
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePPARα agonists based on stilbene and its bioisosteres: biological evaluation and docking studies†
Barbara
De Filippis
*,
Mariangela
Agamennone
,
Alessandra
Ammazzalorso
,
Isabella
Bruno
,
Alessandra
D'Angelo
,
Mauro
Di Matteo
,
Marialuigia
Fantacuzzi
,
Letizia
Giampietro
,
Antonella
Giancristofaro
,
Cristina
Maccallini
and
Rosa
Amoroso
*
Dipartimento di Farmacia, Università degli Studi “G. d'Annunzio”, via dei Vestini 31, 66100 Chieti, Italy
First published on 18th June 2015
Abstract
A new series of gemfibrozil analogues conjugated with trans-stilbene were synthesized and evaluated with the aim of developing new PPARα agonists. The phenyls of stilbene were modified by introducing substituents in the ortho or para position and only the distal ring was substituted with naphthyl or heteroaromatic moieties, keeping the dimethylpentanoic skeleton of gemfibrozil unaltered. Two compounds, 5a and 5d, exhibited good activation of PPARα and were also screened for their activity on PPARα-regulated gene CPT1A. Structure-based studies carried out on the active ligands highlighted the dominant role of ligand solvation energy and hydrophobic effect in determining the PPARα activation.
Introduction
Peroxisome Proliferator-Activated Receptors (PPARs) are transcription factors belonging to the nuclear receptor superfamily, whose members selectively bind lipophilic ligands and transduce chemical signals into specific changes in gene expression. PPAR is a compact molecule, consisting of four functional domains: N-terminal ligand-independent activation domain, DNA binding domain (DBD), co-FBD (site for cofactors coupling) or D domain and C-terminal ligand binding domain (LBD). The binding of PPAR with a ligand induces conformational changes which permit the interaction with Retinoid X Receptor (RXR), thus building an activated heterodimer capable of recognizing the DNA PPAR responsive elements (PPREs). In this process, the nuclear corepressors, often functioning as transcriptional repressors, are replaced with coactivators resulting in transcriptional activation.1There are three PPAR isotypes, designed as PPARα, PPARβ/δ and PPARγ; they are a tightly connected triad and act as regulators of the intermediary metabolism of glucose and lipid homeostasis, adipogenesis, immune response, cell growth, and differentiation.2 Despite the high levels of homology at the protein level, the diversity of PPARs implies diverse distribution in body tissues and different ligand responses. Recently, the development of pan and selective ligands has greatly advanced the understanding of the pathways controlled by PPARs and the therapeutic implications of modulating these receptors.3 The PPARα isoform is highly expressed in the liver, heart, brown adipose tissue, skeletal muscle, and kidney; its activation by natural ligands, fatty acids (FA) and eicosanoids promotes mitochondrial FA oxidation and regulates lipid metabolism. Synthetic agonists of PPARα are hypolipidemic drugs such as fibrates.4
PPARγ, expressed predominantly in adipose tissue, liver, and immune cells, plays crucial roles in fat and glucose metabolism, cell proliferation, differentiation, and apoptosis. PPARγ agonists like thiazolidinediones are used to treat type 2 diabetes, thus improving insulin signaling and glucose uptake by adipose tissue.5 PPARβ/δ is the most widely expressed PPAR subtype, found in liver, skin and large and small intestines; however, the role of PPARβ/δ has been less explored. To date, there are no PPARβ/δ drugs being marketed, but some ligands are currently in clinical trials for metabolic syndrome.6
As a part of the ongoing research to find an effective PPAR-target based drug candidate, we have recently reported PPAR agonists derived from the combination of the antilipidemic moiety of fibrate and natural α-asarone, stilbene, chalcone, and other bioisosteric modifications.7 Good agonistic activity on PPARα was seen with the trans-stilbene derivative (1) of gemfibrozil (EC50 = 1.0 μM). Starting from these studies, we achieved an initial SAR study by systematically varying the structural features of lead 1 to assess the contribution of various changes in the hydrophobic stilbene scaffold to the binding affinity. Particularly, we focused our attention on the distal phenyl group, since this moiety might be critical for the active conformation of PPARα LBD. A suitable structural manipulation might influence the ability of the ligand-bound PPARα to form a transcriptionally active conformation. By varying the nature of the para substituent of distal phenyl or by substituting it with a naphthyl, thiophene or pyridine ring, we sought to estimate the contribution of electronic, steric and hydrogen bonds to the binding affinity. Since the compound containing chlorine was the most active, we also explored the effect of additional chlorine atoms in the ortho positions of the two phenyls of stilbene (Fig. 1).
