
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBisimidazoline arylamides binding to the DNA minor groove: N1-hydroxylation enhances binding affinity and selectivity to AATT sites†
Carlos H.
Ríos Martínez
ab,
Laura
Lagartera
a,
Cristina
Trujillo
 c and
Christophe
Dardonville
*a
c and
Christophe
Dardonville
*a
aInstituto de Química Médica, IQM–CSIC, Juan de la Cierva 3, 28006 Madrid, Spain. E-mail: dardonville@iqm.csic.esz
bINDICASAT-AIP, Edificio 219, Clayton, City of Knowledge, Ciudad de Panamá, República de Panamá
cSchool of Chemistry, Trinity Biomedical Sciences Institute, Trinity College Dublin, 152-160 Pearse St., Dublin 2, Ireland
First published on 8th October 2015
Abstract
Bisimidazoline arylamides and related compounds are high affinity DNA minor groove binders with a preference for AT over GC-rich DNA. However, further selectivity towards different classes of AT-sites (e.g., CGAATTCG, CATATATAT) is not always observed with these series. In this work, we wanted to understand the effect of imidazoline ring N-substitution on binding to DNA AT-sites. The structure–affinity relationships of a series of structurally related bisimidazoline compounds were studied by UV titrations and surface plasmon resonance (SPR) experiments using fish sperm DNA and different hairpin oligonucleotides. We found that in this series, the presence of N1–OH groups enhances the binding affinity to dsDNA CGAATTCG oligonucleotide, resulting in a higher selectivity for dsDNA containing AATT over (AT)4 sequences. The docking models showed that the N-hydroxy derivatives bind in a more planar conformation to the CGAATTCG DNA sequence, display more favorable van der Waals interactions, and show additional H-bonds with the bases and the sugar-phosphate backbone.
Introduction
Research efforts in the past few decades have led to a growing understanding of the DNA structure and drug–DNA interactions. Specific binding to DNA is thought to be achieved either by the formation of hydrogen bonds between the ligand and the base pairs of DNA or the recognition of a specific sequence-dependent shape of the DNA double helix.1 In AT-rich DNA, ligand-induced narrowing of the minor groove and changes in the bending of the DNA helix upon complex formation contribute to the binding of minor groove agents.2–4 Narrow minor grooves, often associated with the presence of A-tracts, strongly enhance the electrostatic potential of the DNA.5 These sequence-specific properties of DNA are used by many DNA-binding proteins1 and minor-groove-targeting compounds as a recognition mechanism. Hence, most of the minor-groove ligands share some structural characteristics (i.e., positive charge(s), linked rather than fused aromatic or heteroaromatic rings, crescent shape –“isohelicity”– matching the curve of the groove) that allow the ideal match between the ligand and the groove through van der Waals and hydrogen bonding (HB) interactions.6Minor groove binders are especially interesting DNA-interacting compounds for antimicrobial drug design because they are sequence selective (in contrast to intercalators which are sequence neutral), principally binding to A/T-rich DNA duplexes. This selectivity is particularly relevant in the case of parasitic pathogens such as trypanosomes whose mitochondrial genome contains a high proportion of A/T-rich DNA sequences.7 Due to their unique structural features,8 the A/T-rich minicircles of mitochondrial kinetoplast DNA (kDNA) appear to be the target for drug interaction.3,4,7
In previous studies, we have discovered diphenyl dicationic compounds 1–5 (Chart 1) that showed excellent in vivo activity against African trypanosomes (T. b. rhodesiense) in mouse models of sleeping sickness.9–12 The binding interaction of 5 with the minor groove of the all-AT DNA sequence d(AAAATTTT)2 and with the self-complimentary nucleotide d(CTTAATTCGAATTAAG)2 was demonstrated by X-ray crystallography.13,14 Nagle et al. showed that arylamide derivatives 1 and 2 bound strongly and selectively to AT oligonucleotides with a slight preference (2- to 3-fold) for dsDNA CGAATTCG vs. CATATATAT sequences.15 The same group observed that N-substitution of the guanidinium groups by hydroxyl radicals abolished almost completely the binding affinity of these ligands.16 Similarly, reduction in DNA binding affinity was observed with diamidine and bisguanidine analogues whose cationic moieties were derivatized with N-alkyl groups.10,17–19
 | ||
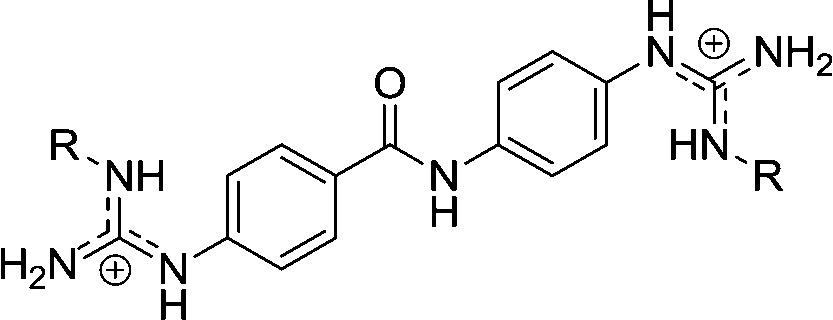
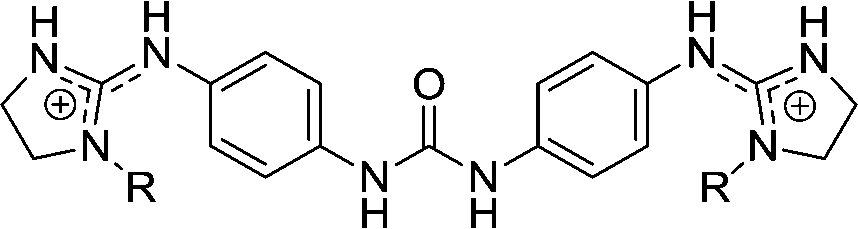
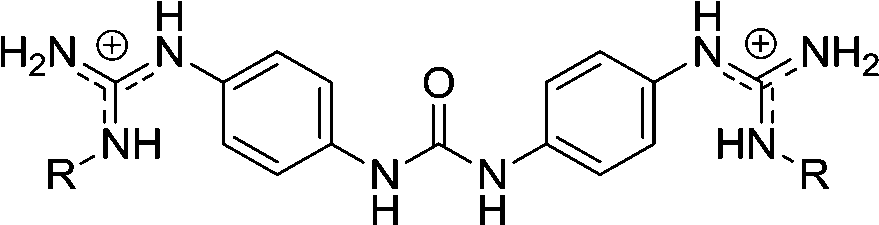
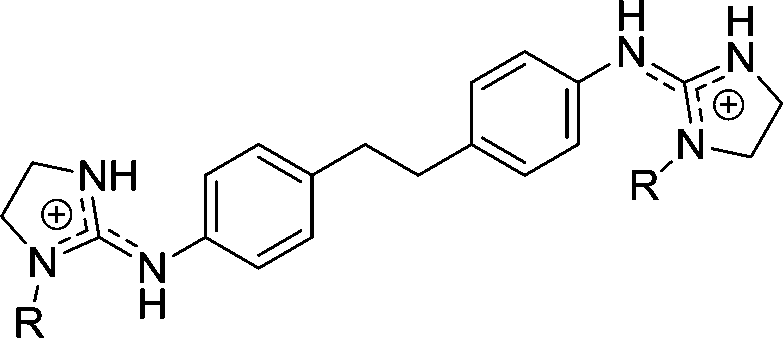
| Chart 1 Structures of the bisimidazoline arylamides (1, 1a, and 1b) and analogues (2–5) used in this study. | ||
The goal of the present study was to understand the effect of imidazoline ring N-substitution on arylamide binding to the minor groove of AT-rich DNA. As the here presented results together with many other studies of minor groove binders point to the involvement of shape-recognition by DNA, it was important to determine the selectivity of binding to different DNA sequences (i.e., DNA shapes). Hence, the binding of a series of ten closely related guanidine and imidazoline compounds (Chart 1) to unspecific fish-sperm DNA and dsDNA containing CGAATTCG, CATATATAT, and CGCGCGCG was studied by UV spectrophotometry and surface plasmon resonance (SPR)-biosensor experiments. Molecular docking studies using representative crystal structures of both dsDNA oligonucleotides CGAATTCG (pdb: 1ENN) and CATATATAT (pdb: 3TED) were carried out to rationalize the findings.
Results and discussion
Spectrophotometric titrations
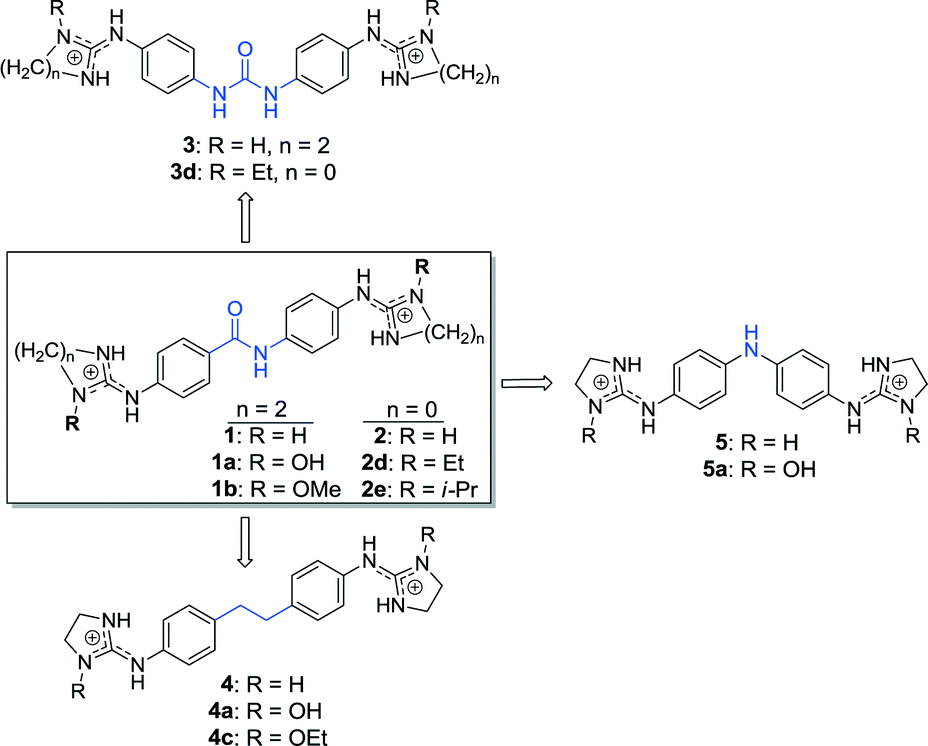
Compounds 1a and 1b have a strong absorption band at 280 nm, compounds 4a and 4c have a strong absorption band at <250 nm and a weak absorption in the 300–360 nm region, and 5a has a strong absorption band at 296 nm. These absorption spectra were strongly perturbed when the bisimidazolines formed a complex with unspecific fish-sperm (FS) DNA (Fig. 1). Compounds 1a and 1b as well as 4a and 4c exhibited isoabsorptive behaviours. For all of the compounds, a clear isosbestic point was observed during titration, indicating a single dominant mode of binding to FS DNA. A weak hypochromicity (9–34%) at the compound peak wavelength was observed upon binding, which is consistent with minor groove binding.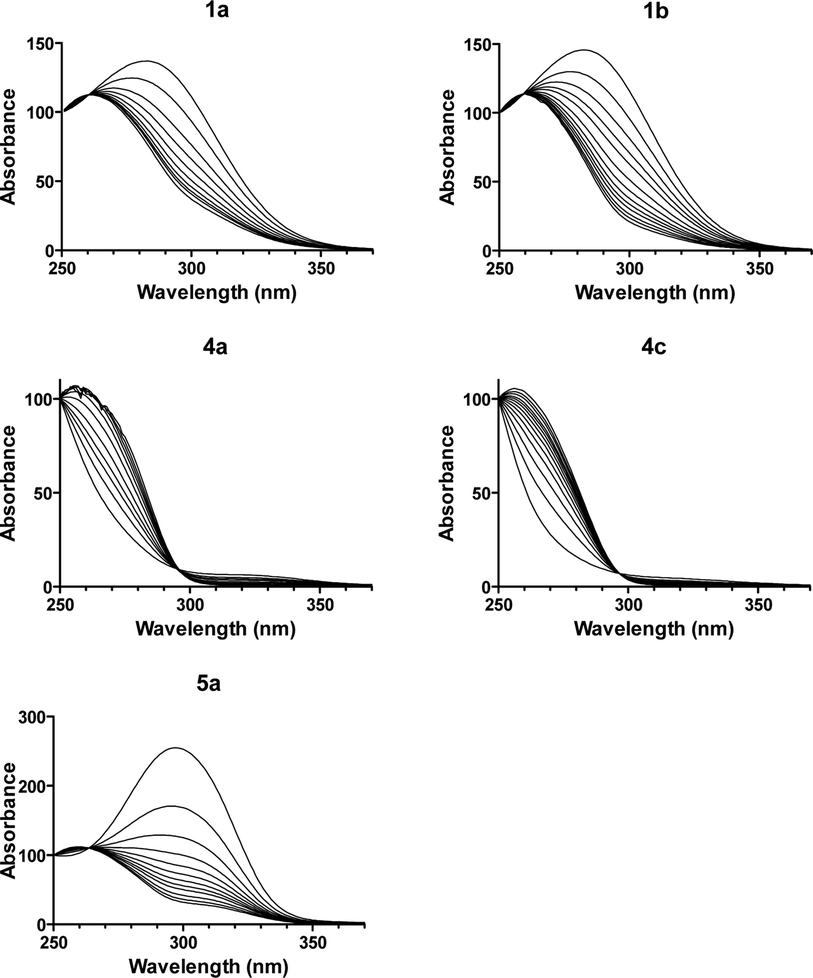 | ||
| Fig. 1 UV titration of 1a, 1b, 4a, 4c, and 5a (30 μM) with FS DNA in 10 mM phosphate buffer (pH 7) at 25 °C. FS-DNA concentrations ranged from 0 to 7.89 × 10−5 M (1a), 0 to 1.8 × 10−4 M (1b), 0 to 2.89 × 10−4 M (4a), 0 to 1.37 × 10−4 M (4c), and 0 to 1.16 × 10−4 M (5a) from top to bottom. | ||
The binding of 1a to the specific dsDNA CGAATTCG hairpin oligonucleotide produced strong alterations in the UV spectra (i.e. similar to that observed with FS DNA), whereas the binding to dsDNA CATATATAT and CGCGCGCG sequences induced much weaker changes (Fig. S1†). These results suggested that 1a binds more specifically to the dsDNA containing AATT. The absorption spectra were strongly perturbed when 5a formed a complex with dsDNA containing AATT (Fig. S1†). An isosbestic point at 325 nm indicated a single mode of interaction with the AATT hairpin. A new band attributed to the compound–DNA complex appeared at approximately 335 nm. Upon interaction with the (AT)4 hairpin, an isosbestic point at 335 nm was observed and a weak band attributed to the compound–DNA complex appeared at 340 nm. In contrast, no isosbestic point or new band was observed during titration with the (CG)4 sequence. This is consistent with the absence of strong interaction of 5a with CG-rich DNA.
SPR-biosensor experiments
The DNA binding affinity and stoichiometry of the compounds were determined using SPR-biosensor experiments with the three DNA hairpin duplexes [i.e., AATT, (AT)4, (CG)4] immobilized on a biosensor chip surface.20,21 The SPR response (RU) at equilibrium in the SPR sensorgrams (i.e., in the plateau region) was converted to r (moles of bound compound per mole of DNA hairpin duplex; r = RU/RUmax) and plotted against the free compound concentration, Cf, flowing on the chip surface (i.e., immobilized DNA hairpin) (Fig. 2). The binding constants were determined by fitting the values to single-site or two-site binding models according to eqn (1) (see the experimental part). When two binding sites exist for a given DNA sequence, only the primary binding constant is given. | ||
| Fig. 2 SPR binding affinity. (1) Sensorgrams for binding of 1a to CGAATTCG and CATATATAT hairpin duplexes using increasing concentrations of the ligand in the range 0.05–57.6 μM (from bottom to top). (2) SPR binding plots of 1a for AATT, (AT)4 and CGCGCGCG hairpins. | ||
Structure–affinity relationships
SPR experiments showed that none of the compounds bind significantly to the dsDNA containing (CG)4 sequences as expected for this class of minor groove binders. The bisimidazoline arylamide derivatives (1, 1a, and 1b) were the strongest DNA binders. However, different binding behaviors and affinities were observed depending on the spacer linking both the phenyl groups and the N1-substituent on the imidazoline rings (Table 1). The main SAR results are presented below.| Structure | Cmpd | R | dsDNA CGAATTCG | dsDNA CATATATAT | dsDNA CGCGCGCG |
|---|---|---|---|---|---|
| K D (×10−6M)b | K D (×10−6M)b | K D (×10−6 M) | |||
a dsDNA hairpins used in the study (the loop is underlined): 5′-biotin-CGAATTCG![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) CGAATTCG-3′ [short name: AATT], 5′-biotin-CATATATATATATATATG-3′ [short name: (AT)4], and 5′-biotin-CGCGCGCGCGCGCGCG-3′ [short name: (CG)4].
b Primary binding constant for fitting to a two-site binding model.
c Nagle et al.15 reported similar binding constants for AATT and (AT)4 oligonucleotides (0.107 × 10−6 M and 0.210 × 10−6 M, respectively).
d There is not enough signal-to-noise ratio to get a binding constant for this hairpin oligonucleotide.
e Non-specific binding.
f Taken from ref. 15.
g Taken from ref. 10.
h Taken from ref. 14. CGAATTCG-3′ [short name: AATT], 5′-biotin-CATATATATATATATATG-3′ [short name: (AT)4], and 5′-biotin-CGCGCGCGCGCGCGCG-3′ [short name: (CG)4].
b Primary binding constant for fitting to a two-site binding model.
c Nagle et al.15 reported similar binding constants for AATT and (AT)4 oligonucleotides (0.107 × 10−6 M and 0.210 × 10−6 M, respectively).
d There is not enough signal-to-noise ratio to get a binding constant for this hairpin oligonucleotide.
e Non-specific binding.
f Taken from ref. 15.
g Taken from ref. 10.
h Taken from ref. 14.
|
|||||
|
1 | H | 0.166c | 0.307c | >10de |
| 1a | OH | 0.077 | 0.273 | 18.5 | |
| 1b | OMe | 0.256 | 0.351 | >10d | |
|
2 | H | 0.141f | 0.556f | >10d |
| 2d | Et | 1.23g | 1.92g | >10e | |
| 2e | iPr | 1.02g | 2.5g | >10e | |
|
3 | H | 11.6 | 8.06 | 400–158.7 |
|
3d | Et | >29 | >29 | |
|
4 | H | 2.78 | 4.35 | 19.6 |
| 4a | OH | 0.870 | 4.0 | 76.9 | |
| 4c | OEt | >29 | >29e | >29 | |
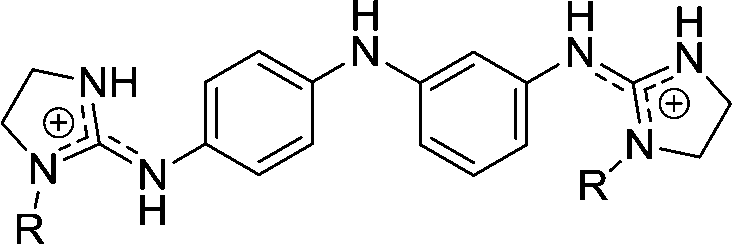
|
5 | H | 2.70 | 2.17h | |
| 5a | OH | 0.568 | 1.79 | 66.7 | |
Effect of linker modification: ethylene, urea and amino analogues
Bisimidazolines (1, 1a, and 1b) and bisguanidine (2, 2d, and 2e) arylamide derivatives showed the highest binding affinities for AT oligonucleotides with a slight preference for dsDNA containing AATT over (AT)4 sequences (1.5 to 2-fold). The binding of all of the compounds except for 3d to AT sequences was adjusted to a two-site binding model. The primary binding constants were about 100 times higher than the secondary binding constants. Compounds with a urea linker (3, 3d) displayed only weak affinity for DNA (KD > 8.06 × 10−6 M) with no sequence selectivity for AT-oligonucleotides and moderate selectivity towards CG for 3 (~10- to 50-fold). These results together with the ratio of moles of bound compound per mole of DNA hairpin duplex (r ≥ 2 for most sequences) are consistent with an intercalation mode of binding and/or external electrostatic interactions for the urea-derived compounds 3 and 3d.The binding affinities of the ethylene linked compounds (4, 4a, and 4c) were 10- to 15-fold weaker than those of the arylamide analogues (1a–b–2d–e), but 2- to 4-times stronger than those of the urea analogues (3, 3d). The primary binding of 4 and 4a to AT sequences was 5 to 10-fold and 30 to 50-fold stronger than the secondary binding, respectively. The amino-linked compounds 5 and 5a bind to dsDNA containing AATT more weakly than the arylamide analogues 1 and 1a (15- and 7-fold, respectively).
All together, these results clearly demonstrate the strong influence of the linker on the DNA binding mode and affinity, and that the arylamide scaffold (“amide linker”) is a privileged scaffold for AT-site DNA binding, independent of the cationic moieties present in the molecule.15
Effect of “imidazoline ↔ guanidine” group modification
Guanidines bind somewhat less strongly than imidazoline analogues. Substitution of the guanidine with alkyl groups (2d, 2e) decreased the binding affinity for AT sequences (6 to 9-fold).10 These results agree with previous studies showing that the bisimidazolines of this class bind more strongly and selectively to AT-rich DNA than their guanidine counterparts, probably due to more favorable van der Waals interactions of the imidazoline rings with the minor groove.11,12,14 Crystallographic studies of 5 bound to dsAT-DNA have shown that these favorable interactions also promote bifurcated hydrogen bonds between the imidazoline endocyclic nitrogens and thymine and adenine atoms in opposite DNA strands that facilitate the recognition of both strands of the DNA within the minor groove.13,14Effect of N1-substituents on AT-site selectivity
The most remarkable effect of the imidazoline N1 substituents was observed with hydroxyl groups. A two-fold increase in affinity towards dsDNA containing AATT was observed for 1b in comparison with the unsubstituted parent compound 1 (KAATT = 0.077 × 10−6 M and 0.166 × 10−6 M, respectively). In contrast, no change in binding affinity to the dsDNA CATATATAT sequence was observed. Thus, N1-hydroxylation resulted in a 3.5-fold selectivity enhancement for dsDNA containing AATT sequences over (AT)4.The same results were observed for the ethylene-linked N-hydroxy derivative 4a (KAATT = 0.87 × 10−6 M vs. 2.78 × 10−6 M for the unsubstituted parent compound 4) and the amino-linked compound 5a (KAATT = 0.568 × 10−6 M vs. 2.70 × 10−6 M for the parent compound 5) showing that this effect may possibly be generalised to other bisimidazolines. On the contrary, the introduction of alkoxy groups (OMe, OEt) was detrimental to the binding to AT-rich DNA, indicating that the OH group of N-hydroxyimidazolines is probably involved in additional stabilization of H-bond interactions with the DNA minor groove.
Noteworthy is the binding behaviour of the N-hydroxy derivative 5a. The kinetics of binding to AT sequences is rather different from the rest of the compounds with a slow dissociation process (Fig. 3). Since the stoichiometry (r value) is > 2, the results suggest a different, more complex, binding mode to AT sequences. We recently made similar observations with a 39 bp dsDNA containing GAATAATCGCGATTATTC which formed a slow-binding (kon = 38 M−1 s−1) long-lasting (koff = 0.00265 s−1) complex with 5a (Dr. L. Campos, personal communication). This result is important as longer drug–target residence time is considered a key driving force for the pharmacodynamic activity and efficacy of many drugs in vivo.22–25
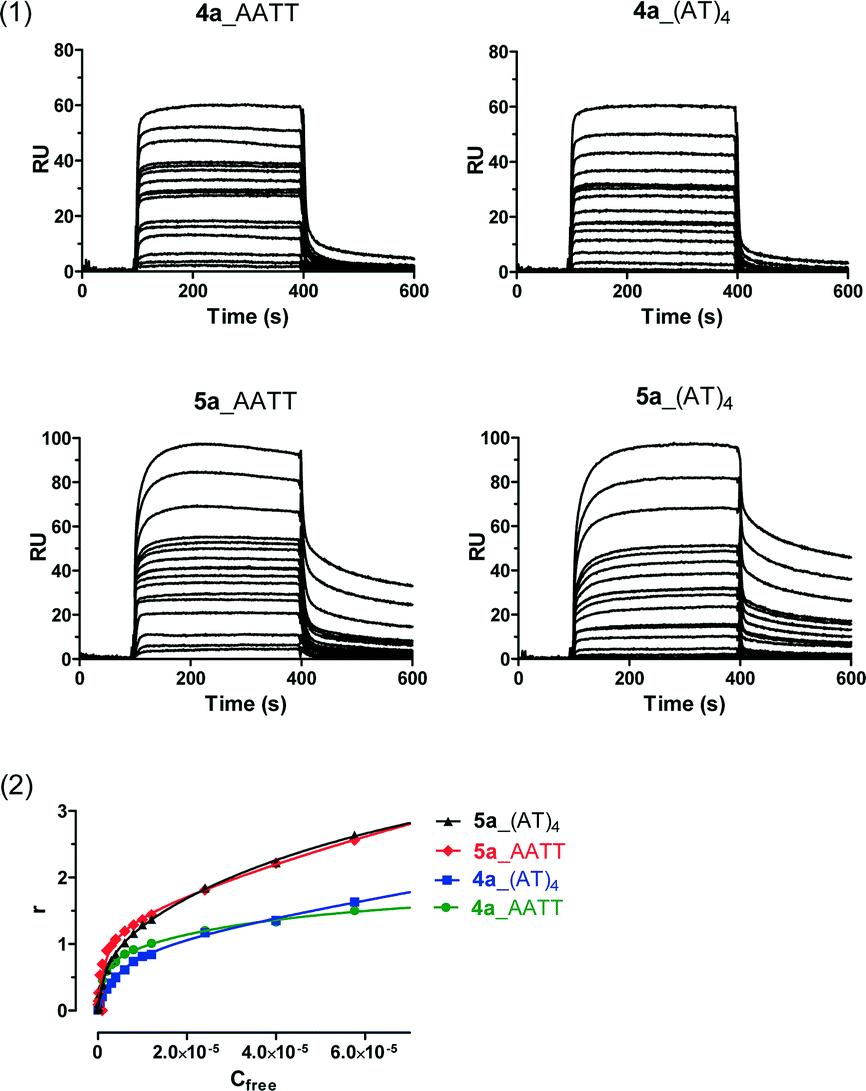 | ||
| Fig. 3 (1) Sensorgrams for binding of 4a and 5a to CGAATTCG and CATATATAT hairpin duplexes using increasing concentrations of the ligand in the range 0.05–57.6 μM (from bottom to top). For 5a, the shape of the sensorgram clearly shows a different profile with a slow binding and dissociation process. (2) SPR binding plots of 4a and 5a for AATT and (AT)4 hairpins. | ||
Molecular docking studies
In order to understand the reasons behind the increased affinity of the N-hydroxy derivatives, docking experiments were run using the crystal structures of d(GCGAATTCG) [1ENN]26 and d(CCATATATATGC) [3TED]27 as templates for the dsDNA containing AATT and (AT)4 sequences, respectively. The docking experiments were able to rank correctly (i.e. in agreement with the experimental values) the ligands into two groups of high (1a > 5a > 1 ≈ 5 ≈ 4a > 4) and low (3 > 4c > 3d) predicted binding affinity, respectively (Table S1†). However, the differences in binding affinities were too small (i.e. within the limits of the standard error of the calculation) and did not allow us to extract useful conclusions in relation to the increased affinity of the N-hydroxy derivatives for the dsDNA containing AATT. Hence, a detailed study of the interactions of lead 1 and its N-hydroxy analogue 1a with both AATT and (AT)4 sequences was carried out.In contrast to the dsDNA containing CATATATAT where the ligands bind in a more twisted conformation, the phenyl rings of 1 and 1a adopt a more coplanar conformation when bound to the dsDNA CGAATTCG (Fig. S3–S6†). These results agree well with the induced-fit observed with this kind of ligand when bound to a narrow minor groove.14 An analysis of the non-covalent interactions28 in the ligand–DNA complexes showed that 1 and 1a form more (and larger) weak attractive interactions with the dsDNA containing CGAATTCG compared to that containing CATATATAT (Fig. S7†), which is consistent with the values of binding affinities measured experimentally.
Finally, the analysis of the HB interactions in the complexes of 1 and 1a with CGAATTCG and CATATATAT oligonucleotides showed distinctive patterns depending on the presence or absence of the N–OH group (Fig. 4 and S8†). For instance, 1 forms two HBs (2.19 and 2.50 Å) with the phosphate groups of the sugar-phosphate backbone and 1 HB (2.35 Å) with the O4′ atom of the A5 deoxyribose of the dsDNA GCGAATTCG (Fig. S8_A†).
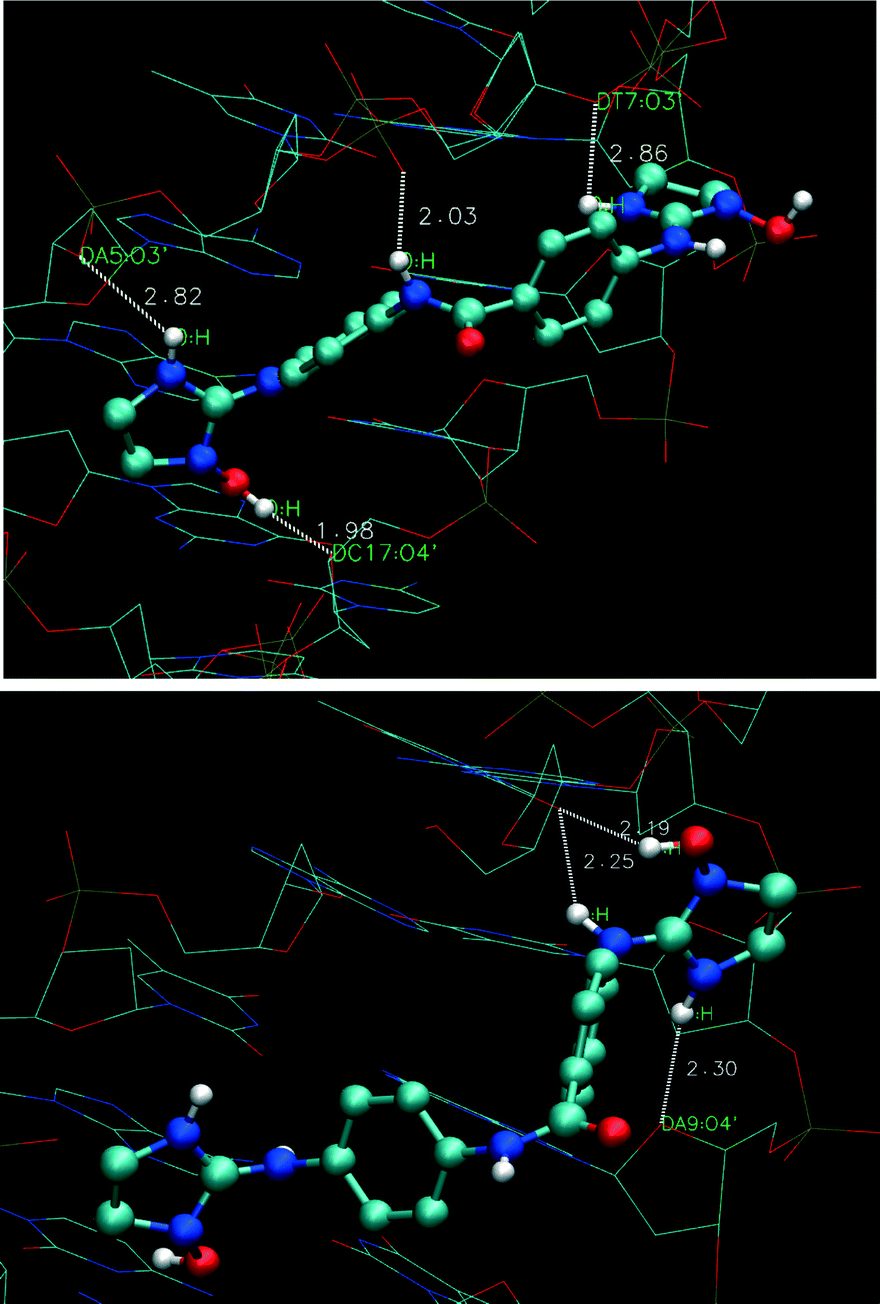 | ||
| Fig. 4 Plot of hydrogen bond (HB) interactions for 1a docked with d(GCGAATTCG) (top) and d(CCATATATATGC) (bottom) oligonucleotides. | ||
In contrast, 1a forms a strong HB between the OH group and the O4′ atom of the C17 deoxyribose (Fig. 4, 1.98 Å) and three HBs (2.03, 2.82, and 2.86 Å) with the phosphate groups of the sugar-phosphate backbone (i.e. with the amide NH and the imidazoline NH from both sides of the molecule, respectively). With the d(CCATATATATGC) oligonucleotide, 1a forms 1 HB (2.30 Å) with the O4′ atom of the A9 deoxyribose and a bifurcated HB between thymine T6 and imidazoline N(2)H (2.25 Å) and N(1)OH (2.19 Å) (Fig. 4) which is absent in the complex with compound 1 (Fig. S8_C†).
As a whole, the docking studies indicate that the better fit of the N-hydroxy derivatives in the narrow minor groove of the dsDNA containing AATT may be due to favorable van der Waals interactions with the walls of the groove, a more planar conformation of the bound ligand, and additional stabilizing H-bonds between the N–OH groups and sugar residues and/or the base edges.
Conclusions
We have shown that N-substituted diphenyl-based bisimidazoline compounds bind strongly to dsDNA, preferentially to A/T containing oligonucleotides. The presence of one hydroxyl substituent on each imidazoline endocyclic nitrogen (N1–OH) results in an increase in binding affinity for the dsDNA containing AATT sequences and no change in affinity for the dsDNA containing (AT)4. This is translated into an increase in sequence selectivity for the dsDNA CGAATTCG (3.5-fold). This effect, which seems to be general for this family of imidazoline compounds (e.g., 1a, 4a, and 5a), differs from that observed with N-hydroxy guanidine analogues which were found to bind poorly to DNA.16 Since the dsDNA sequence CGAATTCG has a very narrow minor groove of 3.5–4.0 Å in the center of the sequence (vs. 5.16–6.79 Å for ATAT) and a concomitant higher electrostatic potential,5 the selective binding of the N-hydroxy bisimidazolines seems to be derived from a tighter fit to this narrower groove.Remarkably, in the case of 5a, the introduction of OH substituents increases the affinity for dsDNA CGAATTCG but also results in a different stoichiometry (i.e. drug/DNA, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vs. 1:1 for 5) and an increased residence time on the DNA. Because the longer half-life of the drug–target complex will minimize the binding to off-target proteins, this kinetic selectivity may improve the therapeutic index of 5a.23,24 Hence, further studies on this compound are warranted.29
:1 vs. 1:1 for 5) and an increased residence time on the DNA. Because the longer half-life of the drug–target complex will minimize the binding to off-target proteins, this kinetic selectivity may improve the therapeutic index of 5a.23,24 Hence, further studies on this compound are warranted.29
Experimental
Syntheses of compounds 1,111a,91b,92,112d,102e,103,113d,94,304a,94c,95,31 and 5a32 have been described previously.UV titrations
The UV-vis spectra were measured on a Perkin-Elmer Lambda 35 UV–vis spectrophotometer in a 1.5 mL quartz cuvette (1 cm pathlength) in 10 mM phosphate buffer (pH 7) previously degassed by sonication. A stock solution of FS-DNA in 5 mL of phosphate buffer was prepared and shaken gently for 1 h. The concentration of the FS-DNA stock solution (C = 780 μM) was worked out from the following equation: Abs260 = 100 × C × d × ε260 using an extinction coefficient of 12800 M(bp)−1 cm−1 at 260 nm for FS-DNA.33
The DNA oligonucleotides 5′-CGAATTCGTCTCCGAATTCG-3′, 5′-CATATATATCCCCATATATATG-3′ and 5′-CGCGCGCGTTTTCGCGCGCG-3′ were purchased from Eurofins MWG Operon with HPLC purification. They were stored at −20 °C as 100 μM stock solutions in phosphate buffer (pH 7) containing 1 mM EDTA + 100 mM NaCl until use. Before titration experiments, the oligonucleotides were diluted 10× with phosphate buffer (pH 7) + 100 mM NaCl, heated at 90 °C in a water bath for 10 minutes, and immediately chilled with ice to favour hairpin formation.
Stock solutions of compounds 1a, 1b, 4a, 4c, and 5a (C = 30 μM and C = 2.5 μM in phosphate buffer) were prepared from 1 mM stock solutions in DMSO by dilution with 10 mM phosphate buffer. The final amount of DMSO in the stock solution was 3%. Spectrophotometric titrations were performed by sequential addition of aliquots of the FS DNA solution (C = 780 μM) to 800 μL of the compound (C = 30 μM) or hairpin oligonucleotide (C = 10 μM) to 500 μL of the compound (C = 2.5 μM) until saturation was observed. The experiments were performed at 25 °C. The spectra were normalized and plotted using GraphPad (Prism).
SPR studies
SPR experiments were performed at 25 °C with a Biacore X-100 apparatus (GE Healthcare, Biacore AB, Uppsala, Sweden) in a MES buffer (10 mM 2-(N-morpholino)ethanesulfonic acid, 1 mM EDTA, 100 mM NaCl, 0.005% surfactant P20, pH 6.25). The 5′-biotin labeled DNA hairpins 5′-CGAATTCGTCTCCGAATTCG-3′, 5′-CATATATATCCCCATATATATG-3′, and 5′-CGCGCGCGTTTTCGCGCGCG-3′ were purchased from Sigma-Aldrich with HPLC purification, dissolved in the experimental buffer and used as such. SPR measurements were carried out as described.10The number of binding sites and the binding constants at equilibrium were obtained from fitting plots of r (r = RU/RUmax) against Cf. The maximum expected response (RUmax) per bound compound at equilibrium was calculated using a refractive index value of 1.4 as reported for similar compounds.34 The results of the binding constants were obtained by fitting the SPR results to a one-site (K2 = 0) or a two-site binding model according to eqn (1):
| r = (K1Cf + 2K1K2Cf2)/(1 + K1Cf + K1K2Cf2) | (1) |
Computational details
All compounds have been optimized using the Gaussian0936 package at the B3LYP37,38 computational level with the 6-311++G(d,p)39 basis sets. The effect of water solvation was then accounted using the SCFR-PCM approach implemented in the Gaussian09 package including the dispersion, repulsion and cavitation energy terms of the solvent in the optimization.A molecular docking study was undertaken using the AutoDock Vina 1.1.2 modelling software.40 The compounds were docked into representative crystal structures of dsDNA containing CGAATTCG [i.e. d(GCGAATTCG)2: pdb 1ENN]26 and CATATATAT [i.e., CCATATATATGC, pdb 3TED].27 The structures were imported into the AutoDock Vina 1.1.2 modelling software and all crystallographic water molecules and other small molecules were removed as they were located away from the ligand-binding regions. AutoDockTools 1.5.6 was used for establishing the Autogrid points as well as for visualization of docked ligand–nucleic acid structures. The target site on the nucleic acid was specified to encompass the entire minor groove site. The grid center was also established by centering the grid box on the minor groove site. The Non-Covalent Interaction (NCI) index, based on the reduced gradient of the electron density, has been calculated to identify attractive and repulsive interactions using the program NCI28 and plotted using the program VMD.41
Acknowledgements
C. R. was a recipient of a PhD fellowship from the government of Panama (SENACYT grant BIDP-2008-030). The assistance of Silvia Soto Alvarez with the SPR experiments and Eden Gebreselassie with the UV experiments is gratefully acknowledged. We thank Dr. N. Jagerovic, Dr. P. Goya, and Dr. Alkorta for logistical collaboration. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- R. Rohs, S. M. West, A. Sosinsky, P. Liu, R. S. Mann and B. Honig, Nature, 2009, 461, 1248–1253 CrossRef CAS PubMed.
- M. Rettig, M. W. Germann, S. Wang and W. D. Wilson, ChemBioChem, 2013, 14, 323–331 CrossRef CAS PubMed.
- D. S. Tevis, A. Kumar, C. E. Stephens, D. W. Boykin and W. D. Wilson, Nucleic Acids Res., 2009, 37, 5550–5558 CrossRef CAS PubMed.
- R. A. Hunt, M. Munde, A. Kumar, M. A. Ismail, A. A. Farahat, R. K. Arafa, M. Say, A. Batista-Parra, D. Tevis, D. W. Boykin and W. D. Wilson, Nucleic Acids Res., 2011, 39, 4265–4274 CrossRef CAS PubMed.
- E. P. Bishop, R. Rohs, S. C. J. Parker, S. M. West, P. Liu, R. S. Mann, B. Honig and T. D. Tullius, ACS Chem. Biol., 2011, 6, 1314–1320 CrossRef CAS PubMed.
- R. Tidwell and D. Boykin, in Small molecule DNA and RNA binder: synthesis to nucleic acid complexes, Wiley-VCH, New York, USA, 2003, pp. 416–460 Search PubMed.
- W. D. Wilson, F. A. Tanious, A. Mathis, D. Tevis, J. E. Hall and D. W. Boykin, Biochimie, 2008, 90, 999–1014 CrossRef CAS PubMed.
- R. E. Jensen and P. T. Englund, Annu. Rev. Microbiol., 2012, 66, 473–491 CrossRef CAS PubMed.
- C. H. Ríos Martínez, F. Miller, K. Ganeshamoorthy, F. Glacial, M. Kaiser, H. de Koning, A. Eze, L. Lagartera, T. Herraiz and C. Dardonville, Antimicrob. Agents Chemother., 2015, 59, 890–904 CrossRef PubMed.
- C. H. Ríos Martínez, L. Lagartera, M. Kaiser and C. Dardonville, Eur. J. Med. Chem., 2014, 81, 481–491 CrossRef PubMed.
- F. Rodríguez, I. Rozas, M. Kaiser, R. Brun, B. Nguyen, W. D. Wilson, R. N. García and C. Dardonville, J. Med. Chem., 2008, 51, 909–923 CrossRef PubMed.
- C. Dardonville, M. P. Barrett, R. Brun, M. Kaiser, F. Tanious and W. D. Wilson, J. Med. Chem., 2006, 49, 3748–3752 CrossRef CAS PubMed.
- F. J. Acosta-Reyes, C. Dardonville, H. P. de Koning, M. Natto, J. A. Subirana and J. L. Campos, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1614–1621 CAS.
- L. S. Glass, B. Nguyen, K. D. Goodwin, C. Dardonville, W. D. Wilson, E. C. Long and M. M. Georgiadis, Biochemistry, 2009, 48, 5943–5952 CrossRef CAS PubMed.
- P. S. Nagle, F. Rodríguez, B. Nguyen, W. D. Wilson and I. Rozas, J. Med. Chem., 2012, 55, 4397–4406 CrossRef CAS PubMed.
- A. Kahvedžić, S.-M. Nathwani, D. M. Zisterer and I. Rozas, J. Med. Chem., 2013, 56, 451–459 CrossRef PubMed.
- R. K. Arafa, M. A. Ismail, M. Munde, W. D. Wilson, T. Wenzler, R. Brun and D. W. Boykin, Eur. J. Med. Chem., 2008, 43, 2901–2908 CrossRef CAS PubMed.
- J. L. Gonzalez, C. E. Stephens, T. Wenzler, R. Brun, F. A. Tanious, W. D. Wilson, T. Barszcz, K. A. Werbovetz and D. W. Boykin, Eur. J. Med. Chem., 2007, 42, 552–557 CrossRef CAS PubMed.
- R. K. Arafa, R. Brun, T. Wenzler, F. A. Tanious, W. D. Wilson, C. E. Stephens and D. W. Boykin, J. Med. Chem., 2005, 48, 5480–5488 CrossRef CAS PubMed.
- B. Nguyen, F. A. Tanious and W. D. Wilson, Methods, 2007, 42, 150–161 CrossRef CAS PubMed.
- W. D. Wilson, Science, 2002, 295, 2103–2105 CrossRef CAS PubMed.
- R. A. Copeland, Expert Opin. Drug Discovery, 2010, 5, 305–310 CrossRef CAS PubMed.
- R. A. Copeland, D. L. Pompliano and T. D. Meek, Nat. Rev. Drug Discovery, 2006, 5, 730–739 CrossRef CAS PubMed.
- G. K. Walkup, Z. You, P. L. Ross, E. K. H. Allen, F. Daryaee, M. R. Hale, J. O'Donnell, D. E. Ehmann, V. J. A. Schuck, E. T. Buurman, A. L. Choy, L. Hajec, K. Murphy-Benenato, V. Marone, S. A. Patey, L. A. Grosser, M. Johnstone, S. G. Walker, P. J. Tonge and S. L. Fisher, Nat. Chem. Biol., 2015, 11, 416–423 CrossRef CAS PubMed.
- J. M. Bradshaw, J. M. McFarland, V. O. Paavilainen, A. Bisconte, D. Tam, V. T. Phan, S. Romanov, D. Finkle, J. Shu, V. Patel, T. Ton, X. Li, D. G. Loughhead, P. A. Nunn, D. E. Karr, M. E. Gerritsen, J. O. Funk, T. D. Owens, E. Verner, K. A. Brameld, R. J. Hill, D. M. Goldstein and J. Taunton, Nat. Chem. Biol., 2015, 11, 525–531 CrossRef CAS PubMed.
- M. Soler-López, L. Malinina and J. A. Subirana, J. Biol. Chem., 2000, 275, 23034–23044 CrossRef PubMed.
- A. Sharma, K. R. Jenkins, A. Héroux and G. D. Bowman, J. Biol. Chem., 2011, 286, 42099–42104 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- The crystal structure of 5a bound to the DNA duplex (AAATTT)2 is currently being solved (Dr. L. Campos and collaborators). This structure will be reported elsewhere..
- F. Rodríguez, I. Rozas, J. E. Ortega, A. M. Erdozain, J. J. Meana and L. F. Callado, J. Med. Chem., 2008, 51, 3304–3312 CrossRef PubMed.
- C. Dardonville, P. Goya, I. Rozas, A. Alsasua, M. I. Martin and M. J. Borrego, Bioorg. Med. Chem., 2000, 8, 1567–1577 CrossRef CAS.
- L. Nieto, A. Mascaraque, F. Miller, F. Glacial, C. Ríos Martínez, M. Kaiser, R. Brun and C. Dardonville, J. Med. Chem., 2011, 54, 485–494 CrossRef CAS PubMed.
- L. A. Mullice, R. H. Laye, L. P. Harding, N. J. Buurma and S. J. A. Pope, New J. Chem., 2008, 32, 2140–2149 RSC.
- S. Mazur, F. A. Tanious, D. Ding, A. Kumar, D. W. Boykin, I. J. Simpson, S. Neidle and W. D. Wilson, J. Mol. Biol., 2000, 300, 321–337 CrossRef CAS PubMed.
- Y. Liu, Y. Chai, A. Kumar, R. R. Tidwell, D. W. Boykin and W. D. Wilson, J. Am. Chem. Soc., 2012, 134, 5290–5299 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. A. J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectrophotometric titration with AATT, (AT)4, and (CG)4 hairpin oligonucleotides (Fig. S1); docking studies: Table S1 and Fig. S2–S8. See DOI: 10.1039/c5md00292c |
| This journal is © The Royal Society of Chemistry 2015 |