
Mechanisms of resistance to aminoglycoside antibiotics: overview and perspectives
Sylvie
Garneau-Tsodikova
a and
Kristin J.
Labby
*b
aUniversity of Kentucky, Department of Pharmaceutical Sciences, 789 South Limestone Street, Lexington, KY, USA. E-mail: sylviegtsodikova@uky.edu; Fax: +859 257 7585; Tel: +859 218 1686
bBeloit College, Department of Chemistry, 700 College Street, Beloit, WI, USA. E-mail: labbyk@beloit.edu; Fax: +608 363 2052; Tel: +608 363 2273
First published on 21st September 2015
Abstract
Aminoglycoside (AG) antibiotics are used to treat many Gram-negative and some Gram-positive infections and, importantly, multidrug-resistant tuberculosis. Among various bacterial species, resistance to AGs arises through a variety of intrinsic and acquired mechanisms. The bacterial cell wall serves as a natural barrier for small molecules such as AGs and may be further fortified via acquired mutations. Efflux pumps work to expel AGs from bacterial cells, and modifications here too may cause further resistance to AGs. Mutations in the ribosomal target of AGs, while rare, also contribute to resistance. Of growing clinical prominence is resistance caused by ribosome methyltransferases. By far the most widespread mechanism of resistance to AGs is the inactivation of these antibiotics by AG-modifying enzymes. We provide here an overview of these mechanisms by which bacteria become resistant to AGs and discuss their prevalence and potential for clinical relevance.
Introduction
This review presents an overview and perspective of the mechanisms of resistance to aminoglycoside (AG) antibiotics. AGs are broad-spectrum antibiotics effective against both Gram-negative and some Gram-positive bacteria. AG structure consists of a 2-deoxystreptamine (2-DOS) ring to which two or more amino-modified sugars are attached via glycosidic bonds (Fig. 1).1 AGs have long been known to exert their antibacterial action by binding to the bacterial ribosome and interfering with bacterial protein translation.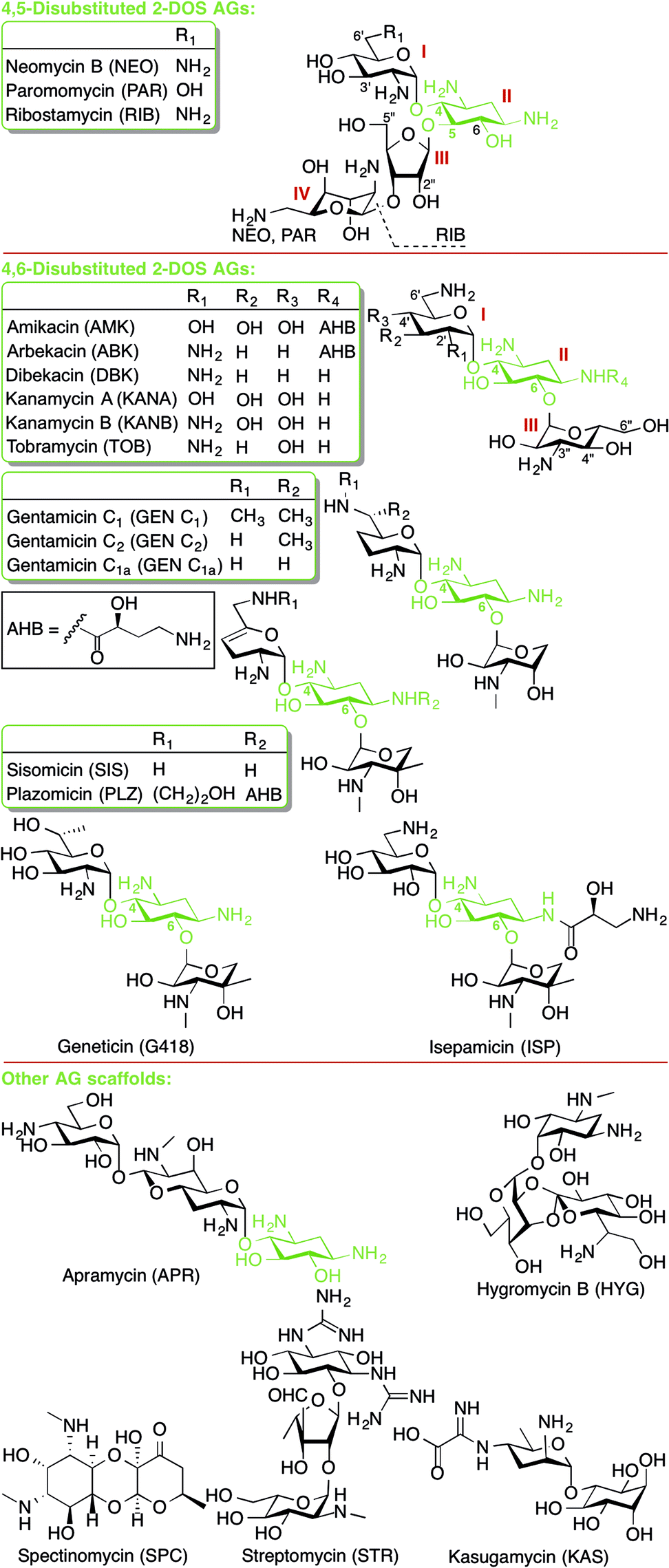 | ||
| Fig. 1 Structures of AGs presented in this review. | ||
Recently, AGs have been examined as potential treatments for fungal infections, Leishmaniasis parasitic infections, and for genetic diseases arising from premature termination codons, such as cystic fibrosis, Rett syndrome and Duchenne muscular dystrophy.2 Currently, however, AGs are typically used to treat Gram-negative infections (Acinetobacter baumannii, Enterobacteriaceae spp, Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa) and as second-line of defence treatment for multidrug-resistant (MDR) tuberculosis (Mycobacterium tuberculosis).3 Therefore, our discussion of mechanisms of resistance to AGs will focus on these two classes of bacteria.
Antibiotic resistance can be classified into three main categories: intrinsic, adaptive, and acquired resistance.4,5 An example of intrinsic antibiotic resistance is the naturally low permeability of the bacterial cell wall, which limits uptake of many antibiotics including AGs. Adaptive antibiotic resistance occurs as a result of an environmental trigger (e.g., nutrient concentration changes or sub-inhibitory levels of antibiotics) that causes temporary changes in gene and/or protein expression levels contributing to the tolerance of antibiotics.5,6 Bacteria growing on surfaces as biofilms maintain an adaptive resistance (often referred to as tolerance) to antibiotics.7 Finally, antibiotic resistance may be acquired by either the incorporation of exogenous genetic material, often a plasmid carrying multiple resistance genes, or via mutation of existing genes.8 While intrinsic and acquired resistance elements are passed on vertically during bacterial reproduction, adaptive resistance is transient and typically reverts upon removal of the environmental trigger. Furthermore, resistance genes on plasmids may be transferred horizontally from one bacterium to another. This is the major cause of the dissemination of antibiotic resistance genes among various bacterial species. Additionally resistance mechanisms may be non-specific (e.g., the cell membrane is impermeable to many toxic small molecules) or specific (e.g., AG-modifying enzymes (AMEs) regioselectively modify only particular AG substrates).
Mechanisms of bacterial resistance to AGs are diverse (Fig. 2). The most common mechanism is inactivation of AGs by a family of enzymes named AMEs. Also, AG resistance can be achieved by mutations of the ribosome target and, increasingly commonly, by modification of the ribosome by a family of ribosomal methyltransferase enzymes.9 The bacterial cell wall serves as an intrinsic barrier, and its impermeability may be increased by acquired lipid modifications that cause repulsion of AGs. Furthermore, even if AGs do enter the bacterial cell, intercellular concentrations may remain low due to the active expulsion of AGs out of the cell by efflux pumps.10
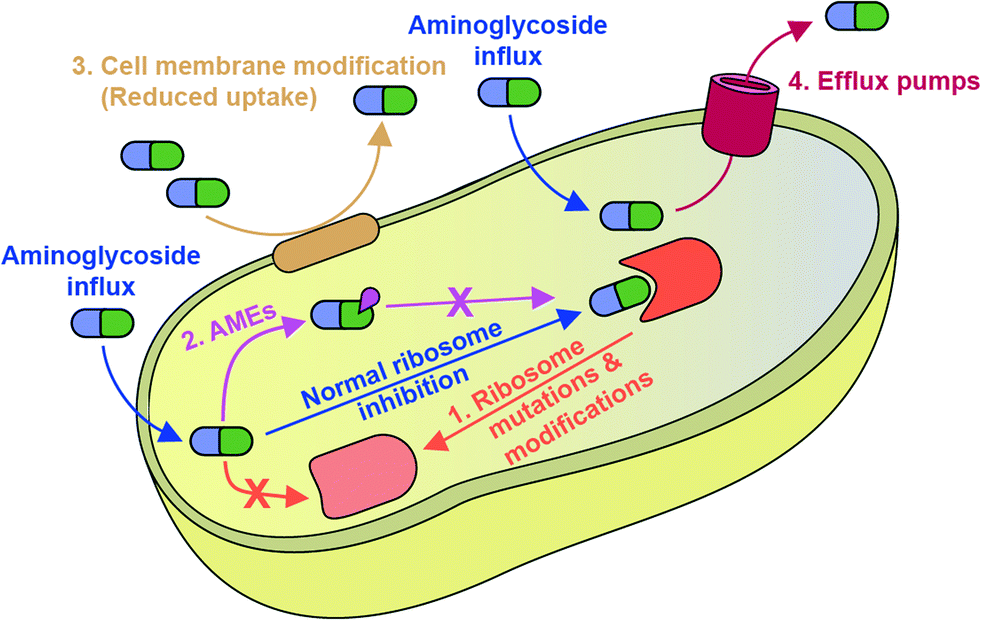 | ||
| Fig. 2 Schematic overview of mechanisms of resistance to AGs discussed in this review. | ||
Bacterial resistance to all classes of antibiotics, including AGs, is becoming a global public health crisis.11,12 It is crucial to understand the mode of action of AGs as well as mechanisms of AG resistance to be able to fight resistance. This review will focus on recent (2010 onward) research towards the understanding of mechanisms of AG resistance, except in the case of cell membrane permeability, an area in which little new literature exists.
Ribosomal mutations and modifications
The classical mechanism of resistance to an antibiotic is bacterial modification of the antibiotic's target. AGs target the A-site of the bacterial ribosome; to evade inhibition by AGs there are two potential, acquired mechanisms of resistance: mutations of the ribosome or enzymatic modifications of the ribosome.Ribosomal mutation
In both eukaryotes and bacteria, protein translation occurs at the ribosome. The bacterial ribosome consists of a large subunit (50S) comprised of 5S and 23S rRNA and 34 proteins and a small subunit (30S) comprised of 16S rRNA and 20 proteins.13 The steps of protein translation occur in three sites at the ribosome, the E-, P-, and A-sites. At the A-site, addition occurs of the amino acyl tRNA complementary to the three base codon of the mRNA. Selectivity for AGs to bind to the bacterial, but not human ribosome, comes from structural differences. Eukaryotic ribosomes contain nucleotide substitutions at two key AG-binding residues (prokaryotic A1408 and G1491 are replaced with guanine and adenine, respectively, Fig. 3A).14 However, human mitochondrial ribosomes contain the prokaryotic residues and, therefore, are susceptible to inhibition by AGs. This is likely responsible for ototoxicity side effects.15 The recent crystal structures of the human mitochondrial ribosome will aid in the design of AGs that avoid this toxicity.16,17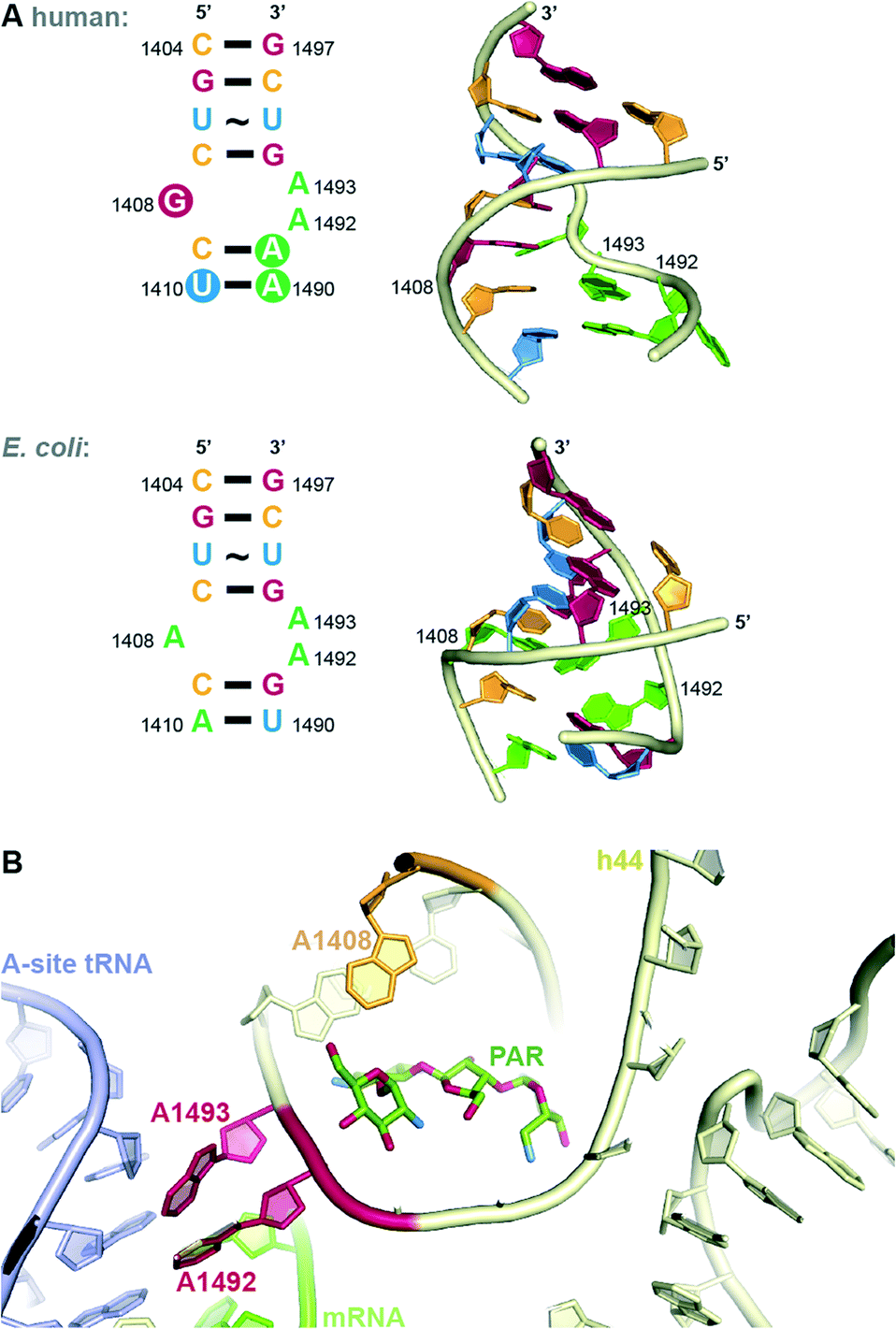 | ||
| Fig. 3 A. Comparison of internal loop of h44 of human and bacterial (E. coli) A-site. B. Structure of PAR bound to h44. | ||
The bacterial A-site, to which AGs bind, is located on the 16S RNA of the 30S bacterial ribosomal subunit.9 In addition to the interactions at the codon, the 16S RNA makes many contacts with the tRNA confirming the correct codon-anticodon match and induces a closed conformation when a match is achieved. Recent crystal structures have elucidated the details of contacts made between various AGs and highly conserved nucleotide bases (A1408, A1492 and A1493 by E. coli numbering) of helix 44 (h44) of the 16S RNA (Fig. 3B). AGs of the 2-DOS scaffold (Fig. 1) may each bind slightly differently to the 16S RNA, but the binding mode of the 2-DOS core is conserved and binding leads to inhibition of tRNA translocation and consequently protein synthesis.9,18 Additionally, AGs have been thought to induce conformational change to mimic the closed, or active, ribosomal conformation.9,13 Specifically, RNA bases A1492 and A1493 flip from intra-helical to extra-helical to accommodate the AG (Fig. 3B). This signals the continuation of translation despite incorrect mRNA-tRNA pairing, resulting in mistranslated proteins. Secondary effects of these mistranslated proteins, such as incorporation into and subsequent disruption of the cell membrane, have been hypothesized to be the true mechanism of AG lethality. Recent work demonstrates that the energetic changes induced by AG binding may be more complex than originally thought.19
Some 2-DOS AGs (neomycin B (NEO), gentamicin (GEN), and paromomycin (PAR)) have been reported to also bind to a secondary site – the major groove of helix 69 (H69) of the 23S RNA of the 50S subunit.20–22 Binding at this allosteric site has been demonstrated to affect the mobility of ribosomal subunits, which interferes with translation and ribosome recycling. Details regarding ribosomal binding of non-2-DOS AGs are beyond the scope of this review and can be found in other recent reviews.1,2,9,13
