
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure–activity relationship study on senktide for development of novel potent neurokinin-3 receptor selective agonists†
Ryosuke
Misu
a,
Koki
Yamamoto
a,
Ai
Yamada
a,
Taro
Noguchi
a,
Hiroaki
Ohno
a,
Takashi
Yamamura
b,
Hiroaki
Okamura
b,
Fuko
Matsuda
c,
Satoshi
Ohkura
c,
Shinya
Oishi
*a and
Nobutaka
Fujii
*a
aGraduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: soishi@pharm.kyoto-u.ac.jp; nfujii@pharm.kyoto-u.ac.jp; Fax: +81 75 753 4570; Tel: +81 75 753 4551
bAnimal Physiology Research Unit, National Institute of Agrobiological Sciences, Tsukuba 305-0901, Japan
cGraduate School of Bioagricultural Sciences, Nagoya University, Chikusa-ku, Nagoya 464-8601, Japan
First published on 5th January 2015
Abstract
Neurokinin B (NKB) regulates the secretion of gonadotropin-releasing hormone (GnRH) in the hypothalamus via activation of the cognate neurokinin-3 receptor (NK3R). The stimulatory effect of NKB and the derivatives on gonadotropin secretion can potentially be used for development of novel regulatory and therapeutic agents for reproductive dysfunctions. Here, we report a comprehensive structure–activity relationship study on the NK3R-selective agonist peptide, senktide. Substitution of the N-terminal succinyl-Asp substructure in senktide with oxalyl-Glu, oxalyl-D-Glu or oxalyl-L-2-aminoadipic acid (Aad) increased receptor binding and NK3R activation. Among these modifications, the oxalyl-D-Glu substructure prevented neutral endopeptidase (NEP) 24.11-mediated degradation, thus providing a novel NK3R agonist peptide with favourable biological and stability properties.
Introduction
Neurokinin-3 receptor (NK3R) belongs to the tachykinin receptor family and is known as a cognate receptor of neurokinin B (NKB, Table 1). Recently, the hypothalamic regulation of reproduction by the NKB–NK3R axis has received considerable attention.1,2 The genetic study of TAC3 and TACR3, which encode NKB and NK3R, respectively, highlighted the prevalence of hypogonadotropic hypogonadism by inactivating mutations of these genes.3,4 Exogenous administration of kisspeptin or gonadotropin-releasing hormone (GnRH) into patients with these genetic mutations restores the circulating level of luteinising hormone (LH),5 suggesting that NKB positively regulates the reproductive hormone cascade via GnRH neuron activation.6 The influence on LH secretion by central administration of an NK3R agonist into ovariectomised and estrogen-primed rats has also been reported previously.7 Because LH secretion from the pituitary gland is implicated in a number of gonadal functions including testosterone production in males, maturation of pre-ovulatory ovarian follicles, and ovulation in females,8,9 selective NK3R modulators have been expected to be a novel class of therapeutic agents for gonadal dysfunctions as well as reproductive disorders. The NK3R modulators would also be applicable to the improvement of the low reproductive efficiency of livestock such as cattle.10| Peptide | Sequence |
|---|---|
| Neurokinin B (NKB) | H-Asp-Met-His-Asp-Phe-Phe-Val-Gly-Leu-Met-NH2 |
| [MePhe7]-NKB | H-Asp-Met-His-Asp-Phe-Phe-MePhe-Gly-Leu-Met-NH2 |
| Senktide | Succinyl-Asp-Phe-MePhe-Gly-Leu-Met-NH2 |
In 2007, it was reported that NKB is colocalised with kisspeptin and dynorphin A (DynA) in a single subpopulation in the hypothalamic arcuate nucleus (ARC) in sheep.11 These peptides cooperatively regulate the secretion of GnRH: NKB and kisspeptin mediate induction, while DynA mediates inhibition of GnRH secretion. Cells containing these three peptides are currently recognised as KNDy (kisspeptin, NKB and DynA) neurons.12 Although NKB is expressed in many other brain regions, the colocalisation of these peptides is unique to ARC, which is conserved among mammalian species including human.13–15
Two NK3R-selective agonist peptides have been reported: [MePhe7]-NKB16 and senktide (Table 1).17 [MePhe7]-NKB was identified through a structure–activity relationship study on NKB(4–10), a short NKB analogue, which was designed based on the sequence of the neurokinin-1 receptor (NK1R)/NK3R agonist peptide, DiMeC7.16 Our previous structure–activity relationship study on naturally occurring tachykinin peptides revealed that substitution of Phe5 with an acidic amino acid (Asp5 or Glu5) and of Val7 with MePhe7 in NKB is indispensable for potent activity and selectivity of [MePhe7]-NKB for NK3R.18 On the basis of these data, a novel potent NK3R agonist with higher NK3R selectivity compared with [MePhe7]-NKB was identified.18 Senktide is the most common NK3R agonist peptide, which was incidentally identified in an N-methylamino acid scanning study on substance P (SP)-related peptides for the development of NK1R-selective agonists.17 It has been used in a number of in vitro and in vivo experiments to investigate NK3R functions.6,7,19–25 For example, senktide-mediated NK3R stimulation evoked dopamine release from dopamine neurons in vitro.19 In addition, the function of the KNDy neurons in the pulsatile secretion of GnRH was demonstrated by in vivo administration of senktide.25 Although senktide is subjected to peptidase-mediated degradation, a novel peptidomimetic derivative of senktide exerts a prolonged effect on the GnRH pulse generator in ovariectomised (OVX) goats.26
In the current study, we report the structure–activity relationships of senktide derivatives with modification of component amino acids or N-terminal functional group to identify a novel potent NK3R agonist. The stability under peptidase-mediated degradation conditions and the in vivo bioactivity of potent NK3R agonist derivatives are also discussed.
