
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of carboxyimidamide-substituted benzo[c][1,2,5]oxadiazoles and their analogs, and evaluation of biological activity against Leishmania donovani†
Leena
Keurulainen
a,
Mikko
Heiskari
a,
Satu
Nenonen
a,
Abedelmajeed
Nasereddin
b,
Dmitry
Kopelyanskiy
b,
Teppo O.
Leino
a,
Jari
Yli-Kauhaluoma
a,
Charles L.
Jaffe
b and
Paula
Kiuru
*a
aFaculty of Pharmacy, Division of Pharmaceutical Chemistry and Technology, University of Helsinki, Viikinkaari 5 E, P. O. Box 56, FI-00014 Helsinki, Finland. E-mail: paula.kiuru@helsinki.fi
bDepartment of Microbiology and Molecular Genetics, IMRIC, Hebrew University-Hadassah Medical School, P. O. Box 12272, Jerusalem 9112102, Israel
First published on 7th August 2015
Abstract
A facile synthesis route to carboxyimidamide-substituted benzoxadiazoles and related derivatives was developed. A total of 25 derivatives were synthesized. They were evaluated for antileishmanial activity by inhibition of Leishmania donovani axenic amastigote growth using a fluorescent viability microplate assay. The most promising derivative (14) demonstrated an antileishmanial EC50 of 4.0 μM, and it also showed activity in infected macrophages (EC50 5.92 μM) without signs of cytotoxicity.
1. Introduction
Leishmaniasis is a spectrum of human diseases caused by at least 20 different species of protozoan parasites. These parasites are responsible for three major diseases: cutaneous, visceral, and mucocutaneous. Over 300 million people are at risk.1 Visceral leishmaniasis (VL), caused primarily by L. donovani (anthroponotic) or L. infantum/L. chagasi (zoonotic), is fatal if left untreated. There are an estimated 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cases of VL per year, and 20000 to 40000 deaths. Over 90% of new cases occur in six countries: Bangladesh, Brazil, Ethiopia, India, South Sudan, and Sudan. Although two new drugs, miltefosine and paromomycin, are currently in use and combination therapy is becoming more common,2 an urgent need for new antileishmanial chemotypes persists. The major issues to be confronted in Leishmania treatment are the toxicity of primary medicines and the development of parasite resistance.
000 cases of VL per year, and 20000 to 40000 deaths. Over 90% of new cases occur in six countries: Bangladesh, Brazil, Ethiopia, India, South Sudan, and Sudan. Although two new drugs, miltefosine and paromomycin, are currently in use and combination therapy is becoming more common,2 an urgent need for new antileishmanial chemotypes persists. The major issues to be confronted in Leishmania treatment are the toxicity of primary medicines and the development of parasite resistance.
In addition, poor efficacy in the case of visceral leishmaniasis/HIV coinfection has raised the need for new antileishmanial treatments.3
In our previous study, 2-arylbenzimidazole and benzo[c][1,2,5]oxadiazole derivatives showed activity against the intracellular Gram-negative bacterium Chlamydia pneumoniae.4 In that study, a similarity-based model of C. pneumoniae dimethyladenosine transferase was used to virtually screen compounds from commercially available databases against C. pneumoniae. The 2-arylbenzimidazoles found were then tested and proven active against L. donovani.5 This prompted us to consider benzo[c][1,2,5]oxadiazole-derived compounds as a new family of antileishmanial agents. In addition, benzoxadiazoles and their N-oxides have been previously studied as antibiotic and antiparasitic agents.6 In this study, a facile synthesis route to carboxyimidamide-substituted benzoxadiazoles was developed, a set of 25 benzo[c][1,2,5]oxadiazole derivatives and other structurally related compounds was synthesized and evaluated as antileishmanial agents.
