
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermoresponsive fluorinated small-molecule drugs: a new concept for efficient localized chemotherapy
Catherine M.
Clavel
,
Patrycja
Nowak-Sliwinska
,
Emilia
Păunescu
and
Paul J.
Dyson
*
Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: paul.dyson@epfl.ch
First published on 8th October 2015
Abstract
Hyperthermia is currently being explored as an adjuvant treatment to conventional therapies with chemotherapeutic agents based on thermoresponsive macromolecules. Although the concept of hyperthermia has existed for many years it has yet to become routinely used in the clinical management of cancer. The development of small thermoresponsive molecules could help to change this paradigm. Temperature-sensitive compounds have recently been developed by covalently modifying drug and drug-like molecules with thermomorphic perfluorinated appendages. Lead thermoresponsive compounds have been validated in a pre-clinical model, displaying high tumor growth inhibition, with strong synergies observed between hyperthermia and the thermomorphic compounds.
Introduction
Surgical removal of solid tumors often fails to result in total remission and is therefore accompanied by chemotherapy, radiotherapy or a combination of the two.1,2 The lack of selectivity of chemotherapy leads to multiple side-effects, such as nephrotoxicity, blood disorders, fatigue, hair loss, nausea and vomiting.3 Therefore, alternative methods that combine chemotherapy with other treatment strategies have been explored in order to improve treatment selectivity, reduce recurrence and improve the quality of life of patients.One approach that may achieve these goals is to combine chemotherapy with hyperthermia (the application of heat). Hyperthermia, delivered at a continued or fractionated dose, can sensitize tumors to chemotherapy, radiotherapy, immunotherapy and immune-based strategies.4 The enhanced sensitivity of tumors to heat and the potential of hyperthermia in cancer treatment has been recognized for many years.5,6 However, the successful application of local hyperthermia (where heat is applied only at the tumor site) in combination with chemotherapeutics, has led to renewed interest in the approach.7 Recent years have witnessed substantial technical improvements in selectively heating superficial and deep-located tumors and the development of thermosensitive macromolecular drug delivery vehicles.8
Hyperthermia sensitizes cells to therapeutic agents and activates drug release from thermoresponsive nanocarriers (usually below 43 °C, referred to as mild hyperthermia) or, at higher temperatures, directly inducing necrosis (above 43 °C, referred to as thermal ablation).9 Importantly, cancer tissue is more thermosensitive than normal tissue between 42 °C and 45 °C,10 with a proportional relationship between cell death and the exposure time/temperature.11 There are various mechanisms by which local hyperthermia affects a cell leading to an enhanced antitumor response, see Fig. 1.12 Hyperthermia disrupts cell membrane function, enhances permeability and modifies the fluidity, stability and shape of the membrane, impeding transmembrane transport proteins and cell surface receptors.13,14 Heat transfer away from tumor tissue depends on the rate and volume of tumor perfusion,15 and this process is usually less efficient in malignant tissue compared to healthy tissues,16,17 enhancing the selectivity of hyperthermia.18
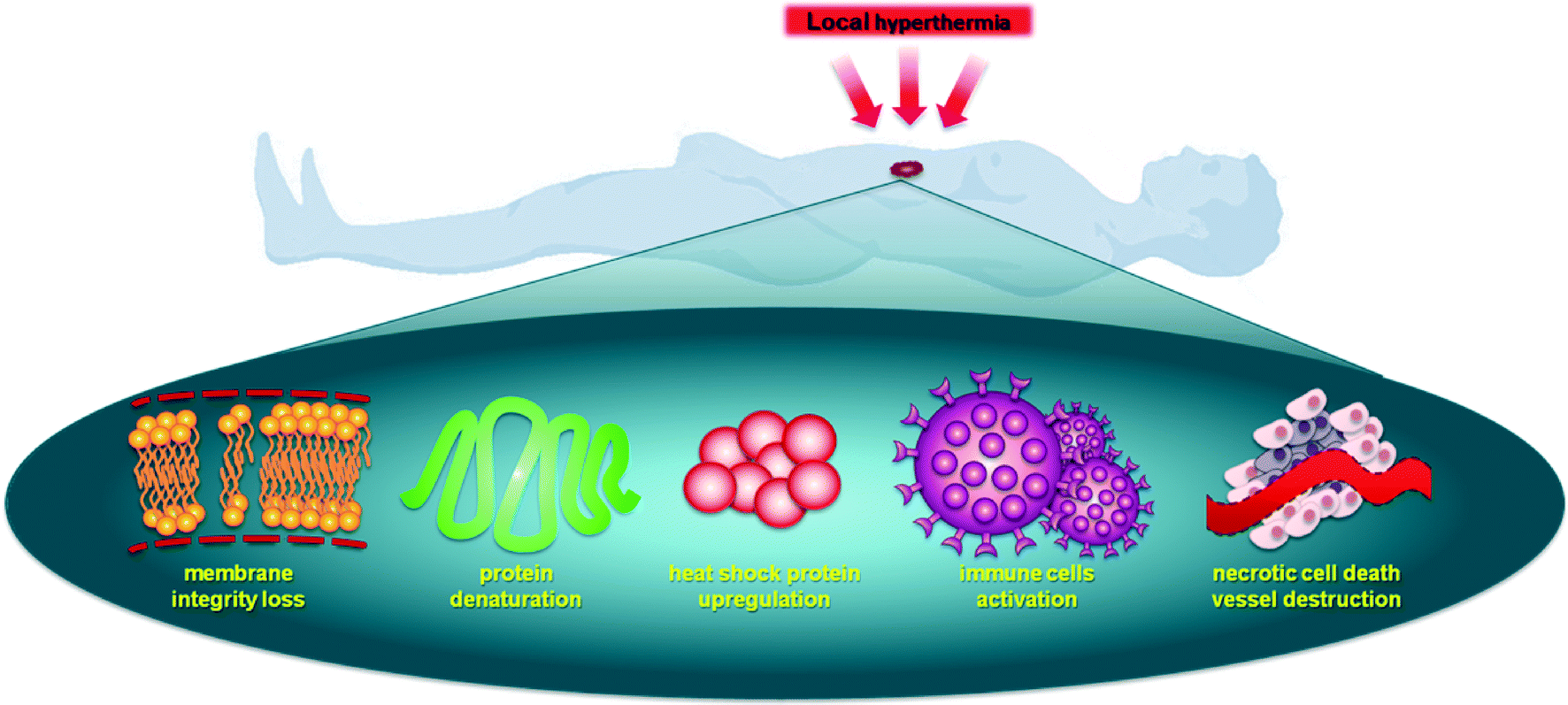 | ||
| Fig. 1 Different mechanisms of antitumor activity induced by local hyperthermia of tumors: loss of membrane integrity, upregulation of heat shock proteins, activation of immune cells, necrotic cell death and vessel destruction. | ||
Hyperthermia can effect cells in many different ways.19 It is known that heat can alter the structure of endogenous molecules such as lipids, nucleotides and proteins. Although the effects on lipids are reversible, the effects on DNA, i.e. the generation of double strand breaks, can be substantial and less easily reversed. However, the largest effects of hyperthermia are believed to be on proteins as they undergo denaturation and aggregation at temperatures >39 °C. This leads to inhibition of many cellular processes such as cell cycle arrest, inactivation of protein synthesis and inhibition of DNA synthesis and repair, resulting in inhibition of proliferation and cell death.20–22 Other important cellular changes induced by hyperthermia include the destruction of the cytoskeleton, making cellular motility difficult, and enhanced degradation of proteins through the proteasome and lysosomal pathways. In addition, changes in cellular metabolism resulting in decreased availability of ATP and enhanced production of reactive oxygen species (ROS) have been described.23,24 All these cellular changes ultimately lead to the loss of cell membrane integrity.