Results and discussion
Synthesis of senktide analogue peptides
All peptide chains were constructed by standard Fmoc-based solid-phase peptide synthesis (Fmoc-SPPS) on Rink-amide resin. Succinylation, carbamoylation, or hydroxycarbamoylation of the peptide's N-terminus was carried out on resin as previously reported.26–28 Modification with oxalyl, aminocarbamoyl, methyl oxalyl, 2-amino-2-oxoacetyl, or sulfamoyl groups at the peptide N-terminus was conducted using appropriate reagents as described in the experimental procedures. The final deprotection and cleavage from the resin with the cocktail TFA/thioanisole/m-cresol/1,2-ethanedithiol/H2O (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5:5:5), followed by RP-HPLC purification afforded the expected peptides as TFA salts. All peptides were identified by ESI-MS or MALDI-TOF-MS and the purity was determined to be more than 98% by analytical HPLC.
:5:5:5:5), followed by RP-HPLC purification afforded the expected peptides as TFA salts. All peptides were identified by ESI-MS or MALDI-TOF-MS and the purity was determined to be more than 98% by analytical HPLC.
Structure–activity relationships of tachykinin consensus sequences in senktide
The C-terminal sequence, Phe-Xaa-Gly-Leu-Met-NH2, is common among mammalian tachykinin peptides including SP, neurokinin A (NKA) and NKB, which are endogenous agonists of NK1R, neurokinin-2 receptor (NK2R) and NK3R, respectively.29–31 In addition, tachykinin peptides share similar sequences among mammalian species, whereas the consensus sequence often contains several amino acid substitutions in other species. For example, insect-derived tachykinin-related peptides with the consensus Phe-Xaa-Gly/Ala-Yaa-Arg-NH2 sequence32 show potent agonistic activity towards the tachykinin peptide receptor, STKR, which has 41% homology with human NK3R. Octopus-derived tachykinin peptides have a variety of C-terminal sequences, including oct-TKRP I with Gly-Thr-Arg-NH2 and oct-TKRP-V with Pro-Pro-Lys-NH2.33 These oct-TKRP peptides show potent agonistic activity towards the oct-TKRP receptor.33 To optimise the amino acids of the consensus sequences in senktide for receptor binding and selectivity, we designed senktide derivatives based on these invertebrate tachykinin-related peptide sequences (see the ESI†). However, these modifications resulted in bioactivity loss, except for the peptide with Gly to Pro substitution, which exhibited weak NK3R binding. Substitution of Phe in the consensus sequence with an aromatic amino acid (Tyr, Trp or His) also led to significantly weak or no NK3R binding. These results suggest that the amino acids in the tachykinin consensus sequence, including the C-terminal Met-NH2, are indispensable for the biological activity of senktide towards NK3R.MePhe is a characteristic amino acid that contributes to the high NK3R selectivity of [MePhe7]-NKB and senktide.16–18 We next designed several senktide derivatives with substitution at the MePhe position with other N-methylamino acids (Table 2). Among derivatives 1a–c with an aliphatic N-methylamino acid, the MeVal- (1a) and MeIle-derivatives (1b) showed slightly lower biological activity than senktide (1a: IC50 = 104 nM, EC50 = 64 pM; 1b: IC50 = 91 nM, EC50 = 50 pM).34–36 In contrast, substitution with MeLeu resulted in loss of binding to NK3R. The MeTyr- (1d) and MeTrp-substituted peptides (1e) also maintained the NK3R binding and activation (1d: IC50 = 28 nM, EC50 = 41 pM; 1e: IC50 = 123 nM, EC50 = 33 pM). Substitution with D-MePhe resulted in loss of NK3R binding, suggesting that NK3R recognises the side chain orientation derived from the L-MePhe chiral centre in senktide. As the highly acidic succinyl-Asp substructure in senktide is likely to interact with the potential basic functional group(s) of NK3R, replacement of MePhe with MeAsp was also attempted; however, the MeAsp derivative (1g) showed no binding to NK3R. Taken together, some appropriate aliphatic or aromatic N-methylamino acids are acceptable at the MePhe position for potent NK3R agonists. Of note, the potent peptides 1a, 1b, 1d and 1e showed no binding affinity to NK1R and NK2R at 10 μM.
| Succinyl-Asp-Phe-Xaa-Gly-Leu-Met-NH2 | |||||
|---|---|---|---|---|---|
| Peptide | Xaa | NK3R | NK1R | NK2R | |
| IC50a (nM) | EC50b (pM) | IC50a (nM) | IC50a (nM) | ||
| a IC50 values indicate the concentration needed for 50% inhibition of receptor binding of ([125I]His3, MePhe7)-NKB to NK3R, [125I]-BH-SP to NK1R, and [125I]-NKA to NK2R. b EC50 values are the concentration needed for 50% of the full agonist activity induced by 100 nM senktide. c Not evaluated. | |||||
| Senktide | MePhe | 45 | 17 | >10000 |
>10000 |
| 1a | MeVal | 104 | 64 | >10000 |
>10000 |
| 1b | MeIle | 91 | 50 | >10000 |
>10000 |
| 1c | MeLeu | >10000 |
—c | —c | —c |
| 1d | MeTyr | 28 | 41 | >10000 |
>10000 |
| 1e | MeTrp | 123 | 33 | >10000 |
>10000 |
| 1f | D-MePhe | >10000 |
—c | —c | —c |
| 1g | MeAsp | >10000 |
—c | —c | —c |
Structure–activity relationships of N-terminal substructures in senktide
Senktide possesses two anionic carboxy groups at the N-terminus, which have been suggested to provide high NK3R selectivity.18 We next investigated the modification of the senktide N-terminal substructure with a number of potential bioisosteres.Initially we substituted the N-terminal succinyl group in senktide with L-Asp or D-Asp derivatives (Table 3). As the succinyl group in senktide is derived from the side chain of L-Asp4 in NKB, regeneration of a chiral centre was expected to allow favourable orientation of negatively charged functional groups.37 Introduction of unmodified L-Asp (2a) resulted in a significant decrease in receptor binding and agonistic activity (2a: IC50 = 175 nM, EC50 = 63 pM). The acetyl capping of the Asp α-amino group (3a) restored the potent bioactivity (3a: IC50 = 68 nM, EC50 = 31 pM), suggesting that a basic functional group at the N-terminal region is unfavourable for NK3R binding and activation. Among derivatives 3a–d with a modification at the second acidic amino acid, the L-Glu-substituted derivative 3c showed 5-fold more potent receptor binding and 3-fold more potent agonist activity compared with those of senktide (3c: IC50 = 7.7 nM, EC50 = 8.1 pM). A similar structure–activity relationship was obtained among the 4a–d series of peptides with an Ac-D-Asp modification at the N-terminus, in which the most potent L-Glu peptide 4c exhibited 3-fold more potent receptor binding and 2-fold stronger agonist activity compared with those of senktide (4c: IC50 = 13 nM, EC50 = 12 pM). The advantage of L-Glu for NK3R activation is consistent with the potent biological activity of PG-KII with an L-Asp–L-Glu substructure at this position, which was derived from the Australian frog, Pseudophryne guentheri.31
| R-Xaa-Phe-MePhe-Gly-Leu-Met-NH2 | ||||||
|---|---|---|---|---|---|---|
| Peptide | R | Xaa | NK3R | NK1R | NK2R | |
| IC50a (nM) | EC50b (pM) | IC50a (nM) | IC50a (nM) | |||
| a IC50 values indicate the concentration needed for 50% inhibition of receptor binding of ([125I]His3, MePhe7)-NKB to NK3R, [125I]-BH-SP to NK1R, and [125I]-NKA to NK2R. b EC50 values are the concentration needed for 50% of the full agonist activity induced by 100 nM senktide. | ||||||
| Senktide | Succinyl | L-Asp | 40 | 23 | >10000 |
>10000 |
| 2a | H-L-Asp | L-Asp | 175 | 63 | >10000 |
>10000 |
| 3a | Ac-L-Asp | L-Asp | 68 | 31 | >10000 |
>10000 |
| 3b | Ac-L-Asp | D-Asp | 83 | 24 | >10000 |
>10000 |
| 3c | Ac-L-Asp | L-Glu | 7.7 | 8.1 | >10000 |
>10000 |
| 3d | Ac-L-Asp | D-Glu | 37 | 18 | >10000 |
>10000 |
| 4a | Ac-D-Asp | L-Asp | 73 | 18 | >10000 |
>10000 |
| 4b | Ac-D-Asp | D-Asp | 55 | 17 | >10000 |
>10000 |
| 4c | Ac-D-Asp | L-Glu | 13 | 12 | >10000 |
>10000 |
| 4d | Ac-D-Asp | D-Glu | 68 | 17 | >10000 |
>10000 |
Taking advantage of these key insights, further investigations were carried out for optimisation of the spatial arrangements of two N-terminal acidic functional groups in senktide (Table 4). Among the senktide derivatives in which L-Asp was substituted with other acidic amino acids including D-Asp, L-Glu, D-Glu or L-2-aminoadipic acid (L-Aad) (5b–e), substitution with D-Asp (5b), L-Glu (5c) or L-Aad (5e) resulted in 2- or 3-fold more potent NK3R agonist activity (5b: IC50 = 16 nM, EC50 = 29 pM; 5c: IC50 = 6.1 nM, EC50 = 30 pM; 5e: IC50 = 9.9 nM, EC50 = 19 pM) compared with that of senktide, whereas peptide 5d with D-Glu showed slightly less potent agonist activity (5d: IC50 = 24 nM, EC50 = 84 pM). Interestingly, significant improvement in bioactivity was observed among the N-terminally oxalyl-modified derivatives, 6a–e. The derivatives with the L-Glu (6c), D-Glu (6d) or L-Aad (6e) substitution exhibited 7- to 9-fold more potent biological activity (6c: IC50 = 0.43 nM, EC50 = 9.1 pM; 6d: IC50 = 0.51 nM, EC50 = 14 pM; 6e: IC50 = 1.4 nM, EC50 = 7.4 pM) compared with that of senktide. Although the peptides with the L-Asp (6a) or D-Asp (6b) substitution showed 10-fold more potent binding inhibition, significant improvement in NK3R activation was not observed (6a: IC50 = 3.6 nM, EC50 = 83 pM; 6b: IC50 = 3.5 nM, EC50 = 22 pM). These results suggest that a combination of carboxy functional groups with a short tether at the N-terminus and with a long side chain at the L-Asp position of senktide would be appropriate for potent agonist activity towards NK3R.
| R-Xaa-Phe-MePhe-Gly-Leu-Met-NH2 | ||||||
|---|---|---|---|---|---|---|
| Peptide | R | Xaa | NK3R | NK1R | NK2R | |
| IC50a (nM) | EC50b (pM) | IC50a (nM) | IC50a (nM) | |||
| a IC50 values indicate the concentration needed for 50% inhibition of receptor binding of ([125I]His3, MePhe7)-NKB to NK3R, [125I]-BH-SP to NK1R, and [125I]-NKA to NK2R. b EC50 values are the concentration needed for 50% of the full agonist activity induced by 100 nM senktide. | ||||||
| Senktide | Succinyl | L-Asp | 43 | 63 | >10000 |
>10000 |
| 5b | Succinyl | D-Asp | 16 | 29 | >10000 |
>10000 |
| 5c | Succinyl | L-Glu | 6.1 | 30 | >10000 |
>10000 |
| 5d | Succinyl | D-Glu | 24 | 84 | >10000 |
>10000 |
| 5e | Succinyl | L-Aad | 9.9 | 19 | >10000 |
>10000 |
| 6a | Oxalyl | L-Asp | 3.6 | 83 | >10000 |
>10000 |
| 6b | Oxalyl | D-Asp | 3.5 | 22 | >10000 |
>10000 |
| 6c | Oxalyl | L-Glu | 0.43 | 9.1 | >10000 |
>10000 |
| 6d | Oxalyl | D-Glu | 0.51 | 14 | >10000 |
>10000 |
| 6e | Oxalyl | L-Aad | 1.4 | 7.4 | >10000 |
>10000 |
Further modifications of the senktide N-terminus with isosteric functional groups were performed in an attempt to improve the biological activity (7–12) (Table 5). All the derivatives maintained the potent binding inhibition and agonist activity toward NK3R (IC50 = 4.1–39 nM; EC50 = 2.5–43 pM). Peptides 10 and 12, with 2-methoxy-2-oxoacetyl and aminosulfonyl groups, respectively, showed slightly more potent biological activity compared with that of senktide [10: IC50 = 4.1 nM, EC50 = 2.9 pM; 12: IC50 = 5.6 nM, EC50 = 2.5 pM], suggesting that a charged anionic functional group is not critical for the N-terminus of senktide. Using the potent peptides, 10 and 12, we further optimised the L-Asp position using acidic amino acids (Table S3†). However, the improvement in agonist activity towards NK3R by modifying the L-Asp position was limited, which was inconsistent with the case of the series of derivatives with an N-terminal oxalyl group. Altogether, we concluded that the oxalyl group is the best N-terminal functional group for NK3R agonists.
| R-Asp-Phe-MePhe-Gly-Leu-Met-NH2 | |||||
|---|---|---|---|---|---|
| Peptide | R | NK3R | NK1R | NK2R | |
| IC50a (nM) | EC50b (pM) | IC50a (nM) | IC50a (nM) | ||
| a IC50 values indicate the concentration needed for 50% inhibition of receptor binding of ([125I]His3, MePhe7)-NKB to NK3R, [125I]-BH-SP to NK1R, and [125I]-NKA to NK2R. b EC50 values are the concentration needed for 50% of the full agonist activity induced by 100 nM senktide. | |||||
| Senktide |

|
29 | 3.4 | >10000 |
>10000 |
| 7 |

|
39 | 13 | >10000 |
>10000 |
| 8 |

|
10 | 43 | >10000 |
>10000 |
| 9 |

|
34 | 6.0 | >10000 |
>10000 |
| 10 |

|
4.1 | 2.9 | >10000 |
>10000 |
| 11 |

|
7.9 | 11 | >10000 |
>10000 |
| 12 |
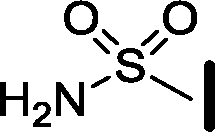
|
5.6 | 2.5 | >10000 |
>10000 |
The highly potent senktide derivatives, 6c–e, were evaluated for agonist activity towards NK3R derived from other mammalian species including rat, goat and cattle (Table 6). All the peptides showed more potent agonist activity towards mammalian NK3R compared with that of senktide. The most potent peptide, 6e, with oxalyl-L-Aad at the N-terminus exhibited 3-, 2- and 4-fold higher potency than senktide in activation of rat, goat and cattle NK3R, respectively.38
Proteolytic degradation of potent NK3R agonist peptides in serum and by endopeptidase treatment
NKB is degraded and inactivated by cleavage at the Gly-Leu peptide bond by membrane-anchored neutral endopeptidase 24.11 (NEP 24.11; also known as neprilysin or enkephalinase).39 Senktide is also inactivated by NEP 24.11-mediated cleavage at the same site.26 To identify potent NK3R agonists with resistance to NEP 24.11-mediated degradation, we assessed the stability of the potent derivatives obtained in the above structure–activity relationship experiments.Peptides 6c and 6d were stable in rat, pig, goat and cattle serum for 24 h similarly to senktide (Fig. S1†),26,40 suggesting that the N-terminal oxalyl capping also prevents the possible degradation by peptidases in the serum, which was observed in the case of [MePhe7]-NKB.26 Next, the resistance of peptides 6c–e to NEP 24.11-mediated degradation was investigated (Fig. 1). More than 70% of 6c and 6e were digested at the Gly-Leu dipeptide bond by 24 h of incubation with NEP 24.11. In contrast, little degradation of 6d by NEP 24.11 was observed after 24 h. These results suggest that substitution of L-Asp in senktide with D-Glu is effective for resistance against NEP 24.11-mediated proteolytic degradation at the distal Gly-Leu cleavage site. Thus, peptide 6d with oxalyl-D-Glu at the N-terminus is a potent NK3R agonist with high biological stability.
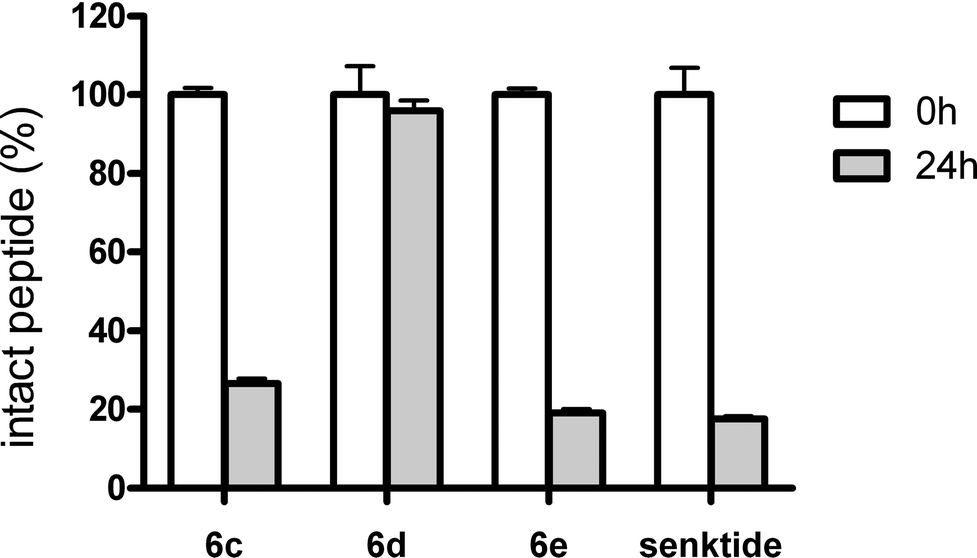 | ||
| Fig. 1 Proteolytic degradation profile of senktide derivatives by NEP 24.11. The peptides were incubated in NEP 24.11 solution for 24 h at 37 °C and were analysed by HPLC (detection at 220 nm). Data represent the mean ± SD (n = 3). | ||
Effect of peripheral administration of peptide 6d on the induction of periodic burst of multiple-unit activity (MUA) volleys in goat
It has been reported that pulsatile GnRH secretion is synchronized with the induction of periodic burst of multiple-unit activity (MUA) volleys.6,41 To assess the in vivo effect on pulsatile secretion of GnRH, we monitored the induction of MUA volleys after intravenous administration of 6d. MUA volleys with intervals less than 80% of the average spontaneous intervolley intervals (T) were defined as ligand-induced, whereas those with intervals higher than 80% of T were spontaneous.26 The action duration (R) was the period from the injection until the onset of the following spontaneous MUA volley, and the number of MUA volleys occurring in this period (V) was counted.26 Peptide 6d (200 nmol) immediately induced several MUA volleys. The R and V values of 6d were similar to those of senktide (Table 7 and Fig. S2†).42 This is inconsistent with the results in our recent study, in which the novel biostable NK3R-selective peptidomimetic agonist 13 [succinyl-Asp-Phe-MePhe-Gly-ψ[(E)-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH]-Leu-Met-NH2] showed prolonged in vivo bioactivity in goat compared with that of senktide.26 These results may suggest that the clearance of the peptide from the circulation should be further optimised to develop NK3R agonists with prolonged in vivo bioactivity.
CH]-Leu-Met-NH2] showed prolonged in vivo bioactivity in goat compared with that of senktide.26 These results may suggest that the clearance of the peptide from the circulation should be further optimised to develop NK3R agonists with prolonged in vivo bioactivity.
| R (min) | V | |
|---|---|---|
| a MUA volley induction in OVX goats by intravenous injection of NK3R ligands (200 nmol, n ≥ 3). Values are represented as the mean ± SD. b R values indicate the duration of the effect of NK3R ligands. c V values indicate the number of ligand-induced MUA volleys. | ||
| Senktide | 47.2 ± 14.2 | 2.6 ± 1.1 |
| 6d | 50.4 ± 23.8 | 2.7 ± 1.5 |
Conclusions
In this study, we investigated the structure–activity relationships of senktide derivatives in relation to agonist activity towards NK3R. The consensus sequence of tachykinin peptides in senktide is important for NK3R binding and activation, while MePhe can be substituted with several hydrophobic N-methylamino acids. Optimisation of the N-terminal substructures resulted in identification of novel potent NK3R agonists (6c–e) with an oxalyl group at the peptide N-terminus. The investigation of the peptide resistance to NEP 24.11-mediated degradation revealed that peptide 6d with D-Glu at the L-Asp position in senktide was highly stable under in vitro conditions. Although further improvement to prevent the possible rapid clearance of senktide and the derivatives is needed, peptide 6d with its enhanced metabolic stability could be an appropriate lead peptide for designing novel NK3R agonists with prolonged in vivo bioactivity.Experimental procedures
General method for peptide synthesis
The protected linear peptides were constructed by Fmoc-based solid-phase synthesis on Rink-amide resin (0.66 mmol g−1, 45.5 mg, 0.025 mmol). Fmoc-protected amino acids (0.075 mmol) were coupled by N,N′-diisopropylcarbodiimide (DIC, 0.0116 cm3, 0.075 mmol) and HOBt·H2O (11.5 mg, 0.075 mmol) in DMF. Coupling of amino acid (0.075 mmol) to N-methylamino acid was carried out with HATU (28.5 mg, 0.075 mmol) and (iPr)2NEt (DIPEA, 0.013 cm3, 0.075 mmol). Completion of each coupling reaction was ascertained using the Kaiser ninhydrin test. The Fmoc-protecting group was removed by treating the resin with 20% piperidine in DMF. The resulting protected peptide resin was treated with TFA/thioanisole/m-cresol/1,2-ethanedithiol/H2O (5 cm3; 80:5:5:5:5) at room temperature for 2 h. After removal of the resin by filtration, the filtrate was poured into ice-cold dry Et2O (40 cm3). The resulting powder was collected by centrifugation and then washed three times with ice-cold dry Et2O (3 × 40 cm3). The crude product was purified by HPLC on a Cosmosil 5C18-ARII preparative column (Nacalai Tesque, 20 × 250 mm). All peptides were characterised by ESI-MS or MALDI-TOF-MS and the purity was calculated to be >98% by HPLC on a Cosmosil 5C18-ARII analytical column (Nacalai Tesque, 4.6 × 250 mm).
General procedures for modification of the peptide N-terminus
:1, 1.0 cm3) for 30 min. After removal of the above reagents, a solution of hydroxylamine hydrochloride (64.3 mg, 0.925 mmol) and DIPEA (0.322 cm3, 1.85 mmol) in DMF (1.0 cm3) was added. The suspension was stirred for 15 min to produce hydroxycarbamoyl-modified peptide resin for peptide 8.
Abbreviations
| Aad | L-2-Aminoadipic acid |
| ARC | Arcuate nucleus |
| BH-SP | Bolton–Hunter labelled substance P |
| DIC | N,N′-Diisopropylcarbodiimide |
| DynA | Dynorphin A |
| GnRH | Gonadotropin-releasing hormone |
| HOAt | N-Hydroxy-7-azabenzotriazole |
| LH | Luteinising hormone |
| MUA | Multiple-unit activity |
| NKA | Neurokinin A |
| NKB | Neurokinin B |
| NK1R | Neurokinin-1 receptor |
| NK2R | Neurokinin-2 receptor |
| NK3R | Neurokinin-3 receptor |
| MePhe | N-Methylphenylalanine |
| OVX | Ovariectomised |
| SP | Substance P |
| SPPS | Solid-phase peptide synthesis |
Acknowledgements
We are grateful to Dr. Akira Hirasawa (Kyoto University) for his generous support in the experiments. This work was supported by a grant from the Ministry of Agriculture, Forestry and Fisheries of Japan (Research Program on Innovative Technologies for Animal Breeding, Reproduction, and Vaccine Development, REP-2001, REP-2003 and REP-2005), by Grants-in-Aid for Scientific Research from JSPS, Japan, and by Platform for Drug Discovery, Informatics, and Structural Life Science from MEXT, Japan. R.M. and T.N. are grateful for JSPS Research Fellowships for Young Scientists.References
- N. E. Rance, S. J. Krajewski, M. A. Smith, M. Cholanian and P. A. Dacks, Brain Res., 2010, 1364, 116–128 CrossRef CAS PubMed.
- R. P. Millar and C. L. Newton, Nat. Rev. Endocrinol., 2013, 9, 451–466 CrossRef CAS PubMed.
- A. K. Topaloglu, F. Reimann, M. Guclu, A. S. Yalin, L. D. Kotan, K. M. Porter, A. Serin, N. O. Mungan, J. R. Cook, M. N. Ozbek, S. Imamoglu, N. S. Akalin, B. Yuksel, S. O'Rahilly and R. K. Semple, Nat. Genet., 2009, 41, 354–358 CrossRef CAS PubMed.
- E. Gianetti, C. Tusset, S. D. Noel, M. G. Au, A. A. Dwyer, V. A. Hughes, A. P. Abreu, J. Carroll, E. Trarbach, L. F. G. Silveira, E. M. F. Costa, B. B. de Mendonça, M. de Castro, A. Lofrano, J. E. Hall, E. Bolu, M. Ozata, R. Quinton, J. K. Amory, S. E. Stewart, W. Arlt, T. R. Cole, W. F. Crowley, U. B. Kaiser, A. C. Latronico and S. B. Seminara, J. Clin. Endocrinol. Metab., 2010, 95, 2857–2867 CrossRef CAS PubMed.
- J. Young, J. T. George, J. A. Tello, B. Francou, J. Bouligand, A. Guiochon-Mantel, S. Brailly-Tabard, R. A. Anderson and R. P. Millar, Neuroendocrinology, 2013, 97, 193–202 CrossRef CAS PubMed.
- Y. Wakabayashi, T. Nakada, K. Murata, S. Ohkura, K. Mogi, V. M. Navarro, D. K. Clifton, Y. Mori, H. Tsukamura, K. Maeda, R. A. Steiner and H. Okamura, J. Neurosci., 2010, 30, 3124–3132 CrossRef CAS PubMed.
- T. Sandoval-Guzmán and N. E. Rance, Brain Res., 2004, 1026, 307–312 CrossRef PubMed.
- W. D. Odell, R. S. Swerdloff, J. Bain, F. Wollesen and P. K. Grover, Endocrinology, 1974, 95, 1380–1384 CrossRef CAS PubMed.
- S. G. Hillier, Mol. Cell. Endocrinol., 2001, 179, 39–46 CrossRef CAS.
- S. W. Walsh, E. J. Williams and A. C. O. Evans, Anim. Reprod. Sci., 2011, 123, 127–138 CrossRef CAS PubMed.
- R. L. Goodman, M. N. Lehman, J. T. Smith, L. M. Coolen, C. V. R. de Oliveira, M. R. Jafarzadehshirazi, A. Pereira, J. Iqbal, A. Caraty, P. Ciofi and I. J. Clarke, Endocrinology, 2007, 148, 5752–5760 CrossRef CAS PubMed.
- M. N. Lehman, L. M. Coolen and R. L. Goodman, Endocrinology, 2010, 151, 3479–3489 CrossRef CAS PubMed.
- M. C. Burke, P. A. Letts, S. J. Krajewski and N. E. Rance, J. Comp. Neurol., 2006, 498, 712–726 CrossRef CAS PubMed.
- V. M. Navarro, M. L. Gottsch, C. Chavkin, H. Okamura, D. K. Clifton and R. A. Steiner, J. Neurosci., 2009, 29, 11859–11866 CrossRef CAS PubMed.
- N. E. Rance, Peptides, 2009, 30, 111–122 CrossRef CAS PubMed.
- G. Drapeau, P. D'Orléans-Juste, S. Dion, N. E. Rhaleb, N. E. Rouissi and D. Regoli, Neuropeptides, 1987, 10, 43–54 CrossRef CAS.
- U. Wormser, R. Laufer, Y. Hart, M. Chorev, C. Gilon and Z. Selinger, EMBO J., 1986, 5, 2805–2808 CAS.
- R. Misu, T. Noguchi, H. Ohno, S. Oishi and N. Fujii, Bioorg. Med. Chem., 2013, 21, 2413–2417 CrossRef CAS PubMed.
- R. Alonso, M. Fournier, P. Carayon, G. Petitpretre, G. Le Fur and P. Soubrié, Eur. J. Neurosci., 1996, 8, 801–808 CrossRef CAS.
- A. J. Stoessl, C. T. Dourish and S. D. Iversen, Br. J. Pharmacol., 1988, 94, 285–287 CrossRef CAS PubMed.
- A. J. Stoessl, C. T. Dourish and S. D. Iversen, Brain Res., 1990, 517, 111–116 CrossRef CAS.
- K. D. Keegan, G. N. Woodruff and R. D. Pinnock, Br. J. Pharmacol., 1992, 105, 3–5 CrossRef CAS PubMed.
- M. A. de Souza Silva, E. L. Mello Jr., C. P. Müller, G. Jocham, R. S. Maior, J. P. Huston, C. Tomaz and M. Barros, Peptides, 2006, 27, 2214–2223 CrossRef CAS PubMed.
- J. S. Kinsey-Jones, P. Grachev, X. F. Li, Y. S. Lin, S. R. Milligan, S. L. Lightman and K. T. O'Byrne, Endocrinology, 2012, 153, 307–315 CrossRef CAS PubMed.
- Y. Wakabayashi, T. Yamamura, K. Sakamoto, Y. Mori and H. Okamura, J. Reprod. Dev., 2013, 59, 40–48 CAS.
- R. Misu, S. Oishi, A. Yamada, T. Yamamura, F. Matsuda, K. Yamamoto, T. Noguchi, H. Ohno, H. Okamura, S. Ohkura and N. Fujii, J. Med. Chem., 2014, 57, 8646–8651 CrossRef CAS PubMed.
- C. Zheng and A. P. Combs, J. Comb. Chem., 2002, 4, 38–43 CrossRef CAS PubMed.
- C. A. Parrish, N. D. Adams, K. R. Auger, J. L. Burgess, J. D. Carson, A. M. Chaudhari, R. A. Copeland, M. A. Diamond, C. A. Donatelli, K. J. Duffy, L. F. Faucette, J. T. Finer, W. F. Huffman, E. D. Hugger, J. R. Jackson, S. D. Knight, L. Luo, M. L. Moore, K. A. Newlander, L. H. Ridgers, R. Sakowicz, A. N. Shaw, C. M. Sung, D. Sutton, K. W. Wood, S. Y. Zhang, M. N. Zimmerman and D. Dhanak, J. Med. Chem., 2007, 50, 4939–4952 CrossRef CAS PubMed.
- K. Kangawa, N. Minamino, A. Fukuda and H. Matsuo, Biochem. Biophys. Res. Commun., 1983, 114, 533–540 CrossRef CAS.
- M. M. Kurtz, R. Wang, M. K. Clements, M. A. Cascieri, C. P. Austin, B. R. Cunningham, G. G. Chicchi and Q. Liu, Gene, 2002, 296, 205–212 CrossRef CAS.
- For a review, see: C. Severini, G. Improta, G. Falconieri-Erspamer, S. Salvadori and V. Erspamer, Pharmacol. Rev., 2002, 54, 285–322 CrossRef CAS.
- H. Torfs, H. B. Oonk, J. V. Broeck, J. Poels, W. Van Poyer, A. De Loof, F. Guerrero, R. H. Meloen, K. Åkerman and R. J. Nachman, Arch. Insect Biochem. Physiol., 2001, 48, 39–49 CrossRef CAS PubMed.
- A. Kanda, K. Takuwa-Kuroda, M. Aoyama and H. Satake, FEBS J., 2007, 274, 2229–2239 CrossRef CAS PubMed.
- Apparent discrepancies between binding competition (IC50 values) and functional assays (EC50 values) may be derived from the different receptor occupancies of the compounds in the concentration range of dose–response curves. In the functional assay, maximal functional responses may be achieved when a small fraction of the receptor population is occupied in the presence of low concentrations of the compounds: see ref. 35 and 36.
- S. Wilson, J. K. Chambers, J. E. Park, A. Ladurner, D. W. Cronk, C. G. Chapman, H. Kallender, M. J. Browne, G. J. Murphy and P. W. Young, J. Pharmacol. Exp. Ther., 1996, 279, 214–221 CAS.
- S. Galandrin and M. Bouvier, Mol. Pharmacol., 2006, 70, 1575–1584 CrossRef CAS PubMed.
- A. D. Ganjiwale, G. S. Rao and S. M. Cowsik, J. Chem. Inf. Model., 2011, 51, 2932–2938 CrossRef CAS PubMed.
- The higher concentrations of ligands were needed for activation of cattle NK3R compared with NK3R activation of other species. This may be due to the less binding affinity of NK3R ligands towards cattle NK3R or the low expression level of cattle NK3R on the cell membrane. This is supported by the less binding of radiolabelled [MePhe7]-NKB towards cattle NK3R (data not shown).
- N. M. Hooper and A. J. Turner, FEBS Lett., 1985, 190, 133–136 CrossRef CAS.
- [MePhe7]-NKB (a reference peptide) was completely degraded in rat and goat sera, and more than 80% of the peptide was degraded in pig and cattle sera.
- S. Ohkura, K. Takase, S. Matsuyama, K. Mogi, T. Ichimaru, Y. Wakabayashi, Y. Uenoyama, Y. Mori, R. A. Steiner, H. Tsukamura, K.-I. Maeda and H. Okamura, J. Neuroendocrinol., 2009, 21, 813–821 CrossRef CAS PubMed.
- No improvement of in vivo bioactivity of peptides 3c, 6c and S3a was observed (data not shown).
- C. A. Mirkin, C. Liu, Y. Wang, A. B. Braunschweig, X. Liao, L. R. Giam, B. Y. Lee, J. S. Fragala and A. K. Henning, PCT Int. Appl., WO2010011398 A2, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4md00514g |
| This journal is © The Royal Society of Chemistry 2015 |