
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and synthesis of potent and selective inhibitors of BRD7 and BRD9 bromodomains†
Duncan A.
Hay‡
ab,
Catherine M.
Rogers‡
bc,
Oleg
Fedorov
bc,
Cynthia
Tallant
bc,
Sarah
Martin
bc,
Octovia P.
Monteiro
bc,
Susanne
Müller
bc,
Stefan
Knapp
bc,
Christopher J.
Schofield
a and
Paul E.
Brennan
*bc
aDepartment of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3TA, UK
bStructural Genomics Consortium, University of Oxford, Old Road Campus Research Building, Roosevelt Drive, Oxford, OX3 7DQ, UK. E-mail: paul.brennan@sgc.ox.ac.uk
cTarget Discovery Institute, University of Oxford, NDM Research Building, Roosevelt Drive, Oxford, OX3 7FZ, UK
First published on 15th June 2015
Abstract
Emerging evidence suggests bromodomain-containing proteins 7 and 9 (BRD7 and BRD9) have roles in the regulation of human transcription and disease including cancer. We describe potent and selective inhibitors of the BRD7 and BRD9 bromodomains intended for use as tools to elucidate the biological roles of BRD7 and BRD9 in healthy and diseased cells.
Compared to other bromodomain- (BRD-) containing proteins, such as bromodomain and extra terminal (BET) family members very little is known about the biological function of BRD-containing protein 9 (BRD9). BRD9 has 598 residues and its BRD is the only functional domain that has been assigned to date.1 Proteomic analysis implies that BRD9 is associated with human BAF-type (BRG1/BRM-associated factors) SWI/SNF (SWItch/Sucrose Non-Fermentable) chromatin remodelling complexes.2 Recently, it has been reported that BRD9 is a core sub-unit of SWI/SNF complexes.3 The BRD9 paralog bromodomain-containing protein 7 (BRD7) is a subunit of PBAF (poly-bromo-associated BAF) SWI/SNF complexes.2,4,5 It is proposed that BRD7 regulates transcription through binding of its BRD to the acetylated form of Lys-14 of histone H3 (H3K14Ac).6 BRD7 and BRD9 are only 36% similar at in terms of their overall residues, but the similarity of their BRDs is much higher (72%).
Evidence is emerging which indicates that BRD9 and BRD7 have roles in oncology and are consequently potential drug targets. BRD9 has been identified as part of a group of genes with copy number variations in non-small cell lung cancer (NSCLC) tumour samples, implying that a chromosomal imbalance in BRD9 could be involved in tumourigenesis.7 Similar analysis of cervical cancer tumour samples also found copy number increases in BRD9.8 The links of BRD7 to oncology are even more compelling. It has been suggested that BRD7 is a possible tumour-suppressor gene, which inhibits G1-S progression by blocking the translocation of β-catenin from the cytoplasm to the cell nucleus and negatively regulating the extracellular signal-regulated kinases (ERK) 1/2 pathway.9,10 Additionally, BRD7 is reported to be a critical regulator of the tumour suppressor protein p53.11–13 BRD7 also regulates estrogen receptor α (ERα) expression through BRCA1 recruitment.14 Analysis of tumour biopsies shows that BRD7 is down-regulated in nasopharyngeal carcinoma, colorectal cancer and epithelial ovarian cancers.9,15,16 In colorectal cancer, BRD7 expression is also negatively correlated with survival time.15 The BRD7 locus is frequently deleted in breast cancer tumours.12 However, it was found that BRD7 mutations represent rare polymorphisms in breast cancer, with no pathogenic effect.17 MicroRNA-200c (miR-200c), which is over-expressed in endometrial carcinoma, inhibits BRD7 expression.18
With little known about the molecular function of BRD7, and even less in the case of BRD9, potent and selective small moleculeinhibitors would be valuable tools to study the roles of these proteins in transcription, elucidate their roles in oncology and other human diseases, and to test them as drug targets. Non-selective BRD9 inhibitors have been reported, but their potent BET inhibition makes them unsuitable to study the function of BRD9 in isolation.19,20 Potent and selective small moleculeinhibitors of BRD7 and BRD9 are presently limited with the first selective BRD7/9 inhibitors LP99 and I-BRD9 having just been reported.21 The availability of inhibitors with similar pharmacology but orthogonal chemotypes lends confidence to the interpretation of results obtained using chemical tools in target discovery; therefore, we sought to develop BRD7/9 inhibitors in a new chemical series.22 Here, we describe the design and synthesis of potent and selective inhibitors of the BRD7 and BRD9 BRDs, based on a series of C-3 keto-indolizines.23
Compound 1 (GSK2801) is a chemical probe for the bromodomain adjacent zinc finger (BAZ) 2A and BAZ2B BRDs (Fig. 1).24 We noted that pyridine-containing analogues of compound 1 inhibited the BRD9 BRD; thus, compounds 2 and 3 were relatively potent inhibitors of BRD9 in a peptide displacement AlphaScreen (amplified luminescent proximity homogeneous assay screen) assay (pIC50 5.6 and 6.3, respectively) (Table 1).25 Notably, we observed that compound 3 was more than 5-fold selective for BRD9 over BAZ2B. The low lipophilicity of these compounds (c log P 2.6, 1.8) made them attractive for further development of a BRD9 chemical probe. Our aim was to investigate the effects of varying the substituents on positions C-1, 3 and 7 of the indolizine ring in variants of compound 3 on BRD9 potency and selectivity while keeping the lipophilicities moderate (c log P 1–3) to ensure good drug-like properties.
 | ||
| Fig. 1 GSK2801 1, a BAZ2A/BAZ2B chemical probe. | ||
| Cmpd | R1 | R2 | R3 | c log Pa | BRD9 ΔTm (°C)b | BRD4(1) ΔTm (°C)b | BRPF1B ΔTm (°C)b | BAZ2B pIC50c |
|---|---|---|---|---|---|---|---|---|
| a Calculated using ChemAxon.31 b Mean ΔTm ± SEM (number of measurements). c Mean pIC50 ± SEM (number of measurements) determined by AlphaScreen. d K d determined by ITC. e Not determined. | ||||||||
| 2 | nPrO- |