While these changes occur at the cellular level in all cells in the heated area, at a higher level vascular disruption also takes place, leading to vascular dysfunction and tissue eradication.25,26 However, as the vasculature is perfused the extent of hyperthermia is more moderate in the vessels, but nevertheless may result in better tumor perfusion. Such increased perfusion facilitates increased trafficking of immune cells including T cells and dendritic cells.27,28 The above-mentioned mechanisms may be differently balanced in specific tumor types as well as from patient to patient. Since heating tumors activates an immune response, at least in part by the upregulation of heat shock proteins, and since activity of some chemotherapeutics depend on heat shock proteins,29 it may suggest that these treatment strategies may act synergistically.
Combining small-molecule anticancer drugs with hyperthermia
Combining hyperthermia with chemotherapy frequently gives contrasting results in vitro and in vivo due to the ways in which hyperthermia effects the tumor microenvironment (see above). Drug exposure to cells remains relatively stable in vitro whereas in malignant tissue it is affected by the changes of the tumor blood flow induced by hyperthermia.30 Nevertheless, early studies demonstrated that alkylating agents such as methyl methanesulfonate (Fig. 2) exhibit superior effects in conjunction with hyperthermia at 41–42 °C,31,32 attributed to higher levels of DNA single strand breaks and reduced DNA repair.33 In other studies comparing melphalan, cisplatin and cyclophosphamide in combination with hyperthermia (41 °C) only cyclophosphamide resulted in an improved therapeutic response.34,35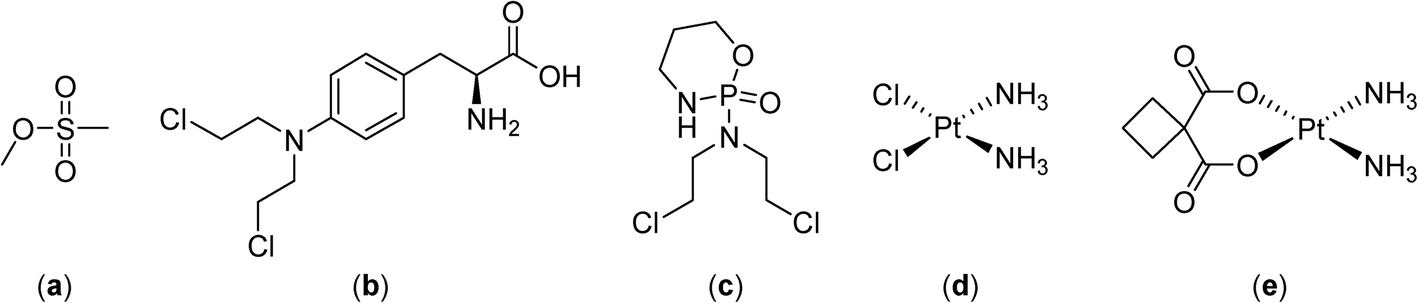 | ||
| Fig. 2 Structures of the alkylating agents methyl methanesulfonate (a), melphalan (b), cyclophosphamide (c), cisplatin (d) and carboplatin (e). | ||
Cisplatin was found to reduce tumor growth more efficiently in mouse mammary and rat glioma tumors when applied simultaneously with hyperthermia,36 although renal damage in rats resulting from the combination treatment were heightened.37 No enhancements in the reduction of tumor growth were detected for the antimetabolites, vinblastine and etoposide (Fig. 3) when combined with hyperthermia, possibly due to drug instability at the elevated temperatures.38
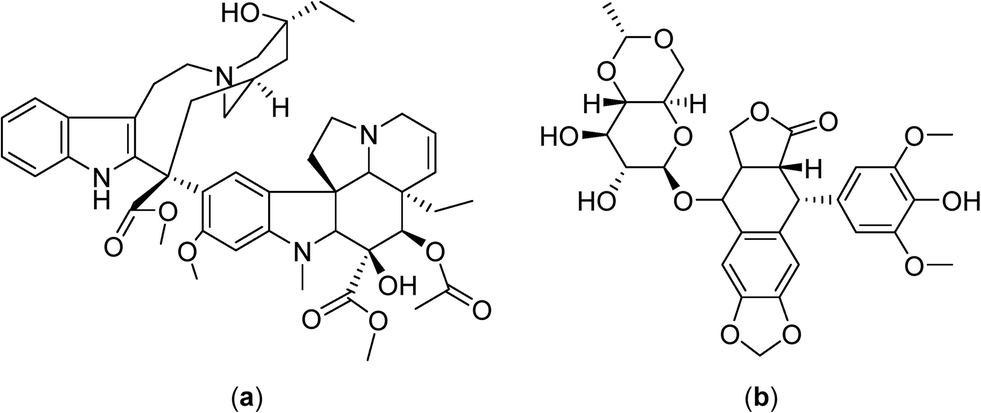 | ||
| Fig. 3 Structures of the antimetabolites vinblastine (a) and etoposide (b). | ||
To improve treatment protocols and reduce the side effects of hyperthermia, heating can be applied regionally or only locally and, furthermore, by careful timing of the treatment, with respect to drug administration, additional enhancements are possible. Indeed, the sequence and timing of chemotherapy and hyperthermia are critical for enhanced tumor reduction and it was shown for several drugs (cisplatin, melphalan and carboplatin, Fig. 2) that simultaneous administration of drug and heat is optimal.39,40 As mentioned above, hyperthermia can modify the tolerance of a tumor to chemotherapy leading to thermotolerance, an adaptive survival response, induced by heat preconditioning, whereby cells become resistant to a subsequent lethal insult. Therefore, the most advantageous treatment schedules involve administration of a chemotherapeutic before application of hyperthermia,41 or the simultaneous application of the two regimens.39,40 For cisplatin, it was shown that the concentration of the drug in the tumor is higher when injected prior to hyperthermia, presumably because hyperthermia induces vasodilatation following administration leading to an initial enhancement of drug retention in the tumor microenvironment with subsequent stabilization of tumor blood flow enhancing entrapment of the drug.42
Continuously circulating a heated solution containing chemotherapeutic agents inside the peritoneal cavity, a technique known as hyperthermic intraperitoneal chemotherapy, allows fast drug delivery to the gastric region. This approach was subsequently applied in clinical trials with cytoreductive surgery in gastric and ovarian carcinomas, in combination with cisplatin (Fig. 2) or mitomycin C (Fig. 4), in the treatment of malignant mesothelioma in combination with mitomycin C in phase II clinical trials,43–45 and in a phase III study on colorectal carcinoma employing 5-fluorouracil and leucovorin (Fig. 4).46–48
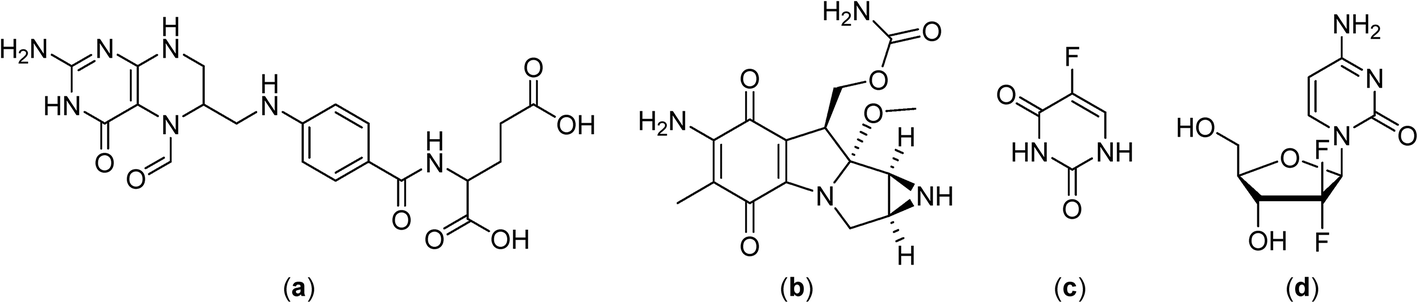 | ||
| Fig. 4 Structures of leucovorin (a) mitomycin C (b), 5-fluorouracil (c) and gemcitabine (d). | ||
Promising results were obtained with gemcitabine (Fig. 4) combined with cisplatin and regional hyperthermia as a second-line treatment for gemcitabine-resistant advanced and metastatic tumors.49 Moreover, patients with cervical cancer that did not respond to radiotherapy were administered weekly with cisplatin and regional hyperthermia in a phase II study with a response rate of 50% observed for hyperthermia-chemotherapy treated patients compared to only 15% for patients treated with chemotherapy alone.50,51
In phase II clinical studies doxorubicin, ifosfamide and etoposide52 or etoposide and ifosfamide (Fig. 5)53 were applied together with regional hyperthermia and shown to improve local control in high-risk, soft-tissue sarcoma compared to chemotherapy alone (a four year overall survival of 59% was achieved with the latter combination compared to 40% for chemotherapy alone). These promising results led to large and randomized phase III clinical trials with the first completed study showing moderate toxicity including skin burns, but with the response rate more than doubling under hyperthermia (28.8 vs. 12.7%), and an increased local progression-free survival at 2 years (76% vs. 61%).7 Another study investigated the chemotherapy combination comprising ifosfamide, carboplatin and etoposide as a second-line treatment with hyperthermia in soft-tissue sarcoma refractory with an objective response rate of 20%.54 In phase III clinical trials hyperthermia led to significant clinical benefits using regional hyperthermia for superficial and deep local advanced tumors such as high-risk soft-tissue sarcoma.
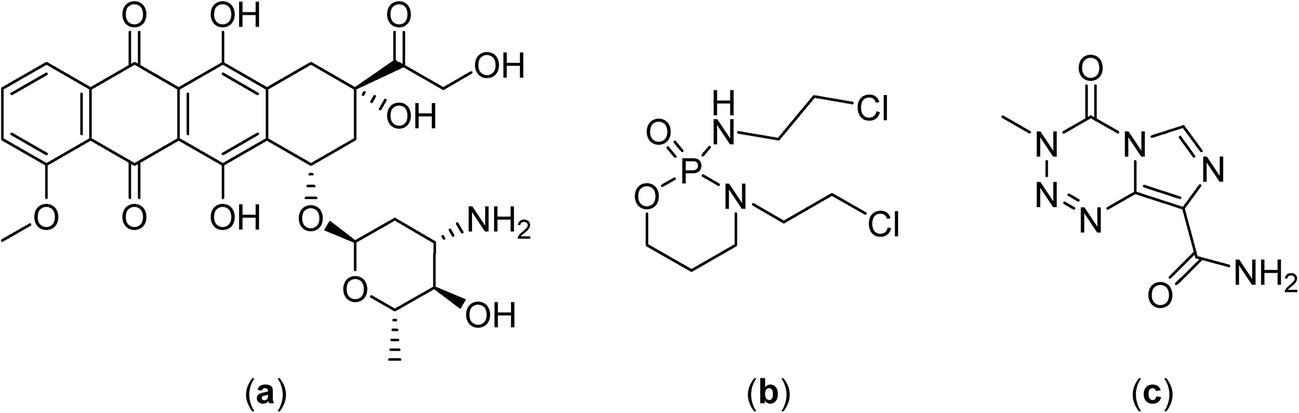 | ||
| Fig. 5 Structures of doxorubicin (a), ifosfamide (b) and temozolomide (c). | ||
Macromolecular thermoresponsive systems
The application of small molecules applied with hyperthermia indicates the considerable potential of the approach in cancer therapy. However, the molecules described above were not originally designed to be applied with hyperthermia, indicating that more effective compounds could be developed. Initial attempts to adapt compounds to hyperthermia were based on the encapsulation of established drugs in liposomes with temperature-dependent drug release characteristics. These macromolecular systems have the added advantage that liposomes also preferentially accumulate in solid tumors due to the enhanced permeation and retention of macromolecules.55Notably, liposomal formulations of doxorubicin (Fig. 5) have been extensively studied in thermotherapy.56,57 A drug delivery system that shows considerable promise is based on a low temperature sensitive liposome containing doxorubicin, termed ThermoDox®, which releases the drug in a few seconds at 42 °C.58,59 The heat-sensitive liposome changes structure as a function of temperature and, as the temperature increases, pores in the liposome are created which release doxorubicin directly into the heated tumor.60 ThermoDox® is currently in phase III clinical trials in combination with hyperthermia for the treatment of hepatocellular carcinoma. However, a significant proportion of the encapsulated doxorubicin in ThermoDox® is lost following intravenous administration61,62 and, consequently, alternative drug delivery systems have been developed. A cationic thermoresponsive liposomal system incorporating doxorubicin and ammonium bicarbonate operates via an alternative mechanism. At the heated tumor site CO2 bubbles are produced that induce the release of the doxorubicin.57 In another variant the doxorubicin is coordinated to manganese ions, which enhances encapsulation without impacting on the temperature-triggered release and pharmacokinetics of the drug delivery system.61 Cisplatin encapsulated in preformed thermoresponsive cholesterol-containing liposomes is stable at 37 °C (<5% released), whereas >95% is released within 5 minutes at 42 °C.63 Nanoparticles have also been explored as thermoresponsive drug nanocarriers, including iron oxide magnetic nanoparticles,64 acid-capped poly(lactic-co-glycolic acid) nanoparticles,65 silica-coated magnetic lanthanum–strontium manganite nanoparticles66 or other surface-modified nanoparticles,67,68 all incorporating doxorubicin as the active drug molecule.
An advantage of certain macromolecular drug delivery systems is that they can traverse the blood–brain barrier.69 For example, a liposomal doxorubicin formulation (Caelyx®) able to cross the blood–brain barrier was found to accumulate in glioblastoma and brain metastases.70 A multimodality treatment comprising radiotherapy, hyperthermia and chemotherapy, i.e. temozolomide (Fig. 5), with added Caelyx® for resistant cases, led to enhanced survival rates in a glioblastoma clinical trial.71 More than 50% patients remained alive after 26 months whereas the median survival following surgery is usually <4 months, which is only slightly improved with radiotherapy.72,73
Heat seeking drug-loaded polypeptide nanoparticles based on a thermally responsive elastin-like polypeptide conjugated to multiple copies of doxorubicin have been reported recently.74 These nanoparticles were able to target tumors that were externally heated to 42 °C.75 Thermal cycling (heating and cooling) of the tumors following injection of the thermally responsive nanoparticles results in a significant enhancement of doxorubicin accumulation in the tumor.74,76
New small-molecule thermoresponsive compounds
Despite the development of macromolecular drug formulations for thermotherapy, notably liposomal formulations, small-molecule drugs not designed for use in combination with hyperthermia continue to be evaluated in clinical trials. There are clinical advantages in using low molecular weight thermosensitive drugs that are selectively activated at the tumor site by the application of hyperthermia. In this context many highly fluorinated compounds have excellent thermomorphic properties77–79 and fluorine compounds already play an important role in medicinal chemistry.80–82 Numerous anticancer drugs such as 5-fluorouracil (Fig. 4),83 rosuvastatin and fluticasone84 and torcetapib85 contain one or more fluorine atoms (Fig. 6). Perfluorinated systems have also been investigated as drug delivery systems and were shown to exhibit prolonged circulation times in the blood.86 Drug absorption and biodistribution rely mainly on the lipophilicity/hydrophilicity of the system and fluorine-containing compounds have unique properties in this regard, with diverging lipophilic and hydrophobic characteristics depending on the fluorine atom content.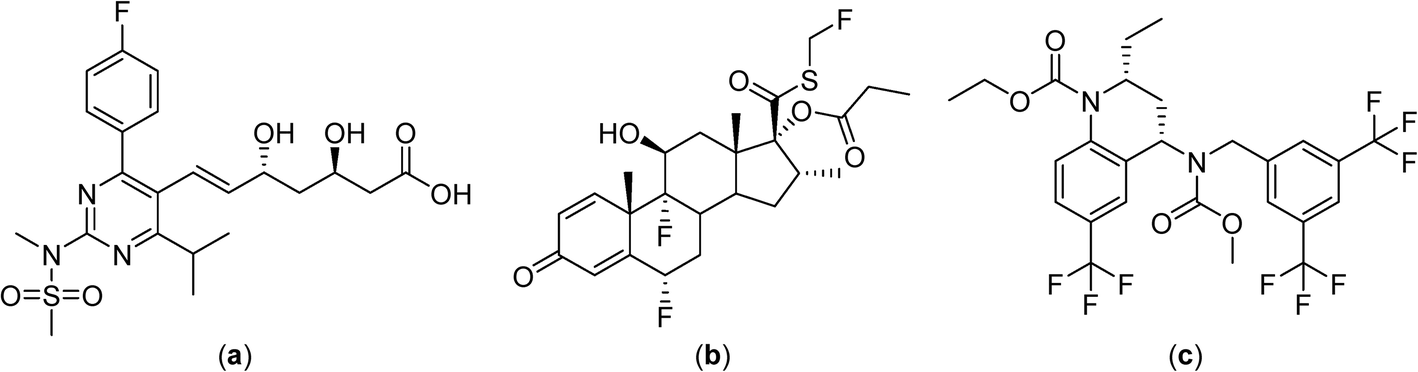 | ||
| Fig. 6 Structures of the fluorine-containing drugs rosuvastatin (a), fluticasone (b) and torcetrapib (c). | ||
Perfluorinated solvents undergo temperature-dependent miscibility with organic solvents and water87,88 and the solubility of certain fluorinated compounds varies considerably as a function of temperature.89,90 Moreover, certain fluoropolymers exhibit biocompatible characteristics and have been evaluated in various biomedical applications.91,92 Based on these observations, the first small-molecule anticancer compounds containing perfluorinated chains attached via a phosphine ligand to bioactive ruthenium(II)–arene moieties were designed and evaluated in vitro (Fig. 7, PTA = 1,3,5-triaza-7-phosphatricyclo[3.3.1.1]decane).93 The solubility of the compounds at 37 °C was low and in some cases increased considerably at 42 °C. Some of the compounds were found to be strongly cytotoxic to human ovarian A2780 and A2780cisR cancer cell lines (the latter having acquired resistance to cisplatin) under normal conditions, i.e. at 37 °C. Including a 42 °C heating cycle for 2 hours during the incubation period increased their cytotoxicity.
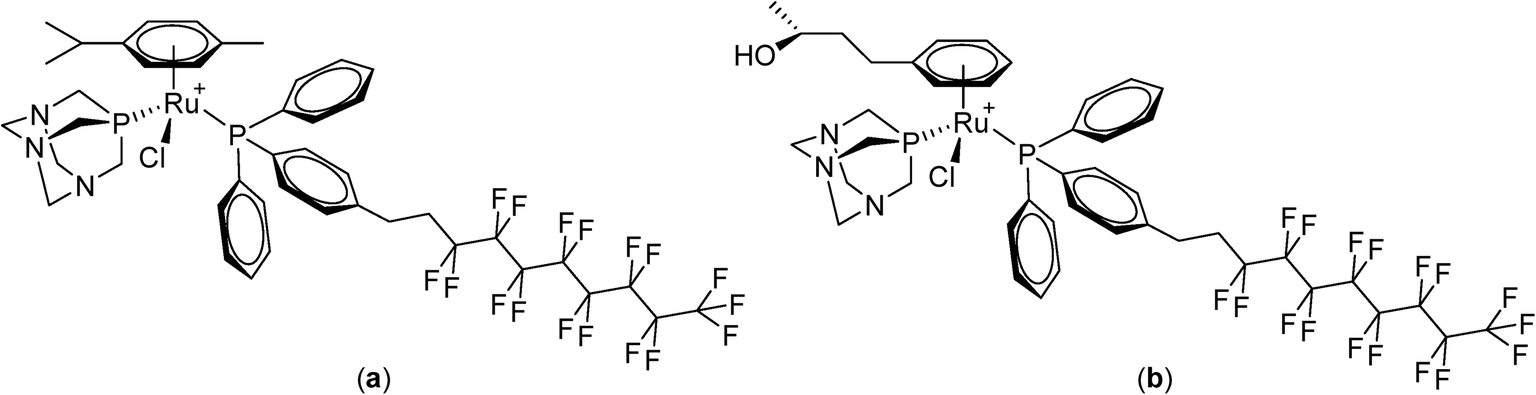 | ||
| Fig. 7 Ruthenium(II)–arene complexes with fluorinated phosphines that modest exhibit thermoresponsive behavior; [Ru(η6-p-cymene)(PTA)Cl(PPh2(p-C6H4(CH2)2(CF2)7CF3))][BF4] (a) and [Ru(η6-p-phenyl-2-butanol)(PTA)Cl(PPh2(p-C6H4(CH2)2(CF2)7CF3))][BF4] (b). The counter anion is not shown. | ||
Although non-fluorinated ruthenium(II)–arene compounds related to those shown in Fig. 7 show encouraging in vivo properties,94–100 as yet, none have progressed to clinical trials. Consequently, the widely explored and clinically approved alkylating agent, chlorambucil, p-(Cl(CH2)2)2N-C6H4-(CH2)3-CO2H, was derivatized with perfluorinated chains via an ester linkage in order to better assess the thermoresponsive potential of the compound. These chlorambucil derivatives were shown to exhibit significant differences when applied to cancer cells under normal conditions and under mild hyperthermia at 42 °C.101 Notably, chlorambucil modified with a long (C10) perfluorous chain, i.e. p-(Cl(CH2)2)2N-C6H4-(CH2)3-CO2-(CH2)2(CF2)9CF3 (Fig. 8), is only cytotoxic following a 2 hour hyperthermia signal. In the various cancer cell lines tested the compound is consistently more cytotoxic following hyperthermia. For example, at 37 °C in the A2780 and A2780cisR cell lines the compound is inactive at the maximum concentration that could be tested (200 μM), whereas with the inclusion of a 2 hour period at 42 °C during the 72 hour incubation period IC50 values of 37 and 40 μM, respectively, were obtained. In the same cell lines chlorambucil was less cytotoxic when applied in combination with hyperthermia, and analogues in which the fluorous chain is replaced by a hydrocarbon chain do not show clear thermoresponsive behavior. Note that the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values of the chlorambucil derivatives with fluorous chains are significantly higher (ca. 9–12) than those with alkyl chains (ca. 6). In the case of the longest chain derivatives (both fluorous and alkyl chains) a significant increase in solubility in water was observed as the temperature changes from 37 to 43 °C.101
P values of the chlorambucil derivatives with fluorous chains are significantly higher (ca. 9–12) than those with alkyl chains (ca. 6). In the case of the longest chain derivatives (both fluorous and alkyl chains) a significant increase in solubility in water was observed as the temperature changes from 37 to 43 °C.101
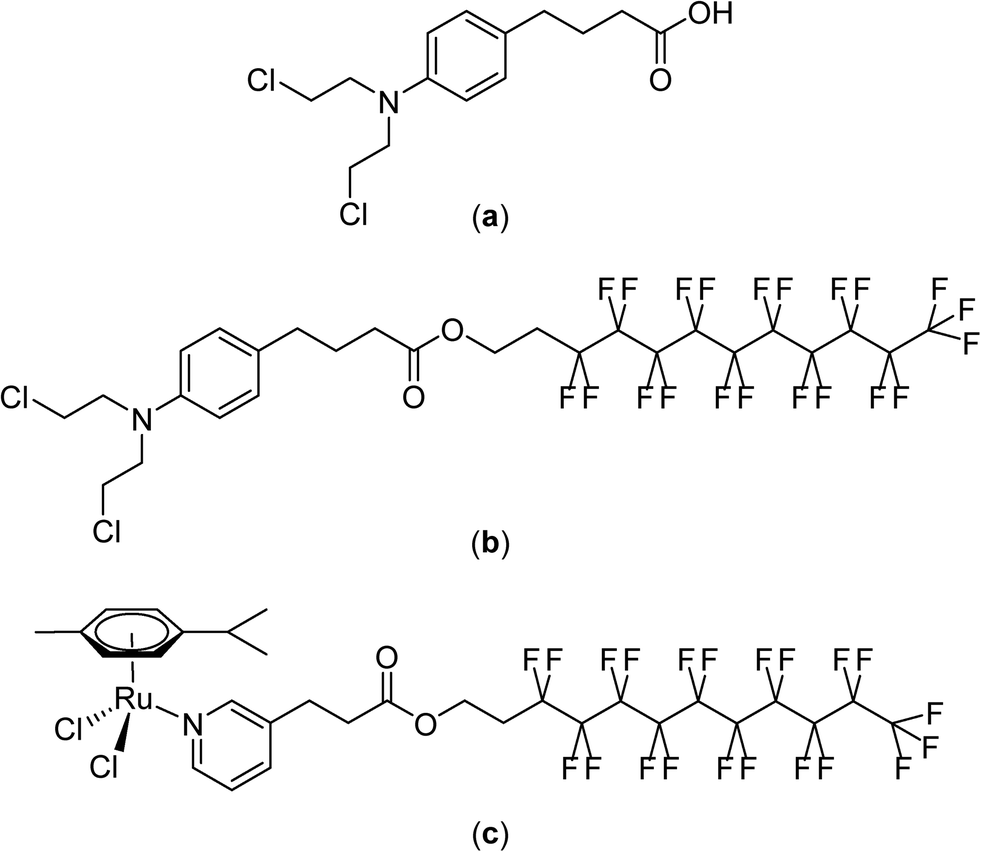 | ||
| Fig. 8 Structures of chlorambucil (a), p-(Cl(CH2)2)2N-C6H4-(CH2)3-CO2-(CH2)2(CF2)9CF3 (b) and [Ru(η6-p-cymene)Cl2(C5H4N-3-(CH2)2-CO2-(CH2)2(CF2)9CF3)] (c). | ||
The thermoresponsive properties of p-(Cl(CH2)2)2N-C6H4-(CH2)3-CO2-(CH2)2(CF2)9CF3 was also demonstrated in vivo using a mouse model bearing the human LS-174 T tumor.102 At a dose of 12.5 mg kg−1, administered a total of three times every four days, a reduction in tumor growth of 59% is observed under normal conditions, i.e. without hyperthermia. When combined with a 30 minute hyperthermia signal a few minutes after injection tumor growth inhibition increases to 79%.
The mechanism of tumor cell death induced by the compound appears to be the same as that of chlorambucil itself and involves DNA damage. However, the mechanism of thermal activation remains unclear, although it would appear to involve increased solubility at the heated tumor site with concomitant cleavage of the fluorous chain at the ester linker. Compounds in which the fluorous chain is covalently linked to the drug via non-cleavable groups do not appear to be endowed with such extensive thermoresponsive activity. Hence, the original thermoresponsive ruthenium complexes mentioned above were redesigned with the fluorous chain tethered to a pyridine ligand via an ester linkage (Fig. 8).103 These ruthenium compounds display a remarkable selectivity to cancer cells in vitro when used in combination with a 2 hour hyperthermia treatment of 41 °C. For the most effective compound, [Ru(η6-p-cymene)Cl2(C5H4N-3-(CH2)2-CO2-(CH2)2(CF2)9CF3)], cytotoxicity was not observed at concentrations <500 μM under normal conditions whereas with the inclusion of a 2 hour heating period at 42 °C during the 72 hour incubation resulted in IC50 values as low as 5 μM (in MCF-7 human breast cancer cells), i.e. two orders of magnitude greater inhibition of cell growth for the combination with hyperthermia. Notably, in non-cancerous human endothelial HEK-293 cells hyperthermia did not lead to such a large increase in cell growth inhibition with an observed IC50 value of 132 μM.
[Ru(η6-p-cymene)Cl2(C5H4N-3-(CH2)2-CO2-(CH2)2(CF2)9CF3)] was evaluated in vivo in the same model used to test the chlorambucil derivative at an equivalent dose and administration/heating regime.102 Tumor growth inhibition of 66% was observed under normal conditions, which increases to 90% when combined with hyperthermia. Based on histochemical analysis tumor growth inhibition was attributed to the inhibition of cell proliferation and, in part, to necrosis, the latter feature having been observed in other combination studies employing hyperthermia.104 The toxicity of the compound appears to be largely limited to the heated tumor region as weight loss and other side effects were not observed. Moreover, the ruthenium distribution in the vital organs is not aggravated by the heat treatment process and the distribution of ruthenium is similar to other ruthenium compounds that are not cytotoxic.102,105,106 It has also been suggested that the selective delivery of the compound to the heat tumor site could also be due to temperature-dependent interactions with certain serum proteins.107
Concluding remarks
Randomized clinical trials have demonstrated overall patient survival prolongation with hyperthermia-drug treatment regimens applied to a wide range of malignancies. However, in order to make a local hyperthermia a more powerful cancer treatment strategy all possible factors of the treatment must be optimized. This optimization process is not trivial due to the difficulties associated with maintaining the optimum intra-tumor temperature,108,109 hyperthermia-induced drug targeting110 and selective drug activation by heat. Nevertheless, multiple studies have shown that hyperthermia complements chemosensitization and the mechanism of action of this dual-therapy appears to be dependent on the particular mechanism of each chemotherapeutic compound.Further improvements of this treatment strategy will undoubtedly involve the development of more efficient heat-responsive drugs. The strategy reported herein, i.e. based on modifying clinically approved drugs or putative drug-like molecules with fluorous chains conjugated via ester linkages, certainly holds promise. A tentative, generic mechanism concerning drug delivery and heat activation is shown in Fig. 9 and, while the approach has thus far only been demonstrated on a limited number of compounds, it is not unreasonable to assume that it can be applied to almost any bioactive (anticancer) molecule. However, further validation of this approach is still needed as fluorous-tagged compounds also display relevant therapeutic properties under normal conditions111–113 and to determine whether it has advantages over the well-established use of thermoresponsive macromolecular drug delivery systems, some of which are progressing through clinical trials.
