DOI:
10.1039/C5MD00242G
(Concise Article)
Med. Chem. Commun., 2015,
6, 1767-1772
Discovery of pyridyl-based inhibitors of Plasmodium falciparum N-myristoyltransferase†
Received
6th June 2015
, Accepted 4th August 2015
First published on 17th August 2015
Abstract
N-Myristoyltransferase (NMT) represents an attractive drug target in parasitic infections such as malaria due to its genetic essentiality and amenability to inhibition by drug-like small molecules. Scaffold simplification from previously reported inhibitors containing bicyclic cores identified phenyl derivative 3, providing a versatile platform to study the effects of substitution on the scaffold, which yielded pyridyl 19. This molecule exhibited improved enzyme and cellular potency, and reduced lipophilicity compared to inhibitor 3. Further structure-based inhibitor design led to the discovery of 30, the most potent inhibitor in this series, which showed single-digit nM enzyme affinity and sub-μM anti-plasmodial activity.
Introduction
Malaria, a parasitic disease in humans primarily caused by Plasmodium falciparum (Pf) and Plasmodium vivax (Pv), was responsible for about 584![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in 2013, mainly of pregnant women and children living in Africa.1 Recent vaccine trials have met with moderate success.2,3 However, the first signs of resistance to artemisinin, currently the first-line antimalarial treatment, have emerged in Southeast Asia.4–6 It is therefore important to develop new antimalarial medications with novel modes of action.7
000 deaths in 2013, mainly of pregnant women and children living in Africa.1 Recent vaccine trials have met with moderate success.2,3 However, the first signs of resistance to artemisinin, currently the first-line antimalarial treatment, have emerged in Southeast Asia.4–6 It is therefore important to develop new antimalarial medications with novel modes of action.7
N-Myristoyltransferase (NMT) is an enzyme ubiquitous in eukaryotes, catalysing co- or post-translational transfer of myristate (C14:0 fatty acid) from myristoyl-coenzyme A (myr-CoA) to the N-terminal glycine of a subset of eukaryotic proteins.8–12 In Plasmodium, numerous essential proteins have been shown to require myristoylation to fulfil their biological functions.13–18 The essentiality and druggability of NMT has been demonstrated in blood stage P. falciparum,19 and in the closely related rodent pathogen P. berghei.20 We recently reported the discovery of several novel series of small molecule parasitic NMT inhibitors,19,21–24 in addition to peptidomimetic inhibitors.25 To date, development of these series has focused on inhibitors containing bicyclic aromatic cores.26–28 In two complementary studies, we report the design and development of a simplified single ring scaffold, which provides a more flexible platform to study substitution patterns on the aromatic core and has enabled targeting of both plasmodial and leishmanial NMTs.29 Crystal structures of these inhibitors in complex with NMT enabled Structure-Activity Relationships (SARs) to be derived; this report describes the discovery of a new highly potent P. falciparum NMT (PfNMT) inhibitor that is active against parasites, and with promising selectivity relative to human NMTs and human cell lines.24
Results and discussion
Following the discovery that single-ring scaffolds displayed good affinity against Leishmania donovani NMT,29 disubstituted (ortho- and meta-) phenyl rings were selected as analogues of the structures of previously reported inhibitors 1 and 2,27,28 and linked through an ester or its bioisosteric oxadiazole to a terminal benzyl substituent (Fig. 1). Among the four different combinations (Table S1, ESI†), compound 3 was found to display comparable enzyme potency and human selectivity to the bicyclic equivalents (Fig. 1).
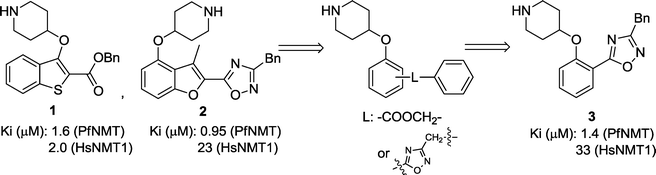 |
| | Fig. 1 Scaffold hopping from previously described inhibitors 1 and 2 containing a bicyclic aromatic core27,28 to identify phenyl 3 as a starting compound. | |
Variation of the single ring core
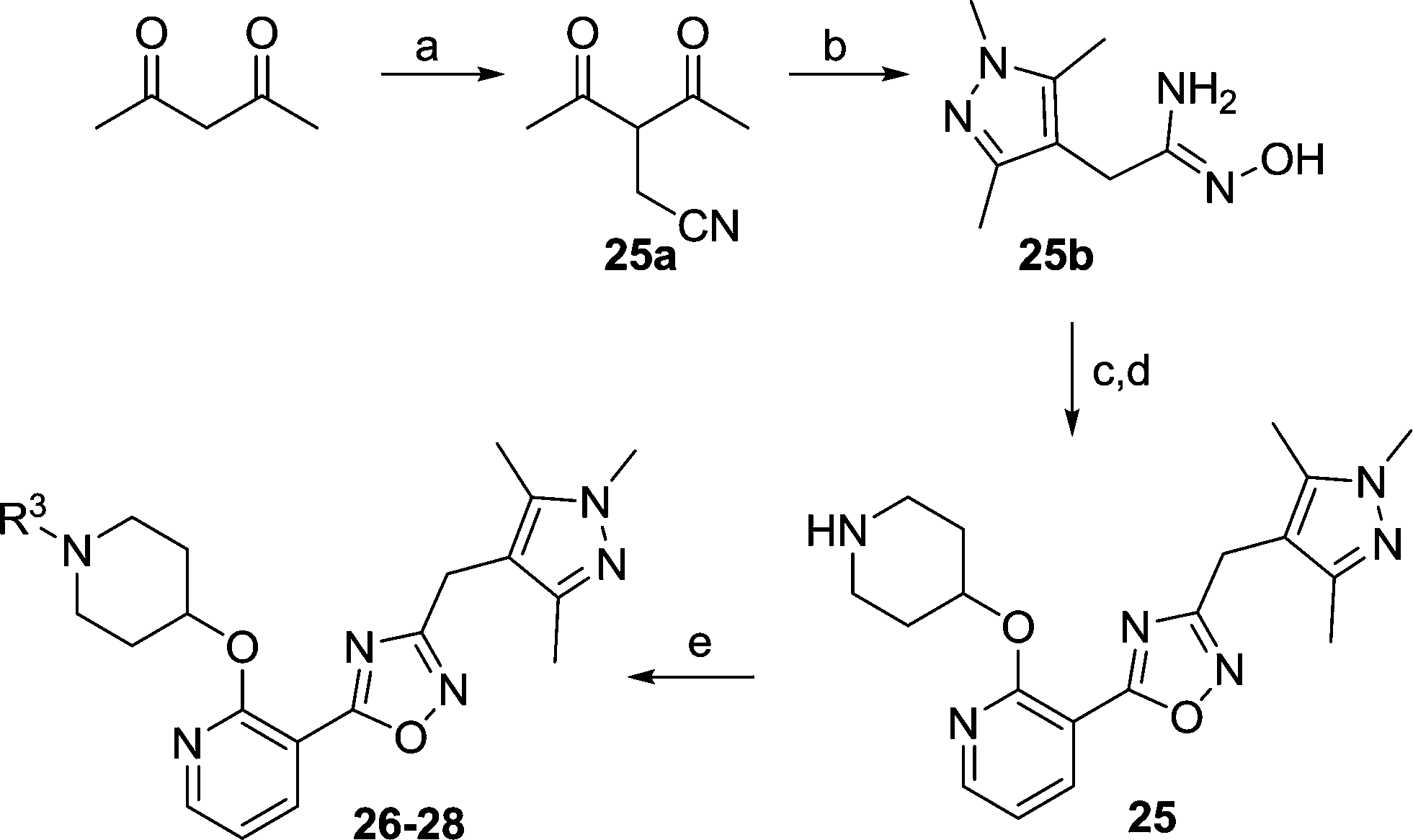
A library of compounds (4–17) with varied substitutions on the single ring scaffold was synthesised (Scheme 1). Initial ester protection of salicylic acid analogues enabled the incorporation of an N-Boc piperidinol side chain into the scaffold via a Mitsunobu reaction, followed by hydrolysis of the ester to form the corresponding carboxylic acid intermediate (4b–17b). The final 1,2,4-oxadiazole compounds were then synthesized using a modification of previously-reported conditions.28
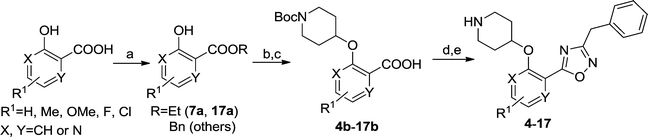 |
| | Scheme 1 Reagents: (a) BnBr, K2CO3, DMF, rt, 2 h, 86%; as for 7a, i). SOCl2, 90 °C, 1 h; ii) EtOH, rt, 1 h, 95%; as for 17a, H2SO4, EtOH/benzene, 80 °C, 2 h, 70%; (b) DIAD, PPh3, t-butyl 4-hydroxypiperidine-1-carboxylate, THF, rt, 4 h; (c)NaOH, MeOH/H2O, 50 °C, 2 h, quantitative over two steps; (d) i). benzylamidoxime, EDCI, HOBt, DIPEA, CH3CN, rt, 4 h; ii). 0.5 N NaOH, rt, 0.5 h, 90%; (e) 10% TFA in DCM (v/v), rt, 2 h, quantitative. | |
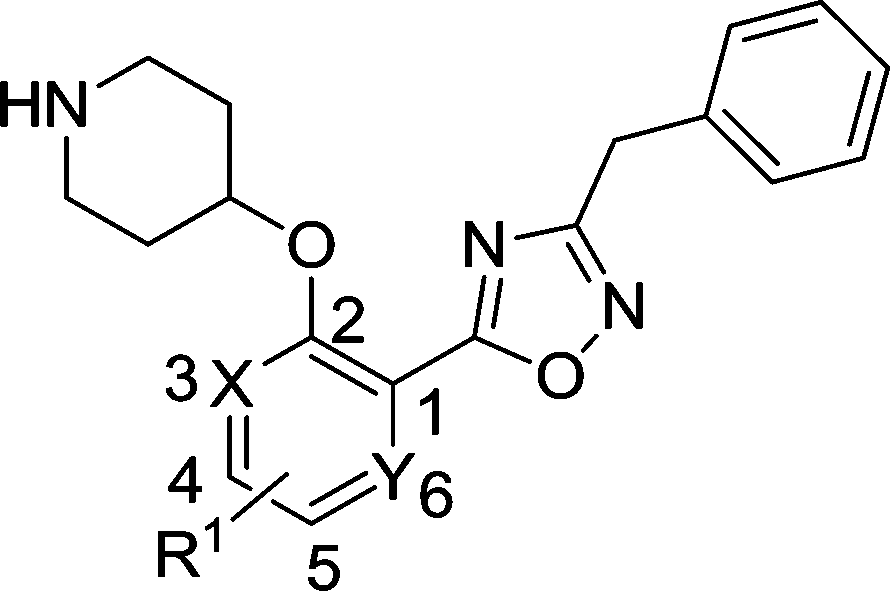
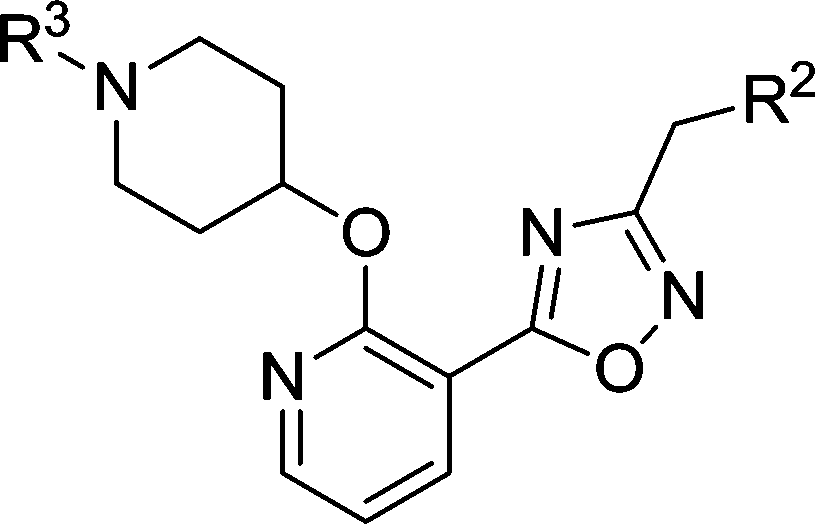
The biological activities of these compounds are detailed in Table 1. Substitution at C-3 was disfavored for inhibition of PfNMT (4 and 5vs.3, Table 1), with only fluorine (6) tolerated at this position by the enzyme. However, the 3-pyridyl analogue of 3 exhibited both reduced lipophilicity and a 10-fold improvement in enzyme affinity (7vs.3). Within the phenyl series, substitution at the C-4 position generally enhanced enzyme inhibition, with methyl (8vs.3) somewhat less effective than halogen (10, 11vs.3) or methoxy (9), with minor variations in selectivity over the human enzyme. Electron-donating substituents at C-5 (12, 13) were disfavored and electron-withdrawing substituents favored (14, 15) with respect to inhibition of PfNMT, but were accompanied by a drop in selectivity over human NMT. Changes at the C-6 position (16, 17) were poorly tolerated.
Table 1 Enzyme activities of compounds with scaffold substitutions
|
|
| Compd no. |
R1 |
X |
Y |
PfNMT Kia (μM) |
HsNMT Kia (μM) |
S.I.b |
|
Apparent inhibitor dissociation constant Ki; half maximal inhibitory concentration (IC50) values were determined by a fluorogenic assay30 and differences in substrate Km accounted for using the Cheng–Prusoff equation to give a value for apparent Ki.
S.I. = selectivity index, calculated as apparent Ki(HsNMT1)/apparent Ki(PfNMT).
|
|
3
|
H |
CH |
CH |
1.4 |
34 |
24 |
|
4
|
3-OMe |
C |
CH |
17 |
n.d. |
— |
|
5
|
3-Me |
C |
CH |
39 |
n.d. |
— |
|
6
|
3-F |
C |
CH |
1.5 |
15 |
10 |
|
7
|
H |
N |
CH |
0.14 |
1.7 |
12 |
|
8
|
4-Me |
CH |
CH |
0.62 |
44 |
70 |
|
9
|
4-OMe |
CH |
CH |
0.18 |
1.2 |
6.8 |
|
10
|
4-F |
CH |
CH |
0.22 |
4.9 |
22 |
|
11
|
4-Cl |
CH |
CH |
0.29 |
17 |
58 |
|
12
|
5-Me |
CH |
CH |
29 |
n.d. |
— |
|
13
|
5-OMe |
CH |
CH |
4.6 |
2.3 |
0.5 |
|
14
|
5-F |
CH |
CH |
0.24 |
1.0 |
4.0 |
|
15
|
5-Cl |
CH |
CH |
0.52 |
0.73 |
1.4 |
|
16
|
6-F |
CH |
C |
4.8 |
n.d. |
— |
|
17
|
H |
CH |
N |
29 |
n.d. |
— |
3-OMe phenyl as the terminal aromatic moiety
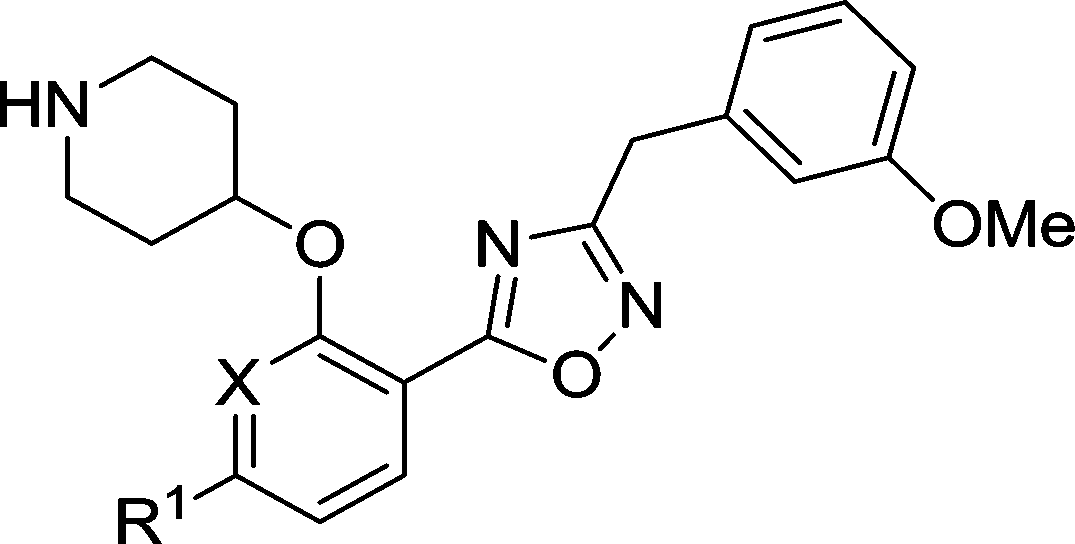
Based on enzyme potency and S.I. values, cores from compounds 7, 9, 10 and 11 were selected for further development, and a terminal 3-OMe phenyl moiety was selected for incorporation via the oxadiazole linker, based on our previous work.28 The corresponding compounds 19–22 along with the unsubstituted phenyl analogue 18 were prepared and it was found that addition of a 3-methoxy group generally enhances PfNMT inhibition activity, although at the expense of some human selectivity (19–22 in Table 2vs.7, 9, 10 and 11 in Table 1). Compound 19 was selected as the frontrunner inhibitor at this stage given its balance of good potency and selectivity; furthermore, its higher LipE value is attractive for further development.31,32
Table 2 Biological activities of 3-OMe phenyl analogues 18–22
Structural studies
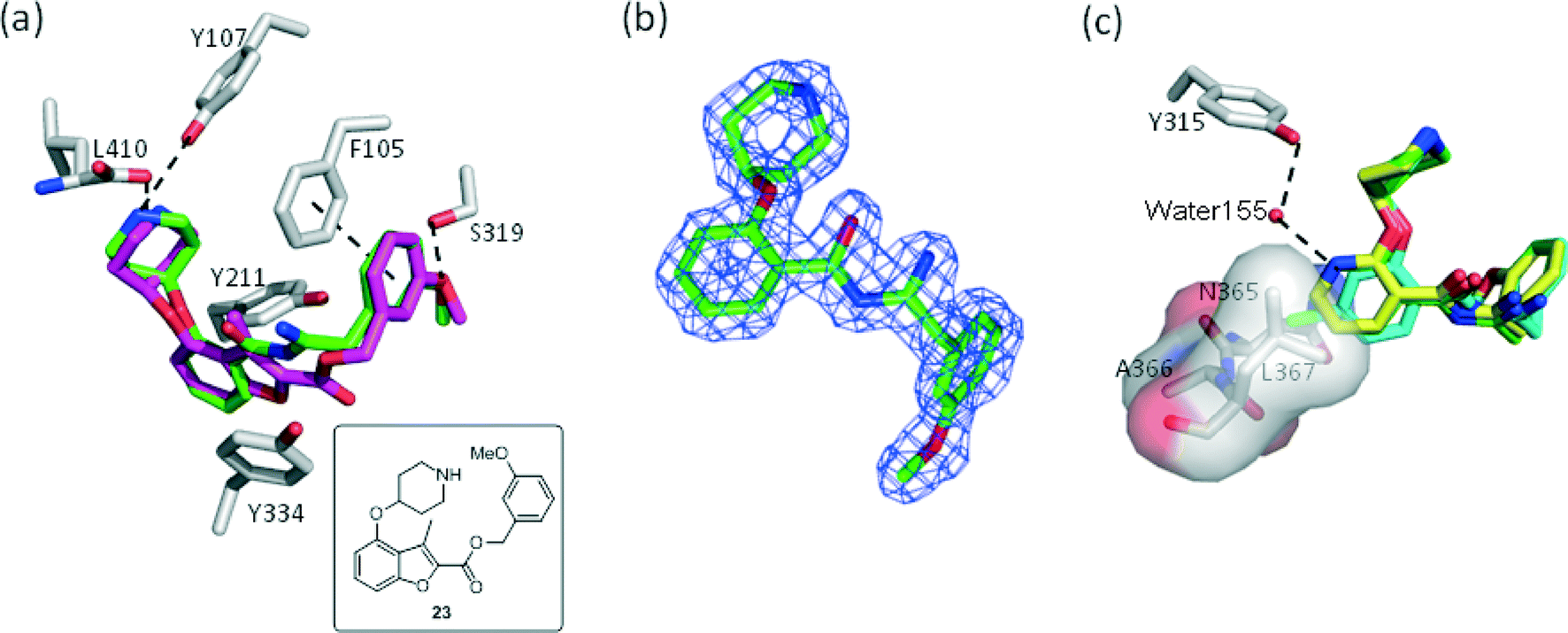
P. vivax (Pv) NMT shares 81% sequence identity with PfNMT, and has been successfully used to guide the discovery of potent PfNMT inhibitors.26–28 In order to interpret the effect of substitution on the scaffold, inhibitors derived from phenyl (18), 4-chloro phenyl (22) and 3-pyridyl (19) were selected for crystallographic studies and co-crystal structures with a non-hydrolysable myr-CoA analogue (NHM)30 and PvNMT were obtained. As expected, inhibitor 18 shows a very similar binding mode to a previously reported inhibitor 23 (Fig. 2a).28 Surprisingly, electron density maps suggest the formation of a ring-open analogue of the oxadiazole for these three test compounds within the crystal (the corresponding analogue for 18 is shown in Fig. 2b), although the desired closed ring was confirmed for the pure isolated inhibitors by both NMR and high resolution mass spectrometry ([M + H]+ = 366.1801, which matches well with its closed ring form), and they were found to be stable under conditions covering a pH range between 1 and 10 (data not shown). This interesting phenomenon was also observed in the related series crystallised in LmNMT,29 and further work will be required to understand its origin; however, it did not impact on our subsequent development of the oxadiazole series. The basic piperidine moiety establishes an ion-pairing interaction with the C-terminal carboxylate (L410) and a cation-dipole interaction with the Y107 hydroxyl, whilst the 3-OMe phenyl forms hydrogen bonding (HB) and π–π interactions with the S319 hydroxyl and F105, respectively (Fig. 2a). The 1,2,4-oxadiazole moiety is sandwiched between Y334 and Y211, forming hydrophobic interactions with these two residues. The observed substitution effects of the core can be clarified by examination of the bound structures (Fig. 2c). Pyridyl 19 is superimposed with phenyl 18, where the pyridyl nitrogen participates in an additional water-mediated HB with the Y315 hydroxyl, providing an explanation for the observed 8-fold enhancement in inhibition (19vs.18, Table 2). 4-Cl in 22 inserts into a hydrophobic pocket formed by N365, A366 and L367; we hypothesise that such hydrophobic interaction provides the observed activity improvement (22vs.18, Table 2).
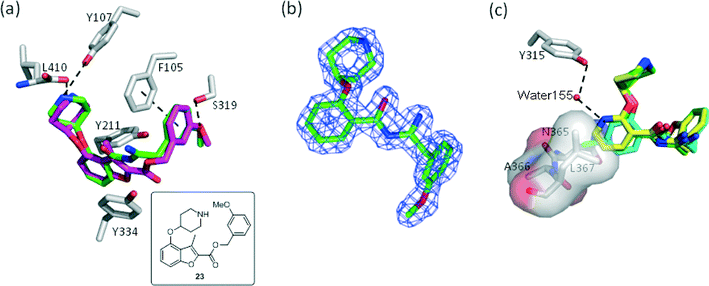 |
| | Fig. 2 Structural overlays help to rationalize the SAR for 18, 19 and 23: (a) 18 (PDB code: 4UFV, 1.80 Å, green) binds to the enzyme (grey) in a similar way to 23 (PDB code: 4B14, magenta); (b) unexpected ring-open oxadiazole observed in the electron density map of 18 (C green); (c) 19 (PDB code: 4UFX, 1.51 Å, yellow) picks up an additional water-mediated hydrogen bond and shares identical binding positions to 18 (green); while insertion of chlorine at C-4 allows the hydrophobic interaction with the pocket comprising of N365, A366 and L367 (22, PDB code: 4UFW, 1.54 Å, cyan). Atoms are colored: N blue, O (non-water) red, O (water) red sphere. | |
Structure-guided development
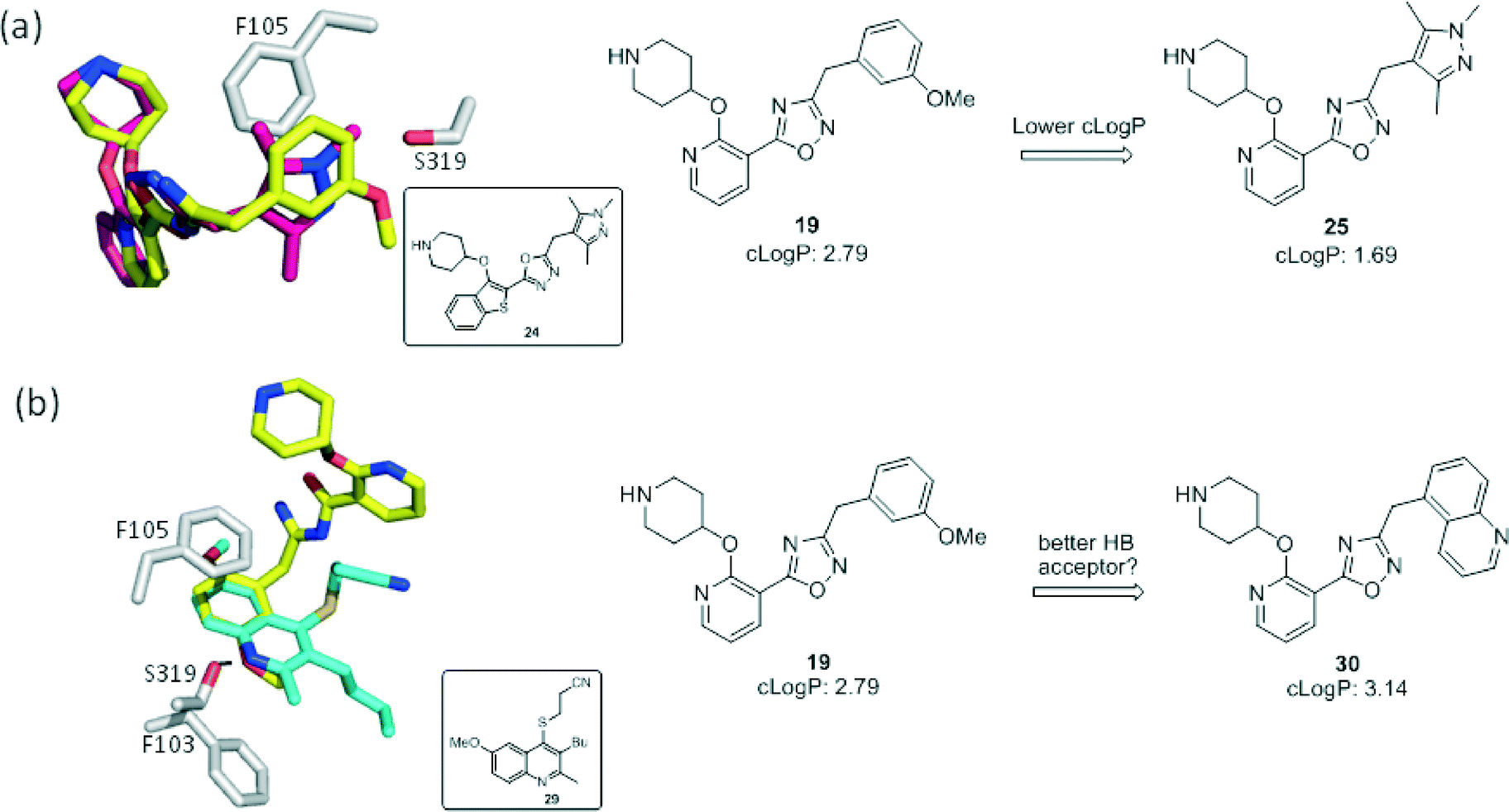
We next considered replacements for the 3-OMe phenyl moiety. A structural overlay with 24, a previously reported Pv/Pf NMT inhibitor,26 suggests that the 1,3,5-trimethylpyrazole shares a similar binding mode to methoxyphenyl (Fig. 3a). Pyrazole 25 was thus prepared (Scheme 2). Although it showed higher LipE than 19, there was no potency boost in either enzyme or cellular level while its N-alkyl piperidine derivatives (26–28) suffered from modest to significant activity drop (Table 3). Another PvNMT inhibitor (29) identified from an earlier screening campaign,23 was found to overlay well with the 3-OMe phenyl moiety in 19 (Fig. 3b). Encouragingly, the combination of quinoline and pyridyl scaffold yielded inhibitor 30 (Scheme 3), which posted a 14-fold increase in both enzyme and cellular potency over 19, with only two heavy atoms added and no compromise in enzyme selectivity over human NMT or acute toxicity against mammalian cells over 48 hours (30vs.19, Table 3). We hypothesise that the improved HB acceptor strength of the quinoline33 contributes to the observed increase in activity.
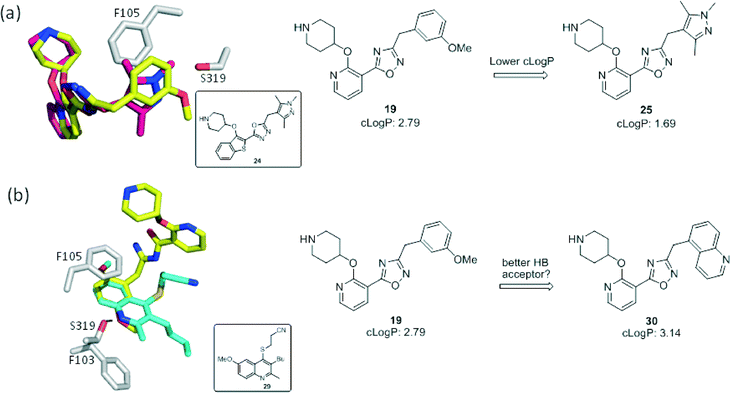 |
| | Fig. 3 Structure-guided design: (a) overlay of 3-OMe phenyl in 19 (PDB code: 4UFX, yellow) with the pyrazole in 24 (PDB code: 4CAF, magenta). Both motifs bind at the S319 pocket; (b) 3-OMe phenyl in 19 superimposes well with the quinoline in 29 (PDB code: 4A95, cyan). Atoms are colored: C (enzyme) grey, N blue, O red, S orange. | |
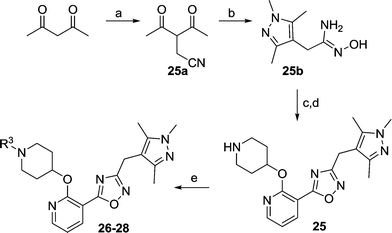 |
| | Scheme 2 Reagents: (a) bromoacetonitrile, NaH, THF, rt, 4 h, 40%; (b) i) mono-methylhydrazine, MeOH, reflux, 4 h; ii) hydroxylamine, MeOH, reflux, 6 h, 95%; (c) i). 7b, EDCI, HOBt, DIPEA, CH3CN, rt, 4 h; ii). 0.5 N NaOH, rt, 0.5 h, 90%; (d) 10% TFA in DCM (v/v), rt, 2 h, quantitative; (e) aldehyde or ketone, NaBH(OAc)3, MeOH, rt, 12 h, 80%. | |
Table 3 Biological activities of inhibitors guided by overlaying crystal structures
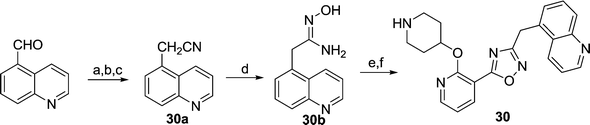 |
| | Scheme 3 Reagents: (a) NaBH4, MeOH, rt, 8 h; (b) MsCl, DCM, 0 °C, 3 h; (c) NaCN, DMSO, 60 °C, 1 h, 22% over three steps; (d) hydroxylamine, MeOH, reflux, 6 h, 97%; (e) i). 7b, EDCI, HOBt, DIPEA, CH3CN, rt, 4 h; ii). 0.5 N NaOH, rt, 0.5 h, 90%; (f) 10% TFA in DCM (v/v), rt, 2 h, quantitative. | |
Conclusion
Scaffold reduction from our earlier PfNMT inhibitors containing bicyclic cores to phenyl-based inhibitors provided a flexible platform to study the effect of scaffold and substituent variations, allowing the discovery of pyridyl as an attractive core, the superiority of which was later explained by structural studies. Further development was achieved through crystallography-guided hybridization of this scaffold with two existing NMT inhibitors, leading to the discovery of 30, a promising PfNMT inhibitor with excellent enzyme affinity and good cellular efficacy. Further lead optimization is underway in an attempt to further improve the cellular potency of this series, with the aim of progressing towards in vivo studies.
Acknowledgements
This work was supported by the Wellcome Trust (grant no. 087792), the Medical Research Council (grant no. 0900278 and U117532067) and the Francis Crick Institute which receives its core funding from Cancer Research UK, the UK Medical Research Council and the Wellcome Trust. The authors are grateful to Andrew Bell, Mark Rackham and Jennie Hutton for valuable discussions. We thank Munira Grainger for providing the parasites and red blood cells used in the in vitro parasite assay, Shirley Roberts for crystal handling and Johan Turkenburg for X-ray data processing. We also acknowledge the Diamond Light Source for synchrotron facilities.
References
-
World Malaria Report 2014, 2014, World Health Organization, Geneva Search PubMed.
- The RTS,S Clinical Trials Partnership, N. Engl. J. Med., 2012, 367, 2284 CrossRef PubMed.
- A. Opar, Nat. Rev. Drug Discovery, 2011, 10, 887 CrossRef CAS PubMed.
- J. Straimer, N. F. Gnadig, B. Witkowski, C. Amaratunga, V. Duru, A. P. Ramadani, M. Dacheux, N. Khim, L. Zhang, S. Lam, P. D. Gregory, F. D. Urnov, O. Mercereau-Puijalon, F. Benoit-Vical, R. M. Fairhurst, D. Menard and D. A. Fidock, Science, 2015, 347, 428 CrossRef CAS PubMed.
- A. P. Phyo, S. Nkhoma, K. Stepniewska, E. A. Ashley, S. Nair, R. McGready, C. ler Moo, S. Al-Saai, A. M. Dondorp, K. M. Lwin, P. Singhasivanon, N. P. J. Day, N. J. White, T. J. C. Anderson and F. Nosten, Lancet, 2012, 379, 1960 CrossRef.
- A. M. Dondorp, F. Nosten, P. Yi and D. Das, N. Engl. J. Med., 2009, 361, 455 CrossRef CAS PubMed.
- J. N. Burrows, E. Burlot, B. Campo, S. Cherbuin, S. Jeanneret, D. Leroy, T. Spangenberg, D. Waterson, T. N. Wells and P. Willis, Parasitology, 2014, 141(Special Issue 01), 128 CrossRef PubMed.
- E. W. Tate, K. A. Kalesh, T. Lanyon-Hogg, E. M. Storck and E. Thinon, Curr. Opin. Chem. Biol., 2015, 24, 48 CrossRef CAS PubMed.
- E. Thinon, R. A. Serwa, M. Broncel, J. A. Brannigan, U. Brassat, M. H. Wright, W. P. Heal, A. J. Wilkinson, D. J. Mann and E. W. Tate, Nat. Commun., 2014, 5, 4919 CrossRef CAS PubMed.
- E. W. Tate, A. S. Bell, M. D. Rackham and M. H. Wright, Parasitology, 2014, 141, 37–49 CrossRef CAS PubMed.
- M. Wright, W. Heal, D. Mann and E. Tate, J. Chem. Biol., 2010, 3, 19 CrossRef PubMed.
- D. R. Johnson, R. S. Bhatnagar, L. J. Knoll and J. I. Gordon, Annu. Rev. Biochem., 1994, 63, 869 CrossRef CAS PubMed.
- D. Guttery, S. Poulin, B. Ramaprasad, A. Wall, R. J. Ferguson, D. J. P. Brady, D. Patzewitz, E. M. Whipple, S. Straschil, M. H. Wright, A. Mohamed, M. A. H. Radhakrishnan, A. S. T. Arold, E. W. Tate, A. A. Holder, B. Wickstead, A. Pain and R. Tewari, Cell Host Microbe, 2014, 16, 128 Search PubMed.
- B. Poulin, E. M. Patzewitz, D. Brady, O. Silvie, M. H. Wright, D. J. P. Ferguson, R. J. Wall, S. Whipple, D. S. Guttery, E. W. Tate, B. Wickstead, A. A. Holder and R. Tewari, Biol. Open, 2013, 2, 1160 CrossRef PubMed.
- W. Leber, A. Skippen, Q. L. Fivelman, P. W. Bowyer, S. Cockcroft and D. A. Baker, Int. J. Parasitol., 2009, 39, 645 CrossRef CAS PubMed.
- J. L. Green, R. R. Rees-Channer, S. A. Howell, S. R. Martin, E. Knuepfer, H. M. Taylor, M. Grainger and A. A. Holder, J. Biol. Chem., 2008, 283, 30980 CrossRef CAS PubMed.
- R. R. Rees-Channer, S. R. Martin, J. L. Green, P. W. Bowyer, M. Grainger, J. E. Molloy and A. A. Holder, Mol. Biochem. Parasitol., 2006, 149, 113 CrossRef CAS PubMed.
- W. H. L. Stafford, R. W. Stockley, S. B. Ludbrook and A. A. Holder, Eur. J. Biochem., 1996, 242, 104 CrossRef CAS PubMed.
- M. H. Wright, B. Clough, M. D. Rackham, K. Rangachari, J. A. Brannigan, M. Grainger, D. K. Moss, A. R. Bottrill, W. P. Heal, M. Broncel, R. A. Serwa, D. Brady, D. J. Mann, R. J. Leatherbarrow, R. Tewari, A. J. Wilkinson, A. A. Holder and E. W. Tate, Nat. Chem., 2014, 6, 112 CrossRef CAS PubMed.
- P. Pino, S. Sebastian, E. A. Kim, E. Bush, M. Brochet, K. Volkmann, E. Kozlowski, M. Llinas, O. Billker and D. Soldati-Favre, Cell Host Microbe, 2012, 12, 824 Search PubMed.
- J. A. Hutton, V. Goncalves, J. A. Brannigan, D. Paape, M. H. Wright, T. M. Waugh, S. M. Roberts, A. S. Bell, A. J. Wilkinson, D. F. Smith, R. J. Leatherbarrow and E. W. Tate, J. Med. Chem., 2014, 57, 8664 CrossRef CAS PubMed.
- J. A. Brannigan, S. M. Roberts, A. S. Bell, J. A. Hutton, M. R. Hodgkinson, E. W. Tate, R. J. Leatherbarrow, D. F. Smith and A. J. Wilkinson, IUCrJ, 2014, 1, 250 CrossRef CAS PubMed.
- V. Goncalves, J. A. Brannigan, D. Whalley, K. H. Ansell, B. Saxty, A. A. Holder, A. J. Wilkinson, E. W. Tate and R. J. Leatherbarrow, J. Med. Chem., 2012, 55, 3578 CrossRef CAS PubMed.
- A. S. Bell, J. E. Mills, G. P. Williams, J. A. Brannigan, A. J. Wilkinson, T. Parkinson, R. J. Leatherbarrow, E. W. Tate, A. A. Holder and D. F. Smith, PLoS Neglected Trop. Dis., 2012, 6, e1625 CrossRef CAS PubMed.
- T. O. Olaleye, J. A. Brannigan, S. M. Roberts, R. J. Leatherbarrow, A. J. Wilkinson and E. W. Tate, Org. Biomol. Chem., 2014, 12, 8132 Search PubMed.
- M. D. Rackham, J. A. Brannigan, K. Rangachari, S. Meister, A. J. Wilkinson, A. A. Holder, R. J. Leatherbarrow and E. W. Tate, J. Med. Chem., 2014, 57, 2773 CrossRef CAS PubMed.
- M. D. Rackham, J. A. Brannigan, D. K. Moss, Z. Yu, A. J. Wilkinson, A. A. Holder, E. W. Tate and R. J. Leatherbarrow, J. Med. Chem., 2013, 56, 371 CrossRef CAS PubMed.
- Z. Yu, J. A. Brannigan, D. K. Moss, A. M. Brzozowski, A. J. Wilkinson, A. A. Holder, E. W. Tate and R. J. Leatherbarrow, J. Med. Chem., 2012, 55, 8879 CrossRef CAS PubMed.
- M. D. Rackham, Z. Yu, J. A. Brannigan, W. P. Heal, D. Pappe, K. V. Barker, A. J. Wilkinson, D. F. Smith, R. J. Leatherbarrow and E. W. Tate, Med. Chem. Commun. 10.1039/c5md00241.
- V. Goncalves, J. A. Brannigan, E. Thinon, T. O. Olaleye, R. Serwa, S. Lanzarone, A. J. Wilkinson, E. W. Tate and R. J. Leatherbarrow, Anal. Biochem., 2012, 421, 342 CrossRef CAS PubMed.
- A. L. Hopkins, G. M. Keseru, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug Discovery, 2014, 13, 105 CrossRef CAS PubMed.
- A. Tarcsay, K. Nyiri and G. M. Keseru, J. Med. Chem., 2012, 55, 1252 CrossRef CAS PubMed.
- H. J. Böhm, S. Brode, U. Hesse and G. Klebe, Chem. – Eur. J., 1996, 2, 1509 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of all intermediates, target compounds and X-ray crystallographic data. The coordinates and structure factor files have been deposited in the Protein Data Bank under the accession codes 4UFV (PvNMT-NHM-18), 4UFW (PvNMT-NHM-22) and 4UFX (PvNMT-NHM-19). See DOI: 10.1039/c5md00242g |
| ‡ Current address: International Discovery Service Unit, WuXi AppTec, Shanghai, 200131, China. |
| § Current address: Department of Chemistry, Kings College London, London SE1 1UL, UK. |
| ¶ Current address: Egerton Court, Liverpool John Moores University, Liverpool L1 2UA, UK. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a