|
–CH3 | 2.6 | 1.1 ± 0.40 (2) pIC50 5.6 ± 0.12 (2)c | −0.35 ± 0.11 (2) | 0.32 ± 0.19 (2) | 5.6 ± 0.12 (2) |
| 3 |
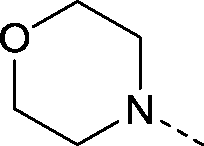
|

|
–CH3 | 1.8 | 5.7 ± 0.071 (2) pIC50 6.3 ± 0.066 (2)c | 1.9 ± 0.27 (2) | 1.3 ± 0.25 | 5.5 ± 0.033 (2) |
| 15 |

|

|
–CH3 | 2.6 | 0.81 ± 0.39 (2) | 0.28 ± 0.28 (2) | 0.34 ± 0.046 (2) | ND e |
| 16 | nPrO- |

|
–CH3 | 3.5 | 0.085 ± 0.60 (2) | −0.60 ± 0.042 (2) | −0.18 ± 0.24 (2) | ND |
| 17 |

|

|
–CH3 | 1.9 | 4.2 ± 0.0 (2) pIC50 6.3 ± 0.12 (2)c | 1.2 ± 0.057 (2) | 0.65 ± 0.060 (2) | 4.8 ± 0.10 (2) |
| 18 |
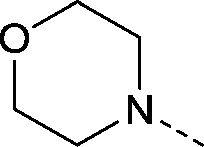
|

|
–CH3 | 2.2 | 1.3 ± 0.11 (2) | 0.82 ± 0.071 (2) | 0.53 ± 0.035 (2) | ND |
| 19 |

|

|
–CH3 | 2.4 | −0.23 ± 0.067 (2) | −0.39 ± 0.0035 (2) | −0.22 ± 0.053 (2) | ND |
| 20 |

|

|
–CH3 | 2.7 | −1.1 ± 0.21 (2) | −4.0 ± 0.47 (2) | −2.5 ± 0.12 (2) | ND |
| 21 |
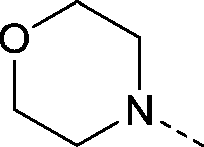
|
|
–CH3 | 1.8 | 0.35 ± 0.046 (2) | 0.075 ± 0.13 (2) | −0.040 ± 0.099 (2) | ND |
| 22 | nPrO- |
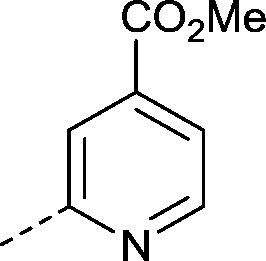
|
–CH3 | 2.6 | −0.030 ± 0.37 (2) | −0.24 ± 0.17 (2) | −0.090 ± 0.021 (2) | ND |
| 23 |

|
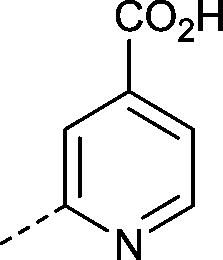
|
–CH3 | 1.5 | −0.060 ± 0.11 (2) | −0.24 ± 0.11 (2) | 0.010 ± 0.12 (2) | ND |
| 24 |

|
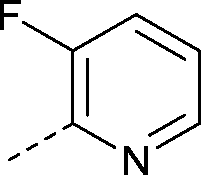
|
–CH3 | 1.9 | 1.3 ± 0.27 (2) | 0.41 ± 0.23 (2) | 0.71 ± 0.32 (2) | ND |
| 25 |
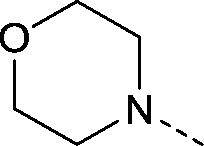
|
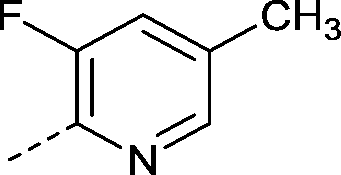
|
–CH3 | 2.5 | −0.28 ± 0.039 (2) | −0.42 ± 0.15 (2) | −0.48 ± 0.11 (2) | ND |
| 26 |
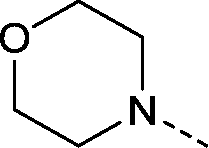
|

|
–CH3 | 1.5 | 0.035 ± 0.053 (2) | 0.43 ± 0.092 (2) | 0.29 ± 0.11 (2) | ND |
| 27 |
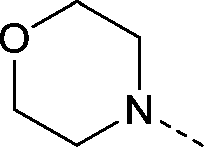
|

|
–CH3 | 3.1 | −0.30 ± 0.12 (2) | 0.55 ± 0.088 (2) | 0.040 ± 0.13 (2) | ND |
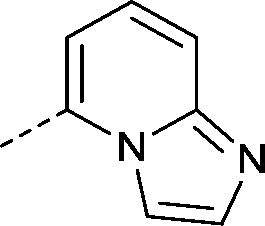
| 28 |
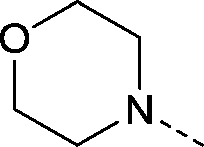
|

|
–CH3 | 1.3 | 4.5 ± 0.19 (2) pIC50 6.9 ± 0.065 (2)cKd 0.068 ± 0.0049 μMd | 1.3 ± 0.11 (2) | 1.7 ± 0.11 (2) | <4.0 (2) |
| 29 |
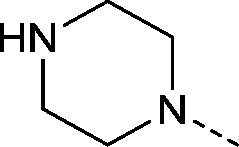
|

|
–CH3 | 1.5 | 5.7 ± 0.20 (2) | 4.0 ± 0.095 (2) | 1.7 ± 0.52 (2) | ND |
| 30 |

|
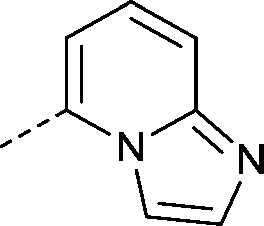
|
–CH3 | 1.0 | 5.1 ± 0.064 (2) | 2.6 ± 0.17 (2) | 2.3 ± 0.30 (2) | ND |
| 31 |

|

|
–CH3 | 1.4 | 7.5 ± 0.16 (2) | 4.1 ± 0.24 (2) | 3.5 ± 0.47 (2) | ND |
| 32 |

|

|
−Et | 2.0 | 3.0 ± 0.14 (2) | 0.98 ± 0.14 (2) | 0.20 ± 0.12 (2) | ND |
| 33 |
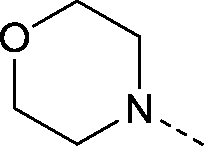
|

|
−CF3 | 2.5 | 0.26 ± 0.099 (2) | −0.40 ± 0.17 (2) | 0.20 ± 0.35 (2) | ND |
| 34 |

|

|
−Et | 2.1 | 1.2 ± 0.035 (2) | 0.51 ± 0.35 (2) | −0.15 ± 0.19 (2) | ND |
| 35 |
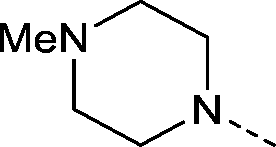
|

|
−iPr | 2.6 | 1.0 ± 0.19 (2) | 0.19 ± 0.70 (2) | 0.71 ± 0.24 (2) | ND |
| 36 |

|

|
−cPr | 2.2 | 0.76 ± 0.98 (2) | −0.65 ± 0.16 (2) | −0.085 ± 0.081 (2) | ND |
| 37 |

|

|
−nBu | 3.0 | 0.27 ± 0.095 (2) | −0.78 ± 0.19 (2) | −0.50 ± 0.067 (2) | ND |
| 38 |
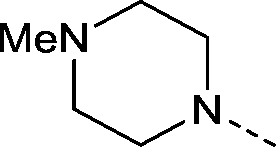
|
|
−CF3 | 2.5 | 0.30 ± 0.028 (2) | −0.70 ± 0.31 (2) | −0.26 ± 0.025 (2) | ND |
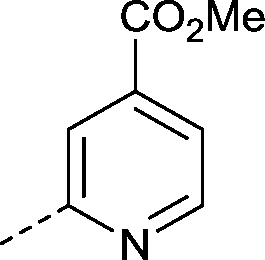
The indolizine target compounds (15–38) were synthesized by quaternisation of substituted pyridines4–14 followed by base-promoted [3 + 2] cycloaddition of the pyridinium salts with alkynes (Scheme 1).26,27 The analogues (15–38) were screened for binding to bromodomain proteins by differential scanning fluorimetry (DSF, ΔTm) (Table 1).28 In order to obtain an initial idea of potency and selectivity the analogues were initially screened against BRD9, BRD4(1) and bromodomain and PHD finger containing (BRPF) 1B. Propoxy analogues 16 and 22 showed significantly less affinity against BRD9 than the corresponding morpholino analogues 15 and 21 in the DSF assay, indicating that the morpholine ring promotes affinity for BRD9. The introduction of substituents onto the 3-, 4-, 5- and 6-positions of the pyridine ring, as in compounds 15, 17–21 and 23–25, generally led to a significant drop in BRD9 potency compared to compound 3. Out of the substituted pyridine analogues tested, compound 17 had the highest affinity for BRD9 (ΔTm 4.2 °C). Analogues were then prepared which replaced the pyridine ring with an alternative heterocycle (26, 27 and 28). The best tolerated of these was the imidazopyridine compound 28 (BRD9 ΔTm 4.5 °C).
 | ||
| Scheme 1 Synthesis of indolizine analogues. Reagents and conditions: (a) R3COCH2X (X = Cl,Br), THF or acetone (15–91%); (b) alkyne (R2CCH), K2CO3, DMF, 90 °C (2.3–40%). | ||
The SAR at the R1 position of the indolizine scaffold was further examined; the C-7 piperazine analogues were highly potent against BRD9, but were not very selective with respect to BRD4. For example, compound 31 was potent against BRD9 (ΔTm 7.5 °C), but was also relatively potent against BRD4(1) (ΔTm 4.1 °C) and BRPF1B (ΔTm 3.5 °C). The Tm shifts for BRD4(1) and BRPF1B would correspond to AlphaScreen IC50 ≤ 1 μM based on benchmarking with similar published compounds such as compound 28a in Hay, et al. (BRD4(1) IC50 0.20 μM, Tm 3.2)29 and compound 34 in Bennett, et al. (BRPF1B IC50 0.34 μM, Tm 6.2).30
The C-3 ketone analogues (32–38) with modifications at R3 generally showed a drop in BRD9 potency with respect to the methyl ketone analogues (e.g. compound 28). Compound 32 was the most encouraging of the larger ketones, which manifested moderate BRD9 affinity (ΔTm 3.0 °C), but no appreciable potency against BRD4(1) or BRPF1B (ΔTm < 1 °C). The pIC50 was determined for the most potent and selective compounds using the AlphaScreen assay (Table 1). Compound 17 was of similar potency to compound 3, with pIC50 6.3. Additionally, compound 17 was ~30-fold selective over BAZ2B in the AlphaScreen assay and showed a weak affinity for BRD4 in the DSF assay (1.2 °C). Compound 28 was highly potent against BRD9 (pIC50 6.9) and was almost completely inactive against BAZ2B. The Kd of compound 28 for BRD7 and BRD9 was measured by isothermal titration calorimetry (ITC), which confirmed that 28 was highly potent against BRD9 (Kd 68 nM) and slightly less potent against BRD7 (Kd 368 nM). For BRD9, the binding is apparently driven by a large enthalpic contribution (ΔH −10.7 kcal mol−1, TΔS −1.32 kcal mol−1) whereas BRD7 binding is entropy dominated (ΔH −2.64 kcal mol−1, TΔS 5.99 kcal mol−1) (ESI,† Table S2). No appreciable binding of compound 28 to BRD4(1) could be detected by ITC at 15 μM.
The broader selectivity of compound 28 for a selection of BRDs was assessed by DSF (Fig. 2A) which has been shown to correlate well with biolayer interferometry and chemoproteomic profiling as a measure of inhibitor selectivity in this series.24 As expected, compound 28 was potent against the BRD9 paralog BRD7 (ΔTm 5.6 °C). The results indicate that compound 28 is selective for BRD7/BRD9, with little affinity across BRD proteins from sub-families 1–3 and 5–8, including BAZ2A and BAZ2B. In addition to the previously discussed affinity against the BET family and BRPF1B, weak affinity was seen for the BRDs from CBP, p300 and FALZ (ΔTm 1.8, 2.0 and 1.1 °C respectively).
 | ||
| Fig. 2 (A) DSF screening of compound 28 against various BRD subfamily members, indicating selectivity for BRD7 and BRD9. (B) View from X-ray crystal structure of compound 28 complexed to BRD9 showing hydrogen-bond from ketone to N216 (3.06 Å) and a watermolecule (2.92 Å). A π–π interaction can also be seen between the indolizine ring of 28 and Y222; (C) As in (B) showing the surface of the binding pocket and selected residues which are in close proximity to 28. (D) Compound 28 decreases FRAP recovery time of a BRD9-GFP construct in a dose dependent manner in SAHA treated U2OS cells. | ||
The X-ray structure of compound 28 complexed to BRD9 was determined (Fig. 2B and C). The ketone of 28 forms the expected KAc mimicking interactions between N216 and Y163.1 Additionally, there is an apparent π–π interaction between the indolizine ring of compound 28 and the phenol of Y222. The C–7 morpholino moiety binds in a more hydrophilic region formed by residues I169 and Y173. The selectivity of compound 28 over BRD4(1) can be rationalised by the orientation of the C–1 imidazopyridine moiety which binds in a lipophilic region, sandwiched between the I169 and F160 side-chains. The corresponding region in BET BRDs is much narrower and cannot accommodate groups in this region.
Compound 28 was next profiled in a fluorescence recovery after photobleaching (FRAP) cellular assay (Fig. 2D) using green fluorescent protein fused to BRD9 (BRD9-GFP).21,32 When U2OS cells were treated with the pan-HDAC inhibitor SAHA to increase global histone acetylation, a clear decrease in recovery time could be seen between chromatin binding BRD9-GFP and the N100F KAc binding site mutant which can not bind chromatin. Increasing concentrations of compound 28 showed equivalent inhibition of BRD9-GFP/chromatin association as the non-binding N100F mutant. The FRAP assay confirms the cell permeability of compound 28 and its ability to inhibit nuclear BRD9/KAc-histone interactions.
In summary, the results reveal the feasibility of preparing potent and selective small moleculeinhibitors of the BRD7/BRD9 BRDs. Analogues of the indolizine lead2 were efficiently prepared via a [3 + 2] cycloaddition reaction. The optimised compound 28 is highly potent in vitro (Kd 68 nM) and selective over most of the other BRD sub-families, with modest affinity only for BRPF1B, CBP/p300 and FALZ. Further characterization of compound 28 in a FRAP assay of BRD9/chromatin showed it to be a potent inhibitor in cells. Used in conjunction with the other, structurally unrelated BRD9 inhibitors LP99 and I-BRD9, compound 28 will serve as a useful chemical probe to elucidate the biological roles of BRD7 and BRD9 in transcription and pathogenesis.
Funding sources
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, Merck, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z]. We also thank the European Union the Biotechnology and Biological Sciences Research Council (BBSRC) and the British Heart Foundation for funding.Acknowledgements
We thank Hiroshi Nara (Takeda) for kind assistance in proof-reading this manuscript.Notes and references
- P. Filippakopoulos, S. Picaud, M. Mangos, T. Keates, J.-P. Lambert, D. Barsyte-Lovejoy, I. Felletar, R. Volkmer, S. Müller, T. Pawson, A.-C. Gingras, C. H. Arrowsmith and S. Knapp, Cell, 2012, 149, 214–231 CrossRef CAS PubMed.
- E. Middeljans, X. Wan, P. W. Jansen, V. Sharma, H. G. Stunnenberg and C. Logie, PLoS One, 2012, 7 Search PubMed.
- C. Kadoch, D. C. Hargreaves, C. Hodges, L. Elias, L. Ho, J. Ranish and G. R. Crabtree, Nat. Genet., 2013, 45, 592–601 CrossRef CAS PubMed.
- M. D. Kaeser, A. Aslanian, M.-Q. Dong, J. R. Yates III and B. M. Emerson, J. Biol. Chem., 2008, 283, 32254–32263 CrossRef CAS PubMed.
- S. Tae, V. Karkhanis, K. Velasco, M. Yaneva, H. Erdjument-Bromage, P. Tempst and S. Sif, Nucleic Acids Res., 2011, 39, 5424–5438 CrossRef CAS PubMed.
- C. Peng, J. Zhou, H. Y. Liu, M. Zhou, L. L. Wang, Q. H. Zhang, Y. X. Yang, W. Xiong, S. R. Shen, X. L. Li and G. Y. Li, J. Cell. Biochem., 2006, 97, 882–892 CrossRef CAS PubMed.
- J. U. Kang, S. H. Koo, K. C. Kwon, J. W. Park and J. M. Kim, Cancer Genet. Cytogenet., 2008, 182, 1–11 CrossRef CAS PubMed.
- L. Scotto, G. Narayan, S. V. Nandula, S. Subramaniyam, A. M. Kaufmann, J. D. Wright, B. Pothuri, M. Mansukhani, A. Schneider, H. Arias-Pulido and V. V. Murty, Mol. Cancer, 2008, 7 Search PubMed.
- J. Zhou, J. Ma, B.-C. Zhang, X.-L. Li, S.-R. Shen, S.-G. Zhu, W. Xiong, H.-Y. Liu, H. Huang, M. Zhou and G.-Y. Li, J. Cell. Physiol., 2004, 200, 89–98 CrossRef CAS PubMed.
- C. Peng, H. Y. Liu, M. Zhou, L. M. Zhang, X. L. Li, S. R. Shen and G. Y. Li, Mol. Cell. Biochem., 2007, 303, 141–149 CrossRef CAS PubMed.
- A. E. Burrows, A. Smogorzewska and S. J. Elledge, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14280–14285 CrossRef CAS PubMed.
- J. Drost, F. Mantovani, F. Tocco, R. Elkon, A. Comel, H. Holstege, R. Kerkhoven, J. Jonkers, P. M. Voorhoeve, R. Agami and G. Del Sal, Nat. Cell Biol., 2010, 12, 380–389 CrossRef CAS PubMed.
- F. Mantovani, J. Drost, P. M. Voorhoeve, G. Del Sal and R. Agami, Cell Cycle, 2010, 9, 2777–2781 CrossRef CAS.
- M. T. Harte, G. J. O'Brien, N. M. Ryan, J. J. Gorski, K. I. Savage, N. T. Crawford, P. B. Mullan and D. P. Harkin, Cancer Res., 2010, 70, 2538–2547 CrossRef CAS PubMed.
- W.-J. Wu, K.-S. Hu, D.-L. Chen, Z.-L. Zeng, H.-Y. Luo, F. Wang, D.-S. Wang, Z.-Q. Wang, F. He and R.-H. Xu, Eur. J. Clin. Invest., 2013, 43, 131–140 CrossRef CAS PubMed.
- Y.-A. Park, J.-W. Lee, H.-S. Kim, Y.-Y. Lee, T.-J. Kim, C. H. Choi, J.-J. Choi, H.-K. Jeon, Y. J. Cho, J. Y. Ryu, B.-G. Kim and D.-S. Bae, Clin. Cancer Res., 2014, 20, 565–575 CrossRef CAS PubMed.
- J. Penkert, B. Schlegelberger, D. Steinemann and D. Gadzicki, Fam. Cancer, 2012, 11, 601–606 CrossRef CAS PubMed.
- Y.-A. Park, J.-W. Lee, J.-J. Choi, H.-K. Jeon, Y. Cho, C. Choi, T.-J. Kim, N. W. Lee, B.-G. Kim and D.-S. Bae, Gynecol. Oncol., 2012, 124, 125–133 CrossRef CAS PubMed.
- O. Fedorov, H. Lingard, C. Wells, O. P. Monteiro, S. Picaud, T. Keates, C. Yapp, M. Philpott, S. J. Martin, I. Felletar, B. D. Marsden, P. Filippakopoulos, S. Müller, S. Knapp and P. E. Brennan, J. Med. Chem., 2014, 57, 462–476 CrossRef CAS PubMed.
- S. Picaud, M. Strocchia, S. Terracciano, G. Lauro, J. Mendez, D. L. Daniels, R. Riccio, G. Bifulco, I. Bruno and P. Filippakopoulos, J. Med. Chem., 2015, 58, 2718–2736 CrossRef CAS PubMed.
- P. G. K. Clark, L. C. C. Vieira, C. Tallant, O. Fedorov, D. C. Singleton, C. M. Rogers, O. P. Monteiro, J. M. Bennett, R. Baronio, S. Müller, D. L. Daniels, J. Méndez, S. Knapp, P. E. Brennan and D. J. Dixon, Angew. Chem., Int. Ed., 2015, 54, 6217–6221 CrossRef CAS PubMed.
- M. E. Bunnage, E. L. P. Chekler and L. H. Jones, Nat. Chem. Biol., 2013, 9, 195–199 CrossRef CAS PubMed.
- C.-w. Chung, A. W. Dean, J. M. Woolven and P. Bamborough, J. Med. Chem., 2011, 55, 576–586 CrossRef PubMed.
- P. Chen, A. Chaikuad, P. Bamborough, M. Bantscheff, C. Bountra, C. W. Chung, O. Fedorov, P. Grandi, D. Jung, R. Lesniak, M. Lindon, S. Muller, M. Philpott, R. Prinjha, C. Rogers, C. Selenski, C. Tallant, T. Werner, T. M. Willson, S. Knapp and D. H. Drewry, J. Med. Chem., 2015 DOI:10.1021/acs.jmedchem.5b00209.
- M. Philpott, J. Yang, T. Tumber, O. Fedorov, S. Uttarkar, P. Filippakopoulos, S. Picaud, T. Keates, I. Felletar, A. Ciulli, S. Knapp and T. D. Heightman, Mol. BioSyst., 2011, 7, 2899–2908 RSC.
- R. S. Kulkarni, B. Padmashali and C. Sandeep, Tetrahedron Lett., 2013, 54, 6411–6414 CrossRef PubMed.
- X. He, K. Ju, Y. Shang, C. Wang, S. Yu and M. Zhang, Tetrahedron Lett., 2009, 50, 6981–6984 CrossRef PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. C. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- D. Hay, O. Fedorov, P. Filippakopoulos, S. Martin, M. Philpott, S. Picaud, D. S. Hewings, S. Uttakar, T. D. Heightman, S. J. Conway, S. Knapp and P. E. Brennan, Med. Chem. Commun., 2013, 4, 140–144 RSC.
- J. Bennett, O. Fedorov, C. Tallant, O. Monteiro, J. Meier, V. Gamble, P. Savitsky, G. A. Nunez-Alonso, B. Haendler, C. Rogers, P. E. Brennan, S. Muller and S. Knapp, J. Med. Chem., 2015 DOI:10.1021/acs.jmedchem.5b00458.
- Calculator Plugins were used for structure property prediction and calculation, Marvin 15.1.26.0, 2015, ChemAxon (http://www.chemaxon.com) Search PubMed.
- M. Philpott, C. M. Rogers, C. Yapp, C. Wells, J. P. Lambert, C. Strain-Damerell, N. A. Burgess-Brown, A. C. Gingras, S. Knapp and S. Muller, Epigenet. Chromatin, 2014, 7, 14 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5md00152h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |