
Nanoarray based “superaerophobic” surfaces for gas evolution reaction electrodes
Zhiyi
Lu†
,
Yingjie
Li†
,
Xiaodong
Lei
,
Junfeng
Liu
and
Xiaoming
Sun
*
State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: sunxm@mail.buct.edu.cn; Fax: +86-10-64425385; Tel: +86-10-64448751
First published on 27th January 2015
Abstract
Electrochemical gas evolution reactions are now of great importance in energy conversion processes and industries. Key to the improvement of catalytic performance lies the development of efficient catalytic electrodes. Besides the exploration of highly active catalysts, the fast removal of the gas products on the electrode surface should be realized because the adhered gas bubbles would block the following catalytic reactions and decrease the efficiency. In this paper, we introduce an ideal structure, a “superaerophobic” surface, to diminish the negative effects caused by the adhered gas bubbles. Several recent works focusing on addressing this issue are presented with the target reactions of hydrogen evolution and oxygen evolution. It is demonstrated that micro/nano-engineering of the catalyst directly on the current collector is a promising approach to minimize the negative effective induced by the gas bubble adhesion. In the last section, we also discuss the promise of this methodology for other energy related systems.
1. Introduction
Gas evolution reactions (GERs), which are processes of converting liquid reactants to gas products accompanied with electron transfer in aqueous media, are of great importance in various industries, covering a number of energy storage and conversion systems. The hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) are two essential components for electrochemical water splitting, which is considered as one of the most effective approaches for future energy solutions.1,2 In addition, the OER is also crucial in the charge process for rechargeable metal–air batteries,3,4 and the HER is necessary for the chlor-alkali industry5 as well as the chlorine evolution reaction (ClER). GERs are also important in anodic reactions in direct liquid fuel cells, such as methanol oxidation and hydrazine oxidation.6–8For the above mentioned GERs, catalysts are required to reduce the overpotentials to achieve high reaction rates (i.e. high current densities). Constructing ideal catalytic electrodes for GERs should follow three principles. Firstly, for each target reaction, the catalyst selected should possess a high intrinsic activity, which is determined by the electronic structure of the active center.9–16 Secondly, the catalyst film should be porous and with high electric conductivity, which permits the penetration of the electrolyte and accelerates the electron transfer throughout the whole electrode.17–19 Thirdly, the surface of the catalytic electrode should easily release the as-formed gas bubble product because severe adhesion of the gas bubble will block the contact between the catalyst and reactant, cause an excessive ohmic drop and thereby decrease the efficiency.20 Although it is reported that inducing ultragravity21 or ultrasonic treatment22 is beneficial to the disengagement process of gas bubbles, they are not cost-effective for industrial production.
This review summarizes several recent works focusing on nanoscale electrode surface engineering to address the third principle of constructing an efficient catalytic electrode, which simultaneously meets the first and second principles. It is demonstrated that micro/nano-engineering of the catalyst directly on the current collector to gain a low-adhesion electrode is a promising approach to minimize the negative effective induced by the gas bubble adhesion. A new concept, the “superaerophobic” surface, is introduced to describe the interaction between gas bubbles and the low-adhesion electrodes. The HER and OER are the target reactions shown here, and the optimized architectures for the various catalysts are presented. In the last section, the promise of this methodology for GERs in other systems is also discussed.