2. Results and discussion
2.1 Chemistry
Initially, benzo[c][1,2,5]oxadiazole-5-carbonitrile 4 was to be synthesized from 5-chloro- or 5-bromobenzo[c][1,2,5]oxadiazole in a microwave-assisted reaction using NaCN, CuCN, or K4[Fe(CN)6],7 but these syntheses failed to produce the target nitrile. Instead the synthesis of benzo[c][1,2,5]oxadiazole derivatives was started from the commercially available 4-amino-3-nitrobenzoic acid (Scheme 1). It was converted to benzo[c][1,2,5]oxadiazole-5-carboxylic acid (1) by using a two-step procedure via the N-oxide intermediate that was produced in the presence of sodium hypochlorite in an alkaline EtOH–H2O solution and subsequently reduced to the carboxylic acid 1.8 Next, compound 1 was converted to the primary amide 3via the acyl chloride 2 in the presence of aqueous ammonia in 1,4-dioxane. The reaction of the obtained primary amide 3 with trifluoroacetic anhydride and Et3N in THF9 gave the corresponding nitrile 4, which was converted to the amidoxime 5 by using hydroxylamine hydrochloride10 in the presence of Et3N in EtOH. The final step to obtain the derivatives (6–18, 24) was carried out in the presence of the substituted phenyl isocyanates in THF or CHCl3. The products 21–23 were synthesized in a similar manner by using a solution of Et3N in THF. Compound 20 was synthesized from the acyl chloride 2via the hydroxamic acid 19. The final products 33–39 were synthesized from the commercially available nitriles by using the methods described above (Scheme 2). Finally, the related benzimidazole derivative 25 was formed from 3,4-diaminobenzonitrile in refluxing formic acid.11 | ||
| Scheme 1 Synthetic procedures and chemical structures of the products 2–24. Reagents and conditions: a) KOH, EtOH, H2O, 70 °C, 2 h, then NaOCl, 0 °C → rt, 21 h, 87%; b) P(OEt)3, EtOH, 70 °C, 17 h, 67%; c) SOCl2, reflux, 4 h, 97%; d) NH3 aq., 1,4-dioxane, 0 °C, 1.5 h, 90%; e) TFAA, Et3N, THF, rt, 3 h, 79%; f) H2NOH·HCl, Et3N, EtOH, rt, 2 h, 94%; g) substituted phenyl isocyanate, THF or CHCl3, rt, 3–21 or 96–120 h, 27–70%; h) MeOH, 0 °C, 0.5 h then H2NOH·HCl, MeOH, rt, 1.5 h, 57%; i) THF, phenyl isocyanate, 57%; j) PhCOCl/PhOCOCl/PhCH2COCl, Et3N, THF, 70 °C, 2–4 h, 14–63%. | ||
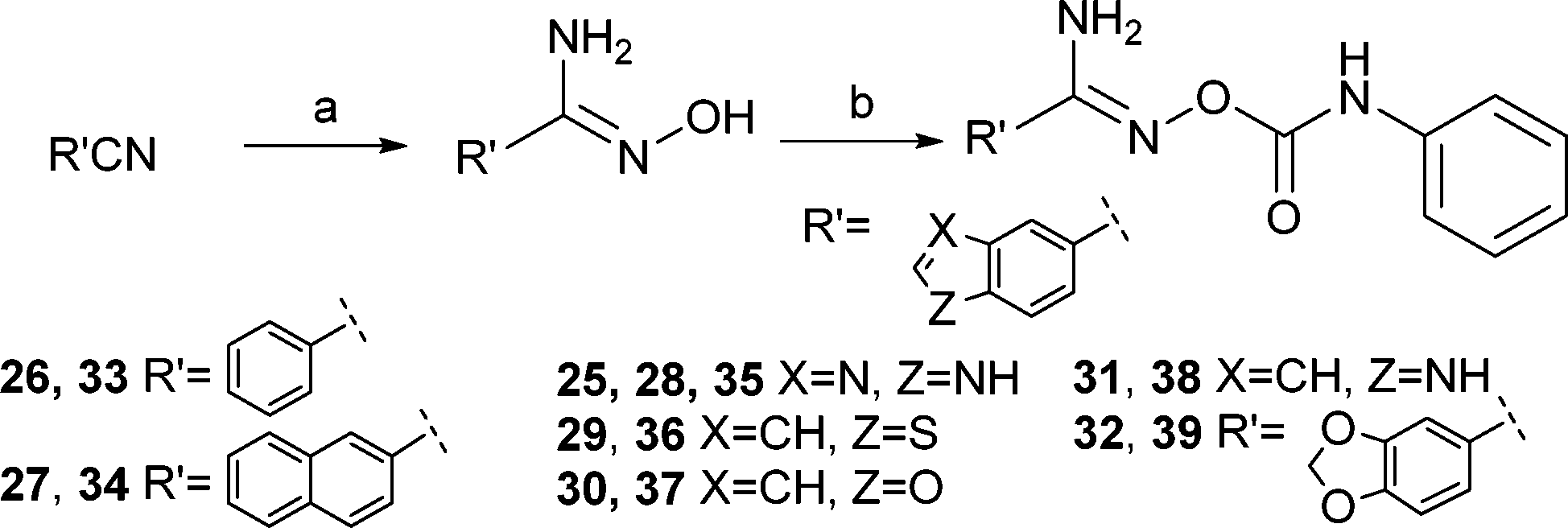 | ||
| Scheme 2 Synthetic procedures and chemical structures of the products 25–39. Reagents and conditions: a) H2NOH·HCl, Et3N or Na2CO3, EtOH or H2O, rt/100 °C, 3.5–24 h, 38–98%; b), phenyl isocyanate, THF, CHCl3 or 1,4-dioxane, rt, 2–5 h or 72 h, 6–85%. | ||
2.2 Evaluation of antileishmanial activity
The activity of the new compounds was determined by using a fluorescent viability microplate assay with L. donovani axenic amastigotes and alamarBlue.12 All compounds were initially screened at 50 μM, and the most active derivatives sequentially tested at lower concentrations (15 and 5 μM).First, a set of benzo[c][1,2,5]oxadiazole derivatives 6–18, with different substitution patterns in the “Eastern” phenyl ring of the structure, were synthesized and assayed. The derivative with no additional substituents (compound 6) showed 70% and 24% inhibition of L. donovani proliferation at 50 μM and 15 μM, respectively, whereas meta-Cl substitution (compound 14) increased growth inhibition to 95% and 87% at 50 μM and 15 μM, respectively (Table 1). Overall, meta substitution including Cl-, OMe-, Me-, and CF3-substituted derivatives seems to be beneficial for antileishmanial activity.
| % inhibitiona | |||
|---|---|---|---|
| Compound | 50 μM | 15 μM | 5 μM |
| a ±SEM: at 50 μM 0.1–2.3%, 15 μM 0.2–1.9%, and 5 μM 2.4–5.9%. b Precipitates at 50 μM. c Amphotericin B is positive control and was tested at 1 μM. | |||
| 6 | 70.1 | 24.2 | — |
| 7 | 42.0 | — | — |
| 8 | 67.2 | 44.6 | — |
| 9 | 64.1 | — | — |
| 10 | 38.0b | — | — |
| 11 | 71.4 | 31.8 | — |
| 12 | 46.0 | — | — |
| 13 | 29.6b | — | — |
| 14 | 94.8 | 87.2 | 43.4 |
| 15 | 44.0 | — | — |
| 16 | 42.1 | — | — |
| 17 | 51.4 | — | — |
| 18 | 51.7 | — | — |
| 20 | 96.2 | 68.2 | 54.6 |
| 21 | 51.1 | — | — |
| 22 | 26.5 | — | — |
| 23 | 22.4 | — | — |
| 24 | 43.9b | — | — |
| 33 | 78.4 | 5.4 | — |
| 34 | 98.8 | 24.9 | — |
| 35 | 72.0 | — | — |
| 36 | 97.9 | 35.2 | — |
| 37 | 95.2 | 48.0 | — |
| 38 | 91.9 | 21.6 | — |
| 39 | 93.0 | 33.9 | — |
| Amphotericin B | — | — | 99.7c |
Replacing HN of the heteroatom containing chain with oxygen or carbon atom (compounds 22 and 23) significantly reduced antileishmanial activity to 27 and 22%, respectively, at 50 μM. Similarly, removal of the aniline NH (compound 21) reduced activity, but this compound still showed more activity (51% growth inhibition) than carboxy 22 or benzylic 23 derivatives. Moreover, the cyclohexane derivative 24 displays leishmanial inhibition in the absence of an aniline moiety. Replacement of the amino group in the imidamide 20 with a carbonyl group increased inhibition to 96% and 68% at concentrations of 50 μM and 15 μM, respectively.
Finally, a second set of related heterocyclic compounds was synthesized with replacements of the benzo[c][1,2,5]oxadiazole moiety. These heterocycles (benzothiophene 36, benzofuran 37, indole 38, and 1,3-benzodioxole 39) showed an increase in antileishmanial activity at 50 μM compared to the benzoxazole derivative 6. This suggests that the benzo[c][1,2,5]oxadiazole ring is not crucial to antileishmanial activity. In addition, the phenyl and naphthalene derivatives 33 and 34 significantly inhibited growth of L. donovani amastigotes (78% and 5% for 33, and 99% and 25% for 34, at concentrations of 50 μM and 15 μM, respectively). Good inhibition of compound 34 can be hypothesized to result from increased lipophilicity of the compound. Moreover, the phenyl derivative 33 is a very interesting compound, because the shorter synthetic route is likely to increase the prospects for further preparation of new antileishmanial derivatives of this chemotype.
Further evaluation of the most promising derivatives 14 and 20 revealed low EC50 values on axenic amastigotes, 4.2 and 8.1 μM respectively; moderate cytotoxicity on the human THP-1 macrophage cell line, 79.9 and 33.7 μM respectively; and no cytotoxicity against murine fibroblasts at the highest concentration tested, 300 μM (Table 2). Values for amphotericin B, a reference compound used to treat leishmaniasis, are given for comparison. When tested at 5 μM on infected macrophages, a concentration non-toxic for the macrophage cell line, both compounds, 14 and 20, were still active, 33.1 and 19.9% amastigote growth inhibition respectively, albeit lower than that observed with axenic amastigotes. The EC50 for 14, the most active and least cytotoxic derivative, on intracellular amastigotes in infected macrophages was 5.92 μM similar to that observed using axenic amastigotes.
| Compound | Axenic amastigotes EC50 ± SEM (n = 2) (μM) | Intracellular amastigotes EC50 ± SEM (n = 3) (μM) | THP-1 EC50 (μM) (n = 2) [SI] | Fibroblasts EC50 (μM) (n = 2) [SI] |
|---|---|---|---|---|
| 14 | 4.2 ± 0.2 | 5.92 ± 1.7 | 79.9 ± 5.4 [19.0] | >300 [>71.4] |
| 20 | 8.1 ± 0.9 | — | 33.7 ± 5.0 [4.2] | >300 [>37.0] |
| Amphotericin B | 0.09–0.7 (ref. 12, 14–16) | 0.11–0.26 (ref. 17) | 2.1–7.6 (ref. 15 and 16) | 2.2 (ref. 18) |
3. Conclusion
The developed synthesis route to carboxyimidamide-substituted benzo[c][1,2,5]oxadiazoles and related derivatives facilitates access to a compound library and determination of the structure-activity relationships for this antileishmanial heterocyclic chemotype. Carboxyimidamide-substituted benzoxadiazole derivative 14 is the most promising compound of this study, demonstrating good antileishmanial inhibition activity in infected macrophages and, remarkably, no signs of cytotoxicity. Although mechanism of action of the compounds is not known yet, it is worth continuing the development of these compounds to explore further prospects of these compounds.4. Experimental section
4.1 Chemistry
Unless otherwise stated, reactions were carried out in oven-dried glassware under an argon atmosphere. All reagents were commercially available and were acquired from Fluka (Buchs, Switzerland), Aldrich (Schnelldorf, Germany), Riedel-de Haën (Seelze, Germany), and Alfa Aesar (Karlsruhe, Germany). THF was distilled over sodium/benzophenone ketyl. The progress of chemical reactions was monitored by thin-layer chromatography on silica gel 60-F254 plates acquired from E. Merck (Darmstadt, Germany). The eluent consisted of EtOAc and n-hexane, or EtOAc and MeOH, and detection was conducted at 254 or 366 nm. The products were purified by flash chromatography on silica gel with a Biotage SP1 purification system (Uppsala, Sweden) using 25+M cartridges or SNAP 10 g, 25 g, or 50 g cartridges, detection at 254 nm. Melting points were measured using an IA9100 digital melting point apparatus (Electrothermal Engineering, Essex, UK) and are uncorrected. IR spectra were recorded on a Bruker Vertex 70 FT-IR spectrometer (Ettlingen, Germany) with ATR technique. The synthesized compounds were analyzed by NMR on a Varian Mercury 300 MHz spectrometer (Palo Alto, CA, USA). 1H and 13C NMR were recorded as solutions in DMSO-d6 (Aldrich). Chemical shifts (δ) are given in parts per million (ppm) relative to the NMR solvent signals (DMSO-d6 2.50 and 39.51 ppm for 1H and 13C NMR, respectively). LC-MS analyses were performed with a HP1100 instrument (Agilent, Palo Alto, USA) with UV detector (λ 210 nm) and an Esquire LC spectrometer (Bruker Daltonik, Bremen, Germany) with ESI ion source. Signal separation was carried out by the use of a Waters XBridge C18 column (2.1 mm × 50 mm, 2.1 μm) with a Waters XBridge C18 guard column (Milford, MA, USA) (2.1 mm × 10 mm, 2.5 μm). The eluent consisted of water (+0.1% HCO2H) and acetonitrile (+0.1% HCO2H) (gradient run 80:20 → 5:95). Purity of all tested compounds was >95%. High resolution mass spectra (HRMS) were measured on a Synapt G2 HDMS Q-TOF-instrument (Milford, MA, USA) with positive mode ESI.
:2) to give 4 (1.10 g, 79%) as a pink solid. 1H NMR (300 MHz, DMSO-d6) δ 8.98 (t, J = 1.2 Hz, 1H), 8.28 (dd, J = 9.3, 1.2 Hz, 1H), 7.82 (dd, J = 9.3, 1.2 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 148.4, 147.9, 131.8, 125.7, 118.3, 117.2, 115.4.
:2) to give 14 (0.10 g, 60%) as a white powder. 1H NMR (300 MHz, DMSO-d6) δ 9.68 (s, 1H), 8.57 (s, 1H), 8.14–8.13 (m, 2H), 7.73–7.71 (m, 1H), 7.52 (dd, J = 8.4, 1.2 Hz), 7.36 (t, J = 8.4 Hz, 1H), 7.18 (s, 2H), 7,12 (dd, J = 8.4, 1.2 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 153.6, 152.1, 148.9, 148.8, 140.0, 134.9, 133.1, 131.4, 130.4, 122.8, 118.8, 117.8, 116.0, 114.6. LC-MS: [M + H]+ 332.0 m/z (tr = 6.9 min). FT-IR (ATR, cm−1): 3440, 3349, 1739, 1647, 1274. HRMS (ESI): m/z calcd. for C14H10ClN5O3 [M + H]+ 332.0550, found 332.0551.
:1) to yield 19 (0.10 g, 57%) as a light solid. 1H NMR (300 MHz, DMSO-d6) δ 11.56 (br s, 1H), 9.40 (br s, 1H), 8.37 (s, 1H), 8.14 (dd, J = 9.4, 1.0 Hz, 1H), 7.88 (dd, J = 9.4, 1.0 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 161.7, 149.0, 148.7, 139.6, 131.0, 116.5, 114.8.
:1) to yield 20 (0.12 g, 57%) as a light brownish solid. 1H NMR (300 MHz, DMSO-d6) δ 12.79 (br s, 1H), 10.38 (br s, 1H), 8.55 (t, J = 1.2 Hz, 1H), 8.22 (dd, J = 9.5, 1.2 Hz, 1H), 7.92 (dd, J = 9.5, 1.2 Hz, 1H), 7.52–7.49 (m, 2H), 7.37–7.32 (m, 2H), 7.11–7.05 (m, 1H). 13C NMR (75 MHz, DMSO-d6) δ 163.0, 152.0, 149.1, 148.6, 138.0, 134.9, 130.6, 129.0, 123.3, 118.5, 117.1, 116.7. LC-MS: [M + H]+ 299.2 m/z (tr = 5.2 min). FT-IR (ATR, cm−1): 3321, 3224, 1759, 1663, 1535, 1203, 888. HRMS (ESI): m/z calcd. for C14H11N4O4 [M + H]+ 299.0780, found 299.0779.
4.2 Biology
:1 parasite/macrophage ratio) used to infect the macrophages. Twenty-four hours later the adherent cells were washed 4–5 times with warm RPMI-1640 alone, and infected macrophages detached by treating with Trypsin EDTA. Infected macrophages (5 × 104 cells/100 μL per well) were dispensed in triplicate into white 96-well flat bottom plates (NUNC, Denmark) and the compounds diluted in complete RPMI-1640 containing 1% DMSO final concentration and added in triplicate (50 μL per well). Cultures were incubated for a further 48 h (37 °C, 5% CO2), lysed by the addition of Bright-Glo Luciferase Assay substrate (100 μL per well, Promega, MT, U.S.A.), and chemiluminescence measured using a microplate reader (Fluoroskan Ascent, Thermo Scientific). Amphotericin B (1 μM, >90% inhibition of parasite growth) was included as a positive control. Complete medium, both with and without DMSO, was used as negative controls. Experiments were repeated three times. Calculation of the EC50's and statistical analysis were carried out using GraphPad Prism Version 6.0b (GraphPad Software, Inc. San Diego, CA).
Acknowledgements
We thank Ms. Anna Keltikangas née Takala for the synthesis assistance. This study was supported by the Academy of Finland (projects 264020 and 265481 to JYK). CLJ holds the Michael and Penny Feiwel Professorial Chair of Dermatology.Notes and references
- J. Alvar, I. D. Vélez, C. Bern, M. Herrero, P. Desjeux, J. Cano, J. Jannin, M. den Boer and the WHO Leishmaniasis Control Team, PLoS One, 2012, 7, e35671 CAS , Sustaining the drive to overcome the global impact of neglegted tropical diseases, Second WHO Report of Neglegted Diseases, 2013.
- M. L. den Boer, J. Alvar, R. N. Davidson, K. Ritmeijer and M. Balasegaram, Expert Opin. Emerging Drugs, 2009, 14, 395–410 CrossRef CAS PubMed.
- S. L. Croft, S. Sundar and A. H. Fairlamb, Clin. Microbiol. Rev., 2006, 19, 111–126 CrossRef CAS PubMed; J. Alvar, P. Aparicio, A. Aseffa, M. D. Boer, C. Cañavate, J.-P. Dedet, L. Gradoni, R. T. Horst, R. López-Vélez and J. Moreno, Clin. Microbiol. Rev., 2008, 21, 334–359 CrossRef PubMed.
- J. K. O. Alvesalo, A. Siiskonen, M. J. Vainio, P. S. M. Tammela and P. M. Vuorela, J. Med. Chem., 2006, 49, 2353–2356 CrossRef CAS PubMed.
- L. Keurulainen, A. Siiskonen, A. Nasereddin, N. Sacerdoti-Sierra, D. Kopelyanskiy, T. O. Leino, P. Tammela, C. L. Jaffe, J. Yli-Kauhaluoma and P. Kiuru, Bioorg. Med. Chem. Lett., 2015, 25, 1933–1937 CrossRef CAS PubMed.
- S. D. Jorge, A. Masunari, C. O. Rangel-Yagui, K. F. M. Pasqualoto and L. C. Tavares, Bioorg. Med. Chem., 2009, 17, 3028–3036 CrossRef CAS PubMed; Y. Cho, T. R. Ioerger and J. C. Sacchettini, J. Med. Chem., 2008, 51, 5984–5992 CrossRef PubMed; L. Boiani, C. Davies, C. Arredondo, W. Porcal, A. Merlino, A. Gerpe, M. Boiani, J. P. Pacheco, M. Á. Basombrío, H. Cerecetto and M. González, Eur. J. Med. Chem., 2008, 43, 2229–2237 CrossRef PubMed; D. Castro, L. Boiani, D. Benitez, P. Hernández, A. Merlino, C. Gil, C. Olea-Azar, M. González, H. Cerecetto and W. Porcal, Eur. J. Med. Chem., 2009, 44, 5055–5065 CrossRef PubMed.
- R. K. Arvela and N. E. Leadbeater, J. Org. Chem., 2003, 68, 9122–9125 CrossRef CAS PubMed; D. Wang, L. Kuang, Z. Li and K. Ding, Synlett, 2008, 69–72 Search PubMed; Y. Ren, W. Wang, S. Zhao, X. Tian, J. Wang, W. Yin and L. Cheng, Tetrahedron Lett., 2009, 50, 4595–4597 CrossRef PubMed.
- R. Mueller and L. J. Street, US Pat., 0122861, 2012 Search PubMed.
- E. A. A. Wallén, J. A. M. Christiaans, M. M. Forsberg, J. I. Venäläinen, P. T. Männistö and J. Gynther, J. Med. Chem., 2002, 45, 4581–4584 CrossRef PubMed.
- H. Käsnänen, M. J. Myllymäki, A. Minkkilä, A. O. Kataja, S. M. Saario, T. Nevalainen, A. M. P. Koskinen and A. Poso, ChemMedChem, 2010, 5, 213–231 CrossRef PubMed.
- M. Muzerelle, A. Quattropani, C. Montage and J. Dorbais, WO Pat., 069949, 2010 Search PubMed.
- O. Shimony and C. L. Jaffe, J. Microbiol. Methods, 2008, 75, 196–200 CrossRef CAS PubMed.
- A. Debrabant, M. B. Joshi, P. F. P. Pimenta and D. M. Dwyer, Int. J. Parasitol., 2004, 34, 205–217 CrossRef PubMed.
- M. Vermeersch, R. Inocencio da Luz, K. Tote, J.-P. Timmermans, P. Cos and L. Maes, Antimicrob. Agents Chemother., 2009, 53, 3855–3859 CrossRef CAS PubMed.
- M. de Rycker, I. Hallyburton, J. Thomas, L. Campbell, S. Wyllie, D. Joshi, S. Cameron, I. H. Gilbert, P. G. Wyatt, J. A. Frearson, A. H. Fairlamb and D. W. Gray, Antimicrob. Agents Chemother., 2013, 57, 2913–2922 CrossRef CAS PubMed.
- S. Oh, B. Kwon, S. Kong, G. Yang, N. Lee, D. Han, J. Goo, J. L. Siqueira-Neto, L. H. Freitas-Junior, M. Liuzzi, J. Lee and R. Song, Med. Chem. Commun., 2014, 5, 142–146 RSC.
- K. Seifert, P. Escobar and S. L. Croft, J. Antimicrob. Chemother., 2010, 65, 508–511 CrossRef CAS PubMed.
- Y. S. Rizk, A. Fischer, M. de Castro Cunha, P. O. Rodrigues, M. C. S. Marques, M. de Fatima, C. Matos, M. C. T. Kadri, C. A. Carollo and C. C. P. de Arruda, Mem. Inst. Oswaldo Cruz, 2014, 109, 1050–1056 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedures for compounds 6–13, 15–18, 21–39. See DOI: 10.1039/c5md00119f |
| This journal is © The Royal Society of Chemistry 2015 |