
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of new hit scaffolds by INPHARMA-guided virtual screening†
Justyna
Sikorska
a,
Luca
Codutti
a,
Lars
Skjærven
a,
Bettina
Elshorst
c,
Rebeca
Saez-Ameneiro
a,
Andrea
Angelini
a,
Peter
Monecke
c and
Teresa
Carlomagno
*ab
aEMBL, Structural and Computational Biology Unit, Meyerhofstraße 1, D-69117 Heidelberg, Germany. E-mail: carlomag@embl.de
bHelmholtz Zentrum für Infektionsforschung, Inhoffenstrasse 7, Braunschweig, Germany
cSanofi-Aventis Deutschland GmbH, R&D LGCR/Structure, Design & Informatics, Industriepark Höchst, Bldg. G877, D-65926 Frankfurt am Main, Germany
First published on 15th June 2015
Abstract
Structure-based drug design (SBDD) relies on the availability of high-quality structures that describe protein–ligand interactions. INPHARMA is an NMR-based method that allows the determination of ligand binding poses to accuracy higher than 2 Å. In this work, we demonstrate that INPHARMA can be used to find novel ligand scaffolds as inhibitors of a model system protein, the cyclin-dependent kinase (Cdk-2). The workflow is given as follows: first, we determine the binding poses to Cdk-2 of six low-affinity fragments and use them to derive a structure-based pharmacophore. Two of the ligands show an unexpected binding mode, which differs from the one observed in crystal structures of other kinases. Second, we use the INPHARMA-generated pharmacophore for virtual screening of the ZINC database; one of the hit compounds is found to bind Cdk-2 in the low μM range and shows selectivity for Cdk-2 against kinases of other families. Our results demonstrate that INPHARMA is an efficient structure-based tool in solution to evolve low-affinity fragments into hit compounds.
Introduction
The quest for new drug leads comprises the in silico screening of ligand libraries with the help of an experimentally derived pharmacophore. In the absence of structural information for the target protein, “ligand based” pharmacophores are generated by aligning a series of ligands with known biological activity. Conversely, “structure based” pharmacophores utilize knowledge of the protein structure to predict key protein–ligand interactions. The “structure-based” approach is powerful; however, its application is limited by the availability of structures of the protein and/or of protein–ligand complexes.1Next to X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy has emerged as a potent technique for the determination of receptor–ligand interactions. NMR offers two strategies to detect ligand binding interfaces: (1) ligand spectra in the presence of the target protein report on the ligand pharmacophore (ligand-observing experiments); (2) protein spectra in the presence of the small molecule binder provide information on the protein interacting residues (protein-observing experiments). The ligand-observing approach is broadly applicable to low-affinity ligands in complex with proteins of any size and has the advantage of not requiring expensive isotope labeling of the target. Among ligand-observing experiments, the STD2 and waterLOGSY3 permit the identification of ligand binding epitopes; tr-NOEs4,5 determine the bioactive conformation of the ligand, while INPHARMA6–8 provides access to the relative binding mode of two low-affinity ligands interacting competitively within the same cavity of the target protein.
The INPHARMA method relies on the interligand, spin-diffusion mediated transfer of magnetization (NOE) between the two ligands L1 and L2. As the ligands are competitive binders, such NOEs do not originate from direct transfer of magnetization between L1 and L2 but rather from a spin–diffusion process, mediated by the protons of the receptor binding pocket. The intensity of the signals depends on the specific interactions of each of the two ligands with the protein; a number of such intermolecular NOEs describe the relative orientation of the two ligands in the receptor-binding pocket (Fig. S1A†). The quantitative analysis of the INPHARMA NOEs enables ranking pairs of binding orientations obtained by docking L1 and L2 to the receptor.6 By using a combination of INPHARMA data sets acquired for multiple ligand pairs, molecular dynamic simulations and ensemble docking, we showed that INPHARMA reliably selects the correct ligand binding modes for a set of four to five ligands. For this selection process, the INPHARMA information is measured for all possible pairwise combinations and the data are fitted simultaneously (INPHARMA-STRING).9
This work describes the implementation of INPHARMA-STRING in a classical structure-based drug-design process, aimed at evolving low-affinity fragment ligands into a hit compound. The pipeline that we develop is applied to the cyclin-dependent kinase Cdk-2. First, we determine the binding orientations of six low-affinity fragments by INPHARMA-STRING and use them to build a three-dimensional, structure-based pharmacophore model. Second, we implement the 3D pharmacophore in virtual screening of the compounds contained in the ZINC database. The procedure enables the discovery of a new scaffold, which binds Cdk-2 with improved affinity (up to two orders of magnitude with respect to the low-affinity fragments) and shows selectivity for Cdks against kinases of other families. The results demonstrate for the first time the applicability of INPHARMA-STRING to the evolution of low-affinity fragments with low specificity into promising hit compounds.
Results and discussion
NMR studies of Cdk-2 ligands
Six weakly binding ligands were used to explore the features of the Cdk-2 binding pocket (Fig. 1). LL4, ZIP and ADO are purine derivatives; M77 and LL6 display different substitutions of an isoquinoline ring, while RSA is a quinazoline derivative. The pairwise combination of the six ligands results in 15 INPHARMA pairs; however, in the analysis, we used INPHARMA experiments from only 11 pairs. The combinations LL6–M77, RSA–LL6 and RSA–M77 were excluded due to extreme overlap of the resonances of the two ligands; the ADO–LL4 pair was eliminated because of the low number of INPHARMA peaks. For each pair, three NOESY spectra were acquired with different mixing times (300 ms, 500 ms and 800 ms), resulting in a total of 33 experiments and 144 correlation peaks (Table S1†).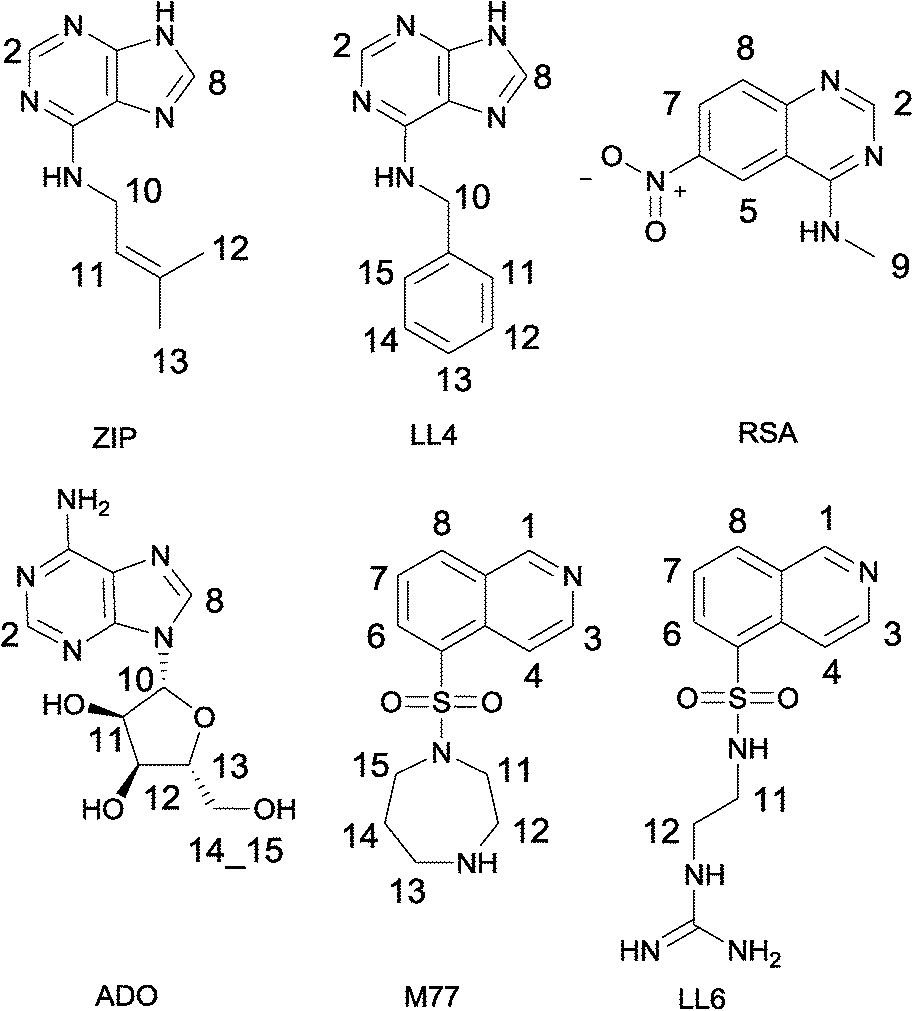 | ||
| Fig. 1 Cdk-2 inhibitors used for the INPHARMA analysis. Kd values were measured by isothermal titration calorimetry and are given as follows: LL4, 62 μM; ZIP, 35 μM; RSA, 208μM; ADO, 184 μM; M77, 460 μM; LL6, 286 μM. | ||
The INPHARMA information content was distributed non-homogeneously, as readily observed for other protein–ligand systems.9 For the purine analogues LL4 and ZIP, the key structural information was contained in INPHARMA-NOEs to purine hydrogens, while the signals from the benzyl and the 3-methyl-2-butenyl fragments were not used due to the extensive degeneracy of the chemical shifts. For LL6 and M77 (Fasudil), the majority of correlations resulted from the 8-isoquinolinesulfonamide fragment; the N-ethyl-guanidine and hexahydro-1,4-diazepine moieties overlapped with protein resonances between 0 and 4 ppm. The most challenging ligand was adenosine (ADO), with INPHARMA peaks stemming exclusively from the H-2 and H-8 of adenine. Finally, for RSA, all protons delivered peaks that could be used for the analysis.
Determination of Cdk-2–ligand interactions by INPHARMA-STRING
According to the workflow proposed in ref. 9, the INPHARMA-STRING data were used to rank in silico generated models of the protein–ligand complexes (Fig. S1B†). For the docking, we collected three representative structures of Cdk-2 (1HCK: ATP-bound; 1H1R: cyclin-bound; 2VTM: cyclin-free) and performed molecular dynamics (MD) simulations to explore the conformational space around the starting models. The resulting ensemble of MD-generated conformers was reduced by clustering analysis, providing 5 receptor structures from each simulation. The six ligands were subsequently docked using Glide10 and Surflex11 to the 15 receptor structures, and the best 10 scoring docking poses of each docking program were retained (generating a total of 150 docking poses per ligand per program = 300 docking poses per ligand). Each of the resulting structures was further subjected to a short MD simulation: five structures per trajectory were selected (ESI†), resulting in 1500 binding modes per ligand. The binding free energy of each complex structure was estimated using MM/GBSA, and the 50% highest energy conformations were excluded from further analysis. The remaining binding poses (750 per ligand) were combined pairwise to calculate the theoretical INPHARMA-NOEs using the in-house program described before9 and the Kd for each ligand measured by calorimetric analysis. The INPHARMA-NOEs predicted for each ligand pair were compared to the experimental ones by means of the Pearson correlation coefficient R (INPHARMA score); subsequently, the INPHARMA-STRING algorithm was implemented for selection of the binding modes that are uniquely in agreement with the experimental data.9Briefly, the INPHARMA-STRING algorithm identifies consensus binding modes among poses with top-ranked INPHARMA scores. It returns a string of six docking modes (one per ligand) with high INPHARMA scores in all 11 combinations used for the selection. During the search, the number of high-ranking docking modes is gradually increased until the consensus string is found.
For our experimental system, the algorithm reached a consensus among the best-ranked 3.6% docking modes (Fig. 2 and S2†). For LL4 and ZIP, the INPHARMA-STRING selected poses are close to those observed by crystallography (all atom root mean square deviation, RMSD, of 0.8–1.4 Å and 1.4 Å, respectively; Fig. 2), thus providing a validation of the algorithm. To verify the stability of the selection, we analyzed the poses selected by INPHARMA-STRING up to 5% of the best-ranked docking modes and found that the poses were similar to each other, with an average RMSD < 1.5 Å (Fig. S3†). LL4 and ZIP showed hydrogen bonding to Glu-81 and Leu-83 in the Cdk-2 hinge region, as well as a hydrophobic interaction with Val-18 (Fig. 3). For LL4, an additional interaction to Lys-33 was occasionally observed (Fig. 3).
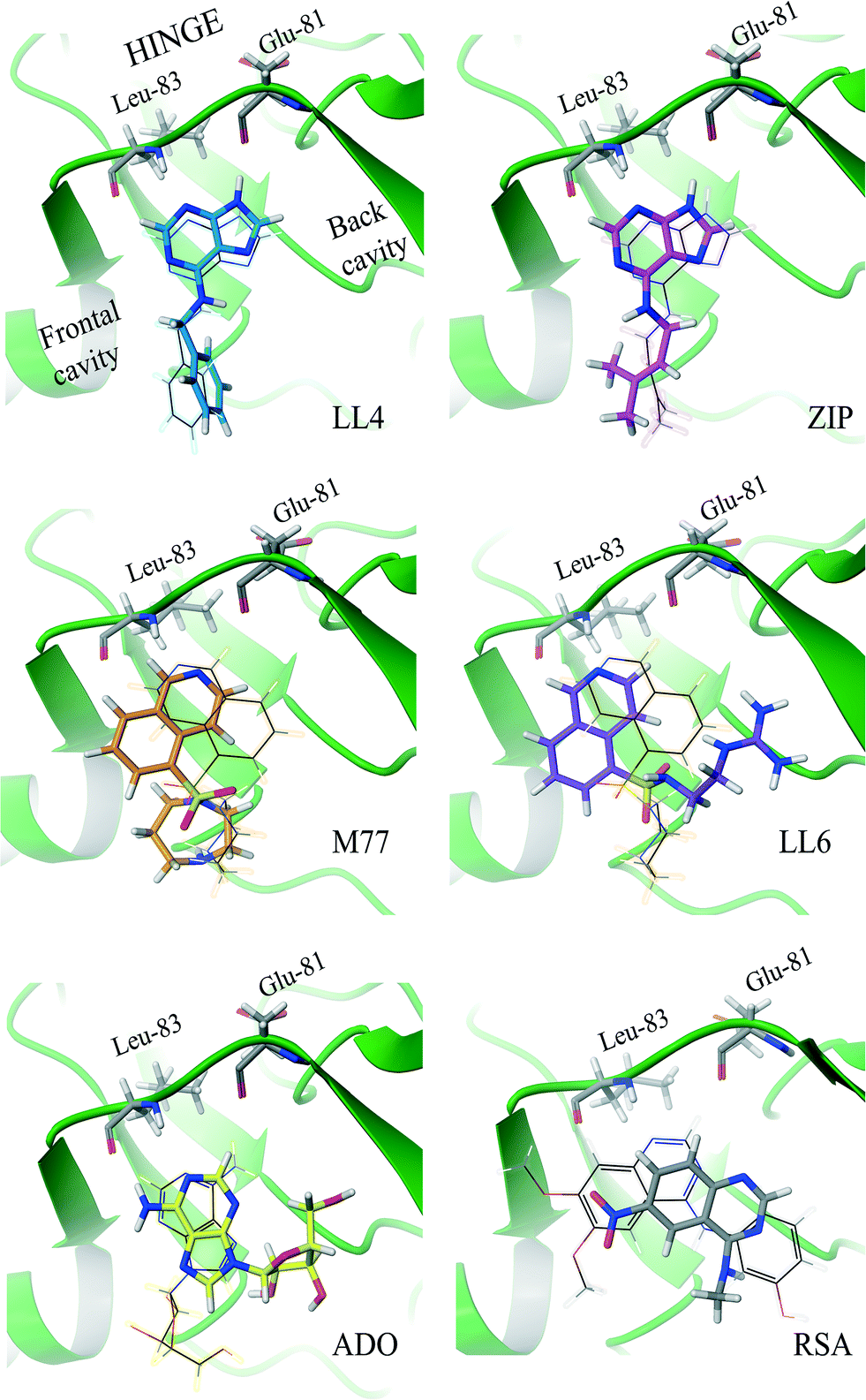 | ||
| Fig. 2 Binding modes of Cdk-2 ligands identified by INPHARMA-STRING (consensus string in the 3.6% best-ranked poses). The INPHARMA-selected binding poses of ZIP, LL4 and ADO are represented as pink, blue and yellow structures (tubes) overlaid to their respective X-ray structures (wires); PDB codes; ZIP, 2EXM; ADO, 1HCK. The M77 and LL6 INPHARMA-selected binding poses are shown in orange and purple and overlaid to the orientation of M77 bound to PKA (1Q8W.pdb). The INPHARMA-selected binding pose of RSA, shown in black, is compared to that of the 4-[3-hydroxyanilino]-6,7-dimethoxyquinazoline in complex with Cdk-2 (1DI8.pdb). | ||
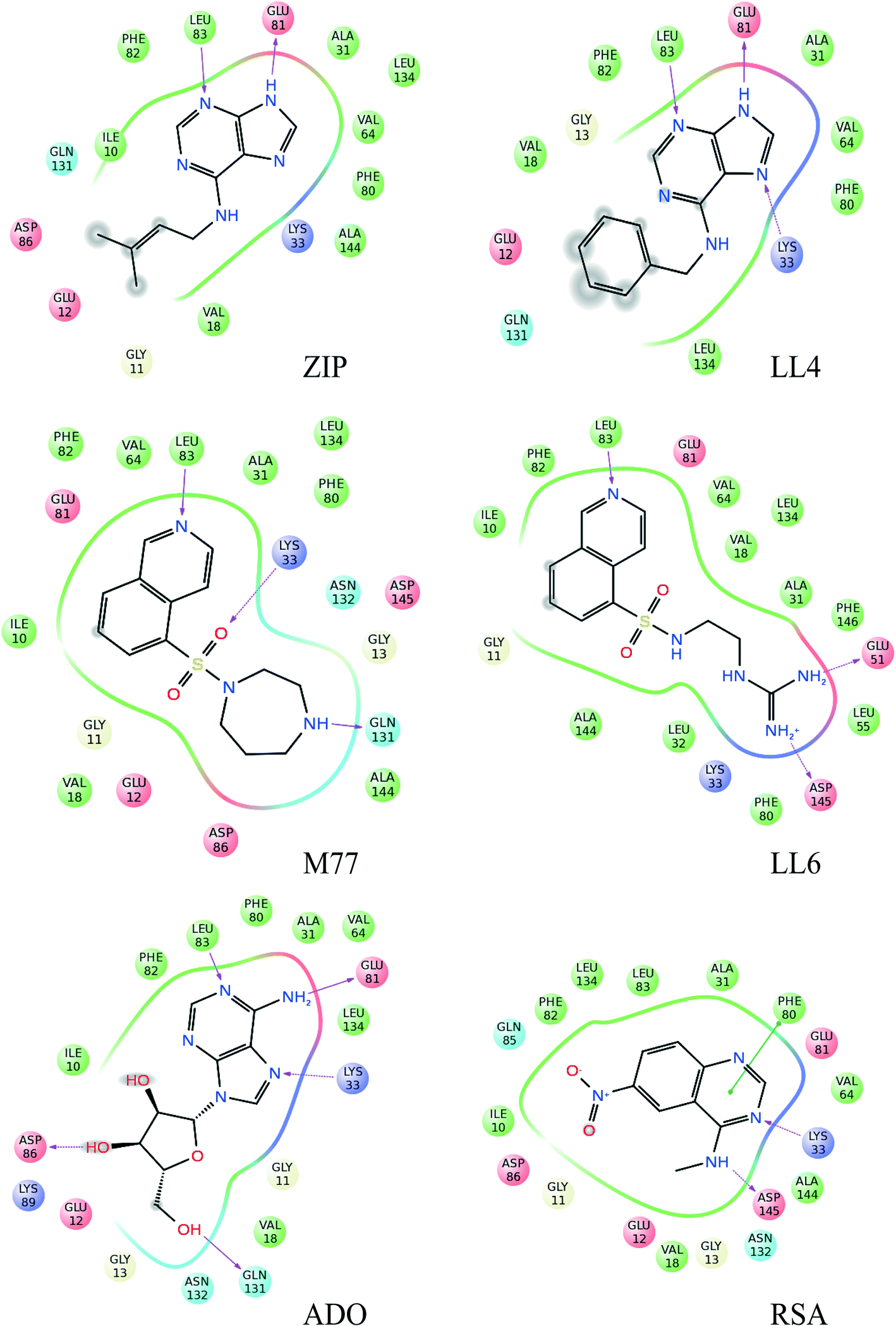 | ||
| Fig. 3 Schematic representation of the protein–ligand interactions for all analyzed compounds. We show all binding interactions observed in the binding poses selected by INPHARMA-STRING in the 5% top-ranked binding modes. The binding site residues within 4 Å from the ligand are indicated as spheres, while hydrogen bonds are represented by red arrows. Blue, hydrogen bond donor; red, hydrogen bond acceptor; green, hydrophobic interactions. | ||
The selection of the M77 and LL6 binding poses was also consistent among the best-ranked 3.5–5% docking modes (Fig. 2 and S3†). For these ligands, no crystallographic structure is available in complex with Cdk-2, probably due to their low affinity. When we compare the INPHARMA-STRING selected poses to the X-ray structure of M77 in complex with protein kinase A (PKA),12 we observe clear differences (Fig. 2).13 Here, M77 and LL6 take advantage of the larger Cdk-2 binding site and occupy the frontal cavity (Fig. S4†), as also confirmed by STD data (Fig. S5†). The 8-isoquinolinesulfonamide fragment of both M77 and LL6 forms a hydrogen bond to Leu-83 as well as hydrophobic interactions with Ile-10 and Phe-82 (Fig. 3). In support of this unexpected interaction mode, binding to the frontal cavity of Cdk-2 had been previously utilized for the design of compounds that, like flavopiridol,14 have higher selectivity for Cdk-2 in comparison to PKA.
For adenosine (ADO), two binding poses were selected by INPHARMA among the 5% best-ranked docking modes (Fig. 2 and S3†). The first binding pose differs by 180° from the binding pose of ATP to Cdk-2 (X-ray structure 1HCK.pdb, Fig. 2), while the second one corresponds to the ATP binding mode. The INPHARMA score did not allow discrimination of the two orientations of ADO; this can be explained by the fact that the INPHARMA NOEs of ADO stem primarily from protons H2 and H8 of the aromatic ring and are therefore insensitive to 180° rotations around the H2–H8 axis.
The pose of RSA selected by INPHARMA-STRING has a prominent hydrogen bond interaction between the N-3 of the quinazoline ring and Lys-33, as well as the methylamine of RSA and Asp-145 (Fig. 3). An additional π–π interaction can be formed between the pyrimidine ring of RSA and Phe-80. However, when a larger population of binding poses was examined (5%), only the hydrogen bond between the N-3 of the quinazoline ring and Lys-33 was consistent through all binding modes. Noticeably, all selected poses are similar to those of the N-phenylquinazolin-4-amine analogues (the closest analogue of RSA that has been co-crystallized with Cdk-2, 1DI8.pdb); however these compounds interact with the Cdk-2 binding site through a hydrogen bond between the N-1 of the quinazoline ring and Leu-83.13
Overall, the INPHARMA-selected poses show receptor–ligand interactions that are in accord with previously published structures of known Cdk-2 inhibitors, thus validating the robustness of the selection.15
Epitope mapping of selected Cdk-2 ligands with saturation transfer difference (STD)
To independently validate the INPHARMA-selected binding modes, we applied saturation transfer difference (STD)2 experiments to ZIP, LL4, LL6, M77 and RSA in the presence of Cdk-2 (Fig. S5†). For LL4, ZIP and RSA, the strongest saturation effects were observed for purine H2 and quinazoline H2, H7, and H8, in good agreement with the INPHARMA-selected poses, which locate these protons in close proximity to Ile-10 or deep inside the binding cavity. For LL6 and M77, the STD data exclude an orientation of the ligand similar to that of M77 in PKA, for which the STD intensities of the buried protons H6 and H7 should be higher than those of the solvent exposed protons H3 and H4. Instead, we observe STD signals of medium intensity for H3 and H4 and of weak intensity for H6 and H7, which is in agreement with the INPHARMA-selected pose.Structure-based pharmacophore generation, validation and virtual screening
With the binding modes of these fragments in our hands, we set out to generate a common structure-based pharmacophore, to be used for virtual screening of the ZINC database.First, we generated the individual pharmacophores of ZIP, LL4, M77, LL6 and RSA using the following protocol. We analyzed all intermolecular interactions observed in the binding poses selected by INPHARMA-STRING in the top-ranking 5% structural pairs with LigandScout3.12.16 Of these interactions, we retained only those that were consistently present in all binding modes of the ligand under consideration. For ZIP and LL4, we decreased the size of the spheres describing the hydrogen bond interaction to Glu-81 and Leu-83 by 0.3 Å to increase pharmacophore specificity. For LL6, we excluded all interactions originating from the guanidinium group, due to the lack of experimental INPHARMA data stemming from this part of the molecule, which contains only exchangeable hydrogens. Furthermore, for M77, which is the weakest binder, we retained only the pharmacophoric features common to LL6.
The common structure-based pharmacophore (CSB) of all five ligands was generated by the alignment of reference points (defined as amino acid residues of the binding site) followed by a merge of all pharmacophoric features. The final pharmacophore consisted of six interactions: two H-bond acceptors (HBA), one H-bond donor (HBD), three hydrophobic contacts (HY) as well as excluded volume spheres representing the surface of the Cdk-2 binding pocket (Fig. 4, S6 and S7†). The HBA1 and HBD represent two known key interactions of Cdk-2 inhibitors with residues of the hinge region: the backbone NH of Leu-83 and the backbone oxygen of Glu-81. The HBA2 feature results from the interaction between the quinazoline nitrogen of RSA and the side chain amino group of Lys-33. Because of the high flexibility of Lys-33 in the Cdk-2 binding site, we also constructed a pharmacophore model where we excluded HBA2 and named it reduced structure-based pharmacophore (RSB). The HY1 and HY2 contacts result from the interaction of the isoquinoline ring of LL6 and M77 with Ile-10 and Phe-82, while HY3 originates from the interaction of the aliphatic and aromatic fragments of ZIP and LL4, respectively, with Val-18.
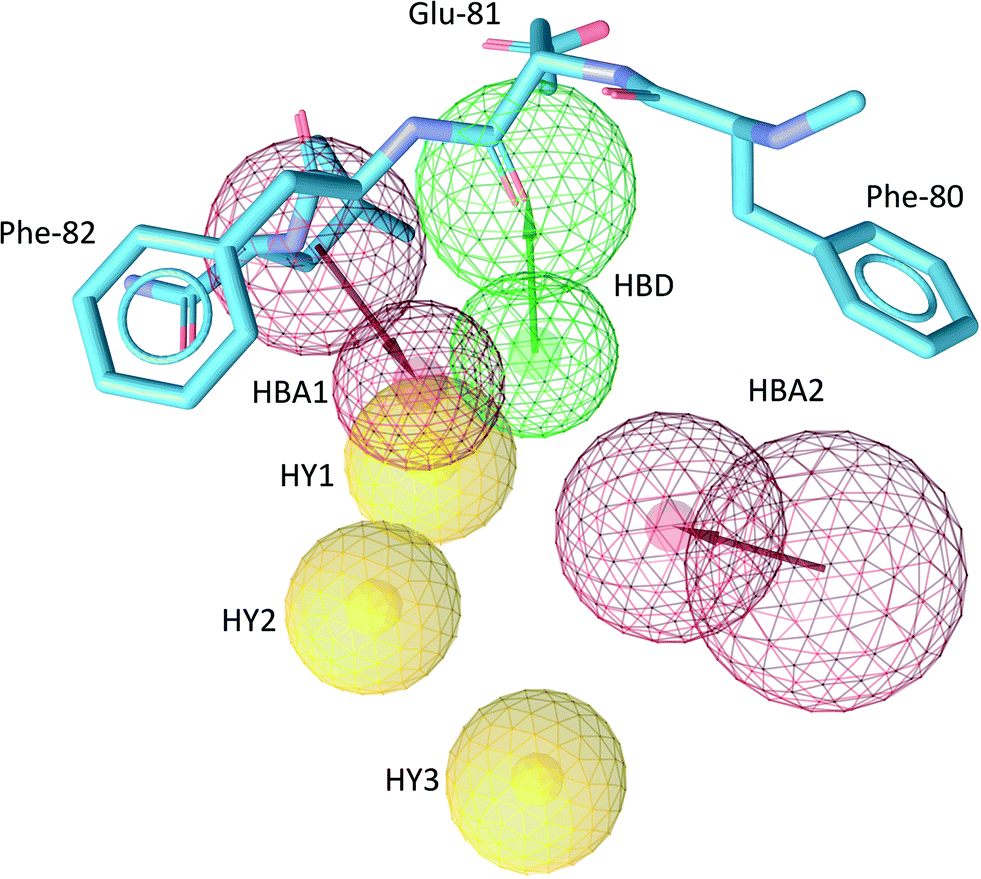 | ||
| Fig. 4 The common structure-based (CSB) pharmacophore used in virtual screening. H-bond acceptors (HBA), red arrows and spheres; H-bond donors (HBD), green arrows and spheres; hydrophobic features (HY), yellow spheres. The radius of the sphere represents the space where the feature can be localized. The excluded volumes are not indicated for clarity. Protein residues 80–82 (hinge region) are shown in cyan sticks. | ||
The pharmacophoric features identified in this study are part of the comprehensive pharmacophore map derived for Cdk-2 from all Cdk-2 binders.15 This confirms the ability of INPHARMA to generate high-quality binding modes for fragment ligands, which can be further used to generate pharmacophore models. Among all six features, HBA1, HBD, HY2 and HY3 are present in >25% of the crystal structures of Cdk-2 in complex with ligands,15 while HBA2 and HY1 are found with 22% occurrence.15
The ability of the CSB and RSB pharmacophore models to identify active compounds was estimated for a collection of entries composed of active and decoy molecules. The pool of active compounds consisted of 115 entries from the ChEMBL database with IC50 below 500 nM, while the 2438 decoys were generated with the program DUD-E (http://dude.docking.org/)17 from 33 active compounds. The performance of the pharmacophores was quantified by the enrichment factor (EF) and the area under the receiver operating characteristic curve (ROC) obtained for the top 1.5% of the scored database.18 The EF represents the ability of the pharmacophore to accurately identify active hits with respect to a random selection.18 The area under the ROC curve (AUC) is a function of both specificity and sensitivity and indicates the probability of ranking a randomly chosen active compound higher than a randomly chosen decoy. For the CSB pharmacophore, LigandScout 3.12 (ref. 16) identified 2 compounds from the 115 active ligands and 2 from 2438 decoys, resulting in an EF1.5% = 11.1 and AUC1.5% = 0.92. With the RSB, we selected 12 out of the 115 active compounds and 5 out of the decoy pool yielding EF1.5% = 14 and AUC1.5% = 0.95. Both pharmacophores were used to screen the 3D ZINC database,19 resulting in 1343 hits for CSB and 1206 hits for RSB (Fig. S8†). The hits were docked to two Cdk-2 structures, and the binding poses were ranked with both GlideScore20 and X-score.21 The two Cdk-2 structures used for the docking had been selected by INPHARMA-STRING in complex with ligands LL6 and RSA and represent two very different orientations of Lys-33 in the binding site. Those compounds were retained, which ranked among the best 100 of both scores and fulfilled at least one H-bond and two hydrophobic interactions of the CSB or RSB pharmacophores (Fig. S9†). This selection procedure resulted in 28 and 16 ligands from the CSB and RSB pharmacophores, respectively (Table S2†).
Evaluation of identified hit compounds by NMR
Of the 44 compounds selected by in silico screening, we purchased 11 commercially available ones, representing 5 distinct chemical skeletons (Table S2, Fig. S8 and S10†). Among them, ZINC20085956 and ZINC06781696 were excluded from further evaluation due to poor solubility. The remaining compounds were subjected to trNOE and STD experiments in the presence of Cdk-2 (Fig. S10†). The 1,2,4-triazole-3-thiones derivatives did not show trNOE. Weak saturation transfer effects were observed only for ZINC02808739.Five compounds exhibited a solubility of <10 μM; for these ligands, we could not record direct trNOE or STD experiments. Instead, we measured competition STD against M77 (Kd = 460 μM) at the highest possible ligand concentration. A 40% decrease in the intensity of M77 H-8 was observed in the presence of 2.5 μM HDS029 (ZINC13682420). Interestingly, HDS029 had been previously identified as a potent inhibitor of the proteins ErbB1, ErbB2 and ErbB4 belonging to the receptor tyrosine kinase family.
Analogously, among the derivatives of 1H-pyrazolo[3,4-B]pyridin-3(2H)-one and benzimidazole, ZINC20085954 caused a 45% decrease in the M77 H-8 peak at 2.5 μM.
Overall, from 11 purchased compounds, only 9 were sufficiently soluble for further analysis. Among those, 3 were identified as Cdk-2 binders through STD experiments, resulting in a hit rate of 33% (defined as a percentage of Cdk-2 binding compounds among all purchased compounds). To independently confirm that ZINC20085954 and ZINC02808739 are kinase inhibitors with a novel chemical skeleton, both compounds were subjected to the kinase phosphorylation assay.
Kinase phosphorylation assay
We tested ZINC20085954 and ZINC02808739 at Reaction Biology Corporation, using their kinase assay platform panel.22 The ability of the ligand to inhibit the catalytic activity of Cdk-2/cyclin A, Cdk-2/cyclin E, PKA, and ALK kinases was evaluated at the highest soluble concentration of the ligands (Table 1). In addition, the binding affinity of ZINC20085954 and ZINC02808739 to Cdk-2/cyclin A was evaluated by measuring the IC50 values. ZINC20085954 showed the strongest inhibition of Cdk-2 activity with IC50 of 2.6 μM. In addition, the compound displayed selectivity for Cdk-2 against PKA, albeit the fact that the fragments (Fig. 1) were not chosen as selective Cdk-2 binders (indeed, M77 binds to PKA stronger than to Cdk-2). ZINC20085954 belongs to the family of the pyrazolo[3,4-B] pyridine derivatives, which were published as inhibitors of the ALK23 during the course of this work (Fig. 5A); however, ALK preserved 87% of activity in the presence of 5 μM ZINC20085954, confirming its selectivity for Cdk-2/cyclin A (Table 1).| Ligand | Single dose concentration | Enzyme activity | IC50 | |||
|---|---|---|---|---|---|---|
| ALK | Cdk-2/cyclin A | Cdk-2/cyclin E | PKA | Cdk-2/cyclin A | ||
| ZINC20085954 | 5 μM | 87% | 35% | 95% | 115% | 2.6 μM |
| ZINC02808739 | 100 μM | — | 69% | 96% | 99% | 1000 μM |
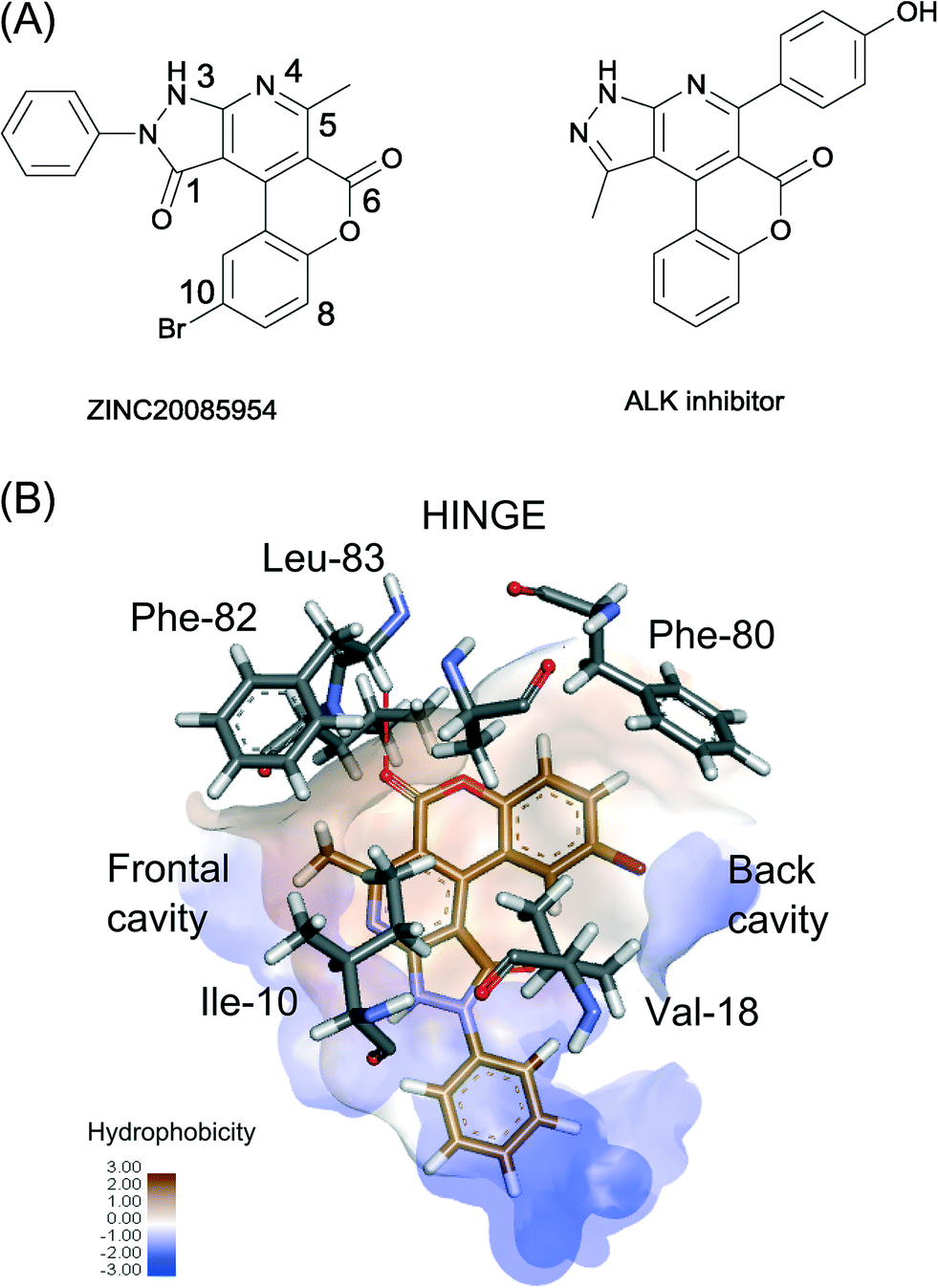 | ||
| Fig. 5 (A) Chemical structure of ZINC20085954 and a representative of ALK inhibitors. (B) The proposed binding of ZINC20085954 with interacting protein residues shown in grey. The hydrophobic surface is represented in a brown/blue scale, and the hydrogen bond to the hinge region is shown as a red dashed line. | ||
Binding mode of ZINC20085954 to Cdk-2
To better understand the selectivity of ZINC20085954 towards Cdk-2, we generated models of the Cdk-2–ZINC20085954 complex by ensemble docking. The top 10 docking results converged to an orientation that fulfils four of the five features of the RSB pharmacophore (Fig. 5B and S9†). ZINC20085954 entertains three hydrophobic interactions with Phe-82/Ile-10, Ala-31 and Val-18 and a hydrogen bond with the backbone nitrogen of the hinge region residue Leu-83. The ligand occupies both the frontal and the back cavities of Cdk-2, which explains its selectivity against PKA, lacking the frontal cavity (Fig. S4†).In addition to the interactions described by the pharmacophore, the bromine atom of ZINC20085954 makes contact with the phenyl ring of the gatekeeper Phe-80, while the C5-methyl forms a CH–π interaction with Phe-82. These non-covalent interactions can contribute significantly to the affinity of ZINC20085954 for Cdk-2 and can rationalize the much lower activity of the compound against ALK, where Phe-80 and Phe-82 are substituted by leucines (Table 1).
Conclusions
In this work, we propose an alternative route for structure-based drug design in the absence of high-resolution X-ray structures of the receptor–ligand complexes. The INPHARMA approach is especially relevant to the design of new leads for proteins that are challenging to crystallize, either by themselves or in complex with the ligands of interest; it also offers an attractive alternative for obtaining high-resolution structural information on protein–ligand interactions without extensive optimization of crystallographic conditions or assignment of NMR protein resonances. In this study, the method allowed us to identify significant differences in the binding mode of purine and isoquinoline derivatives to Cdk-2: while the hydrogen bond between one heterocyclic nitrogen and the NH of Leu-83 is formed by both classes of ligands, the isoquinoline derivatives lack the possibility to form an additional hydrogen bond to the carbonyl of Glu-81. Consequently, the heterocyclic ring flips and occupies the frontal open cavity of Cdk-2 rather than the back cavity. The combination of the pharmacophoric features of both the purine and the isoquinoline derivatives allows for the identification of a new hit molecule, which is selective for Cdk-2 against PKA, lacking the frontal cavity. The novel scaffold can be used for further optimization into a high-activity ligand.Acknowledgements
The authors acknowledge support from the BMBF (grant 0315870B) and the DFG (grant CA 294/6-1). We thank Vladimir Rybin and the EMBL Protein Expression and Purification Core Facility for performing ITC experiments and data analysis.Notes and references
- G. L. Wilson and M. A. Lill, Future Med. Chem., 2011, 3, 735–750 CrossRef CAS PubMed.
- M. Mayer and B. Meyer, Angew. Chem., Int. Ed., 1999, 38, 1784–1788 CrossRef CAS.
- C. Dalvit, P. Pevarello, M. Tatò, M. Veronesi, A. Vulpetti and M. Sundström, J. Biomol. NMR, 2000, 18, 65–68 CrossRef CAS PubMed.
- P. Balaram, A. A. Bothner-By and J. Dadok, J. Am. Chem. Soc., 1972, 94, 4015–4017 CrossRef CAS PubMed.
- G. M. Clore and A. M. Gronenborn, J. Magn. Reson. (1969-1992), 1983, 53, 423–442 CrossRef CAS.
- J. Orts, C. Griesinger and T. Carlomagno, J. Magn. Reson., 2009, 200, 64–73 CrossRef CAS PubMed.
- J. Orts, J. Tuma, M. Reese, S. K. Grimm, P. Monecke, S. Bartoschek, A. Schiffer, K. U. Wendt, C. Griesinger and T. Carlomagno, Angew. Chem., Int. Ed., 2008, 47, 7736–7740 CrossRef CAS PubMed.
- V. M. Sánchez-Pedregal, M. Reese, J. Meiler, M. J. J. Blommers, C. Griesinger and T. Carlomagno, Angew. Chem., Int. Ed., 2005, 44, 4172–4175 CrossRef PubMed.
- L. Skjærven, L. Codutti, A. Angelini, M. Grimaldi, D. Latek, P. Monecke, M. K. Dreyer and T. Carlomagno, J. Am. Chem. Soc., 2013, 135, 5819–5827 CrossRef PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- E. Kellenberger, J. Rodrigo, P. Muller and D. Rognan, Proteins: Struct., Funct., Bioinf., 2004, 57, 225–242 CrossRef CAS PubMed.
- C. Breitenlechner, M. Gaßel, H. Hidaka, V. Kinzel, R. Huber, R. A. Engh and D. Bossemeyer, Structure, 2003, 11, 1595–1607 CrossRef CAS PubMed.
- A. K. Ghose, T. Herbertz, D. A. Pippin, J. M. Salvino and J. P. Mallamo, J. Med. Chem., 2008, 51, 5149–5171 CrossRef CAS PubMed.
- A. Huwe, R. Mazitschek and A. Giannis, Angew. Chem., Int. Ed., 2003, 42, 2122–2138 CrossRef CAS PubMed.
- J. Zou, H.-Z. Xie, S.-Y. Yang, J.-J. Chen, J.-X. Ren and Y.-Q. Wei, J. Mol. Graphics Modell., 2008, 27, 430–438 CrossRef CAS PubMed.
- G. Wolber and T. Langer, J. Chem. Inf. Model., 2004, 45, 160–169 CrossRef PubMed.
- M. M. Mysinger, M. Carchia, J. J. Irwin and B. K. Shoichet, J. Med. Chem., 2012, 55, 6582–6594 CrossRef CAS PubMed.
- N. Triballeau, H.-O. Bertrand and F. Acher, Pharmacophores and Pharmacophore Searches, ed. T. Langer, R. D. Hoffmann, R. Mannhold, H. Kubinyi and G. Folkers, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006 Search PubMed.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, J. Chem. Inf. Model., 2012, 52, 1757–1768 CrossRef CAS PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- R. Wang, L. Lai and S. Wang, J. Comput.-Aided Mol. Des., 2002, 16, 11–26 CrossRef CAS PubMed.
- T. Anastassiadis, S. W. Deacon, K. Devarajan, H. Ma and J. R. Peterson, Nat. Biotech., 2011, 29, 1039–1045 CrossRef CAS PubMed.
- R. Rabot, K. Bedjeguelal, E. B. Kaloun, P. Schmitt, N. Rahier, P. Mayer and E. Fournier, Google Patents, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: computational and experimental procedures, additional figures and tables. See DOI: 10.1039/c5md00116a |
| This journal is © The Royal Society of Chemistry 2015 |