2. Mechanism to design low-adhesion electrodes
As the reactant is a liquid and the product is a gas in GERs, a three-phase interface is formed together with the solid electrode, as shown in Fig. 1A. If the as-formed gas bubbles adhere in large numbers and form a gas film on the electrode surface, the electrolyte diffusion to the catalyst surface will be blocked. Accordingly, the electrode surface should be able to readily release gas products. Thus, how to make the surface dislike the gas product is the main challenge in constructing the architecture of the catalyst film. For a single gas bubble sitting on the surface of the catalyst film (Fig. 1A), it suffers two main forces: one is the buoyant force (Fb) that is proportional to the volume of the gas bubble, assuming that the shape of the gas bubble is a complete sphere (eqn (1)); the other is the adhesion force (Fa) from the underlying catalyst film, which can be expressed as eqn (2). The gravity of the gas bubble is neglected because of the much smaller density of the gas relative to that of the liquid. If the gas bubble is going to be detached from the surface, the buoyant force should become large enough to match the adhesion force (eqn (3)). Therefore, theoretically, the releasing diameter of the gas bubble is dependent on the adhesion force from the catalyst film. Derived from eqn (1)–(3), the radius of the gas bubble is proportional to the product of the root of γ and sine function of α (eqn (4)),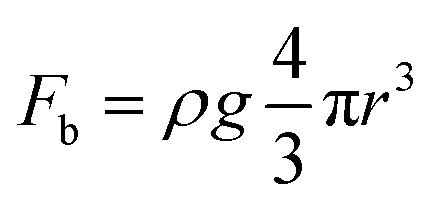 | (1) |
Fa = γ2π(r![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinα)sinα sinα)sinα | (2) |
| Fb = Fa | (3) |
 | (4) |
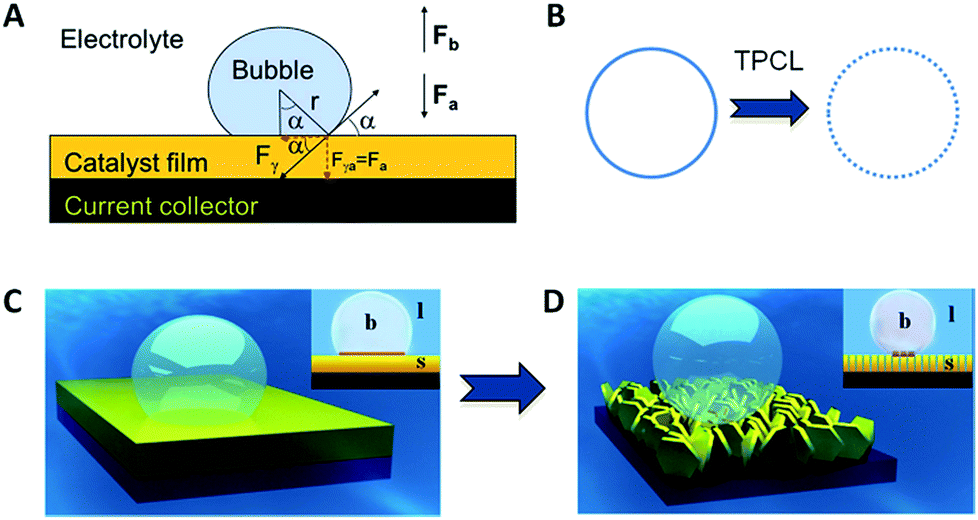 | ||
| Fig. 1 (A) Force analysis of a single gas bubble sitting on the catalyst film; (B) triple-phase contact lines (TPCLs) on the flat film (left) and nanostructured film (right); (C and D) schematic illustration of the adhesion behavior of gas bubbles on the flat film (left) and nanostructured film (right). Inset: side views to show the different intact and discontinuous TPCL on flat and nanostructured films, and the different contact angles. The brown zone is a “dead area” isolated by an as-formed bubble (reprinted with permission from ref. 20, Copyright 2014, Wiley-VCH). | ||
The adhesion force originates from the triple phase (solid–liquid–gas) contact line (TPCL), which is a continuous circle for an absolutely flat film (Fig. 1B, left). Constructing a nanostructured film can offer a much rougher surface, which significantly decreases the surface solid fraction and thus cuts the TPCL into discontinuous dots (Fig. 1B, right). Given that each point of the TPCL possesses the same adhesion force to the gas bubble for the same material, the broken TPCL would show a much smaller accumulated adhesion force relative to the continuous TPCL. Moreover, direct growth of the nanostructures can bring about many other advantages, such as good electron transportation and high porosity, which make the catalyst film more conductive and facilitate the penetration of electrolyte (reactant), leading to the improvement of catalytic performance.
3. Realization and demonstration of low-adhesion electrodes
3.1 Hydrogen evolution reaction (HER)
The HER is a typical example of GERs, as it has a variety of applications for energy conversion processes.23,24 This reaction needs catalysts to reduce the overpotential to operate this reaction, and the activities of the catalysts are highly dependent on their electronic structures.25–27 About a decade ago, MoS2 emerged as an efficient catalyst for the HER due to the suitable binding energy to hydrogen (not too strong or not too weak) at the edge sites, and thus the Mo edges are considered to be the active sites in MoS2.13,28–33 Recently, many compounds have been synthesized and identified with superior HER activities because of their new active sites, such as WS2,34 CoSe2,23 CoS2,35,36 NiP,37 CoP,38,39 Mo2C,40 MoB41 and so on. More recently, metal free catalysts have also been investigated with high HER performance.42,43 To further optimize their HER performance, the rational design of the structures of the catalysts is now being actively pursued.44Our group has employed MoS2 as an electro-catalyst to demonstrate the effectiveness of the low-adhesion surface according to the structural designing.20 Three MoS2 films with different surface architectures (nanostructured, microstructured and flat films) were fabricated by in situ sulfidation and precipitation processes, as shown in Fig. 2A–C. The adhesion force measurements revealed that the flat film exhibited a strong interaction with the gas bubble while the nanostructured film showed a negligible response (Fig. 2D–F), indicating that the construction of a nanoporous surface was an efficient approach to reduce the adhesion force towards gas bubbles. In addition, the contact angles of the gas bubbles on the nanostructured surface were larger than 150°, resulting in a surface with a low adhesion force (eqn (2)) and “superaerophobic” properties.
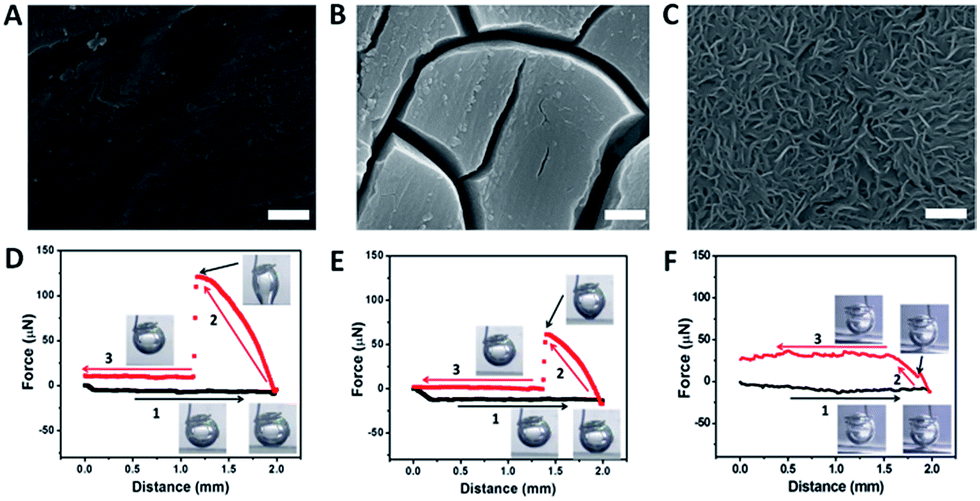 | ||
| Fig. 2 (A–C) The different surface morphologies of MoS2 films; (D–F) the corresponding gas bubble adhesion forces of the above surface structures (reprinted with permission from ref. 20, Copyright 2014, Wiley-VCH). | ||
The difference between the releasing sizes of the as-formed hydrogen bubbles is the consequence of constructing different surface architectures. As shown in Fig. 3A–C, the flat film that possesses the highest adhesion force shows the largest releasing size of the gas product (∼500 μm in diameter), while the microstructured film can reduce the diameter to ∼300 μm. It is found that the releasing size of gas bubbles on the nanostructured surface is as small as 50–100 μm, demonstrating the effectiveness of nano-engineering for reducing the solid–gas interaction. The reduced gas bubble size would give rise to the timely release of the adhered gas bubbles and thus diminishes the negative effects (blocking electrolyte penetration and covering the working area). As a result, the nanostructured film shows the fastest hydrogen evolution current density increase and the most stable working state (Fig. 3D and E). Specifically, for the flat and the nanostructured films, their electrochemical surface areas (which can be correlated with the active site density), Tafel slopes, exchange current densities and series resistances are very similar, and thus the different current density increase can only be correlated with the different bubble evolution behavior.