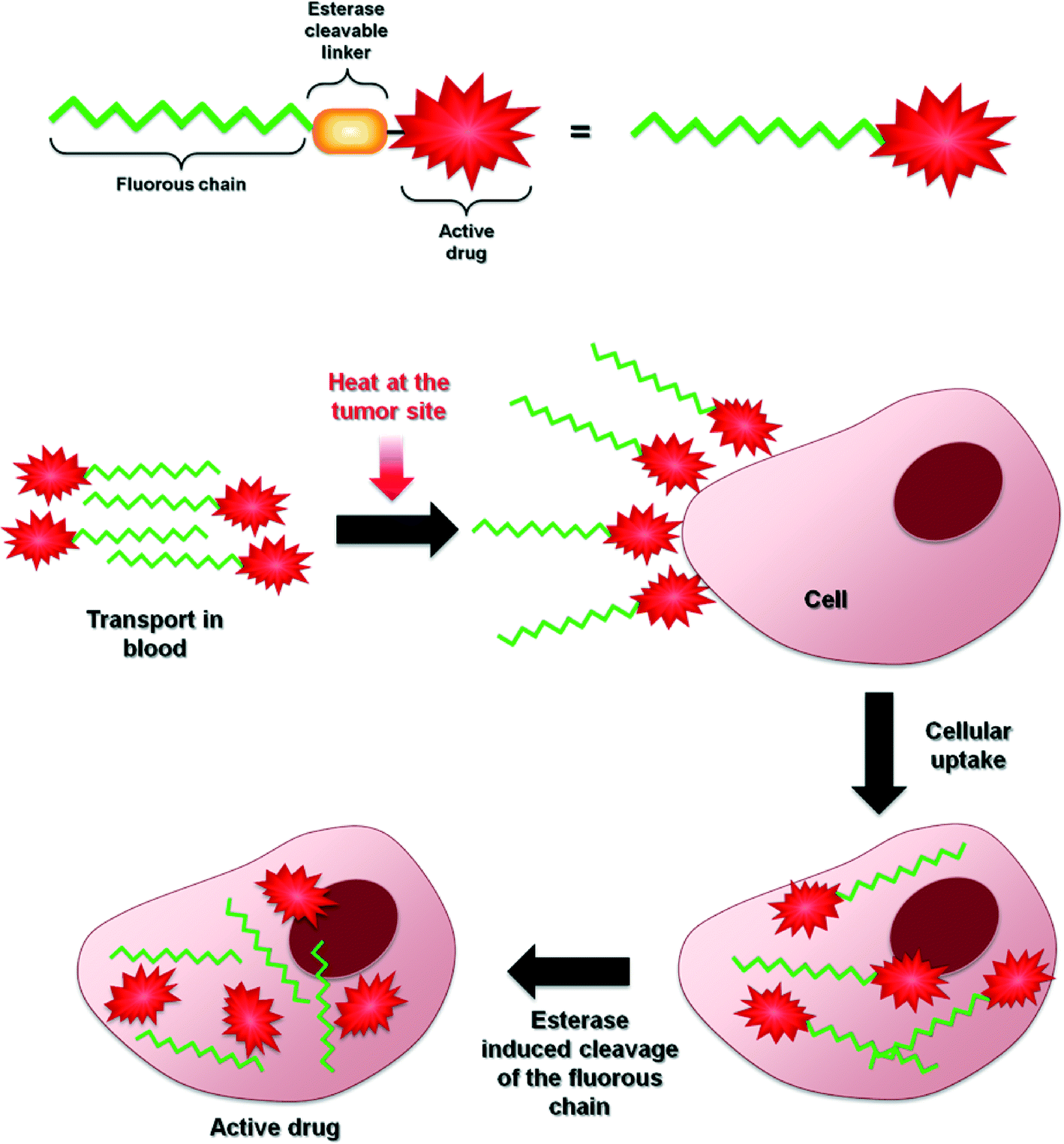 | ||
| Fig. 9 Proposed generic mechanism of action of thermoresponsive fluorous-tagged drugs. | ||
Acknowledgements
We thank the Swiss National Science Foundation and EPFL for financial support.References
- A. J. Breugom, M. Swets, J. F. Bosset, L. Collette, A. Sainato, L. Cionini, R. Glynne-Jones, N. Counsell, E. Bastiaannet, C. B. van den Broek, G. J. Liefers, H. Putter and C. J. van de Velde, Lancet Oncol., 2015, 16, 200–207 CrossRef CAS.
- E. Elimova, H. Shiozaki, R. Wadhwa, K. Sudo, Q. Chen, J. S. Estrella, M. A. Blum, B. Badgwell, P. Das, S. Song and J. A. Ajani, World J. Gastroenterol., 2014, 20, 13637–13647 CrossRef PubMed.
- N. Pabla and Z. Dong, OncoTargets Ther., 2012, 3, 107–111 CrossRef.
- W. Rao, Z. S. Deng and J. Liu, Crit. Rev. Biomed. Eng., 2010, 38, 101–116 CrossRef.
- P. Wust, B. Hildebrandt, G. Sreenivasa, B. Rau, J. Gellermann, H. Riess, R. Felix and P. M. Schlag, Lancet Oncol., 2002, 3, 487–497 CrossRef CAS.
- O. Dahl, in Thermoradiotherapy and Thermochemotherapy, ed. M. H. Seegenschmiedt, P. Fessenden and C. C. Vernon, Springer Verlag, Berlin, 1995, vol. 1, pp. 103–121 Search PubMed.
- R. D. Issels, L. H. Lindner, J. Verweij, P. Wust, P. Reichardt, B. C. Schem, S. Abdel-Rahman, S. Daugaard, C. Salat, C. M. Wendtner, Z. Vujaskovic, R. Wessalowski, K. W. Jauch, H. R. Durr, F. Ploner, A. Baur-Melnyk, U. Mansmann, W. Hiddemann, J. Y. Blay and P. Hohenberger, Lancet Oncol., 2010, 11, 561–570 CrossRef CAS.
- M. H. Falk and R. D. Issels, Int. J. Hyperthermia, 2001, 17, 1–18 CrossRef CAS PubMed.
- E. Vorotnikova, R. Ivkov, A. Foreman, M. Tries and S. J. Braunhut, Int. J. Radiat. Biol., 2006, 82, 549–559 CrossRef CAS PubMed.
- R. Cavaliere, E. C. Ciocatto, B. C. Giovanella, C. Heidelberger, R. O. Johnson, M. Margottini, B. Mondovi, G. Moricca and A. Rossi-Fanelli, Cancer, 1967, 20, 1351–1381 CrossRef CAS.
- B. C. Giovanella, A. C. Morgan, J. S. Stehlin and L. J. Williams, Cancer Res., 1973, 33, 2568–2578 CAS.
- B. Hildebrandt, P. Wust, O. Ahlers, A. Dieing, G. Sreenivasa, T. Kerner, R. Felix and H. Riess, Crit. Rev. Oncol. Hematol., 2002, 43, 33–56 CrossRef.
- R. A. Coss and W. A. Linnemans, Int. J. Hyperthermia, 1996, 12, 173–196 CrossRef CAS.
- A. W. Konings and A. C. Ruifrok, Radiat. Res., 1985, 102, 86–98 CrossRef CAS.
- P. M. Gullino and F. H. Grantham, J. Natl. Cancer Inst., 1961, 27, 1465–1491 CAS.
- G. C. Li, Cancer Res., 1984, 44, 4886s–4893s CAS.
- K. A. Ward and R. K. Jain, Int. J. Hyperthermia, 1988, 4, 223–250 CrossRef CAS.
- C. W. Song, H. Park and R. J. Griffin, in Thermotherapy for Neoplasia, Inflammation, and Pain, ed. M. Kosaka, T. Sugahara, K. Schmidt and E. Simon, Springer, Japan, 2001, ch. 44, pp. 394–407, DOI:10.1007/978-4-431-67035-3_44.
- A. Bettaieb, P. K. Wrzal and D. A. Averill-Bates, in Cancer Treatment - Conventional and Innovative Approaches, ed. L. Rangel, 2013, DOI:10.5772/55795.
- W. C. Dewey, L. E. Hopwood, S. A. Sapareto and L. E. Gerweck, Radiology, 1977, 123, 463–474 CrossRef CAS PubMed.
- J. E. Sisken, L. Morasca and S. Kibby, Exp. Cell Res., 1965, 39, 103–116 CrossRef CAS.
- A. Westra and W. C. Dewey, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1971, 19, 467–477 CrossRef CAS PubMed.
- C. Streffer, Recent Results Cancer Res., 1988, 107, 7–16 CAS.
- D. K. Kelleher, T. Engel and P. W. Vaupel, Int. J. Hyperthermia, 1995, 11, 241–255 CrossRef CAS.
- X. Sun, L. Xing, C. C. Ling and G. C. Li, Int. J. Hyperthermia, 2010, 26, 224–231 CrossRef CAS PubMed.
- R. J. Griffin, R. P. Dings, A. Jamshidi-Parsian and C. W. Song, Int. J. Hyperthermia, 2010, 26, 256–263 CrossRef PubMed.
- C. T. Lee, T. Mace and E. A. Repasky, Int. J. Hyperthermia, 2010, 26, 232–246 CrossRef PubMed.
- E. A. Repasky, S. S. Evans and M. W. Dewhirst, Cancer Immunol. Res., 2013, 1, 210–216 CrossRef CAS PubMed.
- J. Fucikova, P. Kralikova, A. Fialova, T. Brtnicky, L. Rob, J. Bartunkova and R. Spisek, Cancer Res., 2011, 71, 4821–4833 CrossRef CAS PubMed.
- E. G. Mimnaugh, R. W. Waring, B. I. Sikic, R. L. Magin, R. Drew, C. L. Litterst, T. E. Gram and A. M. Guarino, Cancer Res., 1978, 38, 1420–1425 CAS.
- J. A. Dickson and M. Suzangar, Cancer Res., 1974, 34, 1263–1274 CAS.
- K. Suzuki, Nagoya J. Med. Sci., 1967, 30, 1–21 CAS.
- E. Ben-Hur and M. M. Elkind, Radiat. Res., 1974, 59, 484–495 CrossRef CAS.
- D. J. Honess and N. M. Bleehen, Br. J. Radiol., 1985, 58, 63–72 CrossRef CAS PubMed.
- D. J. Honess, J. Donaldson, P. Workman and N. M. Bleehen, Br. J. Cancer, 1985, 51, 77–84 CrossRef CAS PubMed.
- E. B. Douple and R. C. Richmond, Int. J. Radiat. Oncol., Biol., Phys., 1982, 8, 501–503 CrossRef CAS.
- O. Mella, R. Eriksen, O. Dahl and O. D. Laerum, Eur. J. Cancer Clin. Oncol., 1987, 23, 365–373 CrossRef CAS.
- C. E. Ng, A. M. Bussey and G. P. Raaphorst, Int. J. Hyperthermia, 1996, 12, 551–567 CrossRef CAS.
- O. Dahl and O. Mella, Anticancer Res., 1982, 2, 359–364 CAS.
- J. Overgaard, Int. J. Radiat. Oncol., Biol., Phys., 1989, 16, 535–549 CrossRef CAS.
- P. L. Ausmus, A. V. Wilke and D. L. Frazier, Cancer Res., 1992, 52, 4965–4968 CAS.
- A. E.-M. Osman, M. M. Ahmed, M. T. Khayyal and M. M. el-Merzabani, Tumori, 1993, 79, 268–272 CAS.
- C. W. Helm, L. Randall-Whitis, R. S. Martin 3rd, D. S. Metzinger, M. E. Gordinier, L. P. Parker and R. P. Edwards, Gynecol. Oncol., 2007, 105, 90–96 CrossRef CAS PubMed.
- B. W. Loggie, R. A. Fleming, R. P. McQuellon, G. B. Russell, K. R. Geisinger and E. A. Levine, Am. Surg., 2001, 67, 999–1003 CAS.
- S. Fujimoto, M. Takahashi, T. Mutou, K. Kobayashi, T. Toyosawa, E. Isawa, M. Sumida and H. Ohkubo, Cancer, 1997, 79, 884–891 CrossRef CAS.
- V. J. Verwaal, S. Bruin, H. Boot, G. van Slooten and H. van Tinteren, Ann. Surg. Oncol., 2008, 15, 2426–2432 CrossRef PubMed.
- V. J. Verwaal, S. van Ruth, E. de Bree, G. W. van Sloothen, H. van Tinteren, H. Boot and F. A. Zoetmulder, J. Clin. Oncol., 2003, 21, 3737–3743 CrossRef PubMed.
- O. Glehen, G. Passot, L. Villeneuve, D. Vaudoyer, S. Bin-Dorel, G. Boschetti, E. Piaton and A. Garofalo, BMC Cancer, 2014, 14, 183 CrossRef PubMed.
- K. E. Tschoep, S. Boeck, F. Berger, V. Maier, S. Abdel-Rahman, M. Kuhlencordt, C. Salat, M. Schmidt, V. Heinemann and R. D. Issels, J. Clin. Oncol., 2008, 26, 4635 Search PubMed.
- R. C. Rietbroek, M. S. Schilthuis, P. J. Bakker, J. D. van Dijk, A. J. Postma, D. Gonzalez Gonzalez, A. J. Bakker, J. van der Velden, T. J. Helmerhorst and C. H. Veenhof, Cancer, 1997, 79, 935–943 CrossRef CAS.
- R. de Wit, J. van der Zee, M. E. van der Burg, W. H. Kruit, A. Logmans, G. C. van Rhoon and J. Verweij, Br. J. Cancer, 1999, 80, 1387–1391 CrossRef CAS PubMed.
- C. M. Wendtner, S. Abdel-Rahman, J. Baumert, M. H. Falk, M. Krych, M. Santl, W. Hiddemann and R. D. Issels, Eur. J. Cancer, 2001, 37, 1609–1616 CrossRef CAS.
- R. D. Issels, S. W. Prenninger, A. Nagele, E. Boehm, H. Sauer, K. W. Jauch, H. Denecke, H. Berger, K. Peter and W. Wilmanns, J. Clin. Oncol., 1990, 8, 1818–1829 CAS.
- M. Fiegl, M. Schlemmer, C. M. Wendtner, S. Abdel-Rahman, W. Fahn and R. D. Issels, Int. J. Hyperthermia, 2004, 20, 661–670 CrossRef CAS PubMed.
- H. Maeda, J. Controlled Release, 2012, 164, 138–144 CrossRef CAS PubMed.
- G. Kong, G. Anyarambhatla, W. P. Petros, R. D. Braun, O. M. Colvin, D. Needham and M. W. Dewhirst, Cancer Res., 2000, 60, 6950–6957 CAS.
- K.-J. Chen, H.-F. Liang, H.-L. Chen, Y. Wang, P.-Y. Cheng, H.-L. Liu, Y. Xia and H.-W. Sung, ACS Nano, 2012, 7, 438–446 CrossRef PubMed.
- J. K. Mills and D. Needham, Methods Enzymol., 2004, 387, 82–113 CAS.
- M. L. Hauck, S. M. LaRue, W. P. Petros, J. M. Poulson, D. Yu, I. Spasojevic, A. F. Pruitt, A. Klein, B. Case, D. E. Thrall, D. Needham and M. W. Dewhirst, Clin. Cancer Res., 2006, 12, 4004–4010 CrossRef CAS PubMed.
- C. D. Landon, J. Y. Park, D. Needham and M. W. Dewhirst, Open Nanomed. J., 2011, 3, 38–64 Search PubMed.
- G. N. C. Chiu, S. A. Abraham, L. M. Ickenstein, R. Ng, G. Karlsson, K. Edwards, E. K. Wasan and M. B. Bally, J. Controlled Release, 2005, 104, 271–288 CrossRef CAS PubMed.
- B. Banno, L. M. Ickenstein, G. N. C. Chiu, M. B. Bally, J. Thewalt, E. Brief and E. K. Wasan, J. Pharm. Sci., 2010, 99, 2295–2308 CAS.
- J. Woo, G. N. C. Chiu, G. Karlsson, E. Wasan, L. Ickenstein, K. Edwards and M. B. Bally, Int. J. Pharm., 2008, 349, 38–46 CrossRef CAS PubMed.
- S. Purushotham, P. E. Chang, H. Rumpel, I. H. Kee, R. T. Ng, P. K. Chow, C. K. Tan and R. V. Ramanujan, Nanotechnology, 2009, 20, 305101 CrossRef CAS PubMed.
- T. Betancourt, B. Brown and L. Brannon-Peppas, Nanomedicine, 2007, 2, 219–232 CrossRef CAS PubMed.
- S. Louguet, B. Rousseau, R. Epherre, N. Guidolin, G. Goglio, S. Mornet, E. Duguet, S. Lecommandoux and C. Schatz, Polym. Chem., 2012, 3, 1408–1417 RSC.
- N. Andhariya, B. Chudasama, R. V. Mehta and R. V. Upadhyay, J. Nanopart. Res., 2011, 13, 1677–1688 CrossRef CAS.
- A. Akbarzadeh, H. Mikaeili, N. Zarghami, R. Mohammad, A. Barkhordari and S. Davaran, Int. J. Nanomed., 2012, 7, 511–526 CAS.
- K. Hynynen, Methods Mol. Biol., 2009, 480, 175–185 CAS.
- M. I. Koukourakis, S. Koukouraki, I. Fezoulidis, N. Kelekis, G. Kyrias, S. Archimandritis and N. Karkavitsas, Br. J. Cancer, 2000, 83, 1281–1286 CrossRef CAS PubMed.
- G. P. Baronzio, G. Parmar, M. De Santis and A. Gramaglia, in Cancer Treatment - Conventional and Innovative Approaches, ed. L. Rangel, InTech, 2013, ch. 13 Search PubMed.
- K. Nakagawa, Y. Aoki, T. Fujimaki, M. Tago, A. Terahara, K. Karasawa, K. Sakata, Y. Sasaki, M. Matsutani and A. Akanuma, Int. J. Radiat. Oncol., Biol., Phys., 1998, 40, 1141–1149 CrossRef CAS.
- P. J. Miller, R. S. Hassanein, P. G. Giri, B. F. Kimler, P. O'Boynick and R. G. Evans, Int. J. Radiat. Oncol., Biol., Phys., 1990, 19, 275–280 CrossRef CAS.
- J. R. McDaniel, S. R. MacEwan, X. Li, D. C. Radford, C. D. Landon, M. Dewhirst and A. Chilkoti, Nano Lett., 2014, 14, 2890–2895 CrossRef CAS PubMed.
- J. R. McDaniel, J. Bhattacharyya, K. B. Vargo, W. Hassouneh, D. A. Hammer and A. Chilkoti, Angew. Chem., Int. Ed., 2013, 52, 1683–1687 CrossRef CAS PubMed.
- D. E. Meyer, B. C. Shin, G. A. Kong, M. W. Dewhirst and A. Chilkoti, J. Controlled Release, 2001, 74, 213–224 CrossRef CAS.
- M. Wende and J. A. Gladysz, J. Am. Chem. Soc., 2003, 125, 5861–5872 CrossRef CAS PubMed.
- C. Rocaboy and J. A. Gladysz, New J. Chem., 2003, 27, 39–49 RSC.
- C. Gimbert, A. Vallribera, J. A. Gladysz and M. Jurisch, Tetrahedron Lett., 2010, 51, 4662–4665 CrossRef CAS PubMed.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- R. Filler and R. Saha, Future Med. Chem., 2009, 1, 777–791 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- C. Heidelberger, N. K. Chaudhuri, P. Danneberg, D. Mooren, L. Griesbach, R. Duschinsky, R. J. Schnitzer, E. Pleven and J. Scheiner, Nature, 1957, 179, 663–666 CrossRef CAS PubMed.
- J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- J. Osorio, Nat. Rev. Cardiol., 2010, 7, 541 CrossRef PubMed.
- C. Hansch, A. Leo and D. Hoekman, Exploring QSAR: Volume 2: Hydrophobic, Electronic, and Steric Constants, American Chemical Society, 1995 Search PubMed.
- I. T. Horvath, G. Kiss, R. A. Cook, J. E. Bond, P. A. Stevens, J. Rabai and E. J. Mozeleski, J. Am. Chem. Soc., 1998, 120, 3133–3143 CrossRef CAS.
- B. Richter, A. L. Spek, G. van Koten and B. J. Deelman, J. Am. Chem. Soc., 2000, 122, 3945–3951 CrossRef CAS.
- F. O. Seidel and J. A. Gladysz, Adv. Synth. Catal., 2008, 350, 2443–2449 CrossRef CAS PubMed.
- M. Wende, R. Meier and J. A. Gladysz, J. Am. Chem. Soc., 2001, 123, 11490–11491 CrossRef CAS.
- M. P. Krafft, Adv. Drug Delivery Rev., 2001, 47, 209–228 CrossRef CAS.
- S. D. Xiong, L. Li, J. Jiang, L. P. Tong, S. L. Wu, Z. S. Xu and P. K. Chu, Biomaterials, 2010, 31, 2673–2685 CrossRef CAS PubMed.
- A. K. Renfrew, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2010, 49, 2239–2246 CrossRef CAS PubMed.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- A. Bergamo, A. Masi, P. J. Dyson and G. Sava, Int. J. Oncol., 2008, 33, 1281–1289 CAS.
- P. Nowak-Sliwinska, J. R. van Beijnum, A. Casini, A. A. Nazarov, G. Wagnieres, H. van den Bergh, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS PubMed.
- A. Weiss, B. H. Berndsen, M. Dubois, M. Müller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- S. M. Guichard, R. Else, E. Reid, B. Zeitlin, R. Aird, M. Muir, M. Dodds, H. Fiebig, P. J. Sadler and D. I. Jodrell, Biochem. Pharmacol., 2006, 71, 408–415 CrossRef CAS PubMed.
- A. Bergamo, A. Masi, A. F. Peacock, A. Habtemariam, P. J. Sadler and G. Sava, J. Inorg. Biochem., 2010, 104, 79–86 CrossRef CAS PubMed.
- C. M. Clavel, O. Zava, F. Schmitt, B. H. Kenzaoui, A. A. Nazarov, L. Juillerat-Jeanneret and P. J. Dyson, Angew. Chem., Int. Ed., 2011, 50, 7124–7127 CrossRef CAS PubMed.
- C. M. Clavel, P. Nowak-Sliwinska, E. Paunescu, A. W. Griffioen and P. J. Dyson, Chem. Sci., 2015, 6, 2795–2801 RSC.
- C. M. Clavel, E. Paunescu, P. Nowak-Sliwinska and P. J. Dyson, Chem. Sci., 2014, 5, 1097–1101 RSC.
- W. Nie, X. L. Ma, Y. X. Sang, Y. L. Li, X. Gao, G. C. Xu, G. B. Shen, H. S. Shi, X. X. Liu, F. T. Wang and Y. Q. Wei, Clin. Exp. Med., 2014, 14, 203–213 CrossRef CAS PubMed.
- S. Zorzet, A. Sorc, C. Casarsa, M. Cocchietto and G. Sava, Met.-Based Drugs, 2001, 8, 1–7 CrossRef CAS PubMed.
- M. Cocchietto and G. Sava, Pharmacol. Toxicol., 2000, 87, 193–197 CAS.
- P. Nowak-Sliwinska, C. M. Clavel, E. Paunescu, M. T. Te Winkel, A. W. Griffioen and P. J. Dyson, Mol. Pharmaceutics, 2015, 12, 3089–3096 CrossRef CAS PubMed.
- C. D. Kaddi, J. H. Phan and M. D. Wang, Nanomedicine, 2013, 8, 1323–1333 CrossRef CAS PubMed.
- G. M. Hahn, Br. J. Cancer, Suppl., 1982, 5, 238–242 CAS.
- B. M. Dicheva and G. A. Koning, Expert Opin. Drug Delivery, 2014, 11, 83–100 CrossRef CAS PubMed.
- E. Paunescu, C. M. Clavel, P. Nowak-Sliwinska, A. W. Griffioen and P. J. Dyson, ACS Med. Chem. Lett., 2015, 6, 313–317 CrossRef CAS PubMed.
- C. M. Clavel, E. Paunescu, P. Nowak-Sliwinska, A. W. Griffioen, R. Scopelliti and P. J. Dyson, J. Med. Chem., 2014, 57, 3546–3558 CrossRef CAS PubMed.
- E. Paunescu, P. Nowak-Sliwinska, C. M. Clavel, R. Scopelliti, A. W. Griffioen and P. J. Dyson, ChemMedChem, 2015, 10, 1539–1547 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |