
Thioflavin-based molecular probes for application in Alzheimer's disease: from in silico to in vitro models†
C.
Rodríguez-Rodríguez
*a,
M. A.
Telpoukhovskaia
a,
J.
Alí-Torres‡
b,
L.
Rodríguez-Santiago
b,
Y.
Manso§
c,
G. A.
Bailey
a,
J.
Hidalgo
c,
M.
Sodupe
b and
C.
Orvig
*a
aMedicinal Inorganic Chemistry Group, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: crisrod@chem.ubc.ca; orvig@chem.ubc.ca; Fax: +1 604 822 2847; Tel: +1 604 822 4449
bDepartament de Química, Universitat Autònoma de Barcelona, 08913 Bellaterra, Barcelona, Spain
cDepartament de Fisiologia Animal, Universitat Autònoma de Barcelona, 08913 Bellaterra, Barcelona, Spain
First published on 7th October 2014
Abstract
Alzheimer's disease (AD) is a neurological disease of confusing causation with no cure or prevention available. The definitive diagnosis is made postmortem, in part through the presence of amyloid-beta plaques in the brain tissue, which can be done with the small molecule thioflavin-T (ThT). Plaques are also found to contain elevated amounts of metal ions Cu(II) and Zn(II) that contribute to the neurotoxicity of amyloid-beta (Aβ). In this paper, we report in silico, in vitro, and ex vivo studies with ThT-derived metal binders 2-(2-hydroxyphenyl)benzoxazole (HBX), 2-(2-hydroxyphenyl)benzothiazole (HBT) and their respective iodinated counterparts, HBXI and HBTI. They exhibit low cytotoxicity in a neuronal cell line, potential blood–brain barrier penetration, and interaction with Aβ fibrils from senile plaques present in human and transgenic mice AD models. Molecular modelling studies have also been undertaken to understand the prospective ligand–Aβ complexes as well as to rationalize the experimental findings. Overall, our studies demonstrate that HBX, HBT, HBXI, and HBTI are excellent agents for future use in in vivo models of AD, as they show in vitro efficacy and biological compatibility. In addition to this, we present the glycosylated form of HBX (GBX), which has been prepared to take advantage of the benefits of the prodrug approach. Overall, the in vitro and ex vivo assays presented in this work validate the use of the proposed ThT-based drug candidate series as chemical tools for further in vivo development.
Introduction
Thus far, no disease-modifying treatments or early diagnoses exist for neurodegenerative disorders such as Alzheimer's disease (AD). This progressive disorder has a characteristic hallmark, which is the accumulation of extracellular deposits termed amyloid.1,2 This general term denotes aggregates organized in a non-native highly stable structure,1,2 with specific tinctorial properties (binding to thioflavin-T (ThT) and Congo Red (CR)),3–5 high resistance to degradation,6,7 and a fibrillar appearance.6,7 Considerable research efforts have been devoted to develop multifunctional molecules not only to gain knowledge about the molecular mechanisms underlying the neurodegenerative diseases, but also to lead to novel early detection and therapeutic intervention strategies.In particular for AD, the Aβ peptide assembly that comprises the amyloid plaques has been used as a therapeutic target to develop small molecules, nanoparticles, natural products and/or small peptides to interfere with its aggregation.8–15 Similarly, targeting amyloid-containing senile plaques has been used for disease diagnosis and monitoring.16–19 Another key aspect, supported by a large body of clinical and experimental evidence, is that imbalance of metal ions in the brain plays a critical role in Aβ misfolding, contributing to oxidative stress and the formation of senile plaques.10–15
With the purpose of developing multifunctional molecules aiming at multiple targets that must be addressed in AD, we designed and synthesized small molecules able to interact with both metal ions and senile plaques.9,20 The strategy for their rational design was inspired by two well-known molecules in the field: ThT, the traditional histological dye that signals the presence of amyloid fibrils;3 and clioquinol (5-chloro-7-iodo-8-hydroxyquinoline, CQ),21–23 the parent compound of the most successful metal-ion ligand PBT2, which is in phase II clinical trials for AD (Fig. 1).24–26 The preliminary findings confirmed that 2-(2-hydroxyphenyl)benzoxazole (HBX), 2-(2-hydroxyphenyl)benzothiazole (HBT) (Fig. 1) and their respective iodinated derivatives (HBXI and HBTI) showed several favourable functionalities, including binding to metal ions Cu(II) and Zn(II) and dissociation of Aβ aggregates in the presence of these metal ions.20
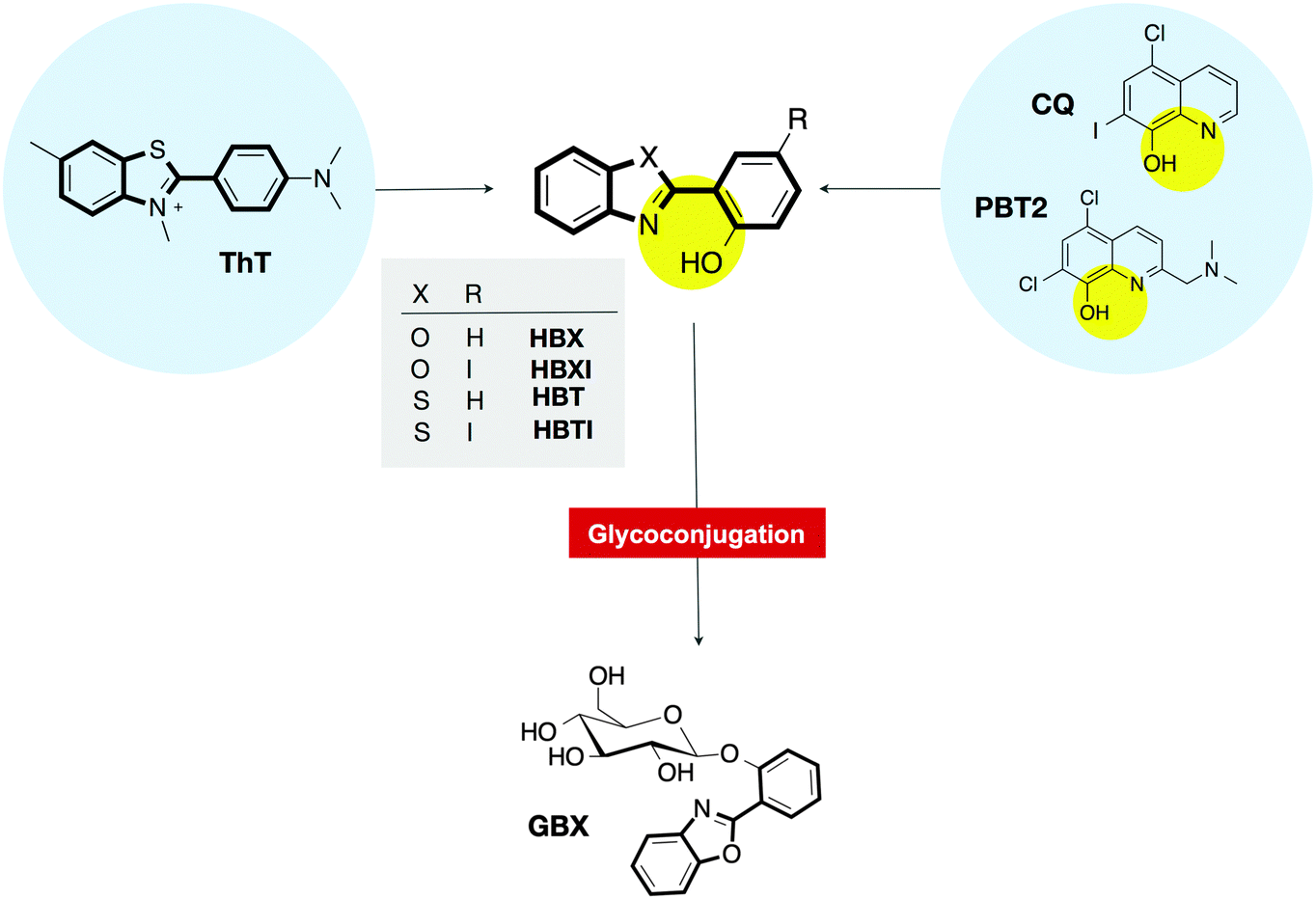 | ||
| Fig. 1 Thioflavin-T (ThT), clioquinol (CQ) and its successor PBT2 are basic molecular scaffolds from which multifunctional molecules HBX, HBXI, HBT and HBTI are derived. The glycosylated prodrug version of HBX is presented as GBX. | ||
In this work, we study the binding of compounds HBX, HBT, HBXI, and HBTI to Aβ1–40 fibrils in vitro as well as using molecular docking studies. Moreover, the ability of HBX and HBXI to interact with amyloid plaques is exhibited ex vivo in mouse models of AD as well as human brain sections using fluorescence microscopy. In addition to this, we add a novel glycosylated prodrug (GBX) to the library of compounds to exploit the benefit of glycosylation in drug design; the glucose moiety is shown to be deprotected by an enzyme, releasing the active compound. Finally, we study the cytotoxicity and permeability of these compounds with the mouse neuronal cell line bEnd.3.
Results and discussion
Binding affinity study using Aβ1–40 fibrils in solution
To quantify the binding affinity of the ThT derivatives to Aβ deposits, a fluorescence titration assay was performed to determine the dissociation constant (Kd) of the compounds. In the presence of pregrown Aβ1–40 fibrils, HBX, HBT, HBXI and HBTI increase their emission intensity (Fig. S1, ESI†), enabling the determination of Kd by titrating a solution containing Aβ1–40 fibrils with the corresponding ligand. The binding profiles obtained are shown in Fig. S2 (ESI†), and this behaviour was best fitted to a one-site binding model to give the respective Kd summarized in Table 1. The observed values range from 4.4 to 5.8 suggesting that among the tested compounds, HBTI (Kd = 4.42 μM) exhibits the highest affinity for the Aβ1–40 fibrils followed by HBXI (Kd = 5.07 μM). This observation implies that the incorporation of an iodine atom at the para position to the OH group may enhance its amyloid binding affinity. Conversely, the non-iodinated derivatives show a weaker binding affinity to Aβ1–40 fibrils with HBT (Kd = 5.24 μM) better than HBX (Kd = 5.84 μM). The observed binding affinities to Aβ1–40 fibrils in solution are comparable to that of the related compound BTA-1 (Kd = 5.23 μM)27 measured by a similar method.27 We set out to study the differences in interactions among these compounds using computations, as detailed below.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP and logBB), experimental cytotoxicity (EC50) in bEnd.3 cell line, dissociation constant (Kd), free binding energy as determined by molecular docking (ΔGbinding), ligand efficiency (LE) and DFT calculations (ΔEDFT), for the ThT-derivatives series (HBX, HBT, HBXI, HBTI, and GBX). ThT, CQ and the anti-cancer agent cisplatin are included for comparison
logP and logBB), experimental cytotoxicity (EC50) in bEnd.3 cell line, dissociation constant (Kd), free binding energy as determined by molecular docking (ΔGbinding), ligand efficiency (LE) and DFT calculations (ΔEDFT), for the ThT-derivatives series (HBX, HBT, HBXI, HBTI, and GBX). ThT, CQ and the anti-cancer agent cisplatin are included for comparison
| Compound |
clogP |
logBBa |
EC50 (μM) | K d (μM) | ΔGbinding (kcal mol−1) | LE (kcal mol−1) | ΔEDFT (kcal mol−1) |
|---|---|---|---|---|---|---|---|
|
a logBB = −0.0148 TPSA + 0.152 clogP + 0.139,34–37 with the TPSA and clogP parameters defined in the text.
|
|||||||
| HBX | 2.74 | −0.13 | 73.28 | 5.84 | −6.81 | 0.43 | −14.8 |
| HBT | 3.42 | 0.17 | 78.93 | 5.24 | −6.64 | 0.41 | −16.7 |
| HBXI | 4.04 | 0.07 | 7.75 | 5.07 | −7.61 | 0.44 | −18.9 |
| HBTI | 4.70 | 0.36 | 21.23 | 4.42 | −7.52 | 0.45 | −22.0 |
| GBX | −0.25 | — | 150.90 | — | — | — | — |
| CQ | 3.73 | 0.21 | 18.58 | — | — | — | — |
| Cisplatin | — | — | 14.97 | — | — | — | — |
| ThT | 0.16 | 0.06 | — | 1.74 | — | — | — |
Molecular modeling on Aβ1–40 fibrils
A combined quantum mechanics and docking approach for the investigation of Aβ–ligand complexes is presented. We will first discuss the results obtained from docking simulations. Secondly, results from electronic structure calculations will be provided.Molecular docking
In order to gain insight into the molecular determinants that modulate the interactions of the ligands bound to Aβ1–40 in fibrillar form, in silico studies have been undertaken. Shedding light on the detailed structural information at the molecular and atomic level of amyloid fibrils is essential, but also an understanding of the intermolecular forces that drive the formation of peptide–ligand complex is necessary for rational design of compounds that bind specifically to Aβ (e.g. therapeutic and diagnostic applications).28 Overall, much effort has been expended to understand the specific binding of several molecules with Aβ at both experimental and computational levels.29–33 Previously, this approach has been found to be fairly successful in Aβ1–40 monomer and also Aβ1–42 fibril models.29–33 Since the binding site in the receptor is not known, the technique of blind docking was used to explore the entire protein surface. To account for protein flexibility, docking was performed against the ten previously minimized structures of Aβ1–40 fibrils that were based on experimentally determined NMR measurements (PDB code 2LMN). The binding energy (ΔGbinding) of each ligand–Aβ1–40 fibril complex obtained for the most stable cluster is indicated in Table 1. All the ligands present similar interactions and consequent conformations. In all cases, the ligands are positioned inside the U-shaped Aβ1–40 in the groove formed by the β-sheet structure, parallel to the long axis of the fibril. The preferred docking site for the benzothiazole derivatives (HBT and HBTI) is located along the central straight groove of the upper layer formed by the residues Ala21-Phe19 and Ile32-Leu34 (pose A, Fig. 2a), and for the benzoxazole framework (HBX and HBXI) their preference is the subsequent cavity formed by Phe19-Leu17 and Leu34-Val36 (pose B, Fig. 2a). In each pose, the difference lies in the orientation of the different rings of the molecule to accommodate the iodine atom and minimize the repulsion with the β-sheet structure. A closer view of the predicted binding sites illustrates that most of the amino acids participating in the formation of these Aβ1–40–ligand complexes are aromatic (Fig. 2b). Due to the shared scaffold of the ligands, the resulting interaction patterns are very similar. As a general trend, it is observed that these molecules occupy hydrophobic cavities formed by Phe residues in the upper layer. In most of the complexes, the benzoxazole/benzothiazole motif and the phenol ring establish CH–π interactions with Phe19 (from H and/or I chains) (Fig. 2b). Additionally, only those complexes formed by HBX, HBXI and HBTI with Aβ1–40 fibrils showed a hydrogen bond interaction between the phenol ring and the carbonyl group from the backbone of the peptide (Fig. 2b). Based on docking results, to estimate the efficiency of compounds, the obtained binding affinity was assessed in relation to the number of heavy atoms in the molecule in order to compare the affinity corrected for their size.38,39 The ligand efficiency parameter (LE) can be calculated using eqn (1)38,39| LE = −ΔGbinding/NHA | (1) |
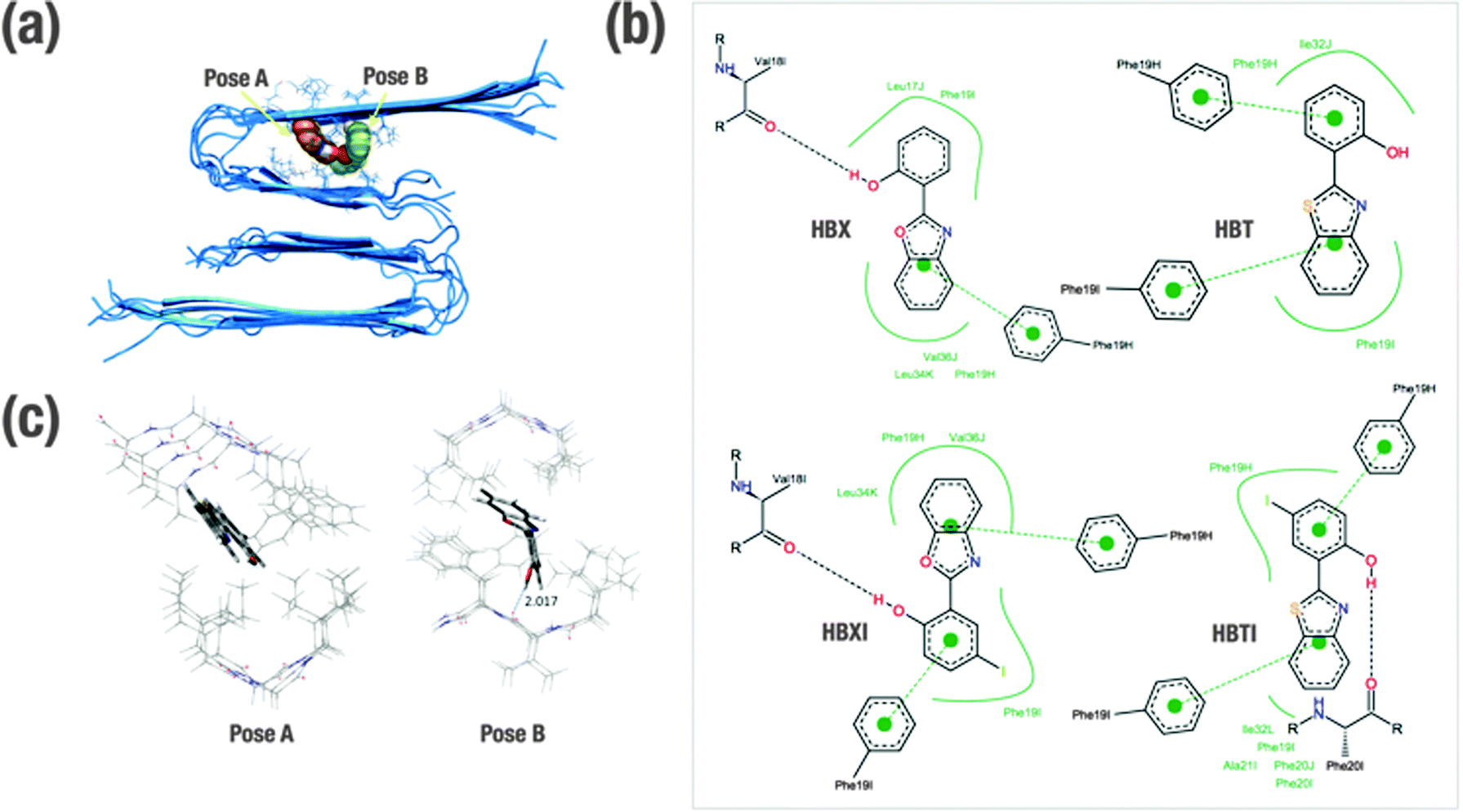 | ||
| Fig. 2 (a) Internal binding sites of HBT/HBTI (pose A) and HBX/HBXI (pose B) bound to Aβ1–40 from molecular docking studies. Both molecules are located in the crevices defined by the bottom residues 32–34 (pose A) or 34–36 (pose B). (b) 2D ligand–peptide interaction diagrams for the indicated ligands. Black dashed lines—hydrogen bonds; green solid lines—hydrophobic interactions, green dashed lines—π–π interactions. (c) ONIOM optimized geometries for HBT (pose A) and HBX (pose B) bound to Aβ1–40. The initial structures were obtained from molecular docking studies. | ||
Electronic structure calculations
Optimized ligand–fibril structures for HBT and HBX are shown in Fig. 2c, whereas those for the analogous HBTI and HBXI compounds are presented in Fig. S3 (ESI†). The orientation of the ligand in pose A is essentially conserved after the ONIOM optimization. However, for the ligand in pose B the final orientation exhibits larger changes to correct initial destabilizing H⋯H interactions between the ligand and the peptide generated in the docking process. These changes, however, cause only slight variations in the relative orientation of the ligand versus the fibril, the internal geometry of the ligand remaining very similar to that obtained in the docking simulations. Analyses of the different structures reveal that all four ligands are mainly stabilized by CH–π interactions between the aromatic rings of the ligands and CH groups of the peptide backbone or side chains. The main distances for these interactions are listed in Table S1 (ESI†). Markedly, hydrogen bonds between the OH group of phenol and a carbonyl group of the fibril are observed for ligands HBX and HBXI (2.017 and 2.211 Å distance, respectively). As a consequence, the intramolecular hydrogen bond of the ligand is disrupted and the ligand becomes non-planar. It can be observed in Table 1 that the presence of iodine and sulphur atoms in the ligand leads to an increase of the interaction energy (in absolute value) due to larger dispersion interactions. Indeed, the computed DFT ligand affinities for the iodine-containing molecules (HBTI and HBXI) are greater than for the non-iodinated molecules. Moreover, sulphur-containing molecules exhibit stronger interactions with the fibrils than the corresponding oxygen-containing molecules, in good agreement with previous observations on ThT-derivative ligands interacting with amyloid fibril models.32 Overall, the observed trend for ligand affinity is HBTI > HBXI > HBT > HBX, which matched the experimental observations (see dissociation constants in Table 1).Ex vivo Aβ staining within brain sections
One of the limitations of using ThT is that it is a charged molecule,17,40 and thus obtaining neutral ThT derivatives may prove advantageous for future in vivo studies. The first step in this endeavour is to establish whether the title compounds are able to selectively interact with Aβ deposits ex vivo. The fluorescent properties of our compounds enable the monitoring of their binding to Aβ plaques by fluorescence imaging. As was previously shown, the tested compounds interact with amyloid fibrils obtained from amylin (IAPP10–29 fragment) and Aβ1–42.20 In the present study, HBX and HBXI were tested for ex vivo Aβ plaque binding on AD human and AD mouse model (Tg2576 and TgCRND8) brain tissues. The staining of sections of paraformaldehyde fixed brains of AD human and TgCRND8/Tg2576 mice with well-known histochemical methods such as Congo Red staining revealed the existence of dense Aβ amyloid deposits (data not shown). To determine the optimal concentration of dye, different solutions were incubated with brain sections. At the lowest concentration (∼0.16 μg ml−1) of dye, no binding to senile plaques was observed (data not shown), whereas at higher concentrations (∼0.05 mg ml−1 and 0.5 mg ml−1) increased fluorescent signal was observed in those areas rich in senile plaques. Fig. 3 shows the positive Aβ plaque targeting ability of the fluorescent HBX and HBXI incubated with brain sections at 0.05 mg ml−1 overnight. These data strongly suggest that these ThT derivatives have potential application in vivo for the diagnosis of AD, especially since these molecules are neutral and may be able to cross the BBB, in contrast with the parent molecule ThT.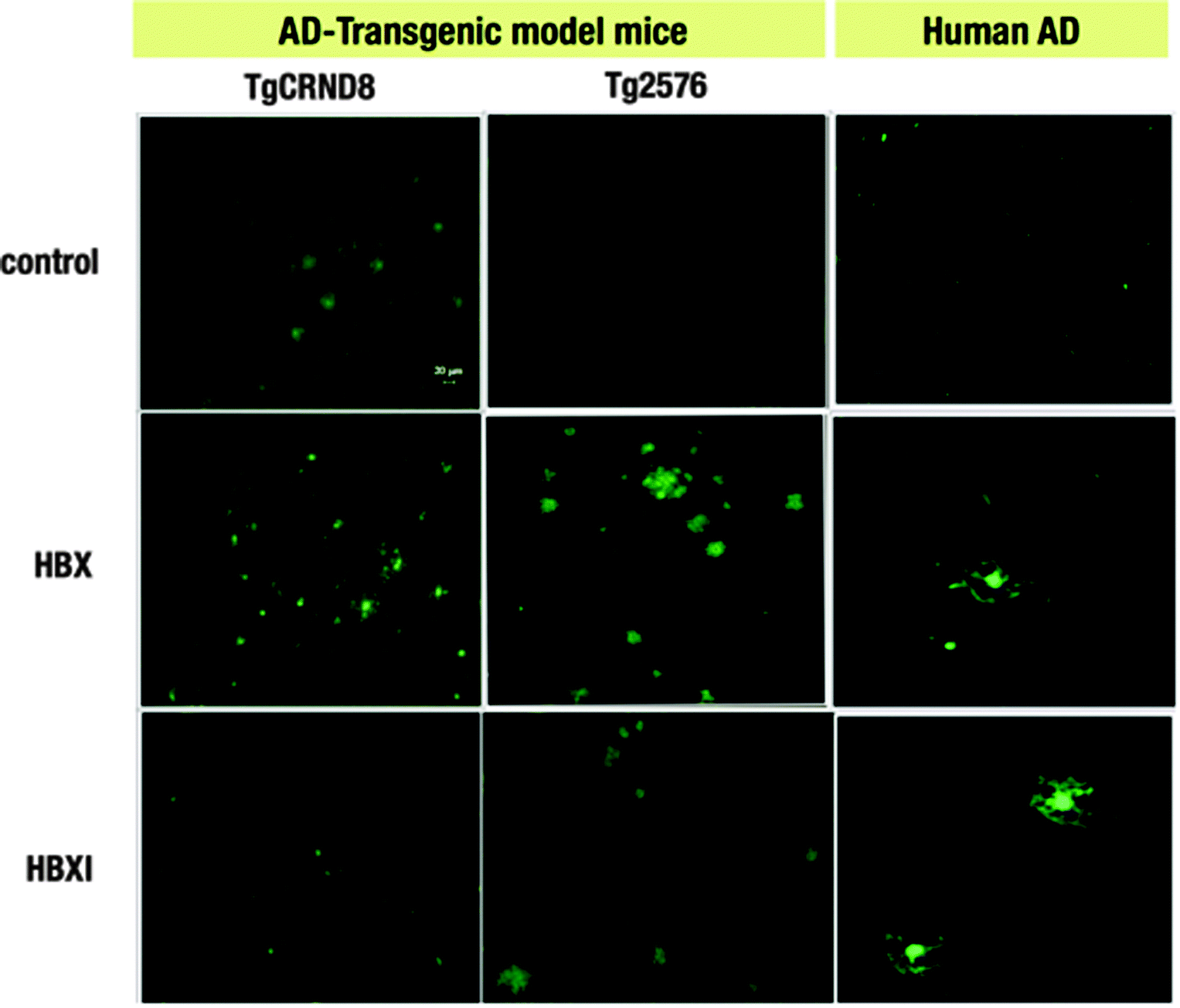 | ||
| Fig. 3 Ex vivo staining of Aβ plaques in two different AD mice models (TgCRND8 and Tg2576) and AD human brain sections by HBX and HBXI. (Green: FITC channel. Scale bar = 20 μm). | ||
Glycosylated prodrug
The term ‘prodrug’ describes a pharmacologically inactive derivative of an active drug that undergoes biotransformation to unmask its therapeutic activity. Usually, enzymes at the specific site of action carry out their activation, thereby releasing the therapeutic agent. In this work, glycosylation has been used to enhance the benefits of HBX. It has been demonstrated that glycoconjugates improve the permeability through the glucose transporter (GLUT1),41,42 which is present in the membrane of the endothelial cells forming the BBB.42 GLUT1 facilitates the transport of glucose to the brain and as such has been an attractive target for drug delivery.43 The motivation in functionalizing HBX by glycosylation to form the prodrug GBX is multifold: it may help the molecule get through the BBB via the active transporter GLUT143 and increase prodrug solubility. Additionally, a functional group at the hydroxyl site could mitigate the undesired binding of systemic metal ions by masking the binding site of HBX. Recently, a series of glycosylated prodrugs obtained from two different scaffolds – tetrahydrosalen and 3-hydroxy-4-pyridinones – have shown promising results in vitro and in vivo.44–49 The synthesis of GBX is performed in a one-step reaction. Briefly, the commercially available HBX, purified by recrystallization, was stirred in methanol in the presence of KOH. The addition of 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide yielded a suspension from which the glycosylated derivative GBX was isolated. The basic conditions allowed the removal of the carbohydrate acetyl protection groups during this one-step reaction. Thin layer chromatography (TLC) was used to monitor the progression of the glycosylated reaction.Cell viability in bEnd.3 cells
An important feature of the investigation of this family of ThT-based molecules is the analysis of their tolerability and toxicity upon interaction with their target cells. Immortalized mouse endothelial bEnd.3 cells50 were used in this study to determine the toxicity of HBX, HBT, their respective iodinated derivatives, HBTI and HBXI; and the prodrug GBX. Clioquinol (CQ), and the well-known anti-cancer drug cisplatin (cis-diamminedichloroplatinum(II)) were included for comparison. Cells were evaluated by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) assay, where the viability of cells is determined by the reduction of the yellow MTT into the purple formazan product in the mitochondria of living cells.The obtained EC50 values, defined as the concentration of compound at which half of the maximal effect is observed with respect to that without the tested molecule, are summarized in Table 1, Table S2 and represented in Fig. S4 (ESI†). HBX and HBT show similar EC50 values of 73.28 and 78.9 μM, respectively. As it was envisaged, the high EC50 value of GBX (150.9 μM) shows the benefit of glycosylation. Amongst the two iodinated-derivatives, HBXI presents the lower EC50 value (7.75 μM), whereas HBTI is less toxic (EC50 value of 21.23 μM) than cisplatin and CQ that present EC50 values of 18.58 and 14.97 μM, respectively. On the basis of present cellular studies performed on the bEnd.3 cells, the MTT assay results confirm that an increase of hydrophobicity in our ligands leads to an increased toxicity, as it is observed with the incorporation of iodine. Overall, the EC50 values of the tested ThT-based compounds, with the exception of HBXI, are above those for cisplatin and CQ, the reference compounds. These data, summarized in Table 1 and Table S2 (ESI†), encourage further in vivo evaluation studies.
Cell-based models for the BBB permeability
As the highly selective blood–brain barrier (BBB) prevents the vast majority of drug candidates for neurodegenerative diseases from reaching the central nervous system (CNS), it is essential to consider early screening for BBB drug permeation in drug development.51 Several models have been developed and suggested to assess the ability of a compound to reach the brain.52,53 Computational approaches are commonly used to screen large molecular databases and predict their permeability through the BBB based on physicochemical features of the molecules.34,35,37 Traditionally, numerous studies have demonstrated that the prediction of drug passage into the CNS via passive diffusion can be estimated using a simple relationship, which establishes the crucial role of polarity and hydrogen bonding capability.34 The molecules presented in this work have already been studied in silico to predict their ability to cross the BBB based on the pharmacokinetic parameter logBB, the brain–blood partitioning coefficient.20,54 The reported logBB values,20 summarized in Table 1, are based on computationally derived physicochemical descriptors such as the calculated octanol–water partition coefficient (clogP) and the topological surface area (TPSA).34 According to this model, compounds with logBB > −0.3 can cross the BBB easily via passive diffusion.20,34
In the present work, we use a cell-based assay as initial confirmation of penetration that is predicted based on the previously calculated logBB values. The immortalized mouse brain endothelial bEnd.3 cell line is used as an in vitro model since it mimics the properties and complexity of the BBB.50 During the experiment, the extent of uptake of compounds into the cell was established, which is indicative of their ability to cross the barrier. Selected representative images in which areas rich in cells showed bright blue fluorescence of variable intensity are shown in Fig. 4 for cells incubated with compounds HBX, HBT, and their iodinated derivatives. For the sake of clarity, the corrected total cell fluorescence has been measured and plotted (Fig. 4a). Some differences were observed in the fluorescence emission vs. time of incubation of the tested compounds that may be indicative of a certain degree of uptake, which is probably related to subtle differences in the physicochemical characteristics of each compound. After ∼5 min of incubation, bEnd.3 cell samples in the presence of HBT and HBTI showed a clear fluorescence intensity whereas HBX and HBXI gave a weaker signal (Fig. 4b), even after 15 min of incubation. No fluorescence signal was observed on negative control cells (data not shown). Unfortunately, the range of excitation and emission wavelengths was not suitable for the study of the permeability of GBX (Fig. S5, ESI†). Also, GBX uptake through the BBB is envisaged to be via active transport, not passive diffusion, and thus the logBB calculation is not applicable. Overall, these observations may confirm the previous in silico prediction, which stated that the small molecules would be able to cross the BBB via passive diffusion at different degrees of uptake.
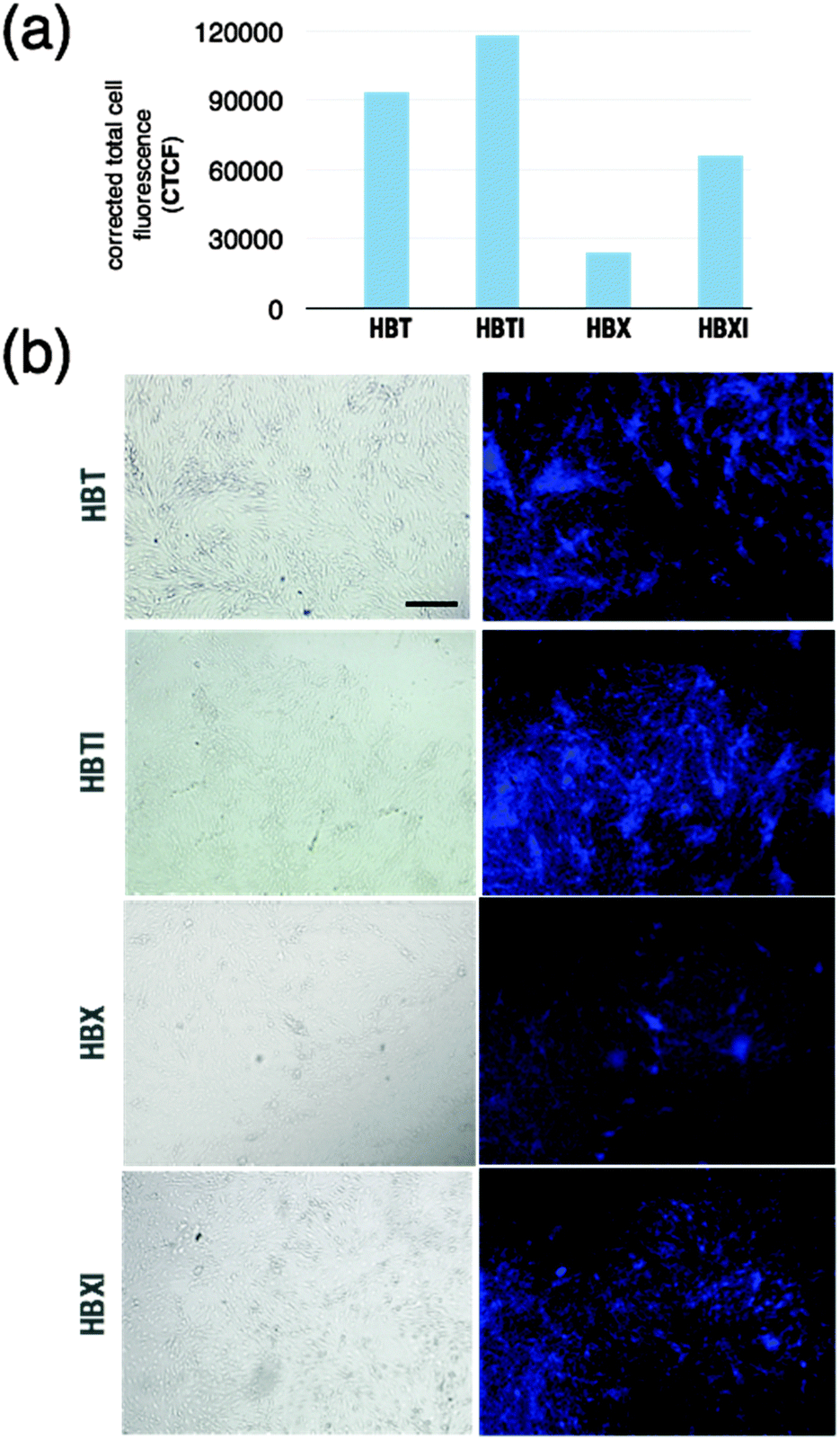 | ||
| Fig. 4 (a) The intensity of fluorescence of bEnd.3 cells treated with the tested compounds is plotted as corrected total cell fluorescence (CTCF) for comparison. (b) Column 1 shows phase-contrast microscopy images of the cells, and column 2 shows fluorescence microscopy images. Fluorescence signal from bEnd.3 cells incubated with the selected compounds. The cell culture was visualised under a fluorescence microscope with a blue DAPI filter. Negative binding control with PBS, indicating the absence of fluorescence, is not shown. Scale bar = 100 μm. | ||
Enzymatic glycoside cleavage. Prodrug activation
To confirm the prodrug activation and the potential of the glycosylated ThT-derivative to act as substrate for glucosidases, GBX was incubated with Agrobacterium sp. β-glucosidase (Abg) which has relatively broad substrate specificity.55 Abg was found to fully deglycosylate GBX within two hours (Fig. S6, ESI†). After this time, the absence of GBX, the presence of HBX, and a free sugar can be identified on a TLC plate under UV-light. In addition to its identification by matching the retention factor (Rf), the free ligand HBX turned a dark color when it comes in contact with Fe(III), and glucose spots were discolored in the presence of H2SO4 in ethanol after being heated (Fig. S6, ESI†). Deglycosylation was also confirmed with mass spectrometry (MS) by comparing the spectrum of the reaction mixture in the presence and the absence of the enzyme (data not shown). Therefore, when the prodrug derivatives cross the BBB, they should be deglycosylated, which is key in order to release the active form of the drug.Experimental
Materials
All reagents and solvents were purchased from commercial suppliers (Aldrich, Alfa Aesar, TCI America) and used without further purification with exception of 2-(2-hydroxyphenyl)benzoxazole (HBX) and 2-(2-hydroxyphenyl)benzothiazole (HBT), which were recrystallized from methanol. Immortalized mouse-brain capillary endothelial cells (bEnd.3) were grown in Gibco® Dulbecco's Modified Eagle Medium (DMEM with Glutamax; Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% v/v fetal bovine serum (FBS), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin antibiotics (Invitrogen Life Technologies, Carlsbad, CA). Confluent cells were released by trypsinization, reseeded into 75 cm2 cell culture flasks (BD Biosciences, MA), and incubated in a humidified incubator at 37 °C supplied with 5% CO2. Total cell count was performed with a haemocytometer (Neubauer Chamber) with trypan blue dye counting the non-stained cells under a light microscope at 10× magnification. Experiments were performed using cells between passages 45 and 50. Lyophilized synthetic human Aβ1–40 peptide was obtained from EZBioLab Inc. Water was purified to 18.2 MΩ cm by an Elga Ultra Pure Lab System. 1H NMR and 13C NMR spectra were recorded at room temperature using a Bruker AV-300 spectrometer. Low-resolution mass spectra were obtained using a Bruker Esquire Ion Trap ESI-MS spectrometer. Elemental analyses (C, H and N) were performed on a Fisons EA-1108 analyzer. High-resolution mass spectra were acquired on a Micromass LCT instrument (electrospray ionization). UV-visible data for cytotoxicity assays were collected using a Thermomax microplate reader (Molecular Devices) and fluorescence spectra with a Varian Eclipse Fluorescence Spectrophotometer.Synthesis
2-(Benzo[d]oxazol-2-yl)-4-iodophenol (HBXI) and 2-(benzo[d]thiazol-2-yl)-4-iodophenol (HBTI) were prepared according to a previously reported method.20 Experimental details are found in the ESI.†Binding assay using Aβ1–40 aggregates in solution
Aggregation was carried out by gently dissolving lyophilized synthetic human Aβ1–40 peptide (1 mg ml−1) in PBE buffer (10 mM sodium phosphate, 1 mM EDTA, pH 7.4). The solution was incubated at 37 °C for 48 h with gentle and constant shaking to promote the formation of mature amyloid fibrils. The presence of fibrils was confirmed by ThT assay (λex/λem = 450/490) and TEM (data not shown); they were used immediately upon production. A single lot of Aβ1–40 was used for all experiments. The equilibrium dissociation constant (Kd) was determined following a previously described procedure with some variations.27 Briefly, the binding assay was performed using a fixed concentration of Aβ1–40 fibril solution (5 μM) in PBE buffer and varying concentration of ligand (0–2.5 μM). All the prepared solutions were incubated at 37 °C overnight before reading. After the incubation period, the fluorescence was recorded at 440 nm and 460 nm (excitation 320 nm) for HBX and HBT respectively, and at 490 and 520 nm (excitation 340 nm) for HBXI and HBTI. The relative intensity changes were plotted vs. the ligand concentration. For each molecule, the experiment was performed at least in triplicate and the results were analysed and fitted to a one-site binding model using GraphPad Prism 5 (Graphpad Software, Inc., La Jolla, CA, USA).Molecular docking
The selected starting conformations for modelling were obtained from the solid-state nuclear magnetic resonance (ssNMR) measurements of Aβ1–40 fibrils (PDB code 2LMN). Energy minimization for each of the ten structures was carried out with the software package NAMD 2.756 using the CHARMM27 force field and solvating following the TIP3P water model.56 Cubic periodic boundary conditions were used. The input file in each case corresponded to the protein solvated in a water box with a minimum distance of 15 Å from any edge of the box to any protein atom. Na+ and Cl− were added to neutralize the net charge of the system and reach the physiological concentration. The amino acids were protonated according to the physiological pH and the N- and C-termini were kept ionic as under physiological conditions for Aβ1–40. Energy minimization was performed on all systems using the steepest descent technique in two subsequent steps. In the first step, protein backbone atoms were constrained to their initial positions allowing the relaxation of solvent and side chains (5000 steps). In the second step, no restraints were applied and all the systems were relaxed (10000 steps). The molecular geometries of HBX, HBXI, HBT and HBTI were optimized from density functional theory (DFT) calculations, with the B3LYP functional employing the 6-31G(d,p) basis57 set using Gaussian 09.57 The 3D structure for each receptor was based on the minimized structure obtained above. To incorporate protein flexibility, molecular docking was performed on the ten conformations (ensemble-based docking). Autodock 4.2 and MGLTools were used to perform docking calculations.58 For each ligand, hydrogens were added and Gasteiger–Hückel partial charges plus proper atomic types were assigned.59 Autotors was then used to define rotatable bonds.58 The receptor was prepared by adding hydrogen atoms to amino acid residues, and partial atomic charges were assigned to all the atoms using the Kollman method.60 Docking calculations were performed increasing default parameters to obtain 2.5 million Lamarckian Genetic Algorithm (LGA) evaluations, with a population of 150. A grid box was defined to cover the whole system including the peptide and the ligand (126 Å × 126 Å × 126 Å) placed on the centre of mass of the peptide (61.87, 20.18, 70.60). Grid spacing 0.703 Å step and 100 docking runs were conducted. After clustering the resultant poses with a tolerance of 2 Å RMSD, results were classified into different groups taking into account scoring and population criteria for each analysis as well as the main interactions between the inhibitor and the binding site of the receptor. Conformations with the lowest binding energy of the most stable cluster for each complex peptide–ligand were further analysed using DFT calculations.
Electronic structure calculations
Before computing the interaction energy, the ligand pose was refined by performing a geometry optimization of the ligand with the fibrils fixed using the ONIOM scheme,61,62 as implemented in Gaussian 09,57 combining the hybrid meta functional M06-2X63 and the standard basis set 6-31+G(d,p) for the ligand with the AM1 semiempirical molecular orbital method64 for the fibril. This relaxation reduces the repulsion between the ligand and the peptide, which appeared to be underestimated in the docking process, and thus, provides better geometries for computing the interaction energies. Due to the size of the system, ONIOM calculations were carried out only for the relevant part, which includes the ligand and all residues of the fibrils up to a distance of 5 Å from the ligand. The unsaturations generated by cutting the peptide were properly capped with hydrogen atoms. In this way all the residues interacting directly with the ligand were considered, while reducing the computational cost substantially. As the non-interacting residues remain unaffected during the binding process, and the geometry of the fibril is kept fixed, this approach does not affect the final DFT interaction energies. Interaction energies of the different molecules considered in the present study were computed by means of single point DFT(M06-2X/6-31+G(d,p)) calculations the in gas phase on the geometry obtained from the ONIOM optimization. M06-2X has been shown to account for dispersion interactions,65 which play a critical role in biological systems. The final energies were calculated as the difference between the energy of the ligand–peptide complex and the energies of the separated fragments. Obtained values have been corrected for basis set superposition error using the counterpoise method66,67 and considering two fragments: the ligand and the fibrils fragment.Ex vivo Aβ staining within brain sections
Brain samples from transgenic mice Tg2576 and TgCRND8 were used in this study. The Tg2576 mouse model (expressing human APP with the Swedish mutations K670N/M671L) develops amyloid plaques in the cortex and hippocampus after 10–12 months68 whereas the TgCRND8 (overexpressing hAPP harbouring the Swedish and Indiana V717F mutations) develops senile plaques at 3 months of age.2 Brain samples from control and AD human brains were obtained from the Institute of Neuropathology Brain Bank. Paraffin-embedded and frozen brain samples from AD individuals, TgCRND8 and Tg2576 mice were cut using either a microtome (8 μm) or a cryostat (15 to 30 μm) and processed for immunohistochemistry. The sections were incubated overnight at 37 °C with different concentrations of HBX or HBXI (0.16 μg ml−1, 0.05 mg ml−1, 0.5 mg ml−1) previously prepared in ethanol. As controls, brain sections were incubated overnight at 37 °C with ethanol. After washing the tissues three times with phosphate buffered saline (PBS) at pH 7.4, the sections were then mounted under coverslips with Dako Cytomation fluorescent mounting medium (Dako Cytomation, CA, USA). Immunofluorescence staining was visualised and imaged with a Nikon Eclipse 90i fluorescent microscope. Fluorescein isothiocyanate (FITC; λex = 465–495, λem = 515–555) was used for fluorescence imaging applications. To further confirm the presence of amyloid plaques and HBX (or HBXI) binding specificity, Congo Red staining was performed following manufacturer's instructions.Cell culture and cytotoxicity studies
BEnd.3 cells were maintained in a humidified incubator at 37 °C under 5% CO2 and cultured for 6 days in DMEM. After dissociation with trypsin, cells were resuspended in fresh culture medium at a density of 1 × 104 cells per ml. Using a Falcon 96-well flat-bottom plate, 100 μl of the cell suspension was added to the wells. Subsequently, cells were cultured for 24 h in DMEM. The cytotoxicity of HBX, HBT, HBXI, HBTI, CQ, GBX and cisplatin was assessed using a colorimetric MTT assay, which measures the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide due to the enzymatic activity of living cells. Growth medium (100 μL) was used as blank in 6 wells for each compound. 100 μL of HBX, HBT, HBXI, HBTI, CQ, GBX and cisplatin solutions prepared in the cultured media with 0.5% v/v DMSO at concentration range from 2 to 1200 μM were added to 48 wells. Growth medium (100 μL) containing 0.5% v/v DMSO was added to the remaining 6 wells containing cells and served as a control. The plate was incubated at 37 °C for 3 days. MTT solution (5 mg ml−1) in phosphate buffer solution (PBS) was freshly prepared and 50 μL added to each well. After 3 h of incubation under normal growth conditions in the dark, the solution was removed and 100 μL of DMSO was added to each well. Plates were placed in an agitator for 5 min and the absorbance at 595 nm was measured using a plate reader. The percentage of cell viability was calculated by dividing the average absorbance of the cells treated with the compounds by that of the control.Cell-imaging experiments
For fluorescence microscopy, bEnd.3 cells were plated on a Falcon 96-well microplate black/clear imaging plate and cultured for 24 h as detailed above. After being washed three times with PBS, pH 7.4, the cells were incubated for 5 and 15 min with ligands dissolved in DMSO and diluted in PBS to a final concentration of 20 μM. As negative control, the cells were incubated with PBS alone. After the incubation time, the cells were washed three times with PBS and analysed on a fluorescence microscope. Images were taken using an Olympus IX70 Inverted Fluorescence Microscope with a DC-330 3CCD MTI colour camera and Image-Pro Plus analysis software. A blue (DAPI, Hoechst) fluorescence filter cube was used throughout (excitation filter 340–380 nm, dichroic mirror 400 nm, emission filter 435–485 nm). The following formula was used to calculate the corrected total cell fluorescence (CTCF) = Integrated density − (Area of selected cell × Mean fluorescence of background readings). All measurements have been performed using ImageJ software v.2.0.0 and the obtained values were plotted for comparison.Enzymatic glycoside cleavage
The enzymatic hydrolysis of the glycosidic bond of GBX was tested with Agrobacterium sp. β-glucosidase (Abg). In a 0.5 ml Eppendorf tube, Abg (15 μM) and GBX (1 mM) were combined in a 20% DMSO/water solution. As a control, the same reaction was run without the enzyme. The progress of the reaction was monitored by thin layer chromatography (TLC) (95% dichloromethane/5% methanol) for two hours at room temperature; the obtained spots were compared to HBX and GBX by visualization under a UV lamp. Furthermore, the free phenol group was distinguished by complexation with Fe(III) and the presence of glucose was confirmed with 5% H2SO4 in ethanol after heating at 120 °C for 15 min.Conclusions
Herein, we present a combination of in vitro, ex vivo and in silico studies to understand the structure-based mechanism involved in the interaction of ThT-derived metal binders HBX, HBT, HBXI and HBTI with amyloid fibrils that are implicated in AD. The motivation behind these studies was to demonstrate the benefits of these molecules as potential imaging and/or therapeutic agents, in terms of high affinity and selectivity for amyloid, adequate brain uptake, and low cytotoxicity. (1) The affinity of the tested ThT-derivative compounds toward Aβ has been assessed, and the dissociation constants of the ligand–Aβ1–40 revealed that HBTI exhibits the highest binding affinity to amyloid fibrils. (2) The experimental observations have been investigated by in silico studies combining molecular docking and electronic structure calculations to present a more detailed description about the binding mode and the electronic properties of the ligand–Aβ1–40 complexes. The obtained trends on the interaction energies are in perfect agreement with the experimentally determined dissociation constants. (3) Importantly in ex vivo studies, HBX and HBXI are able to bind Aβ senile plaques present in brain samples from human AD patients, as well as Tg2576 and TgCRND8 transgenic AD mice. (4) In vitro cell-based assays to assess the cytotoxicity and the cell uptake of the tested molecules established EC50 values for HBX, HBT and HBTI to be above those of cisplatin and CQ. The incubation of the tested compounds with the bEnd.3 neuronal cell line, an in vitro model of BBB permeability, showed bright blue fluorescence in those areas rich in cells under fluorescence microscope. This observation confirms the previous in silico prediction (logBB) which stated that the small molecules would be able to cross the BBB via passive diffusion to different extents. (5) Notably, we have proposed the use of a glycosylated prodrug, GBX, to facilitate the transport to the brain via GLUT1 and enhance other properties of its precursor HBX; specifically, GBX is more soluble in solution and less toxic than its precursor, and can be activated into the active form by an enzyme. This study emphasizes the potential of the application of the ThT-derivative compounds in vivo.
Acknowledgements
We thank Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) from Generalitat de Catalunya for her financial support with the grant Beatriu de Pinós (C.R.R.), the Alzheimer Society of Canada for a doctoral award (M.A.T.), the Natural Sciences and Engineering Research Council of Canada (NSERC) for a Discovery Grant (C.O.), PGSD-2 award (M.A.T.), a summer USRA scholarship (G.A.B.), the Canadian Institutes of Health Research (CIHR) Proof of Principle program (C.O.), the Canada Council for the Arts for a Killam Research Fellowship (C.O.), MICINN (CTQ2011-24847/BQU), Generalitat de Catalunya (SGR2009-638) for financial support (J.A.T., L.R.S. and M.S.), and 2011 ICREA Academia award (M.S.) and Ministerio de Ciencia e Innovación CTQ2011-24847, SAF2008-00435, SAF2011-23272 (J.H.) and AP2005-0588 (Y.M.). The authors thank Prof. K. McNagny (Dept. of Medical Genetics, UBC) for providing the bEnd.3 cell line, Prof. S. Withers (Dept. of Chemistry, UBC) for a gift of Agrobacterium sp. β-glucosidase (Abg), Dr I. Ferrer (Institute of Neuropathology Brain Bank) for providing human brain tissue samples, Dr Paul A. Adlard (The Florey Institute of Neuroscience and Mental Health, University of Melbourne) for generously providing TgCRND8 brain samples, Dr Elena Polishchuk and Jessie Chen from the UBC Bioservices Laboratory for assistance with cell culture maintenance and MTT assays, and WestGrid, Compute/Calcul Canada and Catalonia Supercomputer Centre (CESCA) for computational resources.Notes and references
- D. J. Selkoe, Nature, 2003, 426, 900–904 CrossRef CAS PubMed.
- M. Stefani and C. M. Dobson, J. Mol. Med., 2003, 81, 678–699 CrossRef CAS PubMed.
- H. Levine, Amyloid, 1995, 2, 1–6 CrossRef CAS.
- W. E. Klunk, J. W. Pettegrew and D. J. Abraham, J. Histochem. Cytochem., 1989, 37, 1273–1281 CrossRef CAS PubMed.
- A. A. Reinke and J. E. Gestwicki, Chem. Biol. Drug Des., 2011, 77, 399–411 CAS.
- C. M. Dobson, Nature, 2002, 418, 729–730 CrossRef CAS PubMed.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed.
- A. D. Cohen, M. D. Ikonomovic, E. E. Abrahamson, W. R. Paljug, S. T. Dekosky, I. M. Lefterov, R. P. Koldamova, L. Shao, M. L. Debnath, N. S. Mason, C. A. Mathis and W. E. Klunk, Lett. Drug Des. Discovery, 2009, 6, 437–444 CrossRef CAS PubMed.
- C. Rodríguez-Rodríguez, M. Telpoukhovskaia and C. Orvig, Coord. Chem. Rev., 2012, 256, 2308–2332 CrossRef.
- A. I. Bush, Trends Neurosci., 2003, 26, 207–214 CrossRef CAS PubMed.
- W. R. Markesbery, Arch. Neurol., 1999, 56, 1449–1452 CrossRef CAS PubMed.
- C. A. Rottkamp, A. K. Raina, X. Zhu, E. Gaier, A. I. Bush, C. S. Atwood, M. Chevion, G. Perry and M. A. Smith, Free Radical Biol. Med., 2001, 30, 447–450 CrossRef CAS PubMed.
- G. Multhaup, H. H. Dieter, K. Beyreuther and T. A. Bayer, Handbook of Copper Pharmacology and Toxicology, Humana Press, New York, 1st edn, 2002, pp. 297–317 Search PubMed.
- A. I. Bush, J. Alzheimer's Dis., 2008, 15, 223–240 CAS.
- P. Zatta, D. Drago, S. Bolognin and S. L. Sensi, Trends Pharmacol. Sci., 2009, 30, 346–355 CrossRef CAS PubMed.
- W. E. Klunk, M. L. Debnath and J. W. Pettegrew, Neurobiol. Aging, 1994, 15, 691–698 CrossRef CAS PubMed.
- W. E. Klunk, Y. Wang, G. F. Huang, M. L. Debnath, D. P. Holt and C. A. Mathis, Life Sci., 2001, 69, 1471–1484 CrossRef CAS PubMed.
- Y. Wang, W. E. Klunk, M. L. Debnath, G.-F. Huang, D. P. Holt, L. Shao and C. A. Mathis, J. Mol. Neurosci., 2004, 24, 55–62 CrossRef CAS PubMed.
- E. E. Nesterov, J. Skoch, B. T. Hyman, W. E. Klunk, B. J. Bacskai and T. M. Swager, Angew. Chem., Int. Ed., 2005, 44, 5452–5456 CrossRef CAS PubMed.
- C. Rodríguez-Rodríguez, N. Sánchez de Groot, A. Rimola, A. Álvarez-Larena, V. Lloveras, J. Vidal-Gancedo, S. Ventura, J. Vendrell, M. Sodupe and P. González-Duarte, J. Am. Chem. Soc., 2009, 131, 1436–1451 CrossRef PubMed.
- S. R. Bareggi and U. Cornelli, CNS Neurosci. Ther., 2010, 18, 41–46 CrossRef PubMed.
- B. Regland, W. Lehmann, I. Abedini, K. Blennow, M. Jonsson, I. Karlsson, M. Sjogren, A. Wallin, M. Xilinas and C. G. Gottfries, Dementia Geriatr. Cognit. Disord., 2001, 12, 408–414 CrossRef CAS.
- G. K. Gouras and M. F. Beal, Neuron, 2001, 30, 641–642 CrossRef CAS PubMed.
- L. Lannfelt, K. Blennow, H. Zetterberg, S. Batsman, D. Ames, J. Harrison, C. L. Masters, S. Targum, A. I. Bush, R. Murdoch, J. Wilson and C. W. Ritchie, Lancet Neurol., 2008, 7, 779–786 CrossRef CAS PubMed.
- N. G. Faux, C. W. Ritchie, A. Gunn, A. Rembach, A. Tsatsanis, J. Bedo, J. Harrison, L. Lannfelt, K. Blennow, H. Zetterberg, M. Ingelsson, C. L. Masters, R. E. Tanzi, J. L. Cummings, C. M. Herd and A. I. Bush, J. Alzheimer's Dis., 2010, 20, 509–516 CAS.
- P. J. Crouch, M. S. Savva, L. W. Hung, P. S. Donnelly, A. I. Mot, S. J. Parker, M. A. Greenough, I. Volitakis, P. A. Adlard, R. A. Cherny, C. L. Masters, A. I. Bush, K. J. Barnham and A. R. White, J. Neurochem., 2011, 119, 220–230 CrossRef CAS PubMed.
- A. Lockhart, L. Ye, D. B. Judd, A. T. Merritt, P. N. Lowe, J. L. Morgenstern, G. Hong, A. D. Gee and J. Brown, J. Biol. Chem., 2005, 280, 7677–7684 CrossRef CAS PubMed.
- J. A. Lemkul and D. R. Bevan, ACS Chem. Neurosci., 2012, 3, 845–856 CrossRef CAS PubMed.
- M. R. Jones, E. L. Service, J. R. Thompson, M. C. Wang, I. J. Kimsey, A. S. DeToma, A. Ramamoorthy, M. H. Lim and T. Storr, Metallomics, 2012, 4, 910–920 RSC.
- S. J. Hyung, A. S. DeToma, J. R. Brender, S. Lee, S. Vivekanandan, A. Kochi, J. S. Choi, A. Ramamoorthy, B. T. Ruotolo and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3743–3748 CrossRef CAS PubMed.
- M. A. Telpoukhovskaia, C. Rodríguez-Rodríguez, J. F. Cawthray, L. E. Scott, B. D. Page, J. Alí-Torres, M. Sodupe, G. A. Bailey, B. O. Patrick and C. Orvig, Metallomics, 2014, 6, 249–262 RSC.
- J. Alí-Torres, A. Rimola, C. Rodríguez-Rodríguez, L. Rodríguez-Santiago and M. Sodupe, J. Phys. Chem. B, 2013, 117, 6674–6680 CrossRef PubMed.
- C. Wu, Z. Wang, H. Lei, Y. Duan, M. Bowers and J. Shea, J. Mol. Biol., 2008, 384, 718–729 CrossRef CAS PubMed.
- D. E. Clark, J. Pharm. Sci., 1999, 88, 815–821 CrossRef CAS PubMed.
- D. E. Clark, Drug Discovery Today, 2003, 8, 927–933 CrossRef CAS PubMed.
- D. E. Clark and S. D. Pickett, Drug Discovery Today, 2000, 5, 49–58 CrossRef CAS PubMed.
- J. T. Goodwin and D. E. Clark, J. Pharmacol. Exp. Ther., 2005, 315, 477–483 CrossRef CAS PubMed.
- C. Abad-Zapatero, O. Perisic, J. Wass, A. P. Bento, J. Overington, B. Al-Lazikani and M. E. Johnson, Drug Discovery Today, 2010, 15, 804–811 CrossRef CAS PubMed.
- S. Schultes, C. de Graaf, E. E. J. Haaksma, I. J. P. de Esch, R. Leurs and O. Krämer, Drug Discovery Today: Technol., 2010, 7, e157–e162 CrossRef CAS PubMed.
- R. L. Yona, S. Mazères, P. Faller and E. Gras, ChemMedChem, 2008, 3, 63–66 CrossRef CAS PubMed.
- E. C. Calvaresi and P. J. Hergenrother, Chem. Sci., 2013, 4, 2319–2333 RSC.
- X. Guo, M. Geng and G. Du, Biochem. Genet., 2005, 43, 175–187 CrossRef CAS PubMed.
- T. Halmos, M. Santarromana, K. Antonakis and D. Scherman, Eur. J. Pharmacol., 1996, 318, 477–484 CrossRef CAS PubMed.
- D. E. Green, M. L. Bowen, L. E. Scott, T. Storr, M. Merkel, K. Bohmerle, K. H. Thompson, B. O. Patrick, H. J. Schugar and C. Orvig, Dalton Trans., 2010, 39, 1604–1615 RSC.
- T. Storr, L. E. Scott, M. L. Bowen, D. E. Green, K. H. Thompson, H. J. Schugar and C. Orvig, Dalton Trans., 2009, 3034–3043 RSC.
- L. E. Scott, M. Telpoukhovskaia, C. Rodríguez-Rodríguez, M. Merkel, M. L. Bowen, B. D. G. Page, D. E. Green, T. Storr, F. Thomas, D. D. Allen, P. R. Lockman, B. O. Patrick, M. J. Adam and C. Orvig, Chem. Sci., 2011, 2, 642–648 RSC.
- L. E. Scott, B. D. G. Page, B. O. Patrick and C. Orvig, Dalton Trans., 2008, 6364–6367 RSC.
- H. Schugar, D. E. Green, M. L. Bowen, L. E. Scott, T. Storr, K. Bohmerle, F. Thomas, D. D. Allen, P. R. Lockman, M. Merkel, B. O. Patrick, K. H. Thompson and C. Orvig, Angew. Chem., Int. Ed., 2007, 46, 1716–1718 CrossRef CAS PubMed.
- T. Storr, M. Merkel, G. X. Song-Zhao, L. E. Scott, D. E. Green, M. L. Bowen, K. H. Thompson, B. O. Patrick, H. J. Schugar and C. Orvig, J. Am. Chem. Soc., 2007, 129, 7453–7463 CrossRef CAS PubMed.
- Y. Omidi, L. Campbell, J. Barar, D. Connell, S. Akhtar and M. Gumbleton, Brain Res., 2003, 990, 95–112 CrossRef CAS PubMed.
- W. M. Pardridge, Mol. Interventions, 2003, 3, 90–105 CrossRef CAS PubMed , 151.
- W. M. Pardridge, NeuroRx, 2005, 2, 3–14 CrossRef PubMed.
- W. M. Pardridge, NeuroRx, 2005, 2, 1–2 CrossRef PubMed.
- C. Rodríguez-Rodríguez, A. Rimola, J. Alí-Torres, M. Sodupe and P. González-Duarte, J. Comput.-Aided Mol. Des., 2011, 25, 21–30 CrossRef PubMed.
- J. B. Kempton and S. G. Withers, Biochemistry, 1992, 31, 9961–9969 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
- S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona, S. Profeta and P. Weiner, J. Am. Chem. Soc., 1984, 106, 765–784 CrossRef CAS.
- K. Morokuma, Bull. Korean Chem. Soc., 2003, 24, 797–801 CrossRef CAS.
- T. Vreven and K. Morokuma, J. Chem. Phys., 2000, 113, 2969–2975 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 119, 525 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS.
- K. Hsiao, P. Chapman, S. Nilsen, C. Eckman, Y. Harigaya, S. Younkin, F. Yang and G. Cole, Science, 1996, 274, 99–102 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete ref. 57; synthesis of HBXI and HBTI; Fig. S1–S5 corresponding to fluorescence spectra of the molecules in the presence and absence of synthetic Aβ1–40 fibrils; binding curves of ligand–Aβ1–40 fibrils complexes; ONIOM optimized geometries for HBTI and HBXI bound to Aβ1–40; cytotoxicity curves from MTT assays; TLC plates showing prochelator activation; and Tables S1 and S2. See DOI: 10.1039/c4mt00167b |
| ‡ Present address: Departamento de Química, Universidad Nacional de Colombia, Bogotá, Cundinamarca 111321, Colombia. |
| § Present address: University of Edinburgh, Centre for Neuroregeneration, Chancellor's Building, 49 Little France Crescent, Edinburgh EH16 4SB, UK. |
| This journal is © The Royal Society of Chemistry 2015 |