 | ||
| Fig. 3 (A–C) Different gas evolution behavior on the flat, microstructured and nanostructured MoS2 films; (D) HER performances of the three MoS2 films and commercial 20 wt% Pt/C film fabricated by a drop-casting method; (E) stability results of the three MoS2 films (reprinted with permission from ref. 20, Copyright 2014, Wiley-VCH). | ||
This effect can also be expanded successfully to other electro-catalysts for the HER. Kong et al. have firstly identified a highly-efficient catalyst of CoSe2 with comparable performance to Pt,23 and further they developed a method to grow a CoSe2 nanoparticulate film (Fig. 4A) on a curved substrate to form a three-dimensional electrode, which could deliver a high HER current density at low overpotential and a stable state over a long period (Fig. 4B).45 Faber et al. also demonstrated the enhanced bubble-releasing effect by constructing three different metallic CoS2 films (flat, microwire and nanowire films) and comparing their HER performances (Fig. 4C).35
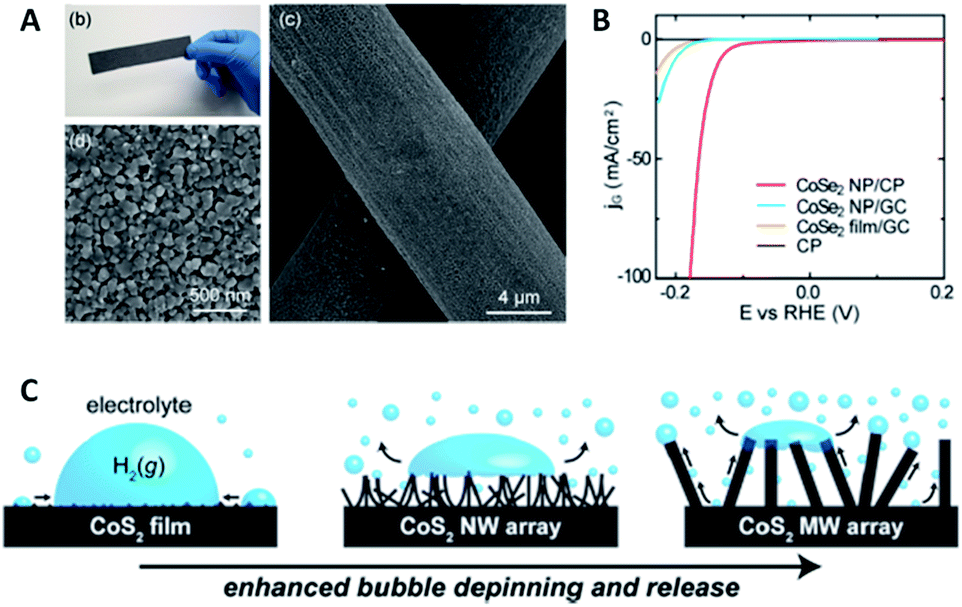 | ||
| Fig. 4 (A and B) Surface characterizations and HER performance of CoSe2 nanostructured film (reprinted with permission from ref. 33, Copyright 2014, ACS); (C) schematic illustration of the evolution behavior of as-formed gas bubbles on CoS2 films with different surface structures (reprinted with permission from ref. 35, Copyright 2014, ACS). | ||
3.2 Oxygen evolution reaction (OER)
Besides the HER, the OER is another important component of the overall water splitting reaction. Since four electrons are involved in the formation of one molecule of oxygen, this reaction possesses more sluggish kinetics relative to the HER, thus more efficient catalysts are required to overcome the high reaction barrier.46 Noble metal oxides (IrO2 and RuO2) are the best catalysts for water oxidation, but their scarcities have greatly restricted their large-scale application.47 Alternatively, since nickel and cobalt are identified to be the active centers for the OER, nickel and cobalt based materials (e.g. mixed transition metal oxides/hydroxides,48–59 cobalt-based perovskites60,61 and cobalt phosphates62,63) have been demonstrated with comparable OER activities. Recently, carbon based metal free catalysts have also been investigated with high OER activities.64,65 Similarly, building oriented nanostructures is an effective approach to further improve the overall performance.Li et al. found that the incorporation of nickel into cobalt oxide nanowire arrays could increase the surface roughness and thereby enhance the OER performance,66 which was also observed by Lu et al.67 Our group also demonstrated that combining the intrinsically active electro-catalysts (NiFe–LDHs and Zn-doped cobalt oxides) with appropriate morphologies (nanoarrays68 and hierarchical nanoarrays69) would readily achieve high OER current densities with low overpotentials. This ultrahigh performance was also partially attributed to the low adhesion force of the electrodes to the gas products, as demonstrated in our study of hierarchical NiCoFe–LDH nanoarrays.70 Although the hierarchical nanoarrays did not show a great improvement for further reducing the adhesion force compared with the nanoarrays, the higher porosity and active site density endowed the electrode with a marginal enhancement in the overall performance. The aforementioned works indicate that the performance of OER electrodes can also be benefited by constructing nanoarrays or hierarchical nanoarrays.
4. Summary and outlook
In this review, several recent works on the engineering of surface structures of electro-catalysts for GERs are summarized. Besides their high porosity and enhanced conductivity, nanoarrays and hierarchical nanoarrays can effectively alleviate the adhesion to the gas bubbles, thus affording the fast release of gas, resulting in a fast current increase and stable working state. This effect has been successfully applied in several non-noble electro-catalysts for both the HER and OER, demonstrating the general efficacy of this concept.Accordingly, as the noble metal catalysts (Pt, IrO2 and RuO2) show the highest intrinsic activities to the corresponding GERs, it is highly expected that ultrahigh performances would be achieved if these catalysts are built into oriented nanoarrays or hierarchical nanoarrays. In addition, since the catalysis reactions only occur at the surface, the noble catalysts can be deposited on the as-prepared conductive nanoarrays or hierarchical nanoarrays to reduce the amount of noble elements without compromising the catalytic performance.
Besides the HER and OER, this approach should also be valid for other GERs (e.g. methanol oxidation, hydrazine oxidation and chlorine evolution reactions, Fig. 5) which are essential in national economies but suffer from severe adhesion of the gas products (CO2, N2 and Cl2). It is well known that methanol oxidation and hydrazine oxidation are both important anodic reactions in direct liquid fuel cells and also suffer from severe bubble adhesion problems.71,72 Therefore, in the near future, progress toward engineering the catalyst materials for these reactions should be made to circumvent the bubble adhesion problem and substantially improve the GER performances.
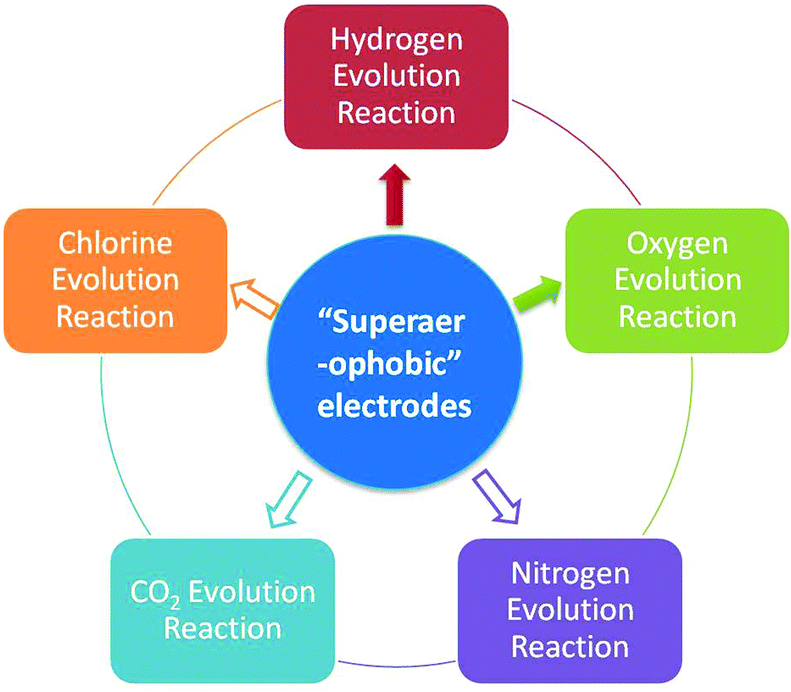 | ||
| Fig. 5 Uses of low adhesion electrodes. The uses directed by solid arrows have been demonstrated and the uses directed by hollow arrows need to be further explored. | ||
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China, the 973 Program (2011CBA00503 and 2011CB932403), the Program for New Century Excellent Talents in Universities, and the Program for Changjiang Scholars and Innovative Research Team in University.Notes and references
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- I. Moussallem, J. Jörissen, U. Kunz, S. Pinnow and T. Turek, J. Appl. Electrochem., 2008, 38, 1177–1194 CrossRef CAS.
- P. Argyropoulos, K. Scott and W. Taama, Electrochim. Acta, 1999, 44, 3575–3584 CrossRef CAS.
- H. Yang, T. Zhao and Q. Ye, J. Power Sources, 2005, 139, 79–90 CrossRef CAS PubMed.
- W. Qian, D. P. Wilkinson, J. Shen, H. Wang and J. Zhang, J. Power Sources, 2006, 154, 202–213 CrossRef CAS PubMed.
- D. K. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6801–6809 CrossRef CAS PubMed.
- P. Du, O. Kokhan, K. W. Chapman, P. J. Chupas and D. M. Tiede, J. Am. Chem. Soc., 2012, 134, 11096–11099 CrossRef CAS PubMed.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dinca, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- H. Wang, Z. Lu, S. Xu, D. Kong, J. J. Cha, G. Zheng, P.-C. Hsu, K. Yan, D. Bradshaw and F. B. Prinz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19701–19706 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013–2036 CrossRef CAS PubMed.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS PubMed.
- H. Wang, Z. Lu, D. Kong, J. Sun, T. M. Hymel and Y. Cui, ACS Nano, 2014, 8, 4940–4947 CrossRef CAS PubMed.
- Y. H. Chang, C. T. Lin, T. Y. Chen, C. L. Hsu, Y. H. Lee, W. Zhang, K. H. Wei and L. J. Li, Adv. Mater., 2013, 25, 756–760 CrossRef CAS PubMed.
- J. Wang, H. x. Zhong, Y. l. Qin and X. b. Zhang, Angew. Chem., Int. Ed., 2013, 125, 5356–5361 CrossRef.
- Z. Lu, W. Zhu, X. Yu, H. Zhang, Y. Li, X. Sun, X. Wang, H. Wang, J. Wang and J. Luo, Adv. Mater., 2014, 26, 2683–2687 CrossRef CAS PubMed.
- M. Wang, Z. Wang and Z. Guo, Int. J. Hydrogen Energy, 2010, 35, 3198–3205 CrossRef CAS PubMed.
- S.-D. Li, C.-C. Wang and C.-Y. Chen, Electrochim. Acta, 2009, 54, 3877–3883 CrossRef CAS PubMed.
- D. Kong, J. J. Cha, H. Wang, H. R. Lee and Y. Cui, Energy Environ. Sci., 2013, 6, 3553–3558 CAS.
- M. Gong, W. Zhou, M.-C. Tsai, J. Zhou, M. Guan, M.-C. Lin, B. Zhang, Y. Hu, D.-Y. Wang and J. Yang, Nat. Commun., 2014, 5, 4695–4701 CrossRef PubMed.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy and G. Eda, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. D. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515–2525 RSC.
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698–702 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- T. Wang, J. Zhuo, K. Du, B. Chen, Z. Zhu, Y. Shao and M. Li, Adv. Mater., 2014, 26, 3761–3766 CrossRef CAS PubMed.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878–3888 CAS.
- T. Wang, L. Liu, Z. Zhu, P. Papakonstantinou, J. Hu, H. Liu and M. Li, Energy Environ. Sci., 2013, 6, 625–633 CAS.