AG resistance may arise from mutations in the rrs gene, which codes for 16S rRNA, that hinder AG-binding. These mutations, however, are not very common, as changes to this vital cellular machinery are often lethal. One viable mutant is A1408G. This mutation disrupts a key hydrogen bonding interaction between 2-DOS AGs and the h44 nucleotide A1408 (Fig. 3B). This mutation, which corresponds to A1401G in M. tuberculosis, as well as C1402T and G1484T have been found in clinically isolated strains of resistant M. tuberculosis.23 Other less clinically prevalent ribosomal mutations have recently been analysed and summarized.24In vitro-selected mutations in Mycobacterium abscessus nucleotides 1406 and 1408 were found to be viable and confer resistance to 2-DOS AGs.25 Recent structural analysis reveals that AGs bearing a 6′-OH group (geneticin (G418), PAR) may evade resistance typically caused by A1408G.26
In addition to contacts made with h44 nucleotides, the non-2-DOS AG streptomycin (STR) interacts with ribosomal protein S12. Mutations in rspL, the gene encoding the S12 protein, lead to high-level STR resistance in M. tuberculosis.27,28 Similarly, spectinomycin (SPC)-resistant Neisseria gonorrhoeae has been determined to contain a mutation in ribosomal protein S5.29
Another ribosomal mechanism of resistance, demonstrated in vitro, is the overexpression of a 16S rRNA fragment resembling helix 34, which sequesters SPC and decreases its binding to the ribosome.30
Ribosomal modification by methyltransferases
In addition to mutations, the AG binding site may be modified enzymatically by 16S ribosomal RNA methyltransferases (RMTases, also commonly referred to as 16S rRNA methylases in the literature).31 RMTases naturally occur in actinomycetes, the bacterial group from which AGs were originally isolated. To protect their ribosomes from inhibition by the AGs they produce, actinomycetes produce RMTases to methylate their own 16S rRNA.RMTases are acquired by other bacterial species most commonly by uptake of a plasmid containing the RMTase gene, and potentially other resistance genes (Table 1). RMTases were only recently discovered; an AG-resistant P. aeruginosa strain isolated in 1997 in Japan was found to contain a plasmid carrying the RMTase RmtA.32 A resistance plasmid isolated from a Citrobacter freundii strain in Poland in 2002 and a multidrug-resistant strain of K. pneumoniae in France in 2003 were found to contain ArmA (aminoglycoside resistance methyltransferase A).33,34 This isolated RMTase displayed from 37 to 47% sequence similarity to intrinsic RMTases from various actinomycetes.34 Low identity between the intrinsic and acquired RMTase genes (less than 30%) suggests that this gene transfer did not occur recently.
| 16S RMTase | Bacterial species | Observed coexisting genesa | Ref. |
|---|---|---|---|
| a Only subsets of these coexisting genes exist in various strains of the specified bacterial species. b In ref. 92, RmtB1 and RmtB2 are reported. | |||
| NpmA | E. coli | bla CTX-M-14, blaSHV-12, blaTEM-1 | 35, 36 |
| ArmA | A. baumannii, C. freundii, K. pneumoniae, E. cloacae, E. coli, S. enterica, S. flexneri, S. marcescens, Providencia sp., P. aeruginosa, P. mirabilis, P. stuartii, Providencia rettgeri | aac(3)-Ia, aac(3)-II, aac(6′)-Ib-cr, aacA4cr, aacC2, aadA1, aadA2, ant3′′9, aphA1, aph(3′)-Ia, aph(3′)-Ib, arr-1, blaADC-30, blaADC-67, blaCMY-2, blaCMY-16, blaCMY-30, blaCTX-M-3, blaCTX-M-14, blaCTX-M-15, blaIMP-1, blaKPC-2, blaNDM-1, blaOXA-1, blaOXA-9, blaOXA-10, blaOXA-23, blaOXA-30, blaOXA-48, blaOXA-51, blaOXA-66, blaOXA-72, blaOXA-82, blaOXA-202, blaPER-1, blaSHV, blaSHV-2, blaSHV-11, blaSHV-12, blaSHV-28, blaSHV-32, blaSHV-33, blaSHV-130, blaSHV-133, blaTEM-1, blaTEM-16, blaVEB-1, blaVIM-1, cmlA1, dfrA12, dfrA14 dfrXII, florR, linF, mel, mph2, qnrA1, qnrB2, qnrS sul1, sul2, tet(A) | 33, 37–61 |
| RmtA | P. aeruginosa | κγ (mercury resistance mobile element)31 | 32 |
| RmtBb | A. baumannii, C. freundii, E. aerogenes, E. amnigenus, E. cloacae, E. coli, K. pneumoniae, L. adecarboxylata, M. morganii, P. aeruginosa, P. mirabilis, S. marcescens | armA, aac(3)-II, aac(6′)-Ib-cr, aadA2, aadA4, aadA5, aphA1-IAB, blaCMY-58, blaCTX-M-3, blaCTX-M-12, blaCTX-M-14, blaCTX-M-15, blaCTX-M-24, blaCTX-M-65, blaDHA-1, blaKPC, blaLAP-1, blaNDM-1, blaNDM-8blaOXA-1, blaOXA-10, blaOXA-23, blaOXA-51, blaSHV, blaTEM-1, blaVIM-1, catA1, catB4, dfrA17, ermB, fosA3, fosC2, qepA, qnrS1, sul1, tetA | 48, 62–78 |
| RmtC | K. pneumoniae, P. mirabilis, S. enterica | aac(3)-II, aac(6′)-Ib, aacA4, aadA1, aadB, aphA1, arr2, blaNDM-1, blaCMY-6, blaCTX-M-15, blaOXA-1, blaOXA-9, blaTEM, blaVEB-6, cmlA7, ereC, sul1 | 57, 79–83 |
| RmtD1 | K. pneumoniae, P. aeruginosa | bla CTX-M-15, blaKPC-2, blaSPM-1 | 84, 85 |
| RmtD2 | E. aerogenes, K. pneumoniae | aadA2, cat, dfrA12, sul1, blaKPC-2 | 85, 86 |
| RmtE | E. coli | aph(3′)-Ia, aphA7, strA, strB | 87 |
| RmtF | C. freundii, E. cloacae, E. coli, K. pneumoniae | aac(6′)-Ib, armA, blaCIT, blaCTX-M, blaDHA, blaNDM-1, blaOXA-1, insEΔ, rmtB, rmtC | 88, 89 |
| RmtG | K. pneumoniae, P. aeruginosa | bla CTX-M-2, blaCTX-M-15, blaCTX-M-59, blaKPC-2, blaSHV-1a, blaTEM-1 | 85, 90, 91 |
| RmtH | K. pneumoniae | bla CTX-M-15, blaSHV-1, blaOXA-1, “ISCR2, an IS91-like transposable element containing resistance genes for sulfonmide, trimethyoprim, and florfenicol, and tetracycline and cephalosporin” | 92 |
RMTases contribute to resistance by methylating a nucleotide in the AG-binding site of the 16S rRNA (the A-site) using S-adenyosyl-L-methionine (SAM) as a cosubstrate. The AG resistance RMTases are divided into two families: those which methylate at the N7 position of nucleotide G1405 (ArmA, RmtA, RmtB, RmtC, RmtD1, RmtD2 RmtE, RmtF, RmtG and RmtH) and those which methylate at the N1 position of A1408 (NpmA). N7-G1405 RMTases confer resistance to 4,6-disubstituted 2-DOS AGs such as amikacin (AMK), GEN, tobramycin (TOB), and kanamycin A (KANA), but not to 4,5-disubstituted 2-DOS AGs (e.g., NEO) or apramycin (APR). The N1-A1408 RMTase NmpA confers pan-AG resistance to both 4,5- and 4,6-disubstituted 2-DOS AGs and to APR. This can be rationalized structurally due to the lack of N7-G1405 hydrogen bond interaction between the ribosome and 4,5-disubstituted 2-DOS AGs or APR; methylation at N7-G1405 will not affect binding for these AGs therefore does not cause resistance to them (Fig. 4A).31 Neither family of RMTases confer resistance to non-A-site binding AGs such STR or SPC.35
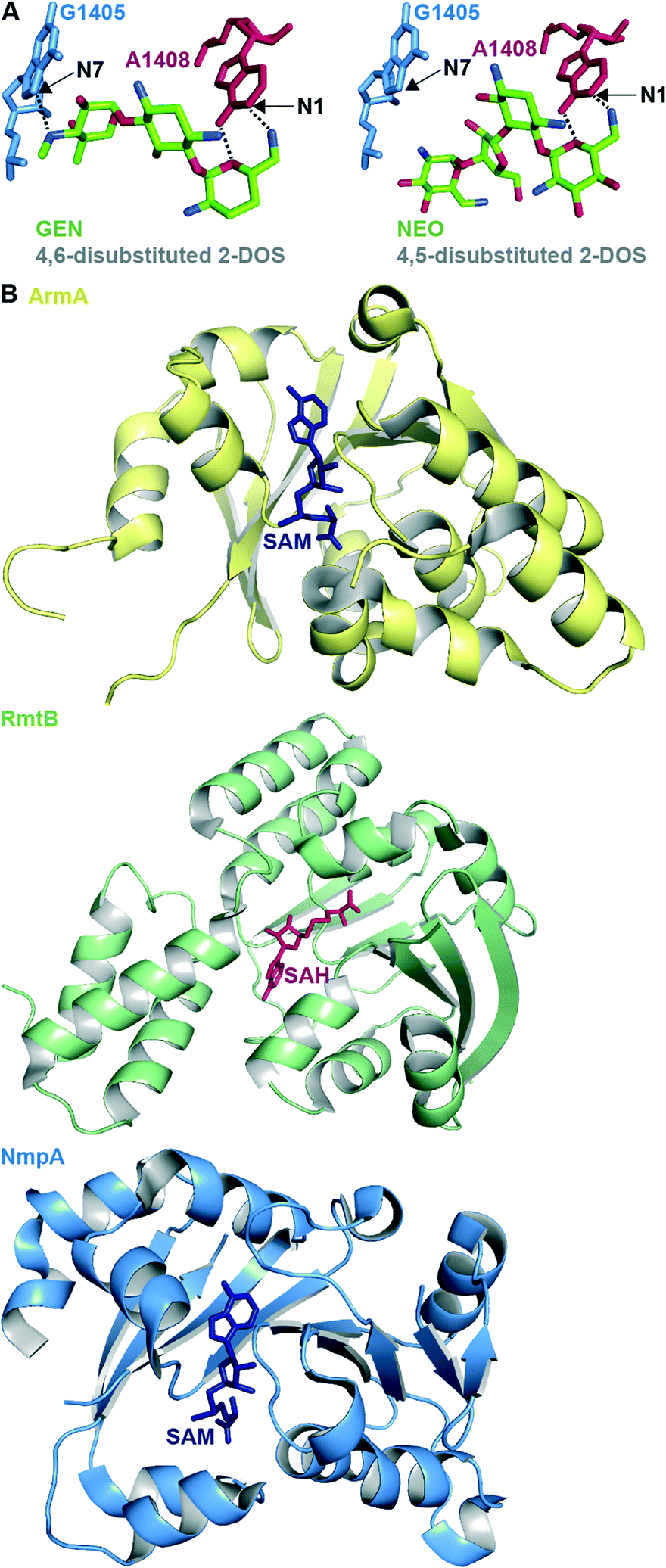 | ||
| Fig. 4 A. Structure of the 4,6-disubstituted 2-DOS AG GEN and 4,5-disubstituted 2-DOS AG NEO in close proximity to ribosome (PDB code 4V53 and 4V52, respectively) with positions methylated by RMTases. B. Representative example of crystal structures of the RMTases ArmA as yellow cartoon with SAM as navy stick (PBD code 3FZG),94 RmtB as green cartoon with SAH as red stick (PBD code 3FRH),94 and NmpA as pale blue cartoon with SAM as navy stick (PBD code 3P2K).97 | ||
The structures of several RMTases have been elucidated using X-ray crystallography (Fig. 4B, Table 2). The three-dimensional structures of intrinsic RMTases when compared with acquired, resistant bacterial RMTases are similar, despite low (~30%) sequence conservation. Furthermore, residues required for enzymatic activity are highly conserved.31
The clinical prevalence of RMTases, while still low, is increasing. This poses a considerable potential threat because RMTases confer resistance to many clinically relevant AGs, including AMK. A noted exception is the structurally rigid APR, which remains resistant to N7-G1405 RMTases, but the N1-A1408 RMTase NmpA can render APR inactive. Also, STR and SPC retain efficacy in the presence of resistance RMTases. Plazomicin (PLZ), an AG currently in clinical trials, was, unfortunately, found to be inactive against Enterobacteriaceae strains containing the RMTase genes armA and rmtC.99,100
RmtB and ArmA are currently the predominant 16S RMTases, and their genes have spread around the world to resistant bacterial strains isolated from humans as well as livestock.31,78 Recently, RMTase resistance genes have been found in food products, suggesting that food may be a possible vehicle for the spread of AG-resistant bacteria.41
Acquired resistance 16S RMTases will coexist with endogenous ribosomal methyltransferases, for example RsmH and RsmI, which methylate C1402 in E. coli for proper ribosome function. Recently, it was found that the presence of resistance-causing N7-G1405 methyltransferase RmtD impedes C1402 methylation.101 ArmA's methylation at G1405 slowed the growth rate of E. coli by blocking methylation at C1402, while NpmA had seemingly little affect on E. coli fitness. Both ArmA and NpmA methylations affected translation fidelity.102 This work will lend insight on predictions of rates of dissemination of 16S RMTases based on fitness. These studies are also crucial for understanding the endogenous methylation at bacterial AG binding sites for the design of improved AGs to evade resistance.
Continued development of improved monitoring of RMTases, as well as other resistance genes, will be crucial to slowing the spread of resistance. These methods include RT-PCR,103 loop-mediated assays,104 and bioinformatics tools105 to detect both previously identified and novel resistance genes. Recent studies have identified putative resistance RMTases in Pyrococcus furiosus106 and four other diverse bacterial species.107
One approach to overcome resistance due to RMTases would be the design of small molecule inhibitors. Working towards this, more structural information is needed for the design of selective RMTase inhibitors. While RMTases unfortunately contribute to resistance to many AGs, the structural similarities among RMTases would lend to the possibility of developing inhibitors that would target multiple RMTases.
AG-modifying enzymes
The most common mechanism of AG resistance is chemical modification by aminoglycoside-modifying enzymes (AMEs). This large family of enzymes contains three subclasses, divided based on the type of chemical modification they apply to their AG substrates: AG N-acetyltransferases (AACs), AG O-nucleotidyltransferases (ANTs), and AG O-phosphotransferases (APHs).108 Each AME modifies an AG at a specific position, and this information is included in the enzyme name (Fig. 5A). Also, bifunctional enzymes, such as AAC(6′)-Ie/APH(2′′)-Ia from Staphylococcus aureus,109–112 AAC(3)-Ib/AAC(6′)-Ib′ from P. aeruginosa,113,114 ANT(3′′)-Ii/AAC(6′)-IId from Serratia marcescens,115–117 and AAC(6′)-30/AAC(6′)-Ib from P. aeruginosa118,119 exist and are capable of multiple types of AG modification. AAC(6′)-Ib is the most prevalent and clinically relevant AME; approximately fifty variants of AAC(6′)-Ib exist in numerous Gram-negative species.120 In Mycobacterium and other bacterial species, upregulation of the enhanced intercellular survival (Eis) protein121 is responsible for resistance to AGs via their multi-acetylation.122–130 In addition to AGs, Eis is also capable of acetylating other anti-tubercular drugs, such as capreomycin.131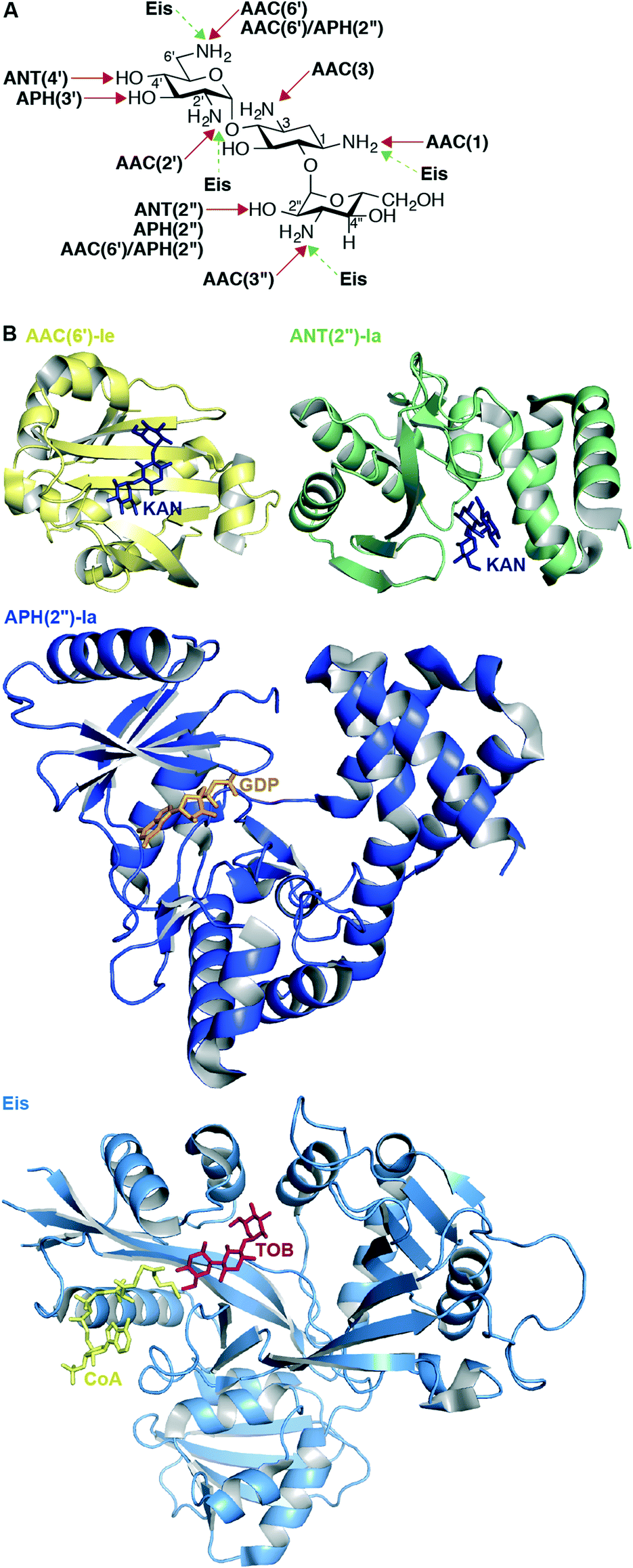 | ||
| Fig. 5 A. Chemical modifications and positions affected by various AMEs on KANB. B. Representative example of recent crystal structures for AACs (AAC(6′)-Ie as yellow cartoon with KANA as navy stick; PDB code 4QC6;148 from the bifunctional enzyme AAC(6′)-Ie/APH(2′′)-Ia), APHs (APH(2′′)-Ia as dark blue cartoon with GDP as orange stick; PDB code 4ORK;144 from the bifunctional enzyme AAC(6′)-Ie/APH(2′′)-Ia), ANTs (ANT(2′′)-Ia as green cartoon with KANA as navy stick; PDB code 4WQL;145 normally a monomer), and Eis as pale blue cartoon with CoA as yellow stick and TOB as red stick (PDB code 4JD6;127 normally an hexamer) resistance enzymes. Note: Only the monomer of each of these enzymes is shown. | ||
The native functions of AMEs remain unclear; they likely had roles in normal cellular metabolism, but have since evolved from their original “proto-resistance genes” to modify AGs upon selective pressure from exposure to these antibiotics.132 AMEs are highly mobile; their genes are transferred on plasmids, integrons, transposons, and other transposable gene elements, often along with other resistance genes (such as RMTases or β-lactamases, “bla” genes, Table 1). Most pathogenic bacteria acquire resistance AMEs through horizontal gene transfer. Eis, however, is natively expressed in M. tuberculosis and mutations in the promoter region or a translational activator cause its upregualtion, leading to resistance.121,133 Also, there is good evidence that another pathogenic mycobacteria, M. abscessus, contains a native AAC(2′) responsible for its intrinsic resistance to AGs.134,135
Over 100 AMEs have been reported, here we highlight those identified and characterized since the publication of a comprehensive list of AMEs in 2010.108 Newly discovered and substrate scope-characterized AACs include several AAC(6′)s: AAC(6′)-Iag from P. aeruginosa,136 AAC(6′)-Iaj from P. aeruginosa,137 AAC(6′)-Iak from Stenotrophomonas maltophilia,138 and AAC(6′)-Ian from S. marcescens.139 A novel AAC(2′), AAC(2′)-IIa confers resistance to the agricultural AG kasugamycin (KAS) in rice pathogenic bacteria Burkholderia glumae and Acidovorax avenae subsp. avenae.140 The Gram-positive Corynebacterium striatum BM4687 bacterial strain has a unique resistance profile (resistant to GEN and TOB, but susceptible to KAN and AMK), due to the presence of what was named AAC(3)-XI.141
A new APH, APH(2′′)-If from Campylobacter jejuni, was found to have the same AG substrate scope as and to have high sequence identity to the APH(2′′) of the bifunctional AAC(6′)-Ie/APH(2′′)-Ia.142
Because of their clinical prominence as a mechanism of AG resistance, an active area of research has been obtaining structural information regarding AMEs. A comprehensive list of resolved crystal structures of AMEs can be found in a recent review.143 Since that publication, new AME crystal structures have been solved and are listed in Table 3 and representative structures shown in Fig. 5B.
| Enzyme | PDB code | Substrate | Cosubstrate | Bacterial species | Oligomeric state | Ref. |
|---|---|---|---|---|---|---|
| a Indicates that there are no reference to confirm the dimeric state, but this is what is observed in the PDB. b Indicates thermostable mutant. Please also see PDB codes 3W0M, 3W0N, 3W0P, 3W0Q, 3W0R, and 3W0S for structures of other thermostable mutants of APH(4)-Ia with hygromycin B (HYG) and various cosubstrates. | ||||||
| AAC(3)-Ib | 4YFJ | — | — | P. aeruginosa | Structural dimera | — |
| AAC(6′)-Ie | 4QC6 | KANA | CoA | Staphylococcus warneri | From bifunctional AAC(6′)-Ie/APH(2′′)-Ia | 144 |
| EisC204A | 4JD6 | TOB | CoA | M. tuberculosis | Hexamer | 127 |
| ANT(2′′)-Ia | 4WQL (4WQK) | KAN (−) | — | K. pneumoniae | Monomer | 145 |
| APH(4)-Iab | 3W0O | HYG | ADP | E. coli | Monomer | 146 |
| APH(3′)-VIII | 4H05 | — | — | Streptomyces rimosus | Structural dimera | — |
| APH(2′′)-Ia | 4ORK | — | GDP | S. aureus | From bifunctional AAC(6′)-Ie/APH(2′′)-Ia | 144 |
| APH(2′′)-IVa | 4N57 | — | ADP | Enterococcus casseliflavus | Structural dimera | — |
Noteworthy is the first structure of an ANT other than ANT(4′);147 the structure of ANT(2′′)-Ia reveals that this enzyme shares molecular features with other nucleotidyltransferases including lincosamide nucleotidyltransferases and DNA polymerase β.145 Augmenting previous studies that suggest that the bifunctional enzyme AAC(6′)-Ie/APH(2′′)-Ia adopts a rigid conformation in solution,112 Vakulenko and co-workers recently constructed an updated homology model of this bifunctional enzyme using small angle X-ray scattering data and their independently resolved structures of each AAC(6′)-Ie and APH(2′′)-Ia.148 During studies of this bifunctional enzyme, the first structure of APH(2′′) variant Ia was determined, revealing that like APH(2′′)-IIIa, APH(2′′)-Ia uses exclusively GTP as a cosubstrate, unlike APH(2′′)-IIa and APH(2′′)-Iva, which both can use either ATP or GTP.144 Also worth mentioning is the first resolved structure of the multiacetylating AME Eis with an AG (TOB), providing structural rationale for the diacetylation of this AG substrate.127
Several strategies are being investigated to overcome resistance caused by AMEs including attempts to regulate AME expression, the design of new AGs that evade AMEs, and the design of AME inhibitors. Recent review articles detail these strategies, but noteworthy advances from the past two years are described here.120,143,149–151 Interestingly, metal cations have been demonstrated to inhibit AAC activity, increasing AG efficacy in resistant strains.152,153 This approach has yet to be explained mechanistically, but provides a potential combined therapy approach to treat AG-resistant strains. Newly designed AGs showing promise as antibacterials that evade AMEs include thioether, alkylated, and acylated AG variants,154–160 AG homo- and hetero-dimers,161–163 and NEO analogues with a fluorinated (S)-4-amino-2-hydroxybutyrate (AHB) side chain that evade resistance of many AMEs as well as ArmA.164
Cell membrane modification
To reach their bacterial target, AGs must traverse the bacterial cell wall. In Gram-negative bacteria the cell envelope consists of an inner cellular membrane, followed by periplasm containing peptidoglycans, and finally a second phospholipid bilayer, the outer membrane (OM) (Fig. 6). Mycobacteria have a unique cellular envelope consisting of an inner membrane followed by a peptidoglycan layer linked to a layer of arabinogalactan, which is linked to high-molecular weight mycolic acids and coated with an outer monolayer of phospholipids (Fig. 7). These multi-layered cell walls act as a barrier and provide an innate mechanism of resistance to AGs for Gram-negative bacteria and mycobacteria. Modifications to cell wall compositions can cause them to be even less permeable.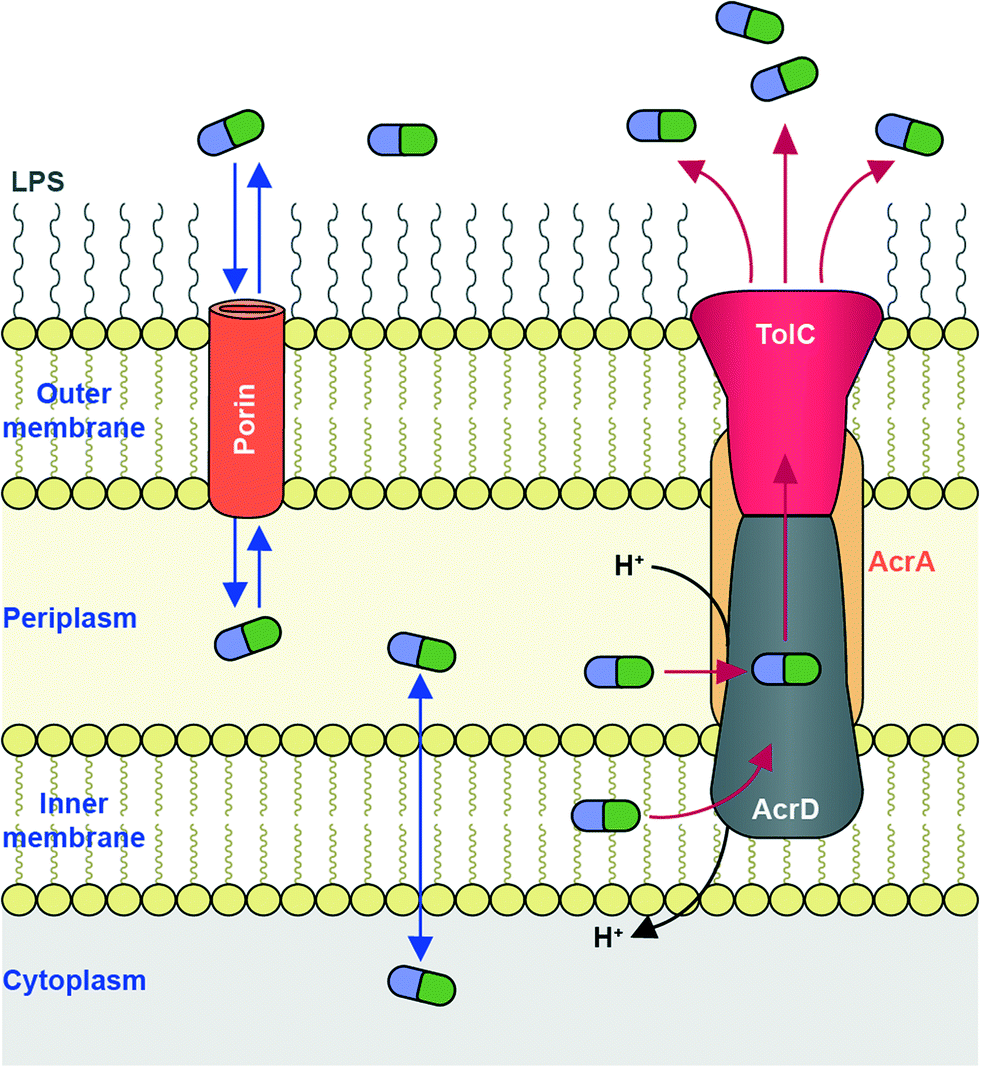 | ||
| Fig. 6 AG transport in Gram-negative bacteria. Influx of AGs occurs through hydrophilic porin protein channels. Efflux of AGs may occur through active transport pumps such as the RND-type efflux pump (e.g., AcrAD-TolC, as shown.) | ||
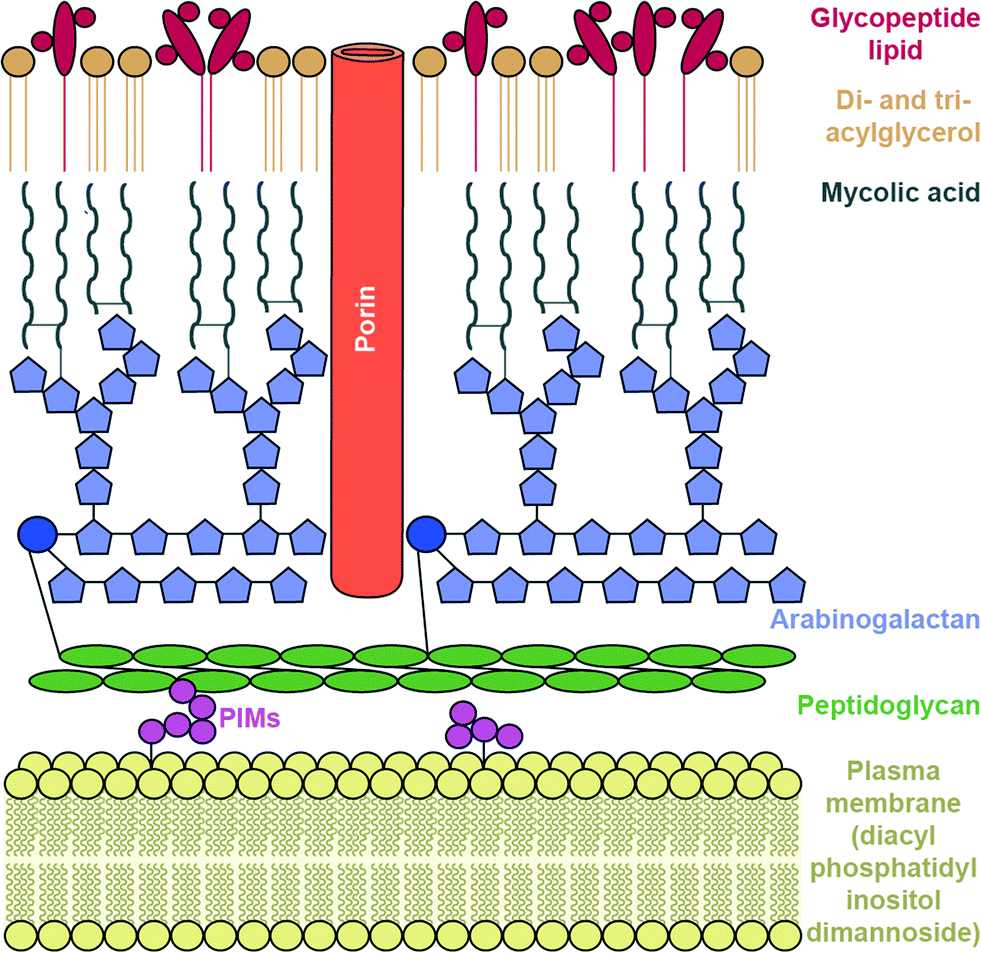 | ||
| Fig. 7 Mycobacterium cell wall. | ||
Due to their cationic, hydrophilic structures, AGs have been hypothesized to penetrate bacterial cell walls through porin channels rather than direct diffusion through the phospholipid bilayer.165,166 However, some experiments have demonstrated that porin-deficient mutants are still capable of AG-uptake.167 Additionally, it has been hypothesized that AGs undergo “self-promoted uptake” in which they interact with and disrupt the OM, allowing their penetration into the bacterial cell.108,168–170 Upon binding to the ribosome AGs interfere with translation, and the aberrant polypeptides synthesized interrupt the cell membrane, further allowing AGs to penetrate the cell wall. AG uptake has been demonstrated to be an energy-consuming process, but details remain unclear.108,168 Bacteria may become resistant to AGs by modification of their OM permeability via alteration of their outermost lipopolysaccharides (LPSs) or down regulation of porins (Fig. 6).
The OM serves as an intrinsic first-line of defence offering Gram-negative bacteria protection from foreign molecules such as AGs. The outward facing half of the OM consists of sugar-functionalized phospholipids (LPSs), which bear a net negative charge, attracting cationic AGs. The most common LPS modification leading to reduced AG uptake is the incorporation of the positively charged 4-amino-4-deoxy-L-arabinose sugar, which effectively reduces the net negative charge of the LPS layer, decreasing affinity for AGs.171–173 Among many Gram-negative bacteria, several two-component systems have been identified (PhoPQ, PmrAB, ParRS, CprRS) to respond to the presence of cationic metals or cationic antimicrobial peptides, or low pH, by attachment of this arabinose onto the phospholipid of the LPS.174,175 Additionally, phosphoethanolamine was reported to be incorporated into the LPS in response to the presence of cationic molecules, under the regulation of the two-component system ColRS (P. aeruginosa) or PmrAB (E. coli, Salmonella enterica).176
The complex, lipid-rich cell wall of mycobacteria is also a very effective barrier for the entry of AGs. The Mycobacterium smegmatis cell membrane lipid profile was recently examined and it was found that the innermost layer of the inner membrane lipid composition consists of an unusual lipid, diacyl phosphatidylinositol dimannoside (Fig. 7).177 This lipid has four hydrocarbon chains, and is proposed to lead to low membrane fluidity and therefore poor drug permeability.
Porin proteins are large water-filled channels allowing the passive diffusion of hydrophilic small molecules. Several types of porins have been identified and studied in Gram-negative bacteria. In E. coli, OmpF is the classical porin – a trimer of β-barrels that quickly transports small, hydrophilic molecules,10 but there is currently no conclusive evidence that AGs are transported through this porin. The relatively narrow size restrictions of porins are hypothesized to limit AG transport. While the transport of beta-lactam antibiotics through porins has been investigated,178 much remains unknown about AG transport through porins in Gram-negative bacteria. OmpF has been hypothesized to be involved in KAN resistance, but experiments remain inconclusive.179
Two types of porins have been identified and characterized in mycobacteria: the MspA-like porins from M. smegmatis and OmpA-like porins from M. tuberculosis.180 Staining and microscopic analysis revealed that the number of MspA pores in M. smegmatis is approximately 50-fold less than pores counted in the outer membranes of Gram-negative bacteria.181 This likely contributes to mycobacteria's characteristically low drug-permeability.182
Numerous studies of the MspA-like porins (MspA, MspB, MspC and MspD) have demonstrated that porin deletion causes a reduction of drug uptake and an increase in MIC.180 However, the MIC value for KANA did not increase significantly in Msp mutants, suggesting that these porins do not transport AGs.183 The crystal structure of MspA has been resolved and while MspA is not directly involved in AG resistance in a pathogenic bacterial species, this structure provides valuable insight of bacterial cell membrane proteins.184M. tuberculosis porin-like molecule “OmpATb”, named as a homologue of E. coli porin OmpA, plays a role in adapting to low pH environments, but does not appear to function as a transport channel.180,181 Furthermore, OmpATb mutants do not show increased drug resistance.185M. tuberculosis protein Rv1698 was thought to function like MspA; initial experiments demonstrated it is capable of uptake of ampicillin, cephaloridine, and chloramphenicol (AGs not examined),186 but this result was later deemed an artifact of overexpression.187 Recently, Rv1698 (now named MctB) was demonstrated to protect M. tuberculosis from high concentrations of copper.188 The newly discovered M. tuberculosis OM protein CpnT plays a role in nutrient uptake and susceptibility of hydrophilic drug molecules.189 The contribution of CpnT to in vitro resistance of large, hydrophilic drugs including STR was only moderate.190
The difference in porin systems, especially that M. tuberculosis does not contain the well-studied MspA-like porins from M. smegmatis, highlights the need for better characterization of AG uptake in the pathogenic species. Unfortunately M. smegmatis may not be accurately serving as a non-pathogenic model organism of M. tuberculosis when it comes to studies of AG influx and resistance. Clearly, much remains to be examined regarding AG-specific uptake by in mycobacteria.
While no documented cases of clinical AG resistance due to porin changes have been reported to our knowledge to date, porin-related resistance to other classes of antibiotics has been reported.10 Possibilities of mutational resistance associated with porins include decreased expression, no expression, or structural mutations that, for example, result in narrowing of the channel size to exclude relatively large AGs. Porin mutations or expression changes, however, may be a limited mechanism of resistance, as they could also lead to decreased uptake of nutrients. Changes in membrane lipids and porin expression are only minor mechanisms of AG resistance – AMEs, RMTases, and efflux pumps are more clinically prominent mechanisms of resistance.
Efflux pumps
An additional bacterial mechanism of resistance to AGs is the active transport of AGs out of cells through efflux pumps. Due to the polycationic structures of AGs, only a few efflux pumps have been demonstrated to remove AGs.191,192The main AG efflux pump in Gram-negative bacteria, AcrAD, is a multidrug transporter and a member of the resistance-nodulation-division (RND) family of efflux pumps. The name AcrAD describes a three-component system that spans the cell envelope; AcrD spans the innermost cellular membrane and functions as a drug-proton antiporter, AcrA is a membrane fusion protein found in the periplasm, and TolC is the outer membrane component of the pump (Fig. 6).193 Intrinsic AcrAD-TolC-type efflux pumps have been identified in many Gram-negative bacterial species (see Li et al. 2015 for a comprehensive review)166 including E. coli,194,195S. enterica,196A. baumannii (AdeABC and AdeDE),197P. aeruginosa (MexXY-OprM),198 and Burkholderia pseudomallei (AmrAB-OprA and BpeAB-OprB).199 AcrAD-TolC homologues also exist in numerous Enterobacteriaceae200 and even in Erwinia amylovora, a plant pathogenic bacteria responsible for causing fire blight in apple, pear and rose species.201 In Stenotrophomonas maltophilia, AGs have also been identified as substrates for RND family efflux pumps SmeIJK and SmeYZ.202,203 Other efflux pumps of which AGs may be (poor) substrates include MexAB-OprM and EmrE (a small multidrug resistance, “SMR” transporter) in P. aeruginosa,204 LmrA (a multidrug ABC transporter) in Lactobacillus lactis,205 and MdfA (a major facilitator superfamily “MFS” transporter) in E. coli, though these are pumps are not likely involved in resistance.206 VcmB and VcmH efflux pumps from in Vibrio cholerea of the multidrug and toxic compound extrusion (MATE) family have also been demonstrated to transport AGs.207
Crystal structures have been solved of AcrB and MexB, RND multidrug transporters whose substrate scope includes mostly hydrophobic molecules and not hydrophilic AGs.208,209 The structures revealed two distinct substrate binding regions – a large proximal substrate channel that accommodates high molecular weight substrates as well as a distal, phenylalanine-containing pocket that accommodates low molecular weight substrates.210 Recently structures of AcrB with an inhibitor present have been solved showing that the inhibitor binds to the hydrophobic phenylalanine trap.209 A homology model of P. aeruginosa MexY was constructed from these structures of AcrB suggesting that the position of a tryptophan residue in MexY, which is a phenylalanine in AcrB, likely prevents the inhibitor from binding MexY and subsequently serving as a universal inhibitor of multidrug efflux transporters.209 In a related study, mutagenesis of residues in the larger, proximal substrate-binding pocket of MexY demonstrated their involvement in AG binding.211 Hopefully soon resolved crystal structures of AcrD or MexY will reveal details of how these pumps accommodate the cationic AG structures and will aide in future work to evade resistance caused by these AG efflux pumps.
The role of MexXY-OprM from P. aeruginosa in AG resistance has been well studied.198 MexXY-OprM accommodates many antibiotic classes, but only AGs, erythromycin, tetracyclines, and glycylcyclines have been demonstrated to induce mexXY expression, and consequently overexpression of MexXY-OprM has been implicated in resistance to these antibiotics.191 The expression of the MexXY-OprM efflux pump has also been shown to be inducible by exposure to reactive oxygen species, contributing to pan-AG resistance.212 The contribution of efflux to AG resistance is currently low. One exception is that the overexpression of MexXY due to a mutation in the repressor gene mexZ is the most common mechanism of AG resistance in lung isolates from cystic fibrosis patients with chronic P. aeruginosa infections.213 In addition to the repressor MexZ, in-depth studies of MexXY have found the two-component system AmgRS to be involved in the regulation pathway of mexXY. AmgRS also protects the cell membrane from damage caused by aberrantly synthesized polypeptides due to AG-ribosomal inhibition.214,215 Despite currently low clinical prominence, efflux pump expression may be used to monitor resistance of other classes of antibiotics; AcrAB (AcrAD homologue) overexpression may be a biomarker for determining the mechanism by which bacteria become MDR.216
AG efflux pumps can also be found in mycobacteria. M. tuberculosis encodes many putative efflux pumps, many of which have yet to be identified and their substrate scopes characterized.187,217 AGs are substrates for mycobacterial Rv1410c (P55) efflux pump.218M. tuberculosis knockout of efflux pump Rv1258c (“Tap”, an MFS transporter) was reported to have decreased MIC values for AGs, suggesting that AGs are substrates for this pumps but not for Rv0849 (MFS transporter) or Rv1218c (an ABC transporter).219
Approaches to combat AG resistance by efflux pumps include devising AGs to evade pumps or adjuvant inhibitors to block them. Spectinamides, semi-synthetic SPC derivatives were found to have increased efficacy against M. tuberculosis, hypothesized to be due to their ability to evade the Rv1218c efflux pump.220In vitro studies show efflux inhibitors to be successful against AMK-resistant M. tuberculosis.221 Other recent work includes a competitive, fluorescent dye assay for studying the efficacy of efflux pump inhibitors.222
At their intrinsic level of expression, efflux pumps contribute little to resistance. It is the overexpression of efflux pumps (usually due to mutations in regulatory genes), the acquisition of mutations within efflux pumps that improve substrate affinity, and synergy with other resistance mechanisms that leads to high levels of antibiotic resistance. The contribution of efflux pumps to resistance also depends on the rate of influx of the drug; in organisms such as P. aeruginosa or M. tuberculosis in which AG influx is slow, increased efflux will be a large contributor to AG resistance. Furthermore, a danger of AG efflux pumps as a mechanism of resistance is that they are multidrug transporters and will also confer resistance to other classes of antibiotics. They have even been shown to remove other biocides such as disinfectants.206,223,224 Further details of work towards the design of AGs to evade efflux pumps and efflux pump inhibitors to block these resistance-causing pumps is well covered in recent reviews.225–230
Other mechanisms of resistance
Though not traditionally considered a mechanism of resistance, membrane proteases may offer protection from AGs. Membrane proteases are part of an intrinsic system of protein biosynthesis quality control that recognizes and degrades misfolded and mistranslated proteins. Because one of the effects of AGs is synthesis of aberrant proteins, which accumulate and perturb cell membrane integrity, any protection from this would provide tolerance to AGs. Mutations in FtsH, a membrane protease in P. aeruginosa lead to increased sensitivity to TOB, demonstrating that FtsH plays a role in intrinsic AG resistance.231,232 Deletion mutations in genes associated with lipid biosynthesis or metabolism (lptA, faoA), phosphate uptake (pstB), and the two-component regulator (amgRS) also result in increased TOB sensitivity.233 In another study of P. aeruginosa, upon exposure to TOB, the expression of asrA, which encodes a Lon-type protease, as well as heat shock genes, was increased.234 This is an example of adaptive resistance, common to chronic P. aeruginosa infections of cystic fibrosis patients.213Adaptive resistance to AGs in these chronic P. aeruginosa infections is associated with the bacteria existing as a biofilm. Biofilms are characterized as bacteria growing aggregately on a surface, surrounded by a matrix of proteins, DNA and carbohydrates. This biofilm matrix has been hypothesized to bind cationic molecules, such as AGs, decreasing their efficacy.213,235 Co-delivery of an AG with a cationic steroid antibiotic, CSA-13, has been demonstrated to overcome this resistance.236 More recently, adaptive resistance in biofilms has been associated with the previously described gene expression changes and the upregulation of MexXY efflux pump expression described in an earlier section of this review.192 Meta-analysis revealed that a variety of genes and proteins undergo expression changes in biofilm when compared to non-biofilm cells, yet, as expected, no smoking gun was revealed.237 Much remains to be understood about adaptive response to AGs.6,238
Perspective and conclusions
The mechanisms discussed here differ in their degree of contribution to bacterial AG resistance. These mechanisms also vary in mobility; some spread quickly to other species via horizontal gene transfer, others are less transferable. Mechanisms also vary in their substrate scope; for example AMEs often have only a few specific AG substrates (e.g., GEN/TOB or AMK), while RMTases work on many AG substrates. In this sense, RMTases are a worrisome mechanism of resistance due to their high mobility and broad substrate scope. The bacterial cell wall and efflux pumps contribute to resistance more broadly. They often work against all AGs and other entire classes of antibiotics too. The less common mechanism of ribosomal mutations can offer either broad resistance to an entire sub-class of AGs (e.g., 2-DOS) or specific resistance to one AG.The most threatening bacterial species harbour multiple mechanisms of resistance, commonly from a single plasmid bearing multiple resistance genes. The effects of multiple mechanisms are additive. For example, the intrinsic nature of the outer membrane as a barrier to restrict AG uptake can act synergistically with other mechanisms of resistance; the combination of minor changes that individually result in only small MIC increases can collectively result in a highly AG-resistant phenotype.239
To combat multi-faceted bacterial resistance, clinicians will need to be able to execute a breadth of therapies. Additionally rapid detection and identification of resistance genes will allow for tailored therapies. This is not only more effective at fighting each uniquely resistant bacterial infection, but also prevents unnecessary use of irrelevant antibiotics. Individualized diagnostics may not be as daunting as it sounds; a recent study demonstrates that in E. coli and K. pneumoniae cultures, resistance to GEN can be pinpointed to just three genes in 97% of the 267 geographically localized isolates.58 Executed efficiently, a combination of periodic complete resistance gene scans and regular screens for resistance genes of known prevalence may be more effective in the long run.
Work to design new AGs that evade resistance continues. Second generation AGs include semi-synthetic derivatives of dibekacin (DBK) such as arbekacin (ABK), bearing an N1-AHB side chain, and isepamicin (ISP) bearing the 1-C shorter (S)-3-amino-2-hydroxypropate N1 side chain (Fig. 1). Both of these compounds were originally synthesized in the 1970s and they were clinically approved in 1990 and 1988, respectively. Despite their structural modifications, which seem to decrease toxicity side effects, ABK and ISP are still susceptible to resistance caused by RMTases and modification from AMEs, though surprisingly sometimes not to modification by AAC(6′)s.116,240,241 The high prominence of AAC(6′) enzymes is likely why ISP is often still quite clinically effective.242
The only new AG currently in the pipeline is plazomicin (PLZ, formerly ACHN-490), a sisomicin (SIS) derivative bearing the N1-AHB side chain (Fig. 1). It has been shown to be effective against methicillin-resistant S. aureus (MRSA) and carbapenem-resistant Enterobacteriaceae (CRE) and to overcome the action of many AMEs, including AAC(6′)s.243,244 PLZ was recently shown to be effective against several strains of Brucella, a zoonotic pathogen,245 and carbapenem-resistant A. baumannii.246 PLZ is currently in Phase 3 clinical trials for patients with bloodstream infections or nosocomial pneumonia due to CRE. PLZ has a few weaknesses; it is not effective against Enterobacteriaceae carrying RMTases ArmA or RmtC, or Providencia stuartii AAC(2′)-I.247 Despite these deficiencies, PLZ is an important new weapon in the pipeline to fight antibiotic resistance.
With better understanding of AG resistance mechanisms, follow novel potential strategies to combat resistance. The discovery of a secondary ribosomal AG-binding site at H69, launches the potential for exploiting this as a bactericidal method.21 Recently it was shown that the binding of NEO induced different ribosomal conformation changes when compared to the structurally similar AG PAR, highlighting the subtleties of AG-ribosome binding.22
Due to their dramatic clinical prevalence, much current research is aimed at blocking AMEs via novel AG derivatives or small molecule AME inhibitors as discussed earlier in this review (AG Modifying Enzymes). Additionally, as resistance due to dissemination of the RMTases increases, inhibitors of these enzymes may also serve as valuable AG adjuvants.
Because multiple resistance genes may coexist within a mobile gene element and therefore are often acquired together, to prevent the spread of resistance, the transfer of these resistance genes must be prevented. Selective pressures, which occur in hospital environments and regions in which antibiotic use is unregulated, must be limited. Ongoing surveillance of resistance genes in humans, animals, and foods will be crucial in delaying the spread of resistance to AGs.
Many of these mechanisms of resistance to AG antibiotics were discovered relatively recently; RMTases in the late 1990s, the first RND pump was characterized in the early 1990s. Due to recent technological advances and increased efforts in scientific discovery, just a few years later we have acquired an in-depth understanding of some of these molecular mechanisms of antibiotic resistance. If our scientific work continues at this pace and is combined with better management of antibiotic use, hopefully bacterial antibiotic resistance will be classified as a manageable medical issue, rather than a global health crisis.
Acknowledgements
We thank all of those working in the field of aminoglycoside drug discovery and apologize if their work is not cited due to the scope of the review presenting examples from 2010–2015. Our work on aminoglycosides and bacterial resistance is supported by the National Institutes of Health (NIH) Grant AI090048 (S. G.-T.) and by startup funds from the College of Pharmacy at the University of Kentucky (S. G.-T.).Notes and references
- B. Becker and M. A. Cooper, ACS Chem. Biol., 2013, 8, 105–115 CrossRef CAS PubMed.
- M. Y. Fosso, Y. Li and S. Garneau-Tsodikova, Med. Chem. Commun., 2014, 5, 1075–1091 RSC.
- K. D. Green and S. Garneau-Tsodikova, Frontiers Microbiol., 2013, 4, 208 Search PubMed.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed.
- L. Fernandez, E. B. Breidenstein and R. E. Hancock, Drug Resist. Updates, 2011, 14, 1–21 CrossRef CAS PubMed.
- A. Skiada, A. Markogiannakis, D. Plachouras and G. L. Daikos, Int. J. Antimicrob. Agents, 2011, 37, 187–193 CrossRef CAS PubMed.
- C. de la Fuente-Nunez, F. Reffuveille, L. Fernandez and R. E. Hancock, Curr. Opin. Microbiol., 2013, 16, 580–589 CrossRef CAS PubMed.
- A. H. van Hoek, D. Mevius, B. Guerra, P. Mullany, A. P. Roberts and H. J. Aarts, Front. Microbiol., 2011, 2, 203 Search PubMed.
- D. N. Wilson, Nat. Rev. Microbiol., 2014, 12, 35–48 CrossRef CAS PubMed.
- L. Fernandez and R. E. Hancock, Clin. Microbiol. Rev., 2012, 25, 661–681 CrossRef CAS PubMed.
- R. Laxminarayan, A. Duse, C. Wattal, A. K. Zaidi, H. F. Wertheim, N. Sumpradit, E. Vlieghe, G. L. Hara, I. M. Gould, H. Goossens, C. Greko, A. D. So, M. Bigdeli, G. Tomson, W. Woodhouse, E. Ombaka, A. Q. Peralta, F. N. Qamar, F. Mir, S. Kariuki, Z. A. Bhutta, A. Coates, R. Bergstrom, G. D. Wright, E. D. Brown and O. Cars, Lancet Infect. Dis., 2013, 13, 1057–1098 CrossRef.
- R. J. Fair and Y. Tor, Perspect. Med. Chem., 2014, 6, 25–64 Search PubMed.
- L. S. McCoy, Y. Xie and Y. Tor, Wiley Interdiscip. Rev.: RNA, 2011, 2, 209–232 CrossRef CAS PubMed.
- Q. Han, Q. Zhao, S. Fish, K. B. Simonsen, D. Vourloumis, J. M. Froelich, D. Wall and T. Hermann, Angew. Chem., Int. Ed., 2005, 44, 2694–2700 CrossRef CAS PubMed.
- S. N. Hobbie, S. Akshay, S. K. Kalapala, C. M. Bruell, D. Shcherbakov and E. C. Bottger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20888–20893 CrossRef CAS PubMed.
- A. Amunts, A. Brown, J. Toots, S. H. Scheres and V. Ramakrishnan, Science, 2015, 348, 95–98 CrossRef CAS PubMed.
- B. J. Greber, P. Bieri, M. Leibundgut, A. Leitner, R. Aebersold, D. Boehringer and N. Ban, Science, 2015, 348, 303–308 CrossRef CAS PubMed.
- M. B. Feldman, D. S. Terry, R. B. Altman and S. C. Blanchard, Nat. Chem. Biol., 2010, 6, 54–62 CrossRef CAS PubMed.
- N. Demeshkina, L. Jenner, E. Westhof, M. Yusupov and G. Yusupova, Nature, 2012, 484, 256–259 CrossRef CAS PubMed.
- M. A. Borovinskaya, R. D. Pai, W. Zhang, B. S. Schuwirth, J. M. Holton, G. Hirokawa, H. Kaji, A. Kaji and J. H. Cate, Nat. Struct. Mol. Biol., 2007, 14, 727–732 CAS.
- L. Wang, A. Pulk, M. R. Wasserman, M. B. Feldman, R. B. Altman, J. H. Cate and S. C. Blanchard, Nat. Struct. Mol. Biol., 2012, 19, 957–963 CAS.
- M. R. Wasserman, A. Pulk, Z. Zhou, R. B. Altman, J. C. Zinder, K. D. Green, S. Garneau-Tsodikova, J. H. Doudna Cate and S. C. Blanchard, Nat. Commun., 2015, 6, 7896 CrossRef CAS PubMed.
- C. E. Maus, B. B. Plikaytis and T. M. Shinnick, Antimicrob. Agents Chemother., 2005, 49, 3192–3197 CrossRef CAS PubMed.
- S. B. Georghiou, M. Magana, R. S. Garfein, D. G. Catanzaro, A. Catanzaro and T. C. Rodwell, PLoS One, 2012, 7, e33275 CAS.
- R. Nessar, J. M. Reyrat, A. Murray and B. Gicquel, J. Antimicrob. Chemother., 2011, 66, 1719–1724 CrossRef CAS PubMed.
- J. Kondo, Angew. Chem., Int. Ed., 2012, 51, 465–468 CrossRef CAS PubMed.
- B. Springer, Y. G. Kidan, T. Prammananan, K. Ellrott, E. C. Bottger and P. Sander, Antimicrob. Agents Chemother., 2001, 45, 2877–2884 CrossRef CAS PubMed.
- L. C. Santos, Open J. Med. Microbiol., 2012, 2, 24–36 CrossRef.
- M. Unemo, D. Golparian, V. Skogen, A. O. Olsen, H. Moi, G. Syversen and S. O. Hjelmevoll, Antimicrob. Agents Chemother., 2013, 57, 1057–1061 CrossRef CAS PubMed.
- G. Thom and C. D. Prescott, Bioorg. Med. Chem., 1997, 5, 1081–1086 CrossRef CAS.
- J. Wachino and Y. Arakawa, Drug Resist. Updates, 2012, 15, 133–148 CrossRef CAS PubMed.
- K. Yokoyama, Y. Doi, K. Yamane, H. Kurokawa, N. Shibata, K. Shibayama, T. Yagi, H. Kato and Y. Arakawa, Lancet, 2003, 362, 1888–1893 CrossRef CAS.
- M. Golebiewski, I. Kern-Zdanowicz, M. Zienkiewicz, M. Adamczyk, J. Zylinska, A. Baraniak, M. Gniadkowski, J. Bardowski and P. Ceglowski, Antimicrob. Agents Chemother., 2007, 51, 3789–3795 CrossRef CAS PubMed.
- M. Galimand, P. Courvalin and T. Lambert, Antimicrob. Agents Chemother., 2003, 47, 2565–2571 CrossRef CAS.
- J. Wachino, K. Shibayama, H. Kurokawa, K. Kimura, K. Yamane, S. Suzuki, N. Shibata, Y. Ike and Y. Arakawa, Antimicrob. Agents Chemother., 2007, 51, 4401–4409 CrossRef CAS PubMed.
- Y. A. Al Sheikh, M. A. Marie, J. John, L. G. Krishnappa and K. H. Dabwab, Libyan J. Med., 2014, 9, 24432 Search PubMed.
- M. Galimand, S. Sabtcheva, P. Courvalin and T. Lambert, Antimicrob. Agents Chemother., 2005, 49, 2949–2953 CrossRef CAS PubMed.
- D. Moissenet, F. X. Weill, G. Arlet, D. Harrois, J. P. Girardet and H. Vu-Thien, J. Clin. Microbiol., 2011, 49, 1676–1678 CrossRef CAS PubMed.
- T. Naas, C. Bentchouala, G. Cuzon, S. Yaou, A. Lezzar, F. Smati and P. Nordmann, Int. J. Antimicrob. Agents, 2011, 38, 135–139 CrossRef CAS PubMed.
- N. Bouzidi, L. Aoun, M. Dekhil, S. A. Granier, L. Poirel, A. Brisabois, P. Nordmann and Y. Millemann, J. Antimicrob. Chemother., 2011, 66, 2180–2181 CrossRef CAS PubMed.
- S. A. Granier, L. Hidalgo, A. San Millan, J. A. Escudero, B. Gutierrez, A. Brisabois and B. Gonzalez-Zorn, Antimicrob. Agents Chemother., 2011, 55, 5262–5266 CrossRef CAS PubMed.
- X. D. Du, D. X. Li, G. Z. Hu, Y. Wang, Y. H. Shang, C. M. Wu, H. B. Liu and X. S. Li, J. Antimicrob. Chemother., 2012, 67, 246–248 CrossRef CAS PubMed.
- H. W. Lee, H. Y. Kang, K. S. Shin and J. Kim, Int. J. Microbiol., 2007, 45, 272–274 CAS.
- K. Karthikeyan, M. A. Thirunarayan and P. Krishnan, J. Antimicrob. Chemother., 2010, 65, 2253–2254 CrossRef CAS PubMed.
- M. Gurung, D. C. Moon, M. D. Tamang, J. Kim, Y. C. Lee, S. Y. Seol, D. T. Cho and J. C. Lee, Diagn. Microbiol. Infect. Dis., 2010, 68, 468–470 CrossRef CAS PubMed.
- J. Y. Sung, K. C. Kwon, H. H. Cho and S. H. Koo, Korean J. Lab. Med., 2011, 31, 265–270 CrossRef CAS PubMed.
- M. A. Islam, M. Huq, A. Nabi, P. K. Talukdar, D. Ahmed, K. A. Talukder, A. Cravioto and H. P. Endtz, J. Med. Microbiol., 2013, 62, 62–68 CrossRef CAS PubMed.
- T. Tada, T. Miyoshi-Akiyama, Y. Kato, N. Ohmagari, N. Takeshita, N. V. Hung, D. M. Phuong, T. A. Thu, N. G. Binh, N. Q. Anh, T. T. Nga, P. H. Truong, P. T. Xuan, T. A. Thu le, N. T. Son and T. Kirikae, BMC Infect. Dis., 2013, 13, 251 CrossRef CAS PubMed.
- T. Tada, T. Miyoshi-Akiyama, K. Shimada, M. Shimojima and T. Kirikae, Antimicrob. Agents Chemother., 2014, 58, 2916–2920 CrossRef PubMed.
- L. Poirel, J. Schrenzel, A. Cherkaoui, S. Bernabeu, G. Renzi and P. Nordmann, J. Antimicrob. Chemother., 2011, 66, 1730–1733 CrossRef CAS PubMed.
- L. Poirel, Z. Al Maskari, F. Al Rashdi, S. Bernabeu and P. Nordmann, J. Antimicrob. Chemother., 2011, 66, 304–306 CrossRef CAS PubMed.
- N. Karah, B. Haldorsen, N. O. Hermansen, Y. Tveten, E. Ragnhildstveit, D. H. Skutlaberg, S. Tofteland, A. Sundsfjord and O. Samuelsen, J. Med. Microbiol., 2011, 60, 515–521 CrossRef CAS PubMed.
- P. McGann, J. Hang, R. J. Clifford, Y. Yang, Y. I. Kwak, R. A. Kuschner, E. P. Lesho and P. E. Waterman, Antimicrob. Agents Chemother., 2012, 56, 1673–1679 CrossRef CAS PubMed.
- L. Dortet, L. Poirel, F. Al Yaqoubi and P. Nordmann, Clin. Microbiol. Infect., 2012, 18, E144–148 CrossRef CAS PubMed.
- Z. Liu, B. Ling and L. Zhou, J. Chemother., 2015, 27, 207–212 CrossRef CAS PubMed.
- S. N. Seiffert, J. Marschall, V. Perreten, A. Carattoli, H. Furrer and A. Endimiani, Int. J. Antimicrob. Agents, 2014, 44, 260–262 CrossRef CAS PubMed.
- S. Shoma, M. Kamruzzaman, A. N. Ginn, J. R. Iredell and S. R. Partridge, Diagn. Microbiol. Infect. Dis., 2014, 78, 93–97 CrossRef CAS PubMed.
- A. N. Ginn, Z. Zong, A. M. Wiklendt, L. C. Thomas, J. Merlino, T. Gottlieb, S. van Hal, J. Harkness, C. Macleod, S. M. Bell, M. J. Leroi, S. R. Partridge and J. R. Iredell, Int. J. Antimicrob. Agents, 2013, 42, 19–26 CrossRef CAS PubMed.
- M. A. El-Sayed-Ahmed, M. A. Amin, W. M. Tawakol, L. Loucif, S. Bakour and J. M. Rolain, Antimicrob. Agents Chemother., 2015, 59, 3602–3605 CrossRef CAS PubMed.
- R. Batah, L. Loucif, A. O. Olaitan, N. Boutefnouchet, H. Allag and J. M. Rolain, Microb. Drug Resist., 2015, 21, 470–476 CrossRef CAS PubMed.
- Z. Belbel, H. Chettibi, M. Dekhil, A. Ladjama, S. Nedjai and J. M. Rolain, Microb. Drug Resist., 2014, 20, 310–315 CrossRef CAS PubMed.
- Y. Doi, K. Yokoyama, K. Yamane, J. Wachino, N. Shibata, T. Yagi, K. Shibayama, H. Kato and Y. Arakawa, Antimicrob. Agents Chemother., 2004, 48, 491–496 CrossRef CAS.
- K. Yamane, J. Wachino, S. Suzuki, K. Kimura, N. Shibata, H. Kato, K. Shibayama, T. Konda and Y. Arakawa, Antimicrob. Agents Chemother., 2007, 51, 3354–3360 CrossRef CAS PubMed.
- L. Poirel, R. A. Bonnin and P. Nordmann, Antimicrob. Agents Chemother., 2011, 55, 4224–4229 CrossRef CAS PubMed.
- B. Perichon, P. Bogaerts, T. Lambert, L. Frangeul, P. Courvalin and M. Galimand, Antimicrob. Agents Chemother., 2008, 52, 2581–2592 CrossRef CAS PubMed.
- Y. J. Park, J. K. Yu, S. I. Kim, K. Lee and Y. Arakawa, Ann. Clin. Lab. Sci., 2009, 39, 55–59 CAS.
- J. Hou, X. Huang, Y. Deng, L. He, T. Yang, Z. Zeng, Z. Chen and J. H. Liu, Antimicrob. Agents Chemother., 2012, 56, 2135–2138 CrossRef CAS PubMed.
- J. J. Li, Z. K. Sheng, M. Deng, S. Bi, F. S. Hu, H. F. Miao, Z. K. Ji, J. F. Sheng and L. J. Li, BMC Infect. Dis., 2012, 12, 373 CrossRef CAS PubMed.
- H. Sun, S. Li, Z. Xie, F. Yang, Y. Sun, Y. Zhu, X. Zhao and S. Jiang, J. Antimicrob. Chemother., 2012, 67, 1635–1638 CrossRef CAS PubMed.
- D. X. Li, S. M. Zhang, G. Z. Hu, Y. Wang, H. B. Liu, C. M. Wu, Y. H. Shang, Y. X. Chen and X. D. Du, J. Antimicrob. Chemother., 2012, 67, 236–238 CrossRef CAS PubMed.
- J. F. Sheng, J. J. Li, S. Tu, Z. K. Sheng, S. Bi, M. H. Zhu, X. M. Shen and L. J. Li, Eur. J. Clin. Microbiol. Infect. Dis., 2012, 31, 1585–1591 CrossRef CAS PubMed.
- Q. Yao, Z. Zeng, J. Hou, Y. Deng, L. He, W. Tian, H. Zheng, Z. Chen and J. H. Liu, J. Antimicrob. Chemother., 2011, 66, 2475–2479 CrossRef CAS PubMed.
- H. Y. Kang, K. Y. Kim, J. Kim, J. C. Lee, Y. C. Lee, D. T. Cho and S. Y. Seol, J. Clin. Microbiol., 2008, 46, 700–706 CrossRef CAS PubMed.
- P. Bogaerts, W. Bouchahrouf, R. R. de Castro, A. Deplano, C. Berhin, D. Pierard, O. Denis and Y. Glupczynski, Antimicrob. Agents Chemother., 2011, 55, 3036–3038 CrossRef CAS PubMed.
- I. Galani, M. Souli, T. Panagea, G. Poulakou, K. Kanellakopoulou and H. Giamarellou, Clin. Microbiol. Infect., 2012, 18, E52–54 CrossRef CAS PubMed.
- T. Tada, T. Miyoshi-Akiyama, R. K. Dahal, M. K. Sah, H. Ohara, T. Kirikae and B. M. Pokhrel, Antimicrob. Agents Chemother., 2013, 57, 2394–2396 CrossRef CAS PubMed.
- X. J. Ma, H. F. Yang, Y. Y. Liu, Q. Mei, Y. Ye, H. R. Li, J. Cheng and J. B. Li, Ann. Lab. Med., 2015, 35, 172–174 CrossRef CAS PubMed.
- Y. T. Deng, Z. L. Zeng, W. Tian, T. Yang and J. H. Liu, Front. Microbiol., 2013, 4, 198 Search PubMed.
- J. Wachino, K. Yamane, K. Shibayama, H. Kurokawa, N. Shibata, S. Suzuki, Y. Doi, K. Kimura, Y. Ike and Y. Arakawa, Antimicrob. Agents Chemother., 2006, 50, 178–184 CrossRef CAS PubMed.
- A. Carattoli, L. Villa, L. Poirel, R. A. Bonnin and P. Nordmann, Antimicrob. Agents Chemother., 2012, 56, 783–786 CrossRef CAS PubMed.
- K. L. Hopkins, J. A. Escudero, L. Hidalgo and B. Gonzalez-Zorn, Emerging Infect. Dis., 2010, 16, 712–715 CrossRef CAS PubMed.
- L. Poirel, G. Revathi, S. Bernabeu and P. Nordmann, Antimicrob. Agents Chemother., 2011, 55, 934–936 CrossRef CAS PubMed.
- Z. Zong, S. R. Partridge and J. R. Iredell, Antimicrob. Agents Chemother., 2008, 52, 794–795 CrossRef CAS PubMed.
- Y. Doi, D. de Oliveira Garcia, J. Adams and D. L. Paterson, Antimicrob. Agents Chemother., 2007, 51, 852–856 CrossRef CAS PubMed.
- M. F. Bueno, G. R. Francisco, J. A. O'Hara, D. de Oliveira Garcia and Y. Doi, Antimicrob. Agents Chemother., 2013, 57, 2397–2400 CrossRef CAS PubMed.
- N. Tijet, P. Andres, C. Chung, C. Lucero, W. H.-A. Group, D. E. Low, M. Galas, A. Corso, A. Petroni and R. G. Melano, Antimicrob. Agents Chemother., 2011, 55, 904–909 CrossRef CAS PubMed.
- M. A. Davis, K. N. Baker, L. H. Orfe, D. H. Shah, T. E. Besser and D. R. Call, Antimicrob. Agents Chemother., 2010, 54, 2666–2669 CrossRef CAS PubMed.
- M. Galimand, P. Courvalin and T. Lambert, Antimicrob. Agents Chemother., 2012, 56, 3960–3962 CrossRef CAS PubMed.
- L. Hidalgo, K. L. Hopkins, B. Gutierrez, C. M. Ovejero, S. Shukla, S. Douthwaite, K. N. Prasad, N. Woodford and B. Gonzalez-Zorn, J. Antimicrob. Chemother., 2013, 68, 1543–1550 CrossRef CAS PubMed.
- L. Poirel, J. Labarca, H. Bello, M. L. Rioseco, S. Bernabeu and P. Nordmann, Antimicrob. Agents Chemother., 2014, 58, 618–619 CrossRef PubMed.
- G. R. Francisco, S. T. Nora, M. F. Bueno, L. V. da Silva Filho and D. de Oliveira Garcia, Antimicrob. Agents Chemother., 2015, 59, 2967–2968 CrossRef CAS PubMed.
- J. A. O'Hara, P. McGann, E. C. Snesrud, R. J. Clifford, P. E. Waterman, E. P. Lesho and Y. Doi, Antimicrob. Agents Chemother., 2013, 57, 2413–2416 CrossRef PubMed.
- N. Husain, K. L. Tkaczuk, S. R. Tulsidas, K. H. Kaminska, S. Cubrilo, G. Maravic-Vlahovicek, J. M. Bujnicki and J. Sivaraman, Nucleic Acids Res., 2010, 38, 4120–4132 CrossRef CAS PubMed.
- E. Schmitt, M. Galimand, M. Panvert, P. Courvalin and Y. Mechulam, J. Mol. Biol., 2009, 388, 570–582 CrossRef CAS PubMed.
- M. Savic, S. Sunita, N. Zelinskaya, P. M. Desai, R. Macmaster, K. Vinal and G. L. Conn, Antimicrob. Agents Chemother., 2015, 59, 2807–2816 CrossRef CAS PubMed.
- R. Macmaster, N. Zelinskaya, M. Savic, C. R. Rankin and G. L. Conn, Nucleic Acids Res., 2010, 38, 7791–7799 CrossRef CAS PubMed.
- N. Husain, S. Obranic, L. Koscinski, J. Seetharaman, F. Babic, J. M. Bujnicki, G. Maravic-Vlahovicek and J. Sivaraman, Nucleic Acids Res., 2011, 39, 1903–1918 CrossRef CAS PubMed.
- J. A. Dunkle, K. Vinal, P. M. Desai, N. Zelinskaya, M. Savic, D. M. West, G. L. Conn and C. M. Dunham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6275–6280 CrossRef CAS PubMed.
- G. G. Zhanel, C. D. Lawson, S. Zelenitsky, B. Findlay, F. Schweizer, H. Adam, A. Walkty, E. Rubinstein, A. S. Gin, D. J. Hoban, J. P. Lynch and J. A. Karlowsky, Expert Rev. Anti-Infect. Ther., 2012, 10, 459–473 CrossRef CAS PubMed.
- D. M. Livermore, S. Mushtaq, M. Warner, J. C. Zhang, S. Maharjan, M. Doumith and N. Woodford, J. Antimicrob. Chemother., 2011, 66, 48–53 CrossRef CAS PubMed.
- B. Gutierrez, S. Douthwaite and B. Gonzalez-Zorn, RNA Biol., 2013, 10, 1324–1332 CrossRef CAS PubMed.
- V. S. Lioy, S. Goussard, V. Guerineau, E. J. Yoon, P. Courvalin, M. Galimand and C. Grillot-Courvalin, RNA, 2014, 20, 382–391 CrossRef CAS PubMed.
- X. Guo, B. B. Dillon, A. N. Ginn, A. M. Wiklendt, S. R. Partridge and J. R. Iredell, Diagn. Microbiol. Infect. Dis., 2014, 80, 29–31 CrossRef CAS PubMed.
- M. Nagasawa, M. Kaku, K. Kamachi, K. Shibayama, Y. Arakawa, K. Yamaguchi and Y. Ishii, J. Infect. Chemother., 2014, 20, 635–638 CrossRef CAS PubMed.
- S. K. Gupta, B. R. Padmanabhan, S. M. Diene, R. Lopez-Rojas, M. Kempf, L. Landraud and J. M. Rolain, Antimicrob. Agents Chemother., 2014, 58, 212–220 CrossRef PubMed.
- A. R. Oany, T. P. Jyoti and S. A. Ahmad, Bioinf. Biol. Insights, 2014, 8, 65–72 CAS.
- M. A. Witek and G. L. Conn, Biochim. Biophys. Acta, 2014, 1844, 1648–1655 CrossRef CAS PubMed.
- M. S. Ramirez and M. E. Tolmasky, Drug Resist. Updates, 2010, 13, 151–171 CrossRef CAS PubMed.
- D. M. Daigle, D. W. Hughes and G. D. Wright, Chem. Biol., 1999, 6, 99–110 CrossRef CAS.
- D. D. Boehr, D. M. Daigle and G. D. Wright, Biochemistry, 2004, 43, 9846–9855 CrossRef CAS PubMed.
- H. Frase, M. Toth and S. B. Vakulenko, J. Biol. Chem., 2012, 287, 43262–43269 CrossRef CAS PubMed.
- S. J. Caldwell and A. M. Berghuis, Antimicrob. Agents Chemother., 2012, 56, 1899–1906 CrossRef CAS PubMed.
- V. Dubois, L. Poirel, C. Marie, C. Arpin, P. Nordmann and C. Quentin, Antimicrob. Agents Chemother., 2002, 46, 638–645 CrossRef CAS.
- C. Kim, A. Villegas-Estrada, D. Hesek and S. Mobashery, Biochemistry, 2007, 46, 5270–5282 CrossRef CAS PubMed.
- D. Centron and P. H. Roy, Antimicrob. Agents Chemother., 2002, 46, 1402–1409 CrossRef CAS.
- C. Kim, D. Hesek, J. Zajicek, S. B. Vakulenko and S. Mobashery, Biochemistry, 2006, 45, 8368–8377 CrossRef CAS PubMed.
- K. D. Green and S. Garneau-Tsodikova, Biochimie, 2013, 95, 1319–1325 CrossRef CAS PubMed.
- R. E. Mendes, M. A. Toleman, J. Ribeiro, H. S. Sader, R. N. Jones and T. R. Walsh, Antimicrob. Agents Chemother., 2004, 48, 4693–4702 CrossRef CAS PubMed.
- K. D. Green, W. Chen and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2011, 55, 3207–3213 CrossRef CAS PubMed.
- M. S. Ramirez, N. Nikolaidis and M. E. Tolmasky, Front. Microbiol., 2013, 4, 121 Search PubMed.
- M. A. Zaunbrecher, R. D. Sikes Jr., B. Metchock, T. M. Shinnick and J. E. Posey, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20004–20009 CrossRef CAS PubMed.
- W. Chen, T. Biswas, V. R. Porter, O. V. Tsodikov and S. Garneau-Tsodikova, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9804–9808 CrossRef CAS PubMed.
- W. Chen, K. D. Green, O. V. Tsodikov and S. Garneau-Tsodikova, Biochemistry, 2012, 51, 4959–4967 CrossRef CAS PubMed.
- W. Chen, K. D. Green and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2012, 56, 5831–5838 CrossRef CAS PubMed.
- R. E. Pricer, J. L. Houghton, K. D. Green, A. S. Mayhoub and S. Garneau-Tsodikova, Mol. BioSyst., 2012, 8, 3305–3313 RSC.
- B. C. Jennings, K. J. Labby, K. D. Green and S. Garneau-Tsodikova, Biochemistry, 2013, 52, 5125–5132 CrossRef CAS PubMed.
- J. L. Houghton, T. Biswas, W. Chen, O. V. Tsodikov and S. Garneau-Tsodikova, ChemBioChem, 2013, 14, 2127–2135 CrossRef CAS PubMed.
- O. V. Tsodikov, K. D. Green and S. Garneau-Tsodikova, PLoS One, 2014, 9, e92370 Search PubMed.
- K. D. Green, R. E. Pricer, M. N. Stewart and S. Garneau-Tsodikova, ACS Infect. Dis., 2015, 1, 272–283 CrossRef CAS.
- K. D. Green, T. Biswas, C. Chang, R. Wu, W. Chen, B. K. Janes, D. Chalupska, P. Gornicki, P. C. Hanna, O. V. Tsodikov, A. Joachimiak and S. Garneau-Tsodikova, Biochemistry, 2015, 54, 3197–3206 CrossRef CAS PubMed.
- J. L. Houghton, K. D. Green, R. E. Pricer, A. S. Mayhoub and S. Garneau-Tsodikova, J. Antimicrob. Chemother., 2013, 68, 800–805 CrossRef CAS PubMed.
- J. A. Perry, E. L. Westman and G. D. Wright, Curr. Opin. Microbiol., 2014, 21, 45–50 CrossRef CAS PubMed.
- A. Z. Reeves, P. J. Campbell, R. Sultana, S. Malik, M. Murray, B. B. Plikaytis, T. M. Shinnick and J. E. Posey, Antimicrob. Agents Chemother., 2013, 57, 1857–1865 CrossRef CAS PubMed.
- F. P. Maurer, V. L. Bruderer, C. Ritter, C. Castelberg, G. V. Bloemberg and E. C. Bottger, Antimicrob. Agents Chemother., 2014, 58, 3828–3836 CrossRef PubMed.
- F. P. Maurer, V. L. Bruderer, C. Castelberg, C. Ritter, D. Scherbakov, G. V. Bloemberg and E. C. Bottger, J. Antimicrob. Chemother., 2015, 70, 1412–1419 CrossRef CAS PubMed.
- K. Kobayashi, I. Hayashi, S. Kouda, F. Kato, T. Fujiwara, S. Kayama, H. Hirakawa, H. Itaha, H. Ohge, N. Gotoh, T. Usui, A. Matsubara and M. Sugai, PLoS One, 2013, 8, e70557 CAS.
- T. Tada, T. Miyoshi-Akiyama, K. Shimada, M. Shimojima and T. Kirikae, Antimicrob. Agents Chemother., 2013, 57, 96–100 CrossRef CAS PubMed.
- T. Tada, T. Miyoshi-Akiyama, R. K. Dahal, S. K. Mishra, K. Shimada, H. Ohara, T. Kirikae and B. M. Pokhrel, Antimicrob. Agents Chemother., 2014, 58, 6324–6327 CrossRef CAS PubMed.
- W. Jin, J. I. Wachino, K. Kimura, K. Yamada and Y. Arakawa, J. Antimicrob. Chemother., 2015, 70, 1331–1337 CrossRef CAS PubMed.
- A. Yoshii, H. Moriyama and T. Fukuhara, Appl. Environ. Microbiol., 2012, 78, 5555–5564 CrossRef CAS PubMed.
- M. Galimand, J. Fishovitz, T. Lambert, V. Barbe, J. Zajicek, S. Mobashery and P. Courvalin, Antimicrob. Agents Chemother., 2015, 59, 5647–5653 CrossRef CAS PubMed.
- M. Toth, H. Frase, N. T. Antunes and S. B. Vakulenko, Antimicrob. Agents Chemother., 2013, 57, 452–457 CrossRef CAS PubMed.
- K. J. Labby and S. Garneau-Tsodikova, Future Med. Chem., 2013, 5, 1285–1309 CrossRef CAS PubMed.
- C. A. Smith, M. Toth, M. Bhattacharya, H. Frase and S. B. Vakulenko, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1561–1571 CAS.
- G. Cox, P. J. Stogios, A. Savchenko and G. D. Wright, mBio, 2015, 6, e02180-14 CrossRef PubMed.
- D. Iino, Y. Takakura, K. Fukano, Y. Sasaki, T. Hoshino, K. Ohsawa, A. Nakamura and S. Yajima, J. Struct. Biol., 2013, 183, 76–85 CrossRef CAS PubMed.
- V. R. Porter, K. D. Green, O. E. Zolova, J. L. Houghton and S. Garneau-Tsodikova, Biochem. Biophys. Res. Commun., 2010, 403, 85–90 CrossRef CAS PubMed.
- C. A. Smith, M. Toth, T. M. Weiss, H. Frase and S. B. Vakulenko, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 2754–2764 CAS.
- K. Shi, S. J. Caldwell, D. H. Fong and A. M. Berghuis, Front. Cell. Infect. Microbiol., 2013, 3, 22 Search PubMed.
- K. Vong and K. Auclair, Med. Chem. Commun., 2012, 3, 397–407 RSC.
- K. D. Green, W. Chen and S. Garneau-Tsodikova, ChemMedChem, 2012, 7, 73–77 CrossRef CAS PubMed.
- D. L. Lin, T. Tran, J. Y. Alam, S. R. Herron, M. S. Ramirez and M. E. Tolmasky, Antimicrob. Agents Chemother., 2014, 58, 4238–4241 CrossRef CAS PubMed.
- Y. Li, K. D. Green, B. R. Johnson and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2015, 59, 4148–4156 CrossRef CAS PubMed.
- M. Y. Fosso, H. Zhu, K. D. Green, S. Garneau-Tsodikova and K. Fredrick, ChemBioChem, 2015, 16, 1565–1570 CrossRef CAS PubMed.
- S. K. Shrestha, M. Y. Fosso, K. D. Green and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2015, 59, 4861–4869 CrossRef CAS PubMed.
- I. M. Herzog, K. D. Green, Y. Berkov-Zrihen, M. Feldman, R. R. Vidavski, A. Eldar-Boock, R. Satchi-Fainaro, A. Eldar, S. Garneau-Tsodikova and M. Fridman, Angew. Chem., Int. Ed., 2012, 51, 5652–5656 CrossRef CAS PubMed.
- P. Shaul, K. D. Green, R. Rutenberg, M. Kramer, Y. Berkov-Zrihen, E. Breiner-Goldstein, S. Garneau-Tsodikova and M. Fridman, Org. Biomol. Chem., 2011, 9, 4057–4063 CAS.
- K. D. Green, W. Chen, J. L. Houghton, M. Fridman and S. Garneau-Tsodikova, ChemBioChem, 2010, 11, 119–126 CrossRef CAS PubMed.
- Y. Berkov-Zrihen, I. M. Herzog, R. I. Benhamou, M. Feldman, K. B. Steinbuch, P. Shaul, S. Lerer, A. Eldar and M. Fridman, Chemistry, 2015, 21, 4340–4349 CrossRef CAS PubMed.
- Y. Berkov-Zrihen, I. M. Herzog, M. Feldman and M. Fridman, Org. Lett., 2013, 15, 6144–6147 CrossRef CAS PubMed.
- D. Watkins, S. Kumar, K. D. Green, D. P. Arya and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2015, 59, 3899–3905 CrossRef CAS PubMed.
- Y. Berkov-Zrihen, K. D. Green, K. J. Labby, M. Feldman, S. Garneau-Tsodikova and M. Fridman, J. Med. Chem., 2013, 56, 5613–5625 CrossRef CAS PubMed.
- A. King, D. Watkins, S. Kumar, N. Ranjan, C. Gong, J. Whitlock and D. P. Arya, Antimicrob. Agents Chemother., 2013, 57, 4717–4726 CrossRef CAS PubMed.
- J. P. Maianti, H. Kanazawa, P. Dozzo, R. D. Matias, L. A. Feeney, E. S. Armstrong, D. J. Hildebrandt, T. R. Kane, M. J. Gliedt, A. A. Goldblum, M. S. Linsell, J. B. Aggen, J. Kondo and S. Hanessian, ACS Chem. Biol., 2014, 9, 2067–2073 CrossRef CAS PubMed.
- H. Nikaido and J. M. Pages, FEMS Microbiol. Rev., 2012, 36, 340–363 CrossRef CAS PubMed.
- X. Z. Li, P. Plesiat and H. Nikaido, Clin. Microbiol. Rev., 2015, 28, 337–418 CrossRef PubMed.
- R. E. Hancock, S. W. Farmer, Z. S. Li and K. Poole, Antimicrob. Agents Chemother., 1991, 35, 1309–1314 CrossRef CAS.
- H. W. Taber, J. P. Mueller, P. F. Miller and A. S. Arrow, Microbiol. Mol. Biol. Rev., 1987, 51, 439–457 CAS.
- R. E. Hancock and A. Bell, Eur. J. Clin. Microbiol. Infect. Dis., 1988, 7, 713–720 CrossRef CAS.
- P. A. Lambert, J. R. Soc. Med., 2002, 95(Suppl 41), 22–26 CAS.
- E. L. Macfarlane, A. Kwasnicka and R. E. Hancock, Microbiology, 2000, 146(Pt 10), 2543–2554 CrossRef CAS PubMed.
- L. Fernandez, W. J. Gooderham, M. Bains, J. B. McPhee, I. Wiegand and R. E. Hancock, Antimicrob. Agents Chemother., 2010, 54, 3372–3382 CrossRef CAS PubMed.
- D. H. Kwon and C. D. Lu, Antimicrob. Agents Chemother., 2006, 50, 1615–1622 CrossRef CAS PubMed.
- J. S. Gunn, Trends Microbiol., 2008, 16, 284–290 CrossRef CAS PubMed.
- B. D. Needham and M. S. Trent, Nat. Rev. Microbiol., 2013, 11, 467–481 CrossRef CAS PubMed.
- E. M. Nowicki, J. P. O'Brien, J. S. Brodbelt and M. S. Trent, Mol. Microbiol., 2015, 97, 166–178 CrossRef CAS PubMed.
- R. Bansal-Mutalik and H. Nikaido, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4958–4963 CrossRef CAS PubMed.
- B. K. Ziervogel and B. Roux, Structure, 2013, 21, 76–87 CrossRef CAS PubMed.
- Y. Fei, V. Ma, N. Maerkl and P. You, J. Exp. Microbiol. Immunol., 2012, 16, 101–107 Search PubMed.
- J. P. Sarathy, V. Dartois and E. J. Lee, Pharmaceuticals, 2012, 5, 1210–1235 CrossRef CAS PubMed.
- M. Niederweis, Mol. Microbiol., 2003, 49, 1167–1177 CrossRef CAS.
- V. Jarlier and H. Nikaido, FEMS Microbiol. Lett., 1994, 123, 11–18 CrossRef CAS PubMed.
- O. Danilchanka, M. Pavlenok and M. Niederweis, Antimicrob. Agents Chemother., 2008, 52, 3127–3134 CrossRef CAS PubMed.
- M. Faller, M. Niederweis and G. E. Schulz, Science, 2004, 303, 1189–1192 CrossRef CAS PubMed.
- C. Raynaud, K. G. Papavinasasundaram, R. A. Speight, B. Springer, P. Sander, E. C. Bottger, M. J. Colston and P. Draper, Mol. Microbiol., 2002, 46, 191–201 CrossRef CAS.
- A. Siroy, C. Mailaender, D. Harder, S. Koerber, F. Wolschendorf, O. Danilchanka, Y. Wang, C. Heinz and M. Niederweis, J. Biol. Chem., 2008, 283, 17827–17837 CrossRef CAS PubMed.
- M. Niederweis, O. Danilchanka, J. Huff, C. Hoffmann and H. Engelhardt, Trends Microbiol., 2010, 18, 109–116 CrossRef CAS PubMed.
- F. Wolschendorf, D. Ackart, T. B. Shrestha, L. Hascall-Dove, S. Nolan, G. Lamichhane, Y. Wang, S. H. Bossmann, R. J. Basaraba and M. Niederweis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1621–1626 CrossRef CAS PubMed.
- O. Danilchanka, J. Sun, M. Pavlenok, C. Maueroder, A. Speer, A. Siroy, J. Marrero, C. Trujillo, D. L. Mayhew, K. S. Doornbos, L. E. Munoz, M. Herrmann, S. Ehrt, C. Berens and M. Niederweis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6750–6755 CrossRef CAS PubMed.
- O. Danilchanka, D. Pires, E. Anes and M. Niederweis, Antimicrob. Agents Chemother., 2015, 59, 2328–2336 CrossRef PubMed.
- K. Poole, Antimicrob. Agents Chemother., 2005, 49, 479–487 CrossRef CAS PubMed.
- K. Poole, in Antibiotic discovery and development, ed. T. J. Dougherty and M. J. Pucci, Springer Science Business Media, LLC, 2012, pp. 349–395 Search PubMed.
- H. Nikaido, Adv. Enzymol. Relat. Areas Mol. Biol., 2011, 77, 1–60 CAS.
- J. R. Aires and H. Nikaido, J. Bacteriol., 2005, 187, 1923–1929 CrossRef CAS PubMed.
- E. Y. Rosenberg, D. Ma and H. Nikaido, J. Bacteriol., 2000, 182, 1754–1756 CrossRef CAS.
- J. M. Blair, H. E. Smith, V. Ricci, A. J. Lawler, L. J. Thompson and L. J. Piddock, J. Antimicrob. Chemother., 2015, 70, 424–431 CrossRef CAS PubMed.
- S. Coyne, P. Courvalin and B. Perichon, Antimicrob. Agents Chemother., 2011, 55, 947–953 CrossRef CAS PubMed.
- Y. Morita, J. Tomida and Y. Kawamura, Front. Microbiol., 2012, 3, 408 Search PubMed.
- R. A. Moore, D. DeShazer, S. Reckseidler, A. Weissman and D. E. Woods, Antimicrob. Agents Chemother., 1999, 43, 465–470 CAS.
- A. Perez, M. Poza, A. Fernandez, C. Fernandez Mdel, S. Mallo, M. Merino, S. Rumbo-Feal, M. P. Cabral and G. Bou, Antimicrob. Agents Chemother., 2012, 56, 2084–2090 CrossRef CAS PubMed.
- D. Pletzer and H. Weingart, BMC Microbiol., 2014, 14, 13 CrossRef PubMed.
- Y. W. Huang, R. S. Liou, Y. T. Lin, H. H. Huang and T. C. Yang, PLoS One, 2014, 9, e111784 Search PubMed.
- Y. T. Lin, Y. W. Huang, S. J. Chen, C. W. Chang and T. C. Yang, Antimicrob. Agents Chemother., 2015, 59, 4067–4073 CrossRef CAS PubMed.
- X. Z. Li, K. Poole and H. Nikaido, Antimicrob. Agents Chemother., 2003, 47, 27–33 CrossRef CAS.
- G. J. Poelarends, P. Mazurkiewicz and W. N. Konings, Biochim. Biophys. Acta, 2002, 1555, 1–7 CrossRef CAS.
- K. Poole, J. Antimicrob. Chemother., 2005, 56, 20–51 CrossRef CAS PubMed.
- A. Begum, M. M. Rahman, W. Ogawa, T. Mizushima, T. Kuroda and T. Tsuchiya, Microbiol. Immunol., 2005, 49, 949–957 CrossRef CAS PubMed.
- M. A. Seeger, A. Schiefner, T. Eicher, F. Verrey, K. Diederichs and K. M. Pos, Science, 2006, 313, 1295–1298 CrossRef CAS PubMed.
- R. Nakashima, K. Sakurai, S. Yamasaki, K. Hayashi, C. Nagata, K. Hoshino, Y. Onodera, K. Nishino and A. Yamaguchi, Nature, 2013, 500, 102–106 CrossRef CAS PubMed.
- R. Nakashima, K. Sakurai, S. Yamasaki, K. Nishino and A. Yamaguchi, Nature, 2011, 480, 565–569 CAS.
- C. H. Lau, D. Hughes and K. Poole, mBio, 2014, 5, e01068 CrossRef PubMed.
- S. Fraud and K. Poole, Antimicrob. Agents Chemother., 2011, 55, 1068–1074 CrossRef CAS PubMed.
- K. Poole, Front. Microbiol., 2011, 2, 65 CAS.
- C. H. Lau, S. Fraud, M. Jones, S. N. Peterson and K. Poole, Antimicrob. Agents Chemother., 2013, 57, 2243–2251 CrossRef CAS PubMed.
- C. H. Lau, T. Krahn, C. Gilmour, E. Mullen and K. Poole, MicrobiologyOpen, 2015, 4, 121–135 CrossRef CAS PubMed.
- M. C. Swick, S. K. Morgan-Linnell, K. M. Carlson and L. Zechiedrich, Antimicrob. Agents Chemother., 2011, 55, 921–924 CrossRef CAS PubMed.
- E. De Rossi, J. A. Ainsa and G. Riccardi, FEMS Microbiol. Rev., 2006, 30, 36–52 CrossRef CAS PubMed.
- P. E. Silva, F. Bigi, M. P. Santangelo, M. I. Romano, C. Martin, A. Cataldi and J. A. Ainsa, Antimicrob. Agents Chemother., 2001, 45, 800–804 CrossRef CAS PubMed.
- M. Balganesh, N. Dinesh, S. Sharma, S. Kuruppath, A. V. Nair and U. Sharma, Antimicrob. Agents Chemother., 2012, 56, 2643–2651 CrossRef CAS PubMed.
- R. E. Lee, J. G. Hurdle, J. Liu, D. F. Bruhn, T. Matt, M. S. Scherman, P. K. Vaddady, Z. Zheng, J. Qi, R. Akbergenov, S. Das, D. B. Madhura, C. Rathi, A. Trivedi, C. Villellas, R. B. Lee, Rakesh, S. L. Waidyarachchi, D. Sun, M. R. McNeil, J. A. Ainsa, H. I. Boshoff, M. Gonzalez-Juarrero, B. Meibohm, E. C. Bottger and A. J. Lenaerts, Nat. Med., 2014, 20, 152–158 CrossRef CAS PubMed.
- T. Coelho, D. Machado, I. Couto, R. Maschmann, D. Ramos, A. von Groll, M. L. Rossetti, P. A. Silva and M. Viveiros, Front. Microbiol., 2015, 6, 330 Search PubMed.
- R. Iyer, A. Ferrari, R. Rijnbrand and A. L. Erwin, Antimicrob. Agents Chemother., 2015, 59, 2388–2397 CrossRef CAS PubMed.
- S. B. Levy, Soc. Appl. Microbiol. Symp. Ser., 2002, 31, 65S–71S CrossRef CAS.
- H. N. Tumah, J. Chemother., 2009, 92, 5–15 Search PubMed.
- M. Askoura, W. Mottawea, T. Abujamel and I. Taher, Libyan J. Med., 2011, 6 Search PubMed.
- G. P. Tegos, M. Haynes, J. J. Strouse, M. M. Khan, C. G. Bologa, T. I. Oprea and L. A. Sklar, Curr. Pharm. Des., 2011, 17, 1291–1302 CrossRef CAS.
- A. K. Bhardwaj and P. Mohanty, Recent Pat. Anti-Infect. Drug Discovery, 2012, 7, 73–89 CrossRef CAS.
- C. Kourtesi, A. R. Ball, Y. Y. Huang, S. M. Jachak, D. M. Vera, P. Khondkar, S. Gibbons, M. R. Hamblin and G. P. Tegos, Open Microbiol. J., 2013, 7, 34–52 CrossRef CAS PubMed.
- T. J. Opperman and S. T. Nguyen, Front. Microbiol., 2015, 6, 421 Search PubMed.
- H. Venter, R. Mowla, T. Ohene-Agyei and S. Ma, Front. Microbiol., 2015, 6, 377 Search PubMed.
- M. A. Kohanski, D. J. Dwyer, J. Wierzbowski, G. Cottarel and J. J. Collins, Cell, 2008, 135, 679–690 CrossRef CAS PubMed.
- A. Hinz, S. Lee, K. Jacoby and C. Manoil, J. Bacteriol., 2011, 193, 4790–4797 CrossRef CAS PubMed.
- T. Krahn, C. Gilmour, J. Tilak, S. Fraud, N. Kerr, C. H. Lau and K. Poole, Antimicrob. Agents Chemother., 2012, 56, 5591–5602 CrossRef CAS PubMed.
- K. N. Kindrachuk, L. Fernandez, M. Bains and R. E. Hancock, Antimicrob. Agents Chemother., 2011, 55, 1874–1882 CrossRef CAS PubMed.
- I. Sadovskaya, E. Vinogradov, J. Li, A. Hachani, K. Kowalska and A. Filloux, Glycobiology, 2010, 20, 895–904 CrossRef CAS PubMed.
- C. Nagant, M. Tre-Hardy, M. El-Ouaaliti, P. Savage, M. Devleeschouwer and J. P. Dehaye, Appl. Microbiol. Biotechnol., 2010, 88, 251–263 CrossRef CAS PubMed.
- P. S. Stewart, M. J. Franklin, K. S. Williamson, J. P. Folsom, L. Boegli and G. A. James, Antimicrob. Agents Chemother., 2015, 59, 3838–3847 CrossRef CAS PubMed.
- K. Poole, J. Antimicrob. Chemother., 2012, 67, 2069–2089 CrossRef CAS PubMed.
- F. El'Garch, K. Jeannot, D. Hocquet, C. Llanes-Barakat and P. Plesiat, Antimicrob. Agents Chemother., 2007, 51, 1016–1021 CrossRef PubMed.
- M. P. Mingeot-Leclercq, Y. Glupczynski and P. M. Tulkens, Antimicrob. Agents Chemother., 1999, 43, 727–737 CAS.
- M. Tod, C. Padoin and O. Petitjean, Clin. Pharmacokinet., 2000, 38, 205–223 CrossRef CAS PubMed.
- S. Maraki, G. Samonis, D. E. Karageorgopoulos, M. N. Mavros, D. Kofteridis and M. E. Falagas, Antimicrob. Agents Chemother., 2012, 56, 3067–3073 CrossRef CAS PubMed.
- I. Galani, M. Souli, G. L. Daikos, Z. Chrysouli, G. Poulakou, M. Psichogiou, T. Panagea, A. Argyropoulou, I. Stefanou, G. Plakias, H. Giamarellou and G. Petrikkos, J. Chemother., 2012, 24, 191–194 CrossRef CAS PubMed.
- R. Almaghrabi, C. J. Clancy, Y. Doi, B. Hao, L. Chen, R. K. Shields, E. G. Press, N. M. Iovine, B. M. Townsend, M. M. Wagener, B. Kreiswirth and M. H. Nguyen, Antimicrob. Agents Chemother., 2014, 58, 4443–4451 CrossRef PubMed.
- S. C. Olsen and S. A. Carlson, Int. J. Antimicrob. Agents, 2015, 45, 76–78 CrossRef CAS PubMed.
- C. Garcia-Salguero, I. Rodriguez-Avial, J. J. Picazo and E. Culebras, Antimicrob. Agents Chemother., 2015, 59, 5959–5966 CrossRef PubMed.
- J. B. Aggen, E. S. Armstrong, A. A. Goldblum, P. Dozzo, M. S. Linsell, M. J. Gliedt, D. J. Hildebrandt, L. A. Feeney, A. Kubo, R. D. Matias, S. Lopez, M. Gomez, K. B. Wlasichuk, R. Diokno, G. H. Miller and H. E. Moser, Antimicrob. Agents Chemother., 2010, 54, 4636–4642 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |