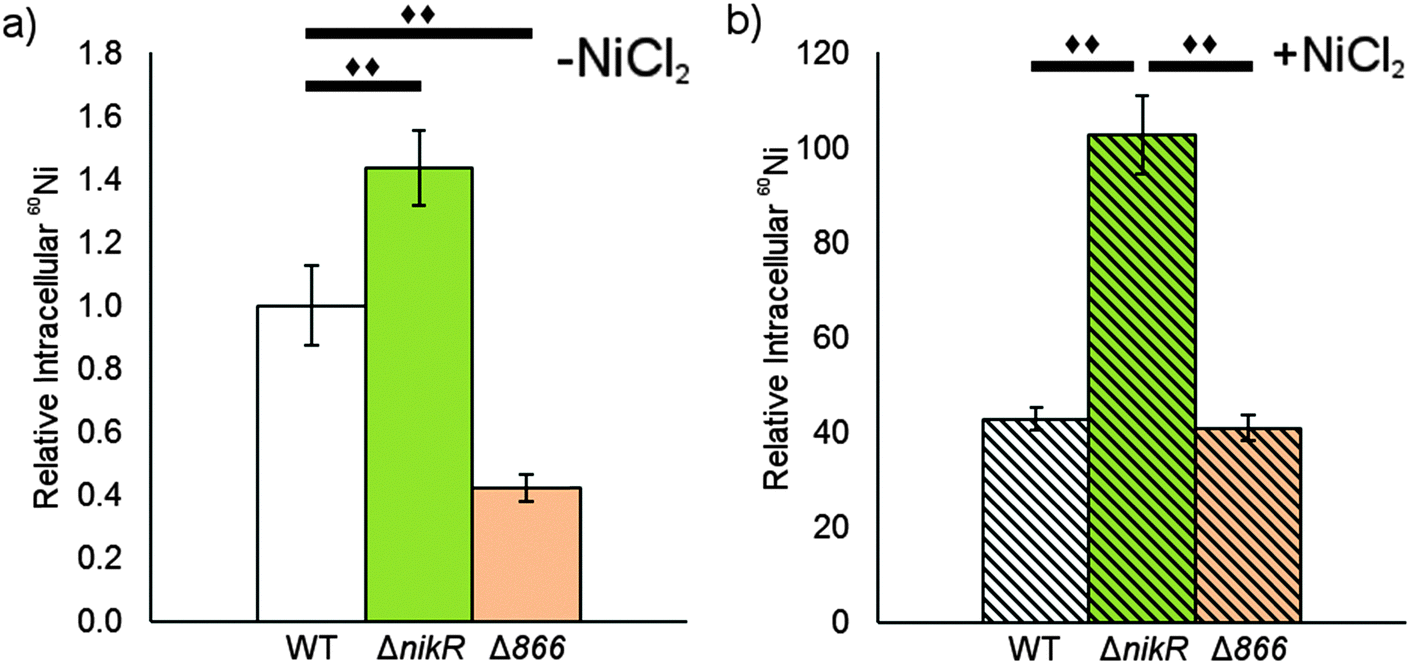 | ||
| Fig. 8 The impact of HPG27_866 on total nickel. The levels of total cellular nickel was measures in wild-type, ΔnikR and Δ866 H. pylori G27 using ICP-MS. Each strain was grown overnight without (a) or with (b) a media supplement of 10 μM NiCl2. Counts from 24Mg were used as an internal control for differences in cell number for each sample. The data represent the average values from five biological replicates and significant changes (P < 0.01) between conditions are marked with a double diamond. | ||
Discussion
H. pylori requires a reliable supply of nickel to activate several enzymes that are critical for colonization, and nickel uptake and distribution is primarily regulated by the pleiotropic transcriptional regulator HpNikR (Fig. 9).23,45 Given the extensive network of nickel homeostasis factors in this organism,14,16,18 as well as the unusually low number of annotated transcription factors in the H. pylori genome compared with similar bacteria,60 it is perhaps not surprising that HpNikR has such expansive responsibilities and as such, it has been dubbed a master regulator. However, it is not clear how broadly HpNikR exerts its control, so we initiated a search for new nickel-regulated HpNikR targets to provide insight into the mechanism of action of HpNikR in H. pylori.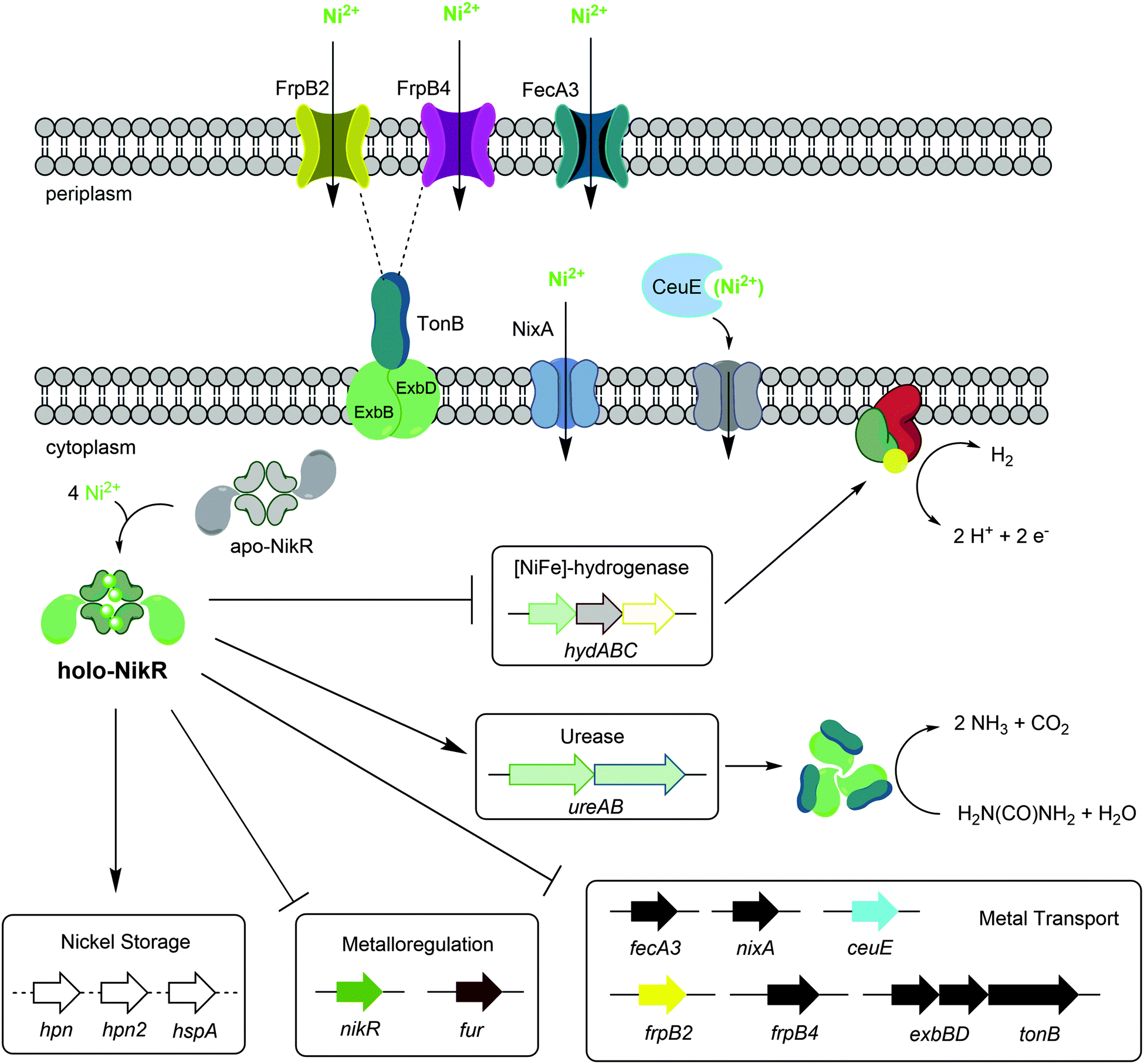 | ||
| Fig. 9 Regulation by Ni(II)-HpNikR in vivo. The transmembrane components of the CeuE nickel transporter have not been explicitly defined, although there is some evidence that FecD/FecE fill these roles. The TonB–ExbB–ExbD complex is believed to provide energy to the FrpB2 outer membrane protein,33 and may also power the FrpB4 homolog as well. Note that although urease is depicted as a cytoplasmic protein, it is present in the cytoplasm, outer membrane and is also excreted outside the cell.72 The exact nature of the nickel transported is not clear for all systems, and only genes regulated by HpNikR in response to nickel are shown. | ||
Our pilot ChIP-sequencing experiment revealed the HPG27_866 and HPG27_1499 genes encoding the iron-responsive outer membrane protein FrpB2 and the nickel-binding periplasmic protein CeuE, respectively. These genes were previously implicated as possible targets of HpNikR via a microarray study,45 but given that direct regulation of these genes by HpNikR had not been demonstrated and that the ChIP-seq strategy tends to produce false positives through non-specific binding,61 we examined whether HpNikR regulates these two genes in response to nickel. In vitro DNA-binding assays were used to demonstrate nickel-responsive HpNikR binding to both promoter sequences, the recognition sequences were mapped out, and in vivo transcriptional analysis indicated that HpNikR represses transcription of both genes in response to an increase in the levels of environmental nickel. Altogether, these results suggest that both 866 and 1499 are components of the HpNikR regulon.
Analysis of the Ni(II)-HpNikR–DNA complexes by using both EMSAs and DNAse footprinting assays revealed half-maximal saturation with ∼100 nM protein on the promoter sequences of both HPG27_866 and HPG27_1499, a value comparable to the affinities of HpNikR for other DNA recognition sites.20,21,26,37,48 Some HpNikR–DNA complexes are significantly weaker, and it was proposed that the promoters regulated by HpNikR can be divided into two classes, with the tighter complexes being observed for the genes that encode proteins that directly utilize nickel (Kd values of low-to-mid nM) and weaker complexes on the promoters of genes of other types of proteins (Kd values of low μM and weaker).20 The affinity of HpNikR for 866 and 1499 promoter sequences places these genes in the former class, along with the genes encoding other nickel membrane transporters as well as urease. However, it is unclear how this two-tiered mode of recognition observed by the in vitro studies translates into the physiological activity of HpNikR.22
DNase footprinting assays revealed that the sequences of the HpNikR binding sites on the 866 and 1499 promoters are 37 bp, AT-rich sequences (Fig. 5). Although neither is an exact match for the initially proposed consensus sequence TATTATT-N11-AATTATA,28 some resemblance is clear, and the 866 site is in full agreement with the more inclusive and recently derived version TRWYA-N15-TRWYA.29 This newer version of the HpNikR consensus was generated by analysing known NikR binding sites from several Helicobacter species. It was then applied to predict NikR targets in Helicobacter genomes, and one of the operators identified was that of the 866 analog in H. pylori 26695, frpB2 (HP0916) (Fig. S4, ESI†).29 It has also been suggested that several specific DNA bases in the HpNikR recognition sequences are more crucial for protein binding than others, with thymine 10 at the 3’ half-site serving a role in distinguishing the tight binders from the weak ones.62 In support of this proposal, both the 1499 and 866 recognition sequences have thymine at position 10, consistent with the relatively tighter affinity of these HpNikR–DNA complexes.
In the H. pylori strain 26695, the gene orthologous to 866 is split by a stop codon caused by a frame-shift deletion, forming frpB2 and the pseudogene frpB3 (HP0916 and HP0915, respectively), which are repressed by the iron metalloregulator HpFur in an iron-dependent manner.32 However, iron-responsive regulation of this gene is not always observed. Iron-dependent changes in 866 expression were not observed in the G27 wild-type or ΔnikR strains under the conditions used in this study, a result consistent with previous experiments with H. pylori G2754 as well as with array-based studies of other strains.35,55 Furthermore, neither of the promoters of HPG27_866 and HP0916/HP0915 contain the putative HpFur recognition sequence and evidence that HpFur binds directly to these promoters is lacking.54,63 In contrast, the promoters in both strains contain a HpNikR binding site (as observed in this work and previously29), albeit with slight variations in the bridging and flanking sequences of each (Fig. S4, ESI†), and the in vitro and in vivo experiments reported here demonstrate that regulation of this gene is directly controlled by HpNikR in response to nickel. Although iron-responsive regulation of 866 is not consistently observed, it is possible that frpB2 expression is modulated indirectly by iron under some conditions through HpNikR activity. It has been previously suggested that the HpNikR and HpFur regulons overlap;18,28 for example both proteins have been demonstrated to directly regulate common genes such as the hydABC cassette22,63 and HpNikR directly represses the transcription of HpFur.20,22
Deletion of HPG27_866 negatively affects nickel accumulation in H. pylori grown under standard laboratory conditions, suggesting that this outer membrane transporter may be involved in nickel uptake. No significant impact was observed on the expression of the ureA or 1499 genes in the Δ866 mutant strain, indicating that HpNikR activity is not affected. The lack of impact on regulation despite lower nickel levels could arise from partitioned pools of nickel within H. pylori, as proposed for E. coli.64 It has also been suggested that FrpB2 contributes to iron uptake as a haemoglobin receptor. Overexpression of the FrpB2 protein encoded by H. pylori J99 allows E. coli to survive in an iron-deficient medium that was supplemented with Hb, and analysis of the crude E. coli lysates suggested that FrpB2 could bind human haemoglobin.31 A connection between nickel and heme uptake was also made with the observation that E. coli NikA, the periplasmic component of the NikABC transporter, can bind heme,65 although how this activity contributes to heme utilization is not clear.66 Future work on the biochemistry and cell biology of FrpB2 will be necessary to pinpoint the specific role that it plays in metal uptake in H. pylori.
HPG27_1499 was also originally annotated as an iron transport protein, possibly due to the homology with the Campylobacter coli periplasmic iron-binding protein.67,68 Despite the weak binding affinity of HpFur to the 1499 binding site,56 iron-responsive regulation of the corresponding gene in H. pylori SS1 was observed only in the stationary growth phase and not in other strains.32,35,55,56 Furthermore, mutation of the genes encoding the putative transmembrane components of this ABC transporter, fecD and fecE, did not dramatically impact the iron uptake in H. pylori 11637,67 suggesting that this transporter is involved in the uptake of a different metal. In this study, weak but significant upregulation of HPG27_1499 was observed upon a brief exposure to high concentrations of iron in the wild-type bacteria (P = 0.049) but not in the ΔnikR mutant strain. As discussed above for HPG27_866, it is possible that iron-responsive regulation of HPG27_1499 is a result of cross-regulation between HpFur and HpNikR. As previously suggested, the sigma factor RpoD (σ80) could also be affecting the transcription of 1499 through an unknown indirect iron-dependent pathway.69 While not yet validated, the rpoD gene was identified as a putative target of HpNikR by our ChIP-seq results and contains the HpNikR consensus binding sequence in its promoter, but further experiments would be required to test this hypothesis.
The assignment of HPG27_1499 as a component of the HpNikR regulon is consistent with the recent re-classification of this protein as a factor involved in nickel acquisition. Interruption of the corresponding ceuE or fecD genes in H. mustelae resulted in decreased total nickel and cobalt.33 Furthermore, recent structural analysis of H. pylori CeuE revealed that it matches the fold of a periplasmic-binding-protein component of an ABC metal transporter and that it binds Ni(L-His)2, similar to E. coli NikA,70 but not heme, vitamin B12 or enterobactin.34 These observations suggest a model for a biological connection between HpNikR and CeuE that is analogous to E. coli NikR and the NikABCDE inner membrane nickel transporter, where HpNikR represses transcription of CeuE to halt nickel import in response to high concentrations of extracellular nickel.19,71 The genes encoding FecD and FecE, HPG27_842 and HPG27_841, respectively, are in a separate operon from 1499 and seemingly lack an HpNikR recognition sequence in the promoter region. However, the fecE gene was also identified as an HpNikR target in our ChIP-Seq data (Table S2, ESI†), suggesting that the transmembrane and periplasmic components of the CeuE/FecD/FecE transporter could be regulated by HpNikR in concert.
Conclusions
Ni(II)-HpNikR binds to the promoters of HPG27_1499 (ceuE) and HPG27_866 (frpB2) in vitro with strong, sub-micromolar binding affinities, and DNase I footprinting revealed binding sites within both promoters that resemble the established pseudo consensus sequences. Furthermore, the transcription of both genes is significantly downregulated when H. pylori is exposed to media supplemented with NiCl2, a response that is dependent on the presence of HpNikR, linking the in vivo transcription of 866 and 1499 to the H. pylori nickel response. These results allow these genes to be assigned to the HpNikR regulon, and the impact of the 866 deletion on nickel accumulation is consistent with a role of the gene product in nickel transport. Future work will establish the mechanism of how these proteins contribute to metal homeostasis in H. pylori. Finally, these experiments provide a snapshot of the HpNikR regulon under a single set of growth conditions, but it is clear that multiple variables impact the activity of this protein, including time and pH.21,22,24,25 Analysis of bacteria exposed to different conditions will provide details of how HpNikR supports H. pylori as it adapts to the challenges of its niche environment.Acknowledgements
The authors would like to thank Dr Lisa Willis for her advice on the construction of H. pylori knockouts. This work was supported in part by funding from the Natural Sciences and Engineering Research Council (NSERC) of Canada and the Canadian Institutes of Health Research. M.D.J. is partially funded through an Ontario Graduate Scholarship.Notes and references
- Y. Li and D. B. Zamble, Chem. Rev., 2009, 109, 4617–4643 CrossRef CAS PubMed.
- S. W. Ragsdale, J. Biol. Chem., 2009, 284, 18571–18575 CrossRef CAS PubMed.
- J. L. Boer, S. B. Mulrooney and R. P. Hausinger, Arch. Biochem. Biophys., 2014, 544, 142–152 CrossRef CAS PubMed.
- J. S. Williamson, Curr. Pharm. Des., 2001, 7, 355–392 CrossRef CAS PubMed.
- B. E. Dunn, H. Cohen and M. J. Blaser, Clin. Microbiol. Rev., 1997, 10, 720–741 CAS.
- M. Sibony and N. L. Jones, Curr. Opin. Gastroenterol., 2012, 28, 30–35 CrossRef PubMed.
- G. Sachs, D. L. Weeks, K. Melchers and D. R. Scott, Annu. Rev. Physiol., 2003, 65, 349–369 CrossRef CAS PubMed.
- K. Stingl, E. M. Uhlemann Em, G. Deckers-Hebestreit, R. Schmid, E. P. Bakker and K. Altendorf, Infect. Immun., 2001, 69, 1178–1180 CrossRef CAS PubMed.
- D. R. Scott, E. A. Marcus, Y. Wen, J. Oh and G. Sachs, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7235–7240 CrossRef CAS PubMed.
- S. B. Mulrooney and R. P. Hausinger, FEMS Microbiol. Rev., 2003, 27, 239–261 CrossRef CAS PubMed.
- M. Pflock, S. Kennard, N. Finsterer and D. Beier, J. Biotechnol., 2006, 126, 52–60 CrossRef CAS PubMed.
- W. Fischer, S. Prassl and R. Haas, in Molecular Mechanisms of Bacterial Infection via the Gut, ed. C. Sasakawa, Springer-Verlag, Berlin, Heidelberg, 2009, pp. 129–171 Search PubMed.
- J. W. Olson and R. J. Maier, Science, 2002, 298, 1788–1790 CrossRef CAS PubMed.
- R. J. Maier, S. L. Benoit and S. Seshadri, BioMetals, 2007, 20, 655–664 CrossRef CAS PubMed.
- K. Schauer, C. Muller, M. Carriere, A. Labigne, C. Cavazza and H. De Reuse, J. Bacteriol., 2010, 192, 1231–1237 CrossRef CAS PubMed.
- C. Belzer, J. Stoof and A. H. van Vliet, BioMetals, 2007, 20, 417–429 CrossRef CAS PubMed.
- J. M. Whitmire, H. Gancz and D. S. Merrell, Curr. Med. Chem., 2007, 14, 469–478 CrossRef CAS PubMed.
- A. Danielli and V. Scarlato, FEMS Microbiol. Rev., 2010, 34, 738–752 CrossRef CAS PubMed.
- M. D. Jones, A. M. Sydor and D. B. Zamble, in Metals in Cells, ed. R. S. Scott and V. Culotta, John Wiley & Sons, Ltd, New York, 2013, pp. 277–288 Search PubMed.
- N. S. Dosanjh, A. L. West and S. L. J. Michel, Biochemistry, 2009, 48, 527–536 CrossRef CAS PubMed.
- Y. Li and D. B. Zamble, Biochemistry, 2009, 48, 2486–2496 CrossRef CAS PubMed.
- C. Muller, C. Bahlawane, S. Auber, C. M. Delay, K. Schauer, I. Michaud-Soret and H. De Reuse, Nucleic Acids Res., 2011, 39, 7564–7575 CrossRef CAS PubMed.
- A. H. van Vliet, S. W. Poppelaars, B. J. Davies, J. Stoof, S. Bereswill, M. Kist, C. W. Penn, E. J. Kuipers and J. G. Kusters, Infect. Immun., 2002, 70, 2846–2852 CrossRef CAS PubMed.
- A. H. van Vliet, F. D. Ernst and J. G. Kusters, Trends Microbiol., 2004, 12, 489–494 CrossRef CAS PubMed.
- S. Bury-Moné, J. M. Thiberge, M. Contreras, A. Maitournam, A. Labigne and H. De Reuse, Mol. Microbiol., 2004, 53, 623–638 CrossRef PubMed.
- E. L. Benanti and P. T. Chivers, J. Biol. Chem., 2007, 282, 20365–20375 CrossRef CAS PubMed.
- F. D. Ernst, E. J. Kuipers, A. Heijens, R. Sarwari, J. Stoof, C. W. Penn, J. G. Kusters and A. H. M. van Vliet, Infect. Immun., 2005, 73, 7252–7258 CrossRef CAS PubMed.
- I. Delany, R. Ieva, A. Soragni, M. Hilleringmann, R. Rappuoli and V. Scarlato, J. Bacteriol., 2005, 187, 7703–7715 CrossRef CAS PubMed.
- J. Stoof, E. Kuipers and A. van Vliet, BioMetals, 2010, 23, 145–159 CrossRef CAS PubMed.
- E. L. Benanti and P. T. Chivers, J. Biol. Chem., 2011, 286, 15728–15737 CrossRef CAS PubMed.
- M. A. González-López and J. J. Olivares-Trejo, BioMetals, 2009, 22, 889–894 CrossRef PubMed.
- A. H. M. Van Vliet, J. Stoof, R. Vlasblom, S. A. Wainwright, N. J. Hughes, D. J. Kelly, S. Bereswill, J. J. E. Bijlsma, T. Hoogenboezem, C. M. J. E. Vandenbroucke-Grauls, M. Kist, E. J. Kuipers and J. G. Kusters, Helicobacter, 2002, 7, 237–244 CrossRef CAS PubMed.
- J. Stoof, E. J. Kuipers, G. Klaver and A. H. M. van Vliet, Infect. Immun., 2010, 78, 4261–4267 CrossRef CAS PubMed.
- M. M. Shaik, L. Cendron, M. Salamina, M. Ruzzene and G. Zanotti, Mol. Microbiol., 2014, 91, 724–735 CrossRef CAS PubMed.
- D. S. Merrell, L. J. Thompson, C. C. Kim, H. Mitchell, L. S. Tompkins, A. Lee and S. Falkow, Infect. Immun., 2003, 71, 6510–6525 CrossRef CAS PubMed.
- E. L. Benanti and P. T. Chivers, J. Bacteriol., 2009, 191, 2405–2408 CrossRef CAS PubMed.
- L. O. Abraham, Y. Li and D. B. Zamble, J. Inorg. Biochem., 2006, 100, 1005–1014 CrossRef CAS PubMed.
- A. M. Maxam and W. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 560–564 CrossRef CAS.
- J. Sambrook and D. W. Russell, Molecular Cloning: A laboratory Manual, Cold Spring Harbor Press, Cold Spring Harbor, New York, 3rd edn, 2001, vol. 2 Search PubMed.
- D. E. Smith and P. A. Fisher, J. Cell Biol., 1984, 99, 20–28 CrossRef CAS PubMed.
- R. Jothi, S. Cuddapah, A. Barski, K. Cui and K. Zhao, Nucleic Acids Res., 2008, 36, 5221–5231 CrossRef CAS PubMed.
- R. Haas, T. F. Meyer and J. P. M. van Putten, Mol. Microbiol., 1993, 8, 753–760 CrossRef CAS PubMed.
- P. J. Franham, Nat. Rev. Genet., 2009, 10, 605–616 CrossRef PubMed.
- E. R. Mardis, Nat. Methods, 2007, 4, 613–614 CrossRef CAS PubMed.
- M. Contreras, J. Thilberge, M. Mandrand-Berthelot and A. Labigne, Mol. Microbiol., 2003, 49, 947–963 CrossRef CAS PubMed.
- G. S. Davis, E. L. Flannery and H. L. T. Mobley, Infect. Immun., 2006, 74, 6811–6820 CrossRef CAS PubMed.
- F. D. Ernst, J. Stoof, W. M. Horrevoets, E. J. Kuipers, J. G. Kusters and A. H. van Vliet, Infect. Immun., 2006, 74, 6821–6828 CrossRef CAS PubMed.
- N. S. Dosanjh, N. A. Hammerbacher and S. L. J. Michel, Biochemistry, 2007, 46, 2520–2529 CrossRef CAS PubMed.
- B. E. Dunn, G. P. Campbell, G. I. Perez-Perez and M. J. Blaser, J. Biol. Chem., 1990, 265, 9464–9469 CAS.
- L. T. Hu and H. L. Mobley, Infect. Immun., 1990, 58, 992–998 CAS.
- K. A. Eaton, C. L. Brooks, D. R. Morgan and S. Krakowka, Infect. Immun., 1991, 59, 2470–2475 CAS.
- A. H. van Vliet, E. J. Kuipers, B. Waidner, B. J. Davies, N. de Vries, C. W. Penn, C. M. Vandenbroucke-Grauls, M. Kist, S. Bereswill and J. G. Kusters, Infect. Immun., 2001, 69, 4891–4897 CrossRef CAS PubMed.
- Y. Wen, E. A. Marcus, U. Matrubutham, M. A. Gleeson, D. R. Scott and G. Sachs, Infect. Immun., 2003, 71, 5921–5939 CrossRef CAS PubMed.
- O. Q. Pich, B. M. Carpenter, J. J. Gilbreath and D. S. Merrell, Mol. Microbiol., 2012, 84, 921–941 CrossRef CAS PubMed.
- F. D. Ernst, S. Bereswill, B. Waidner, J. Stoof, U. Mader, J. G. Kusters, E. J. Kuipers, M. Kist, A. H. van Vliet and G. Homuth, Microbiology, 2005, 151, 533–546 CrossRef CAS PubMed.
- I. Delany, A. B. F. Pacheco, G. Spohn, R. Rappuoli and V. Scarlato, J. Bacteriol., 2001, 183, 4932–4937 CrossRef CAS PubMed.
- C. E. Outten and T. V. O'Halloran, Science, 2001, 292, 2488–2492 CrossRef CAS PubMed.
- S. L. Benoit, S. Seshadri, R. Lamichhane-Khadka and R. J. Maier, Microbiology, 2013, 159, 136–146 CrossRef CAS PubMed.
- J. R. Dean, Practical Inductively Coupled Plasma Spectroscopy, John Wiley & Sons, Ltd, New York, 2005, pp. 89–122 Search PubMed.
- V. Scarlato, I. Delany, G. Spohn and D. Beier, Int. J. Med. Microbiol., 2001, 291, 107–117 CrossRef CAS PubMed.
- P. J. Park, Nat. Rev. Genet., 2009, 10, 669–680 CrossRef CAS PubMed.
- S. E. Evans and S. L. J. Michel, Dalton Trans., 2012, 41, 7946–7951 RSC.
- B. M. Carpenter, J. J. Gilbreath, O. Q. Pich, A. M. McKelvey, E. L. Maynard, Z.-Z. Li and D. S. Merrell, J. Bacteriol., 2013, 195, 5526–5539 CrossRef CAS PubMed.
- J. S. Iwig, J. L. Rowe and P. T. Chivers, Mol. Microbiol., 2006, 62, 252–262 CrossRef CAS PubMed.
- M. Shepherd, M. D. Heath and R. K. Poole, Biochemistry, 2007, 46, 5030–5037 CrossRef CAS PubMed.
- S. Létoffé, P. Delepelaire and C. Wandersman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12891–12896 CrossRef PubMed.
- J. Velayudhan, N. J. Hughes, A. A. McColm, J. Bagshaw, C. L. Clayton, S. C. Andrews and D. J. Kelly, Mol. Microbiol., 2000, 37, 274–286 CrossRef CAS PubMed.
- E. Gasteiger, A. Gattiker, C. Hoogland, I. Ivanyi, R. D. Appel and A. Bairoch, Nucleic Acids Res., 2003, 31, 3784–3788 CrossRef CAS PubMed.
- D. Beier, G. Spohn, R. Rappuoli and V. Scarlato, Mol. Microbiol., 1998, 30, 121–134 CrossRef CAS PubMed.
- P. T. Chivers, E. L. Benanti, V. Heil-Chapdelaine, J. S. Iwig and J. L. Rowe, Metallomics, 2012, 4, 1043–1050 RSC.
- P. T. Chivers and R. T. Sauer, J. Biol. Chem., 2000, 275, 19735–19741 CrossRef CAS PubMed.
- P. R. Hawtin, A. R. Stacey and D. G. Newell, J. Gen. Microbiol., 1990, 136, 1995–2000 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Primer sequences, ChIP-Seq peak targets, additional DNA assay gels and qPCR data. See DOI: 10.1039/c4mt00210e |
| This journal is © The Royal Society of Chemistry 2015 |