- D. Kong, H. Wang, J. J. Cha, M. Pasta, K. J. Koski, J. Yao and Y. Cui, Nano Lett., 2013, 13, 1341–1347 CrossRef CAS PubMed.
- L. Cheng, W. Huang, Q. Gong, C. Liu, Z. Liu, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2014, 53, 7860–7863 CrossRef CAS PubMed.
- M. S. Faber, R. Dziedzic, M. A. Lukowski, N. S. Kaiser, Q. Ding and S. Jin, J. Am. Chem. Soc., 2014, 136, 10053–10061 CrossRef CAS PubMed.
- Y. Sun, C. Liu, D. C. Grauer, J. Yano, J. R. Long, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 17699–17702 CrossRef CAS PubMed.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- Z. Pu, Q. Liu, P. Jiang, A. M. Asiri, A. Y. Obaid and X. Sun, Chem. Mater., 2014, 26, 4326–4329 CrossRef CAS.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- L. Liao, S. Wang, J. Xiao, X. Bian, Y. Zhang, M. D. Scanlon, X. Hu, Y. Tang, B. Liu and H. H. Girault, Energy Environ. Sci., 2014, 7, 387–392 CAS.
- H. Vrubel and X. Hu, Angew. Chem., Int. Ed., 2012, 124, 12875–12878 CrossRef.
- Y. Zhao, F. Zhao, X. Wang, C. Xu, Z. Zhang, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2014, 53, 13934–13939 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783–3790 Search PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- D. Kong, H. Wang, Z. Lu and Y. Cui, J. Am. Chem. Soc., 2014, 136, 4897–4900 CrossRef CAS PubMed.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- A. Di Blasi, C. D'Urso, V. Baglio, V. Antonucci, R. Ornelas, F. Matteucci, G. Orozco, D. Beltran, Y. Meas and L. Arriaga, J. Appl. Electrochem., 2009, 39, 191–196 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949–3956 CAS.
- Z. Lu, H. Wang, D. Kong, K. Yan, P.-C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345–4351 CAS.
- R. D. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- R. D. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- J. Landon, E. Demeter, N. İnoğlu, C. Keturakis, I. E. Wachs, R. Vasić, A. I. Frenkel and J. R. Kitchin, ACS Catal., 2012, 2, 1793–1801 CrossRef CAS.
- T. Alexis, Energy Environ. Sci., 2014, 7, 682–688 Search PubMed.
- J. B. Gerken, S. E. Shaner, R. C. Masse, N. J. Porubsky and S. Stahl, Energy Environ. Sci., 2014, 7, 2376–2382 CAS.
- Y. Xie, Y. Sun, S. Gao, F. Lei, J. Liu and L. Liang, Chem. Sci., 2014, 5, 3976–3982 RSC.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439–2445 Search PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- H. S. Ahn and T. D. Tilley, Adv. Funct. Mater., 2013, 23, 227–233 CrossRef CAS.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390–2396 Search PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2014, 53, 7281–7285 CrossRef CAS PubMed.
- Y. Li, P. Hasin and Y. Wu, Adv. Mater., 2010, 22, 1926–1929 CrossRef CAS PubMed.
- B. Lu, D. Cao, P. Wang, G. Wang and Y. Gao, Int. J. Hydrogen Energy, 2011, 36, 72–78 CrossRef CAS PubMed.
- Z. Lu, W. Xu, W. Zhu, Q. Yang, X. Lei, J. Liu, Y. Li, X. Sun and X. Duan, Chem. Commun., 2014, 50, 6479–6482 RSC.
- X. Liu, Z. Chang, L. Luo, T. Xu, X. Lei, J. Liu and X. Sun, Chem. Mater., 2014, 26, 1889–1895 CrossRef CAS.
- Q. Yang, T. Li, Z. Lu, X. Sun and J. Liu, Nanoscale, 2014, 6, 11789–11794 RSC.
- P. Argyropoulos, K. Scott and W. M. Taama, Electrochim. Acta, 1999, 44, 3575–3584 CrossRef CAS.
- H. Yang, T. S. Zhao and Q. Ye, J. Power Sources, 2005, 139, 79–90 CrossRef CAS PubMed.
Footnote |
| † These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |