
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The discovery of a novel antibiotic for the treatment of Clostridium difficile infections: a story of an effective academic–industrial partnership
John
Mann
a,
Peter W.
Taylor
a,
Colin R.
Dorgan
b,
Peter D.
Johnson
b,
Francis X.
Wilson
b,
Richard
Vickers
b,
Aaron G
Dale
a and
Stephen
Neidle
*a
aUCL School of Pharmacy, University College London, London WC1N 1AX, UK. E-mail: s.neidle@ucl.ac.uk
bSummit Therapeutics, Abingdon, OX14 4RY, UK
First published on 1st July 2015
Abstract
Academic drug discovery is playing an increasingly important role in the identification of new therapies for a wide range of diseases. There is no one model that guarantees success. We describe here a drug discovery story where chance, the ability to capitalise on chance, and the assembling of a range of expertise, have all played important roles in the discovery and subsequent development of an antibiotic chemotype based on the bis-benzimidazole scaffold, with potency against a number of current therapeutically challenging diseases. One compound in this class, SMT19969, has recently entered Phase 2 human clinical trials for the treatment of Clostridium difficile infections.
 John Mann | John Mann spent 25 years at Reading University rising to a Personal Chair in Organic Chemistry in 1990. In 1999 he accepted the McClay Chair of Biological Chemistry at Queen's University Belfast. The Mann Group developed expertise in the synthesis of natural products including carbohydrates, steroids, terpenoids, nucleosides, alkaloids, lignans, and prostanoids. Much of this research was concerned with the design and synthesis of novel products for use in cancer chemotherapy in collaboration with Michael Jarman and Stephen Neidle. The research resulted in over 130 papers, and he is also the author of five textbooks and five popular science books, and most recently a work of ‘science in fiction’ under the title “Drugs for all Reasons”. |
 Peter Taylor | Peter Taylor has spent a large part of his career in Big Pharma and is currently Professor of Microbiology at UCL. His research interests involve novel approaches to the treatment of infectious disease, with a focus on the development of antibacterial therapeutics that suppress or abrogate the emergence of drug resistant variants or compromise the virulence properties of systemic pathogens such as methicillin-resistant staphylococci (MRSA), Bacillus anthracis (the causative agent of inhalation anthrax) and the neonatal neuropathogen Escherichia coli K1. He also researches the impact of spaceflight on the fitness of pathogenic bacteria that may contaminate space vessels. |
 Colin Dorgan | Colin Dorgan graduated with a first class MChem from the University of Liverpool in 2005. As part of the course he took a 12 month placement at Pfizer PGRD and upon completion of his undergraduate studies he returned to the drug discovery industry working for Summit Plc. He undertook research in several therapeutic areas and most notably contributed to the discovery of 2 clinical candidates; Utrophin upregulator C1100 for Duchenne Muscular Dystrophy and antibiotic SMT019969 for C. Difficile infections. He now works outside of the drug discovery industry for Carbosynth Ltd in Oxfordshire, UK. |
 Pete Johnson | Pete Johnson completed his DPhil in Organic Chemistry at the University of Oxford in 2004. He spent a year as a Research Associate at the University of Chicago before returning to the UK and joining Summit (then VASTox) in 2005. Research work over four years covered the design of molecules to upregulate utrophin for the treatment of DMD, and two antibiotic programs including the use of bis-benzimidazoles for treatment of CDAD. Since 2009 he has worked for Evotec Ltd. in the field of neurodegenerative disorders. |
 Francis Wilson | Francis Wilson is Director of Chemistry at Summit Therapeutics. He has been involved in drug discovery for over 30 years working in a wide range of therapeutic areas. At Summit he has played leading roles in the discovery and early development of a number of clinical candidates, including SMT C1100 for treatment of Duchenne muscular dystrophy and SMT19969 for the treatment of C. Difficile infection. He was formerly Director of Chemistry at Cellzome, prior to which he worked with Xenova and Roche, and has authored/co-authored over 100 patents and publications. |
 Richard Vickers | Richard Vickers completed his PhD from Reading University before undertaking postdoctoral studies with Professor Steve Davies, and taking up a stipendiary lectureship at St Catherine's College, at the University of Oxford. He is currently CSO — Antimicrobials at Summit Therapeutics, a UK based biotech company focused on the development of agents in areas of high unmet medical need. His research interests are in the development of new antimicrobial having worked on novel agents for tuberculosis, a project that was partnered with the Eli-Lilly TB Drug Discovery Initiative, and more recently on the development of agents to treat C. difficile infections. |
 Aaron Dale | Aaron Dale (http://www.twitter.com/NotDrDalePhD) is a graduate-entry medicine student at Oxford University. He holds a PhD in drug discovery from University College London for work on the symmetric bis-benzimidazole series of compounds and has carried out post-doctoral research into small molecule inhibitors of the hypoxia inducible factor pathway. His research interests include development of novel antimicrobial and anti-cancer agents, and the use of evidence-based medicine in patient care. He is a keen cyclist and drummer for the Klezmer group Black Shabbat. |
 Stephen Neidle | Stephen Neidle received his undergraduate and graduate education at Imperial College London. He is Professor of Chemical Biology in the School of Pharmacy, University College London, where he has also served as Director of Research. Previously he was Professor of Biophysics at the Institute of Cancer Research UK, where he was also Dean (1997–2002). His research interests focus on the discovery and development of novel nucleic acid-targeted small molecules of potential use in the treatment of those human cancers, neurodegenerative disorders and infective diseases that are of most unmet clinical need. |
The background
The Neidle laboratory, which from 1986–2002 was at the Institute of Cancer Research in the UK (ICR), has long been involved in the study of small molecule–nucleic acid interactions, and in the use of X-ray crystallography and molecular modelling for the rational design of sequence- and structure-recognition agents. In the late 1980s Neidle and his colleagues had started on a programme of X-ray analyses of complexes between double-helical oligonucleotides and small molecules that were known to bind in the minor groove of B-form DNA. Many of these compounds were well-established to have biological activity, especially as anti-infective and anti-parasitic agents. From 1990 onwards a number of crystal structures of DNA complexes involving for example, the bis-(phenyl amidinium) triazene compound berenil,1,2 the analogous aliphatic-linked compound pentamidine3 and the bis-benzimidazole Hoechst 33258,4 had been determined with a PhD student, David Brown (later at Pfizer UK, now at the University of Kent) and a senior post-doctoral associate, Mark Sanderson, (now at King's College London), both playing prominent roles. Much of this structural work was (and still is) in close collaboration5,6 with David Wilson and David Boykin (Georgia State University, USA), who have pioneered the medicinal chemistry-led discovery and development of low-molecular-weight minor-groove binding compounds for the treatment of infective diseases. Their work on tropical parasitic diseases such as trypanosomiasis has resulted in one such compound entering advanced clinical trials.7Earlier crystallographic studies of the dipyrrolic polyamide natural product netropsin by Richard Dickerson and colleagues8 at UCLA had shown that its binding to AT-rich regions of duplex DNA was in the minor groove. Importantly the convex shape of netropsin complemented the concave surface of the groove, giving rise to the concept of isohelicity9 to describe the recognition of AT-rich narrow minor groove DNA sequences. Isohelicity suggests that a particular radius of curvature for the ligand is required in order for optimal binding to take place. However combined biophysical and crystallographic studies had shown that this is not necessarily the case since a water molecule can bridge between an amidinium group and a base edge in the groove even if the ligand itself has a linear or near-linear shape.10
The crystal structures being determined in the Neidle laboratory were being used as starting-points in several qualitative and semi-quantitative molecular modelling studies with the initial intent of rationalising patterns of ligand-DNA hydrogen bonding and deciphering the role of water molecules in the recognition process.11,12 Around this time, several chemistry-based groups, most prominently that of Peter Dervan at CalTech, were developing chemical ways of recognising increasingly long sequences of duplex DNA, ultimately as gene targeting agents.13 This approach uses the concept of stringing together simple small building-blocks, (mostly amido-substituted small heterocyclics which could recognise individual bases by hydrogen-bonding to base edges in the minor groove), into larger polyamide molecules, some of which can recognise more than a complete turn of duplex DNA. The approach has subsequently been extremely successful and is now capable of selective recognition of individual genes within a genome such that it is now being studied in animal models as a potential direct therapy for down-regulating the expression of deleterious genes driving a particular disease state (see for example, a description of targeting the estrogen receptor element in cells and in vivo14). The Neidle group was interested in exploiting its accumulated structural information in an analogous manner, and to using parts of these established minor-groove binding compounds as recognition building-blocks. Typical compounds studied at that time included a number of diamidines such as berenil and pentamidine, the asymmetric bis-benzimidazole DNA stain Hoechst 33258 and symmetric furan-based bis-diamidines from the Boykin/Wilson laboratory.
A chance meeting
John Mann and his group (then at the University of Reading) had collaborated with the Neidle group on a number of occasions, primarily in their work with Michael Jarman and his chemistry group at ICR, directed towards new anticancer agents, such as inhibitors of the enzyme aromatase as potential drugs for the treatment of hormone-dependent breast cancer.15 A totally unexpected meeting of the Mann and Neidle families took place in the supermarket of the small Tuscan town of Montaione in August 1990, when both families were on their summer vacations and chemistry was far from everyone's mind.16 This led to the families meeting together at a local pizzeria, then inevitably to chemical discussions over drinks, and followed by the resolve to develop a common project together.Crystal structures had provided structural knowledge of the minor groove binding for the well-studied asymmetric bis-benzimidazole compound Hoechst 33258.4,17 We decided that since the benzimidazole group was also a well-established pharmacophore with drug-like attributes, it was a suitable scaffold for our joint studies, which were aimed at eventually developing a non-polyamide approach to the recognition of long DNA sequences (perhaps up to 12 base pairs). Molecular modelling used the Hoechst 33258-d(CGCAAATTTGCG)2 complex crystal structure as a starting-point. The application of simple steric principles (by ensuring that there were no non-bonded close contacts at less than van der Waals separation between the joined-up benzimidazole units and atoms in the DNA minor groove) led to the concept of linking two benzimidazole units in a head-to-head symmetric manner, which was predicted to have the complementary shape needed for effective AT base recognition. The modelling indicated that this would be distinct from Hoechst 33258, would recognise four contiguous AT base pairs, and chemical functionality would be achieved via para substitution on a phenyl ring directly attached to each benzimidazole group.
After the summer break, the Mann group embarked on the synthesis of the new series of bis-benzimidazoles (termed BBZs) suggested by the modelling, which had not been previously reported in the literature, and this research continued uninterrupted until 2008, firstly at the University of Reading and later at Queen's University Belfast. The initial series of compounds were prepared18 by a very able Ghanaan PhD student, Yaw Oboku-Boahen, through the oxidative condensation between 3,3′,4,4′-tetraaminobiphenyl and various simple 4-substituted aromatic aldehydes (e.g. 4-methoxy and 4-hydroxy). This reaction (Scheme 1) required heating the compounds in nitrobenzene at 150 °C for 8–10 hours and although the required bis-benzimidazoles were obtained the yields were 25–40% at best. Although the yields were improved by a French PhD student, Anne Baron, thus allowing initial biological evaluation to be carried out, a new milder method was required to provide greater quantities of products. This was discovered and optimised by two excellent postdoctoral fellows, Xiao-Wen Sun and Christine Le Sann.19,21 This involved treating the tetraamine and aryl aldehydes in DMF with the oxidant Oxone at room temperature and typical reaction times of 18 hours. With this reaction method even complex bis-benzimidazoles such as a chlorambucil mustard derivative could be prepared in yields of up to 80%.
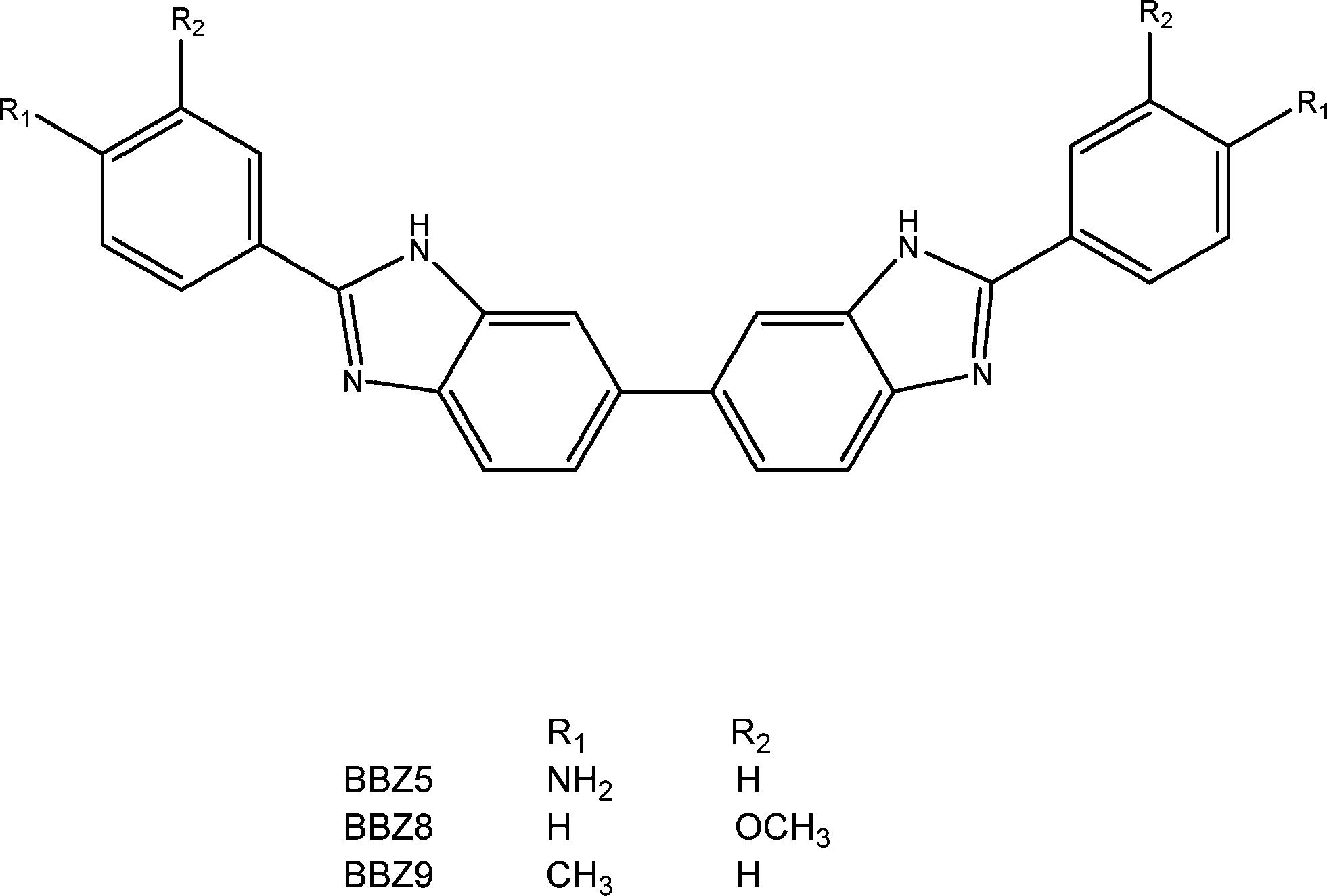
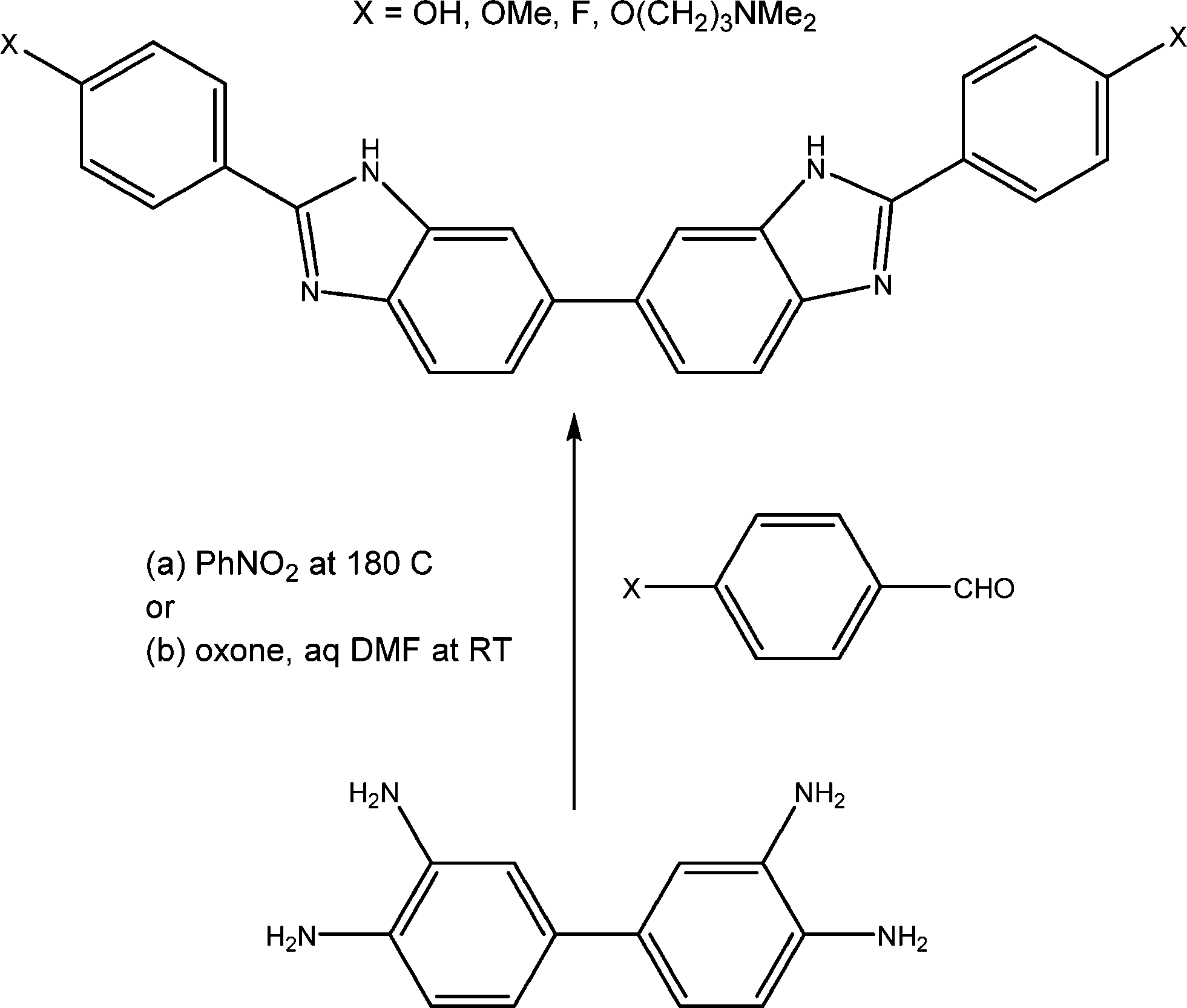 | ||
| Scheme 1 | ||
Once a number of substituted BBZs became available, a technical officer (Emma Rayner), and two PhD students (Ian Simpson and Eric Johansson) in the Neidle group undertook crystallographic studies of the bis-(3′′-hydroxy) and bis-(3′′-dimethylamino-1′′-propyloxy) derivatives (Scheme 1), bound to dodecanucleotide DNA duplexes (Protein Data Bank ids 453D and 1FTD).18,20 The prediction of four base-pair recognition was verified by these crystallographic studies as well as by DNA foot-printing studies conducted by Keith Fox (University of Southampton). Studies by the late Lloyd Kelland (ICR) and John Hartley (University College London) showed that several compounds in the series showed sub-μM inhibition of cell growth in a panel of ovarian cancer cell lines, with the bis-(3′′-dimethylamino-1′′-propyloxy) derivative being the most active whereas substituents with uncharged and more hydrophobic end-groups resulted in a loss of activity.20,21 The bis-(3′′-dimethylamino-1′′-propyloxy) derivative showed some modest anti-tumour activity in human cancer xenograft models,20 although this was insufficient to convince external funders. The anti-cancer project was finally put on hold in 2006, even though a patent had been granted to Stephen Neidle and John Mann in 2003 (ref. 22) in respect of the potential anticancer activity of BBZs.
Microbiological studies
Although both the Neidle and Mann laboratories were then largely focussed on other projects, subsequent events resurrected the BBZ area. Stephen Neidle had moved his group to the School of Pharmacy in London during 2002 and by 2003/4 had started discussing a possible collaboration with his microbiologist colleague Peter Taylor.Peter Taylor had joined the School of Pharmacy in November 1998 after a long period in commercial drug discovery and development. He established a team at the School dedicated to the discovery of new compounds and novel therapeutic modalities for the treatment of life-threatening systemic infections caused by multidrug resistant pathogens such as methicillin resistant Staphylococcus aureus (MRSA), vancomycin resistant enterococci (VRE) and drug resistant Mycobacterium tuberculosis (MDR-TB).
The discussions between Peter Taylor and Stephen Neidle about the BBZ compounds culminated in a suggestion that they might conceivably be inhibitors of bacterial gyrase enzymes, so a preliminary evaluation was undertaken of two BBZs against a selection of medically important bacterial clinical isolates. This work was undertaken by Diana Lisa Ramos Francisco, a Portuguese undergraduate gaining work experience in London in 2003/4 under the European Erasmus exchange programme. She found in early 2004 that compounds CLS549 and CLS 260B possessed no significant in vitro activity against Gram-negative bacteria such as Pseudomonas aeruginosa, Proteus mirabilis and Klebsiella species (strains responsible for many European and Stateside cases of bacteraemia, septicaemia and wound infections) but they elicited potent antibacterial effects against a range of Gram-positive pathogens, including EMRSA-15 and EMRSA-16 (representatives of two major MRSA clones isolated from U.K. hospitals) and group A and group B streptococci. The minimum drug concentrations required to prevent overnight bacterial growth (MIC) against these Gram-positive bacteria were impressive, ranging from 0.03–1 μg ml−1, and there was no evidence of cross-resistance with any other group of antibacterial agents – strongly suggestive of a novel mechanism of antibacterial action.
This promising data led to two successful grant applications that provided an opportunity to assess the antibacterial potential of a wider collection of BBZs. In 2007, an Academic Excellence Award from the Royal Pharmaceutical Society, achieved through national competition, enabled an investigation of the potency and antibacterial spectrum of activity of these compounds. A Capacity Building Award from the Medical Research Council facilitated insights into the mode of action of these putative drugs. A graduate student on the Royal Pharmaceutical Society award, Joao Moreira, profiled 21 symmetric and asymmetric BBZs against a comprehensive panel of Gram-positive clinical isolates, including ten from the initial series synthesised by John Mann and co-workers. This in-depth study23 confirmed the lack of activity against a wide range of Gram-negative pathogens but established that three compounds in particular possessed potent anti-staphylococcal activity against four methicillin-susceptible S. aureus and 61 MRSA strains from Peter Taylor's extensive international collection of clinical isolates, with clinically attractive MIC90 values (MIC required to inhibit 90% of the isolates) in the sub-μg ml−1 range. Similar potent effects were recorded against vancomycin intermediate-susceptible S. aureus (VISA; clinically troublesome MRSA partially resistant to the drug of last resort), Streptococcus pyogenes, Streptococcus agalactiae and β-haemolytoc streptococci. Data from the 21 BBZ compounds facilitated a detailed understanding of structure–activity relationships with respect to their anti-staphylococcal activity. Encouragingly, the key compounds displayed low cytotoxicity towards human fibroblasts and were well-tolerated by zebrafish embryos following injection into the yolk sac. A subsequent detailed investigation of the anti-staphylococcal mode of action was undertaken by Aaron Dale, a graduate student supported by the MRC funding. He showed that BBZs, in common with many conventional, clinically indispensable antibiotics, have a complex, multifactorial antibacterial mechanism which includes the capacity to inhibit the binding of DNA gyrase to DNA and to promote the accumulation of single-stranded DNA breaks.24
Of particular interest was the relatively strong and previously unreported activity of several BBZs against Mycobacterium tuberculosis (Mtb); this was manifest against both the standard laboratory Mtb strain H37RV and 10:104, a recently isolated drug-hetero-resistant Mtb bacterium. Further, the compounds were active against logarithmic phase cultures and latent cultures induced by hypoxia. This latter activity, together with the fact that they were active against resistant clinical isolates, suggested that they represent promising lead compounds for development of an anti-Mtb agent active against latent forms of the pathogen through a novel mechanism of action. Treatment of latent tuberculosis is a vital component of elimination of the disease but is not efficiently implemented with currently available drugs.
Summit entered the picture
John Mann's initial interactions with Summit Therapeutics (http://www.summitplc.com) in 2007 resulted in an initial small-scale project in which they independently agreed to evaluate the MRSA activity of a set of BBZ compounds. This took place soon after the first set of data became available from Peter Taylor's laboratory, and involved outsourcing the screening work to a contract research organisation (CRO), with Richard Vickers leading the Summit activity. This new data set fully confirmed the findings from the Taylor laboratory and at this point all involved decided to work closely together and jointly develop a strategy for the future development of the BBZ compounds. A major early issue was the commercial potential of the MRSA activity. A patent granted in 1998 to Roche in Basle with inventors Walter Häsler, Werner Leupin and Yu-Hau Ji25 had come to light, which covered antibacterial activity for a number of uncharged BBZs. Although the patent focussed on E. coli, a few S. aureus strains were mentioned. Summit undertook some medicinal chemistry in order to attempt to improve on the existing activity and move into novel chemical space. However it soon became apparent that it would be more fruitful to focus on alternative bacterial targets, and Summit suggested in particular working on Clostridium difficile infections. C. difficile is a serious hospital-associated bacterial disease which especially affects the elderly and is hard to treat with existing drugs. In 2011 it was responsible for over 2000 deaths in the UK, and is estimated to cause over 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in the USA. As the School of Pharmacy was not initially set up to work on this pathogen, Summit commissioned C. difficile compound evaluation at several CROs. Again potent activity was observed in several of the uncharged initial BBZ compounds. Summit then embarked on a comprehensive medicinal chemistry programme to optimise activity with appropriate physicochemical properties.
000 deaths in the USA. As the School of Pharmacy was not initially set up to work on this pathogen, Summit commissioned C. difficile compound evaluation at several CROs. Again potent activity was observed in several of the uncharged initial BBZ compounds. Summit then embarked on a comprehensive medicinal chemistry programme to optimise activity with appropriate physicochemical properties.
The medicinal chemistry effort found that the structure–activity requirements were exceptionally narrow and that significant deviations from the original BBZ concept were not tolerated in that the overall curvature and shape of the BBZ scaffold are absolute requirements for retention of activity. This work culminated in the discovery of the novel antibiotic SMT19969,26 which is very closely related to the original BBZ compounds invented by Stephen Neidle and John Mann, and represents a fundamental repositioning of particular BBZ compounds from generalised cell proliferation inhibitors to targeted anti-infective agents. A subsequent Wellcome Trust Seeding Drug Discovery funded project funded some further chemistry, together with extensive animal and pre-clinical toxicology work on the lead compound, with its biological properties being characterized in collaboration with the Neidle laboratory. Table 1 summarises anti-bacterial data for three representatives of the early BBZ compounds, together with data on SMT19969. In terms of mechanism of action, although this has not yet been definitively determined for SMT19969, it is clear from the gyrase and other data that it differs from the other BBZs and is for example not a gyrase or topoisomerase inhibitor in C. difficile, nor does it bind to duplex DNA. SMT19969 does not obey 4/5 of Lipinski's rules, which is precisely why it looks to be such an effective drug for C. difficile infections: as a consequence it is not orally bio-available and is retained in the gut, in close physical proximity to C. difficile bacteria.
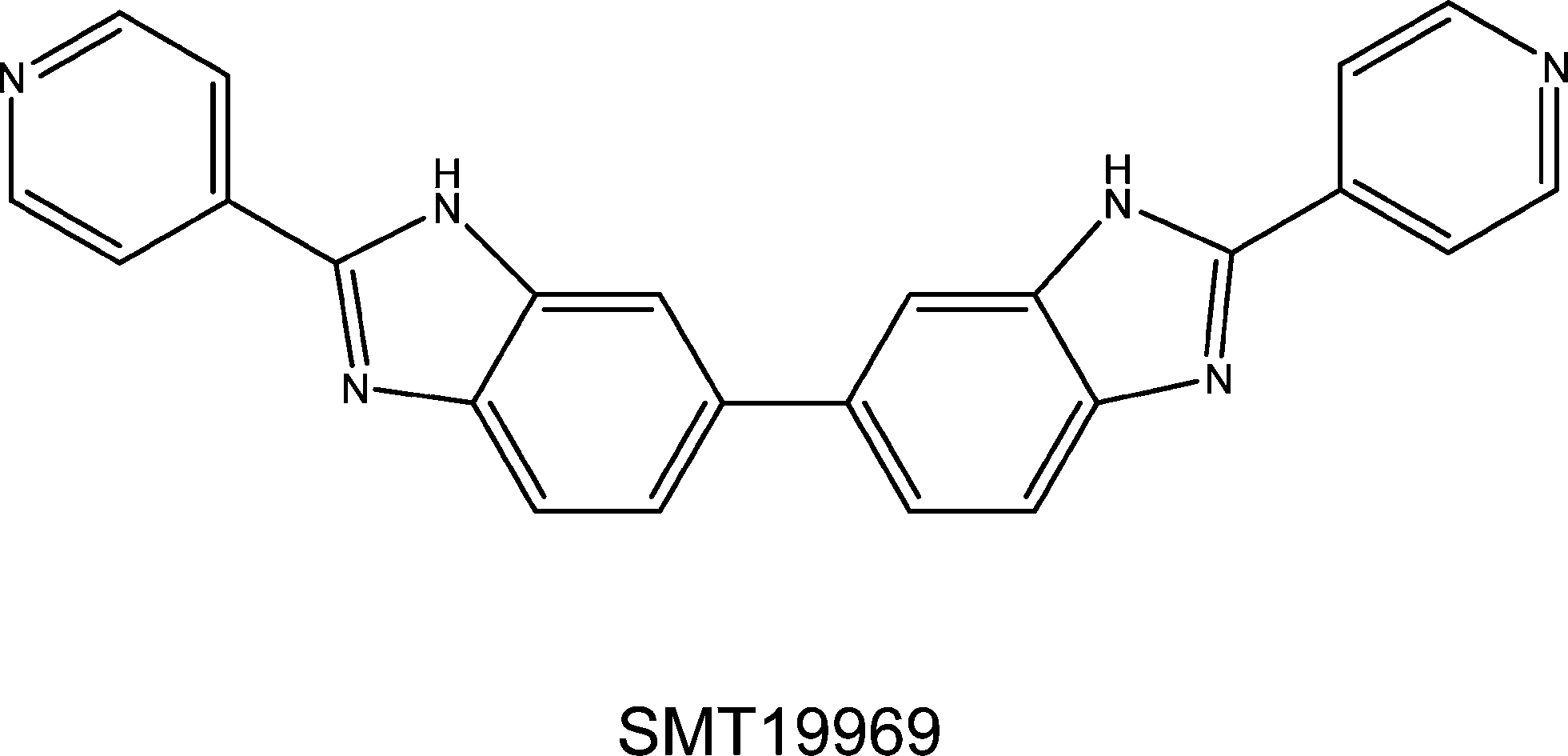
| Escherichia coli | Staphylococcus aureus | Clostridium difficile | Mycobacterium tuberculosis H37Rvc | |||||
|---|---|---|---|---|---|---|---|---|
| NCTC10418 | MC1061 | ATCC 29213 | EMRSA-16 | CD630 | R20291 | (Logarithmic phase) | (Hypoxia-induced latency) | |
| a Dale et al., to be published. b Reference 24. c Reference 23. | ||||||||
| MIC90 (μg ml−1) | ||||||||
| BBZ5 | >64 | 0.06 | 0.125 | 0.5 | 0.03–0.06 | 0.125 | 16 | 8 |
| BBZ8 | >64 | >64 | >128 | >128 | >32 | >32 | >16 | NA |
| BBZ9 | >64 | 0.06 | 0.03–0.06 | 0.06 | 0.06 | 0.06 | 1 | 2 |
| SMT19969 | >16 | NA | >128 | >128 | 0.06–0.125 | 0.125 | NA | NA |
| Response to BBZ5 and BBZ9 | ||||||||
| Bacteriostatic/bacteriocidal? | Bacteriostatic | Bactericidal | Bactericidal | NA | ||||
| Morphological response | Filamentation | Swelling | Filamentation | NA | ||||
| Topoisomerase inhibition IC50 (μM) | Escherichia coli | Staphylococcus aureus | Clostridium difficile | Mycobacterium tuberculosis | ||||
| DNA gyrase supercoiling | ||||||||
| BBZ5 | >50 | 0.3 | 8.4 | 11.3 | ||||
| BBZ8 | >50 | 8.8 | NA | 29 | ||||
| BBZ9 | >50 | 5.6 | NA | 2 | ||||
| SMT19969 | >50 | >50 | >50 | ND | ||||
| Topoisomerase IV decatentation | ||||||||
| BBZ5 | 8.0 | 15.5 | NA | NA | ||||
| BBZ8 | >50 | 70.3 | NA | NA | ||||
| BBZ9 | >50 | 50.9 | NA | NA | ||||
| SMT19969 | >50 | >50 | NA | NA | ||||
| Topoisomerase IV relaxation | ||||||||
| BBZ5 | 6.5 | NA | NA | NA | ||||
| BBZ8 | >50 | NA | NA | NA | ||||
| BBZ9 | >50 | NA | NA | NA | ||||
| SMT19969 | NA | NA | NA | NA | ||||
Summit Therapeutics has subsequently undertaken the further development of the series leading to the advancement of SMT19969 into current clinical trials in humans. This compound shows exceptional selectivity and potency for the bacterium27–29 and pre-clinical data indicated that it has remarkably low toxicity, which was confirmed by a Phase I clinical trial in 2012. The trial found that the novel orally-administered small-molecule antibiotic is selective for the treatment of C. difficile infections and did not affect other pathogens. It was announced in April 2013 that the Phase I trial had been concluded, with very successful results. A Phase 2 trial is currently underway in C. difficile patients and initial results will become available in 2015. In July 2015 the US Food and Drug Administration granted Fast Track designation to SMT19969 for the treatment of C. difficile.
What does the BBZ story tell us about academic drug discovery?
One could argue that the discovery of SMT19969 was accidental, with the key events being the evaluation of the existing BBZ compounds for anti-bacterial activity. However such “accidents” do not occur randomly – they require that those involved are smart enough to spot something of interest, worthy of an investment in time and money. Similarly the initial invention of the BBZ series by John Mann and Stephen Neidle, even though it was initiated by a chance meeting, was based on a great deal of prior knowledge of the structural features of minor groove-binding agents. The BBZ compounds came about, not because Mann and Neidle saw the project as an opportunity to write a grant proposal, but simply because they were enthused by the science and its possibilities, and that they wished to work together on a project of mutual interest. The fact that the original intent of creating novel DNA recognition units based on the BBZ scaffold has been superseded by a quite distinct goal, of creating novel anti-infective agents, is to our minds, a powerful argument for fostering basic science and ensuring that opportunistic links are put in place to assess the therapeutic potential of any outcomes.All of this would not have come to a focus, and SMT19969 would not have existed without the involvement of a commercial organisation, nimble enough to spot a potential opportunity. Whether a larger organisation would have become involved so quickly, or at all, is an interesting question (the original patent assigned to Roche, was abandoned by them before SMT19969 existed). Any early-stage therapeutic project requires substantial funding in order to progress and the Wellcome Trust's enthusiastic support via its Seeding Drug Discovery Programme was crucial. It is notable that this support was not based on the normal requirement of a validated target (as stated above the target(s) are still unclear), but on a robust case focussed around unmet clinical need and the properties of SMT19969 to effectively meet this need.
Acknowledgements
The work describes here has been supported, through its many stages, by a variety of funding sources. The original studies on DNA minor groove recognition in the Neidle laboratory were supported by programme grants from the Cancer Research Campaign and its successor, Cancer Research UK and by a Professorial Life Fellowship to Stephen Neidle. Work in the Taylor laboratory has been supported by grants from the Medical Research Council, who also supported a capacity-building award held jointly by Stephen Neidle and Peter Taylor. They jointly held an Academic Excellence Award from The Royal Pharmaceutical Society. The Wellcome Trust Seeding Drug Discovery Programme supported work at Summit Therapeutics and in the Neidle laboratory, and the Wellcome Trust have continued to support the current clinical trials on SMT19969.Notes and references
- D. G. Brown, M. Sanderson, J. V. Skelly, T. C. Jenkins, T. Brown, E. Garman, D. I. Stuart and S. Neidle, EMBO J., 1990, 9, 1329–1334 CAS.
- D. G. Brown, M. R. Sanderson, E. Garman and S. Neidle, J. Mol. Biol., 1992, 226, 481–490 CrossRef CAS PubMed.
- K. J. Edwards, T. C. Jenkins and S. Neidle, Biochemistry, 1992, 32, 7104–7109 CrossRef.
- N. Spink, D. G. Brown, J. V. Skelly and S. Neidle, Nucleic Acids Res., 1994, 22, 1607–1612 CrossRef CAS PubMed.
- D. Guo, W. D. Wilson and S. Neidle, J. Am. Chem. Soc., 2013, 135, 1369–1377 CrossRef PubMed.
- Y. Chai, M. Munde, A. Kumar, L. Mickelson, S. Lin, N. H. Campbell, M. Banerjee, S. Akay, Z. Liu, A. A. Farahat, R. Nhili, S. Depauw, M.-H. David-Cordonnier, S. Neidle, W. D. Wilson and D. W. Boykin, ChemBioChem, 2014, 15, 68–79 CrossRef CAS PubMed.
- M. N. Soeiro, K. Werbovetz, D. W. Boykin, W. D. Wilson, M. Z. Wang and A. Hemphill, Parasitology, 2013, 140, 929–951 CrossRef CAS PubMed.
- M. L. Kopka, C. Yoon, D. Goodsell, P. Pjura and R. E. Dickerson, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 1376–1380 CrossRef CAS.
- D. Goodsell and R. E. Dickerson, J. Med. Chem., 1986, 29, 727–733 CrossRef CAS PubMed.
- L. Yang, K. Arvind, S. Depauz, R. Nhili, M.-H. David-Cordonnier, M. Lee, M. Ismail, F. M. Farahat, S. Neidle, D. W. Boykin and W. D. Wilson, J. Am. Chem. Soc., 2011, 133, 10171–10183 CrossRef PubMed.
- C. A. Laughton, T. C. Jenkins, K. R. Fox and S. Neidle, Nucleic Acids Res., 1990, 18, 4479–4488 CrossRef CAS PubMed.
- P. A. Greenidge, C. A. Laughton, T. C. Jenkins and S. Neidle, Phys. Chem. Chem. Phys., 1993, 89, 2651–2657 RSC.
- P. B. Dervan and B. S. Edelson, Curr. Opin. Struct. Biol., 2003, 13, 284–299 CrossRef CAS PubMed.
- N. G. Nickols, J. O. Szablowski, A. E. Hargrove, B. C. Li, J. A. Raskatov and P. B. Dervan, Mol. Cancer Ther., 2013, 12, 675–694 CrossRef CAS PubMed.
- R. McCague, M. Jarman, M. G. Rowlands, J. Mann, C. P. Thickitt, D. W. Clissold, S. Neidle and G. Webster, J. Chem. Soc., Perkin Trans. 2, 1989, 196–198 RSC.
- The two families, with their children, were both staying at holiday villas in villages close to this town. Both were shopping when (Mrs) Andrea Neidle called out to Stephen Neidle over the supermarket shelves that she needed help in understanding the Italian for washing detergent. Back came an English voice with the answer, followed by amazement on all sides.
- P. E. Pjura, K. Grzeskowiak and R. E. Dickerson, J. Mol. Biol., 1987, 197, 257–271 CrossRef CAS PubMed.
- S. Neidle, J. Mann, E. L. Rayner, A. Baron, Y. Opoku-Boachen, I. J. Simpson, N. J. Smith, K. R. Fox, J. A. Hartley and L. R. Kelland, Chem. Commun., 1999, 929–930 RSC.
- J. Sun, J. Mann and S. Neidle, Tetrahedron Lett., 2002, 43, 7239–7241 CrossRef.
- J. Mann, A. Baron, Y. Opoku-Boahen, E. Johansson, G. Parkinson, L. R. Kelland and S. Neidle, J. Med. Chem., 2001, 44, 138–144 CrossRef CAS PubMed.
- C. Le Sann, A. Baron, J. Mann, H. van den Berg, M. Gunaratnam and S. Neidle, Org. Biomol. Chem., 2006, 4, 1305–1312 CAS.
- J. Mann and S. Neidle, US Pat., No. US6589971B1, 2003 Search PubMed.
- J. B. Moreira, J. Mann, S. Neidle, T. D. McHugh and P. W. Taylor, Int. J. Antimicrob. Agents, 2013, 42, 361–366 CrossRef CAS PubMed.
- A. G. Dale, J. Hinds, J. Mann, P. W. Taylor and S. Neidle, Biochemistry, 2012, 51, 5860–5871 CrossRef CAS PubMed.
- W. Häsler, Y.-H. Ji and W. Leupin, US Pat., No. US5824698, 1998 Search PubMed.
- P. J. Davies, O. De Moor, C. R. Dorgan, P. D. Johnson, A. G. Roach, R. Storer, R. J. Vickers, F. X. Wilson and G. M. Wynne, US Pat., No. US8975416B2, 2015 Search PubMed.
- E. J. Goldstein, D. M. Citron and K. L. Tyrrell, Antimicrob. Agents Chemother., 2014, 58, 1187–1191 CrossRef PubMed.
- W. Weiss, M. Pulse and R. Vickers, Antimicrob. Agents Chemother., 2014, 58, 5714–5718 CrossRef PubMed.
- A. Sattar, P. Thommes, L. Payne, P. Warn and R. J. Vickers, J. Antimicrob. Chemother., 2015, 70, 1757–1762 CAS.
| This journal is © The Royal Society of Chemistry 2015 |