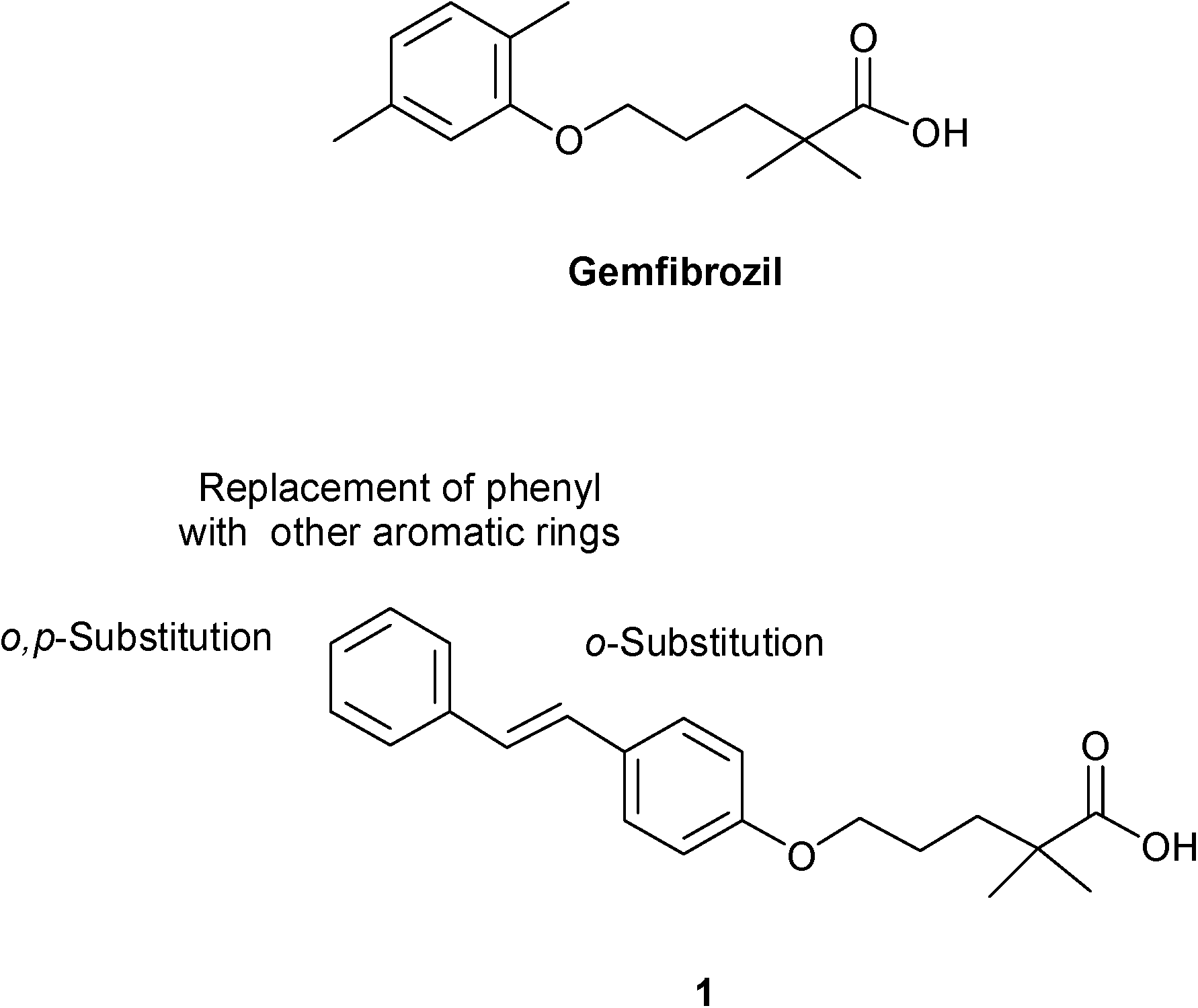 | ||
| Fig. 1 Gemfibrozil, compound 1 and chemical modifications. | ||
The in vitro transactivation assay was used to test the PPAR activation of the new compounds. Ligands with the best EC50 were also selected for the in vitro evaluation of CPT1A gene activation by real-time quantitative PCR (RTqPCR) analysis. To provide an explanation for the observed activity data, docking and post-docking calculations of the studied ligands on the PPARα receptor were carried out, eliciting the most relevant energy contributions to the binding process.
Results and discussion
The reaction of 4-hydroxybenzaldehyde with the appropriate arylacetic acid (2a–l and 2n–p) in the presence of piperidine, at 130 °C, fournished the phenols 3a–l and 3n–p.8 Phenol 3g (Ar = p-NO2C6H4) was reduced to p-aminoderivative 3m and the potassium salts of 3a–p, except 3g, were then reacted with commercially available isobutyl 5-chloro-2,2-dimethylpentanoate, in refluxing DMF, to give the esters 4a–f and 4h–p. Ester 4g showed instability in these reaction conditions thus it has not been possible to synthesize the corresponding acid. The acids 5a–f and 5h–p were finally obtained by hydrolysis of 4a–f and 4h–p in the presence of NaOH in EtOH (Scheme 1).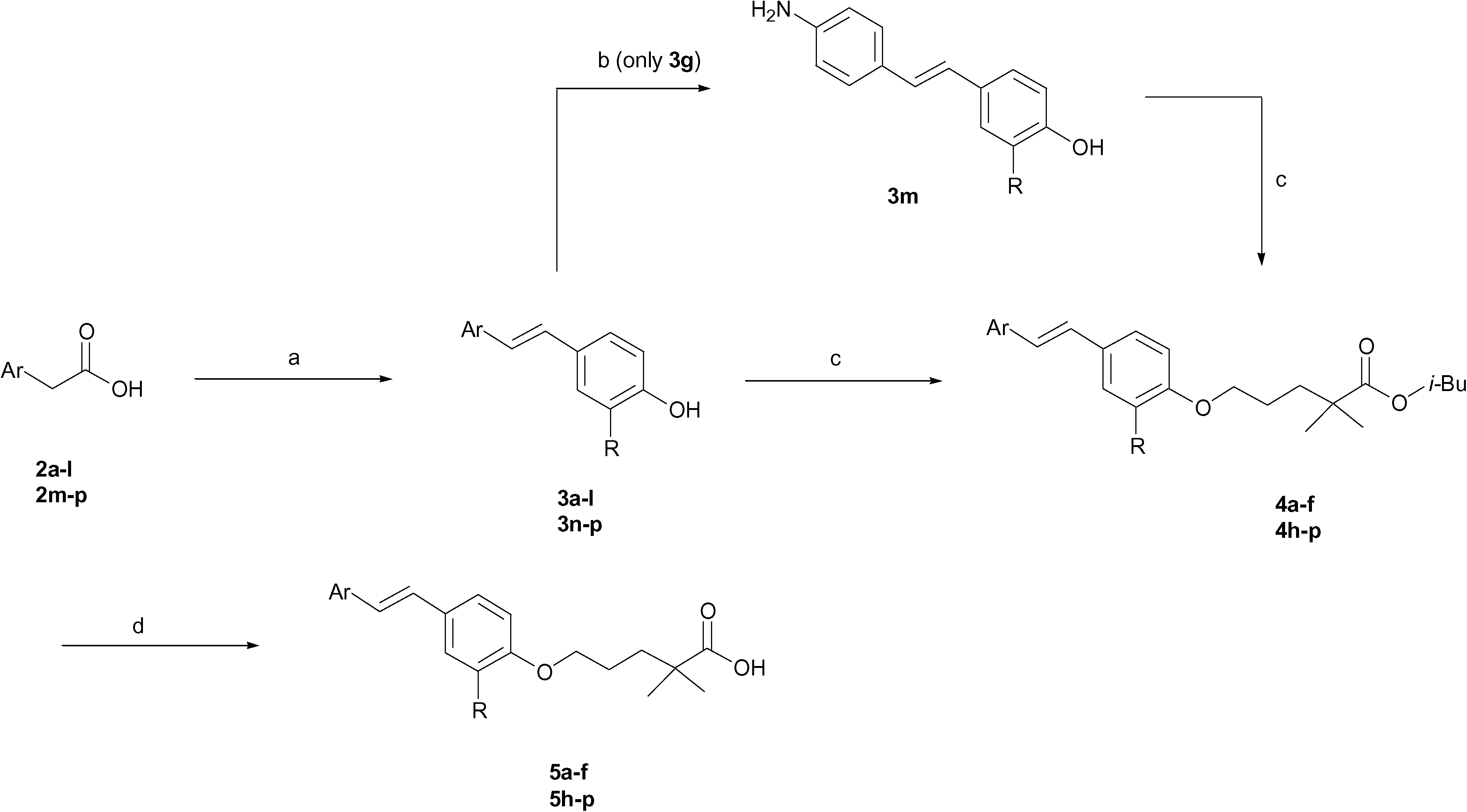 | ||
| Scheme 1 Reagents and conditions: (a) 4-hydroxybenzaldehyde, piperidine, 130 °C, 4–24 h; (b) SnCl2, EtOH, HCl, reflux, 3.5 h; (c) isobutyl 5-chloro-2,2-dimethylpentanoate, K2CO3, DMF, reflux, 5–12 h; (d) NaOH 1%, EtOH, reflux, 8–12 h. | ||
In order to evaluate PPARα agonistic potency and subtype selectivity, we performed an in vitro screening of the new compounds in a cell-based transactivation assay in eukaryotic cells;9 our reporter system utilizes firefly luciferase reporter gene technology to provide optimal assay sensitivity, dynamic range when quantifying nuclear receptor activity, and good correlation with in vivo activity.10 Compounds 5a–f and 5h–p were evaluated for their agonist activity on the human PPARα, PPARγ, and PPARδ subtypes. The results obtained were compared with the corresponding data for gemfibrozil, rosiglitazone, and L-165,041, which were used as reference compounds in the PPARα, PPARγ, and PPARδ transactivation assays, respectively. As the first step, we evaluated the ability of the new compounds to activate PPAR at a concentration of 150 μM and the results are expressed as efficacy (E, %), calculated as the percentage of the maximum obtained fold induction with the reference compounds, normalized to 100%. On the basis of this preliminary study, compounds with E (%) higher than the reference compounds were selected for the determination of the EC50 values. The in vitro PPAR activity results are summarized in Table 1.
| Compound | Ar | R | PPARα | PPARγ | PPARδ | |
|---|---|---|---|---|---|---|
| E a (%) | EC50 (μM)b | E (%) | E (%) | |||
| a E (%), efficacy values calculated as percentage of the maximum obtained fold induction with the reference compounds (gemfibrozil for PPARα, rosiglitazone (EC50 0.039 ± 0.003 μM) for PPARγ, and L-165,041 (EC50 0.021 ± 0.001 μM) for PPARδ); compounds were tested in at least three separate experiments at 150 μM. The results are expressed as mean ± SEM. b Compounds were tested in at least three separate experiments at five concentrations ranging from 1 to 150 μM. The results are expressed as mean ± SEM. c Not determined. | ||||||
| 5a | p-ClC6H4 | H | 227 | 0.8 ± 0.07 | 25 | 20 |
| 5b | p-MeC6H4 | H | 118 | 3.3 ± 0.02 | 34 | 18 |
| 5c | p-i-PrC6H4 | H | 17 | —c | 38 | 42 |
| 5d | p-CF3C6H4 | H | 205 | 0.76 ± 0.02 | 55 | 40 |
| 5e | p-CNC6H4 | H | 23 | — | 89 | 36 |
| 5f | p-OMeC6H4 | H | 164 | 28.4 ± 0.1 | 65 | 18 |
| 5h | Naphthyl | H | 123 | 90.0 ± 1.15 | 22 | 26 |
| 5i | Thienyl | H | 152 | 3.8 ± 0.15 | 78 | 17 |
| 5l | Pyridinyl | H | 115 | 103.2 ± 1.15 | 42 | 39 |
| 5m | p-NH2C6H4 | H | 29 | — | 54 | 59 |
| 5n | C6H5 | Cl | 46 | — | 44 | 40 |
| 5o | o,p-ClC6H4 | H | 32 | — | 30 | 60 |
| 5p | o,p-ClC6H4 | Cl | 20 | — | 18 | 12 |
| 1 | 223 | 1.0 ± 0.02 | 25 | 17 | ||
| Gemfibrozil | 100 | 59.0 ± 3.9 | — | — | ||
| Rosiglitazone | — | — | 100 | — | ||
| L-165,041 | — | — | — | 100 | ||
All compounds showed activation of PPARα, with good efficacy values, except 5c (p-iPrC6H4), 5e (p-CNC6H4), and 5m (p-NH2C6H4), which were inactive. Good efficacies on PPARα were achieved with derivatives 5a (p-ClC6H4) and 5d (p-CF3C6H4), which exhibited values 2 fold of gemfibrozil, comparable with stilbene derivative 1. Compounds 5b (p-MeC6H4), 5f (p-OMeC6H4), 5h (naphthyl), 5i (thienyl), and 5l (pyridinyl) showed PPARα affinity comparable to gemfibrozil and lower than stilbene derivative 1. Based on the above results, compounds 5a, 5b, 5d, 5f, 5h, 5i, and 5l were subjected to EC50 determination. The best results were obtained with 5a and 5d, which contain the electron-withdrawing substituents Cl and CF3; they showed approximately the same potency towards PPARα (EC50 0.8 μM and 0.7 μM, respectively) as compared to 1 (EC50 1.0 μM), and were more active than gemfibrozil. Compounds 5b and 5i showed improved agonistic activity (EC50 3.3 μM and 3.8 μM, respectively) compared to gemfibrozil, but were found to be less active as compared to unsubstitued stilbene 1. At last, 5f, 5h, and 5l showed a decrease in PPARα activity as compared to the parent compound 1, even worse than gemfibrozil. PPARα seems to be able to accommodate the different substituents without greatly affecting the activity except for analogs 5o–p which show very low activity. The insertion of a chlorine atom in the ortho position of the proximal aromatic ring caused a drastic reduction in activity on PPARα (5n and 5pvs.5a) as well as the introduction of a second chlorine atom in the ortho position of the proximal one. These results showed that the effect of the substitution pattern of the aromatic moiety on the binding affinity is strongly dependent on the respective substituent, indicating that the effect of different substitutions on the binding affinity towards the PPARα-LBD is not merely additive. At last, none of the tested compounds was able to exhibit full agonist activity on PPARγ and PPARδ, showing lower potency than rosiglitazone and L-165,041, regardless of the substituent introduced on stilbene.
On the basis of these results, we decided to confirm the capability of 5a and 5d (with the best EC50 values) to act as PPARα ligands by conducting gene expression experiments in HepG2 cells, which express PPARα, and the results were compared with those obtained from gemfibrozil. The enzyme carnitine palmitoyltransferase 1A (CPT1A) catalyzes the long-chain fatty acid translocation into the mitochondrial matrix, an essential step for β-oxidation in the liver,11 and PPARα is known to up-regulate CPT1A expression through ligand-dependent transcriptional activation. For this reason, the expression pattern of CPT1A is a well established in vitro model to study PPARα activation.12 The human hepatocellular carcinoma cell line HepG2 was used to analyze the impact of 5a and 5d on PPARα expression; measurement of mRNA concentration was done as a quantitative analysis by RTqPCR. HepG2 cells were stimulated with increasing amounts of selected compounds (from 1 to 100 μM) and compared with gemfibrozil and 1 at EC50 concentration; control cells were treated with only DMSO. The stimulation of HepG2 cells with gemfibrozil showed a slight increase of mRNA levels, while incubation of cells with increasing amounts of 1 led to a significant and concentration-dependent increase of mRNA levels; indeed, at an intermediate concentration of 10 μM, the mRNA induction was doubled with respect to gemfibrozil and at 100 μM the relative mRNA was about 6.0 (Fig. 2).
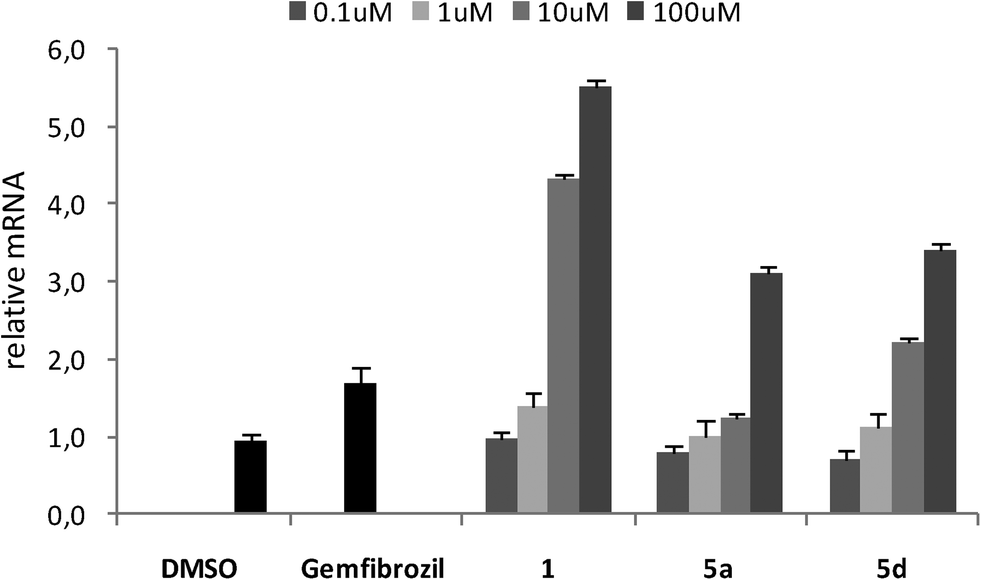 | ||
| Fig. 2 CPT1A expression in HepG2 cells after treatment. RTqPCR was performed to measure the CPT1A mRNA levels. Values shown represent the mean ± SEM of four independent determinations performed in duplicate. Cyclophilin was used as reference gene, and values were normalized to the data obtained from vehicle treated cells. | ||
Compounds 5a and 5d did not affect the expression of CPT1A gene in the same manner as 1, even if a dose-dependent activation of gene expression was observed for both compounds. At a maximum concentration of 100 μM, the mRNA induction for 5a and 5d was approximately doubled with respect to gemfibrozil; moreover, compound 5d also showed a value of induction greater than gemfibrozil at an intermediate concentration of 10 μM.
Docking calculations on PPARα-LBD were carried out to gain more insight into their putative binding mode. All stilbene derivatives, in the deprotonated form, were docked in the crystal structure of PPARα with PDB ID: 2P54 (details of the docking protocol are reported in the ESI†). This X-ray structure was chosen among available crystallographic complexes because of its good resolution (1.79 Å) and the structural similarity between the co-crystallized ligand and the studied compounds. Glide's13 docking protocol was validated by the reproduction of the crystallographic pose of X-ray ligands. This preliminary analysis allowed us to evaluate the ability of the receptor structure to correctly reproduce the X-ray ligands' binding mode.
To further assess the reliability of our model, docked poses of each ligand were subjected to a post-docking refinement using the MM-GBSA module in Prime. This approach minimizes docked complexes, allowing a more accurate calculation of the binding energy. Energy contributions calculated by this method were used to build a customized scoring function (1) that allowed the correlation of the computational and experimental EC50 for the active ligands. To this aim, a multiple linear regression approach was applied to generate a model with good statistical parameters (R2 = 0.98, Std Dev = 0.13, q2loo = 0.96, F = 109.2, P = 2.72 × 10−4), underpinning the reliability of the predicted docking poses (Fig. 3).
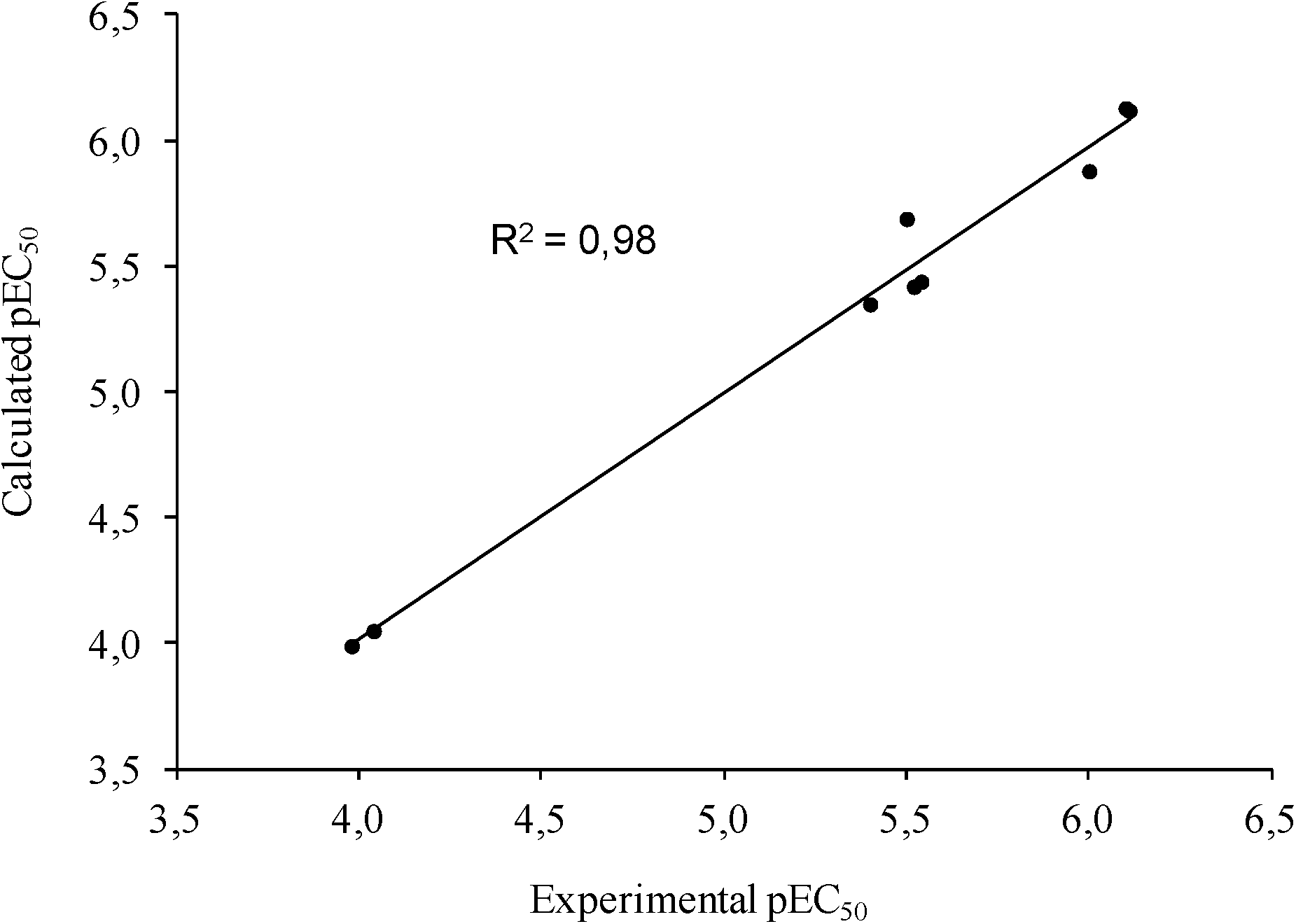 | ||
| Fig. 3 The predicted versus experimental pEC50 values for the activation of PPARα. | ||
It is worth noting that the pEC50 values come from a cell-based assay evaluating the activation of PPARα, and is not directly related to the binding of the ligands to the receptor. However, as exhaustively explained by Lannutti et al.,14 pEC50 and binding ΔG can be considered linearly related.
| pEC50 = 2.96 + 101 + (9.72 × 10−1 × Ligand Solv GB) + (4.32 × 10−2 × Rec Strain Lipo) − (2.04 × 10−1 × Rec Strain SelfCont) | (1) |
The analysis of energy parameters mostly contributing to the ligand potency showed the relevant role of ligand solvation energy (Ligand Solv GB). Interestingly, by evaluating a single correlation with separate parameters, we found that Ligand Solv GB has a strong correlation with pEC50 (R2 = 0.89), contributing significantly to discriminate the relative binding energy and indicating that the ligand affinity is predominantly influenced by the desolvation effect of the active ligands. Besides this aspect, the ligand binding effect on the receptor also plays a relevant role, in particular due to improved hydrophobic contacts (Rec Strain Lipo) and the conformational rearrangement affecting the receptor residue self-contacts (Rec Strain SelfCon). The accurate structure-based protocol, applied to all active stilbene derivatives, afforded docked poses very close to that of the co-crystallized ligand, occupying the L-shaped PPARα pocket. Putative binding poses of the representative compound 5d are stabilized by a combination of H-bonds and hydrophobic interactions (Fig. 4).
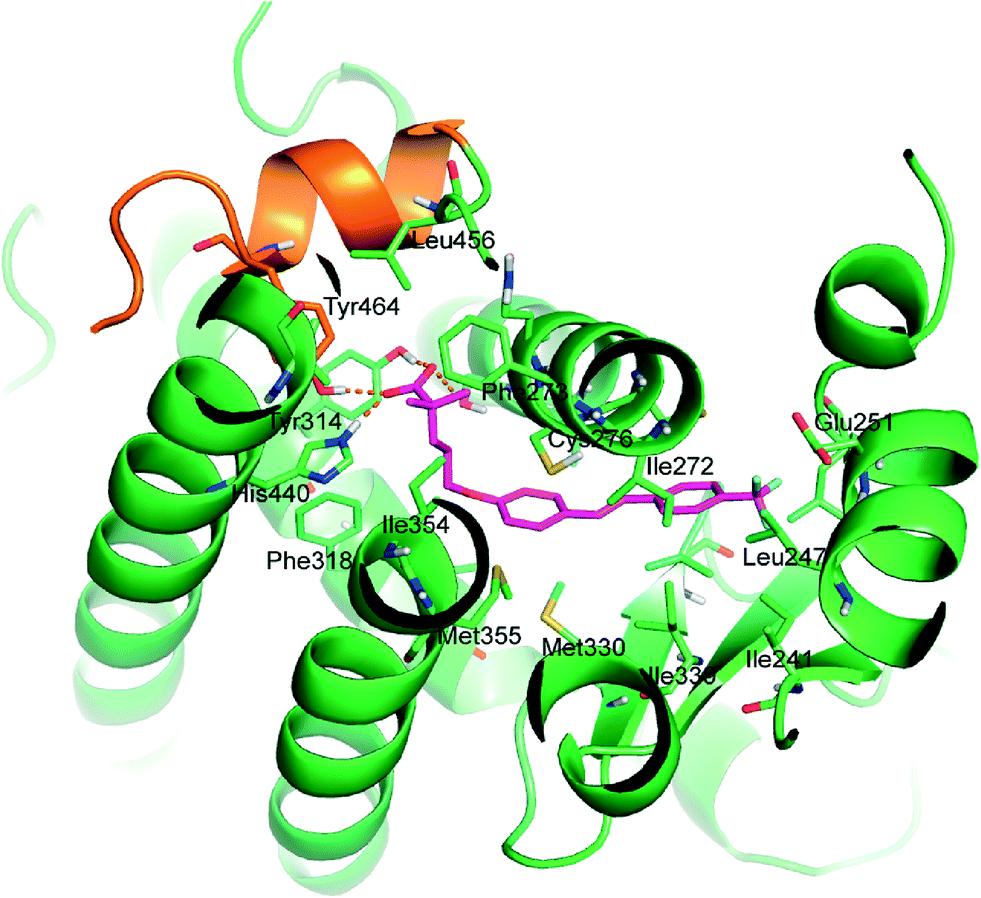 | ||
| Fig. 4 Binding mode of 5d (magenta C atoms) in the PPARα biding site represented as green cartoon. AF2 helix is reported as an orange cartoon. Interacting residues are represented as sticks. Carboxylate H-bonds are reported as orange dashed lines. | ||
The carboxylate group forms the well-recognized H-bond network with Ser280 (H3), Tyr314 (H5), His440 (H11), and Tyr464 (AF2 helix), the latter being essential for the recruitment of co-activators. The remaining interactions between ligands and protein are hydrophobic: the two methyl substituents in α provide positive contacts with Phe273, Val444, Leu456, Gln277, Ile354, and Cys276. The alkyl chain forms good interactions with Phe318, Ile354, His440, and Cys276. The stilbene function interacts with Leu231, Met330, Cys276, Ile272, Cys275, Leu247, Ile339, Val332, Thr279, Met355, Met330, and Leu321. The substituent in the para position, responsible for the different activities among the studied analogues, occupies the bottom of the hydrophobic pocket and engages in favourable lipophilic interactions with Ile241, Leu254, Leu247, and Cγ of Glu251.
Conclusions
In this paper new gemfibrozil analogues conjugated with trans-stilbene were synthesized and evaluated as PPAR agonists. Stilbene was modified by introducing various substituents on the two aromatic rings or replacing the distal phenyl with naphthyl or heteroaromatic moieties, keeping the dimethylpentanoic skeleton of gemfibrozil unaltered. Two compounds, 5a and 5d, exhibited good activation of PPARα at submicromolar concentration, selectivity towards PPARγ and β/δ and a dose dependent activation of PPARα target gene CPT1A, and were also screened for their activity on PPARα-regulated gene CPT1A. Considering the degree of the isoform selectivity displayed by these compounds, we identified new ligands with good potency on PPARα, but low potency and partial agonism on PPARγ and PPARδ. Moreover, structure-based studies carried out on the active ligands highlighted the dominant role of the ligand solvation energy and the hydrophobic effect in determining the PPARα activation.Acknowledgements
The authors gratefully acknowledge the University “G. d'Annunzio” of Chieti for financial support and Prof. Antonio Moschetta (University of Bari, Italy) for his helpful comments and suggestions.References
- (a) B. P. Kota, T. H. Huang and B. D. Roufogalis, Pharmacol. Res., 2005, 51, 85 CrossRef CAS PubMed; (b) J. Berger and D. E. Moller, Annu. Rev. Med., 2002, 53, 409 CrossRef CAS PubMed.
- (a) M. Moreno, A. Lombardi, E. Silvestri, R. Senese, F. Cioffi, F. Goglia, A. Lanni and P. de Lange, PPAR Res., 2010, 435689 Search PubMed; (b) T. Li and J. Y. L. Chiang, PPAR Res., 2009, 501739 Search PubMed; (c) L. la Cour Poulsen, M. Siersbæk and S. Mandrup, Semin. Cell Dev. Biol., 2012, 23, 631 CrossRef CAS PubMed.
- (a) C. Pirat, A. Farce, N. Lebègue, N. Renault, C. Furman, R. Millet, S. Yous, S. Speca, P. Berthelot, P. Desreumaux and P. Chavatte, J. Med. Chem., 2012, 55, 4027 CrossRef CAS PubMed; (b) P. L. Feldman, M. H. Lambert and B. R. Henke, Curr. Top. Med. Chem., 2008, 8, 728 CrossRef CAS PubMed; (c) A. Ammazzalorso, B. De Filippis, L. Giampietro and R. Amoroso, ChemMedChem, 2013, 10, 1609 Search PubMed.
- (a) J. C. Fruchart, Atherosclerosis, 2009, 205, 1 CrossRef CAS PubMed; (b) B. Staels, M. Maes and A. Zambon, Nat. Clin. Pract. Cardiovasc. Med., 2008, 5, 542 CrossRef CAS PubMed.
- (a) M. Ahmadian, J. Myoung Suh, N. Hah, C. Liddle, A. R. Atkins, M. Downes and R. M. Evans, Nat. Med., 2013, 99, 557 CrossRef PubMed; (b) B. Cariou, B. Charbonnel and B. Staels, Trends Endocrinol. Metab., 2012, 23, 205 CrossRef CAS PubMed.
- (a) K.-D. Wagner and N. Wagner, Pharmacol. Ther., 2010, 125, 423 CrossRef CAS PubMed; (b) L. S. Mackenzie and L. Lione, Life Sci., 2013, 93, 963 CrossRef CAS PubMed.
- (a) B. De Filippis, A. Giancristofaro, A. Ammazzalorso, A. D'Angelo, M. Fantacuzzi, L. Giampietro, C. Maccallini, M. Petruzzelli and R. Amoroso, Eur. J. Med. Chem., 2011, 46, 5218 CrossRef CAS PubMed; (b) B. De Filippis, P. Linciano, A. Ammazzalorso, C. Di Giovanni, M. Fantacuzzi, L. Giampietro, A. Laghezza, C. Maccallini, P. Tortorella, A. Lavecchia, F. Loiodice and R. Amoroso, Eur. J. Med. Chem., 2015, 89, 817 CrossRef CAS PubMed.
- (a) E. S. Lazer, H. C. Wong, G. J. Possanza, A. G. Graham and P. R. Farina, J. Med. Chem., 1990, 33, 1892 CrossRef CAS PubMed; (b) S. Hang, M. P. Oya, C. Kung, D. L. Hou, H. F. Maier and J. Kung, Med. Chem., 2005, 48, 5980 CrossRef PubMed.
- I. G. Schulman and R. A. Heyman, Chem. Biol., 2009, 11, 639 CrossRef PubMed.
- L. H. Naylor, Biochem. Pharmacol., 1999, 58, 749 CrossRef CAS PubMed.
- J. D. McGarry and N. F. Brown, Eur. J. Biochem., 1997, 244, 1 CAS.
- J. W. Lawrence, Y. Li, S. Chen, J. G. De Luca, J. P. Berger, D. R. Umbenhauer, D. E. Moller and G. Zhou, J. Biol. Chem., 2001, 276, 31521 CrossRef CAS PubMed.
- Glide, version 5.8, Schrödinger, LLC, New York, NY, 2012.
- F. Lannutti, A. Marrone and N. Re, J. Mol. Graphics Modell., 2011, 29, 865 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5md00151j |
| This journal is © The Royal Society of Chemistry 2015 |