
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGenotype–phenotype modeling considering intermediate level of biological variation: a case study involving sensory traits, metabolites and QTLs in ripe tomatoes†
Huange
Wang
*a,
Joao
Paulo
a,
Willem
Kruijer
a,
Martin
Boer
a,
Hans
Jansen
a,
Yury
Tikunov
b,
Björn
Usadel
c,
Sjaak
van Heusden
b,
Arnaud
Bovy
b and
Fred
van Eeuwijk
a
aBiometris, Wageningen University and Research Centre, PO Box 16, 6700AA Wageningen, The Netherlands. E-mail: hw428@cam.ac.uk
bPlant Research International, PO Box 386, 6700AJ Wageningen, The Netherlands
cInstitute for Biology I, RWTH Aachen University, Worringer Weg 3, 52074 Aachen, Germany
First published on 2nd September 2015
Abstract
Modeling genotype–phenotype relationships is a central objective in plant genetics and breeding. Commonly, variations in phenotypic traits are modeled directly in relation to variations at the DNA level, regardless of intermediate levels of biological variation. Here we present an integrative method for the simultaneous modeling of a set of multilevel phenotypic responses to variations at the DNA level. More specifically, for ripe tomato fruits, we use Gaussian graphical models and causal inference techniques to learn the dependencies of 24 sensory traits on 29 metabolites and the dependencies of those sensory and metabolic traits on 21 QTLs. The inferred dependency network which, though not essentially representing biological pathways, suggests how the effects of allele substitutions propagate through multilevel phenotypes. Such simultaneous study of the underlying genetic architecture and multifactorial interactions is expected to enhance the prediction and manipulation of complex traits.
1 Introduction
Elucidating the genetic architecture of complex traits is a key objective in plant genetics. Existing methods mainly directly identify genomic regions associated with phenotypic variation through single- or multi-trait quantitative trait locus (QTL) analysis. However, between DNA and final phenotype, there exist multiple levels of intermediate substances such as proteins and metabolites, which possess a quantitative nature and vary among individuals within populations. Successfully linking variations at intermediate levels to DNA variations on the one hand and to phenotypic variations on the other hand should enhance the prediction and manipulation of sets of interacting and possibly complex traits.Interactions between and within multilevel phenotypic and omics traits can be learnt by probabilistic graphical models (PGMs), which typically unravel probabilistic conditional independence structures of multiple variables. A particular type of PGMs, namely Gaussian graphical models (GGMs, also known as “covariance selection” or “concentration graph” models),1 has become popular in computational systems biology. GGMs are claimed to be superior to the well-known correlation networks (also called “relevance networks”), as they are based on partial correlations and thereby distinguish between direct and indirect associations.2
The metabolome is of great importance in crop plants, as metabolite concentrations reflect the developmental stage of plants and determine to a great extent many quality traits such as nutritional value and sensory attributes. Recent advances in plant metabolite profiling, including gas chromatography-mass spectrometry (GC-MS), liquid chromatography-mass spectrometry (LC-MS) and nuclear magnetic resonance (NMR), have enabled large-scale analyses that reveal quantitative variation in the metabolic content of various species.3 Accordingly, it has become feasible to investigate associations between metabolites.
Beyond associations, dependencies among metabolites are of interest to plant biologists for understanding adaptation and survival in relation to primary and secondary metabolism. The metabolome is recognized as a highly interactive system, where a metabolite variation may lead to a chain reaction: changes in the concentration of a metabolite alter the concentrations of some other metabolites through specific regulatory pathways. A few methods have been presented to uncover dependencies among associated traits, using previously determined QTLs.4–9 All these approaches require at least one unique QTL for each trait studied. In practice, however, this prerequisite is often not satisfied. To cope with more general scenarios where some of the traits come without QTL or unique QTLs, a QTL + phenotype supervised orientation (QPSO) algorithm was recently proposed.10 This algorithm looks promising for learning dependencies between metabolites whose profiling is still expensive and time-consuming, so that sample sizes are typically small and the power to detect QTLs is subsequently limited.
In this paper, we combine three GGM approaches with the QPSO algorithm to model genotype–phenotype relationships with consideration for the intermediate metabolite variations. Our integrative method is demonstrated through a practical case study, in which we obtain a dependency network involving 24 sensory traits, 29 metabolites and 21 QTLs identified for those sensory traits and metabolites in ripe tomato fruits. In the first place, a high-confidence true positive undirected network, which represents direct associations within and between metabolites and sensory traits, is learnt by the three GGM approaches including: (i) lasso-based neighborhood selection11 (LBNS) in combination with a stability approach to regularization selection12 (StARS), (ii) the PC-skeleton algorithm13 and (iii) the lasso14 in combination with stability selection15 (SS). In the second place, given the undirected network and QTLs previously identified for the sensory traits and metabolites, edge directions (i.e., the directions of associations) are inferred by the QPSO algorithm. In the third place, each sensory trait and metabolite is regressed on its QTLs and inferred parent nodes (i.e., nodes with outgoing edges pointing to this sensory trait or metabolite). The fitted regression coefficients provide more details regarding the estimated dependencies: “+” – positive, “−” – negative, and their absolutes values – the strength of dependencies.
It is known that tomato sensory traits are co-determined by metabolites.16–18 A major concern of plant breeders and physiologists is, thus, how to first identify metabolites and corresponding genomic regions that are responsible for variations in sensory traits, and next develop a strategy for the genetic improvement of certain sensory traits jointly. Our proposed method provides a way to investigate the dependencies within and between metabolites and sensory traits. The estimated dependencies which, though not equal to biological pathways, suggest how the effects of allele substitutions propagate through metabolites to sensory traits. This information should help breeders and physiologists to predict and manipulate the target sensory traits.
2 Materials
2.1 Tomato populations and phenotypic data
The data were collected on ripe fruits of four F2 segregating populations developed in the tomato program of a consortium that was called the Centre for BioSystems Genomics (CBSG; http://www.cbsg.nl/tomato.aspx). Four contrasting tomato cultivars were selected as parental lines, namely C074 (cherry fruit type), C085 (cherry fruit type), R075 (round fruit type) and R104 (round fruit type). Crosses between the parental lines were made following a half-diallel mating design. The F1 plants were selfed and the subsequent F2 generation included four cherry × round populations: C074 × R075, C074 × R104, C085 × R075 and C085 × R104. For each cherry × round population, plants of 48 offspring genotypes were grown.On all plants, 29 metabolites and 24 sensory traits were scored on ripe fruits, which were harvested and prepared as described in ref. 19. Metabolic profiling was carried out in two ways: volatiles were measured using a head space Solid Phase Microextraction-Gas Chromatography-Mass Spectrometry (SPME-GC-MS);19 sugars and acids were quantified using the method of GC-MS of trimethylsilyl ester derivatives.20 All metabolites were identified at level 1 annotation21 using authentic chemical standards analyzed at identical experimental conditions, except beta-damascenone, which has a level 2 identity: NIST mass spectral library 2010 (Mainlib) match 911 (0-1000) and the library retention index deviation of 4 (http://www.nist.gov/srd/nist1a.cfm). All metabolites have corresponding CAS ID numbers (Sheet 1 of Supplementary Dataset S1, ESI†). Sensory profiles were obtained by a trained panel of judges for just 16 out of the 48 genotypes for each cherry × round population. The judges evaluated each genotype for a set of sensory traits including smell, taste, aftertaste, and mouthfeel experience. All sensory attributes were scored on a scale of 0 to 100. In addition to the metabolites and sensory traits, brix was measured for each genotype using a refractometer (GMK-701R; Nie-Co Products, Aalsmeer, NL). Metabolite abundances were transformed to log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 scale for statistical analysis. Prior to network reconstruction, genotypic means for the sensory traits, brix and metabolites were calculated using mixed models, which contained corrections for measurement time (for brix and metabolites), judge (for the sensory traits), population (for all traits) and the presence/absence of the Rin mutation (for all traits). Rin is the recessive ripening-inhibitor mutation that inhibits ripening,22 and was present in all crosses involving parent R075. Fruits from plants that are homozygous for Rin do not ripen and have lower concentrations of metabolites. The corrected genotypic means (Sheet 2–4 of Supplementary Dataset S1, ESI†) were used for further analysis.
10 scale for statistical analysis. Prior to network reconstruction, genotypic means for the sensory traits, brix and metabolites were calculated using mixed models, which contained corrections for measurement time (for brix and metabolites), judge (for the sensory traits), population (for all traits) and the presence/absence of the Rin mutation (for all traits). Rin is the recessive ripening-inhibitor mutation that inhibits ripening,22 and was present in all crosses involving parent R075. Fruits from plants that are homozygous for Rin do not ripen and have lower concentrations of metabolites. The corrected genotypic means (Sheet 2–4 of Supplementary Dataset S1, ESI†) were used for further analysis.
2.2 Genotypic data and QTL analysis
A set of 6000 SNP markers was available from the Infinium BeadArray. A selection of the markers was used to produce a high quality genetic linkage map. The obtained linkage map contained 600 SNP markers, 50 markers per chromosome, evenly spread at about 2 cM.A multi-trait QTL mapping strategy was implemented following the idea described in ref. 23 and 24. This strategy assumes that a single biparental offspring population was present. We turned the four cherry × round F2 populations into a single biparental F2 population by interpreting the two cherry parents to represent a first single parent and the two round tomato parents to represent a second single parent. Phenotypes were then regressed on genetic predictors, i.e. independent variables expressing molecular marker information. Genetic predictors were based on the expected number of alleles coming from the round parents, i.e. conditional QTL probabilities given flanking marker information using a Hidden Markov model.25 Parametrization was such that positive regression coefficients, QTL allele substitution effects, would point to the round allele as increasing the level of the trait, whereas negative QTL effects would imply that the cherry allele increased the trait. In comparison to ref. 23 and 24, for the current multi-trait QTL model we took care to allow for population specific intercepts for each trait. Another deviation from ref. 23 and 24 was that we included a trait specific correction for the presence/absence of the Rin mutation. Our multi-trait QTL model for a vector of trait responses was therefore as follows: traits = population specific trait intercepts + trait specific RIN corrections + trait specific QTLs + trait specific residuals. The trait specific residuals were modeled with trait specific variances and correlations. Multi-trait QTL models were fitted on each of three groups of traits: (1) volatiles; (2) sugars and acids; (3) sensory attributes. The multi-trait QTL modeling was done in GenStat 16 (http://www.vsni.co.uk/software/genstat/). Positions of QTLs identified for the traits studied are summarized in Table 1, and the QTL data are provided in the two spreadsheets of Supplementary Dataset S2 (ESI†).
| SNP/QTL | Chromosome | Position | Metabolite | SNP/QTL | Chromosome | Position | Metabolites | SNP/QTL | Chromosome | Position | Sensory traits |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs8314 | 1 | 10.35 | cis-3-Hexenol | rs2050 | 5 | 39.26 | 6-Methyl-5-hepten-2-one | rs7448 | 2 | 63.5 | Scent_aromaintensity |
| rs6454 | 1 | 128.97 | trans-2-Heptenal | rs2050 | 5 | 39.26 | Geranylacetone | rs7448 | 2 | 63.5 | Scent_tomato |
| rs6454 | 1 | 128.97 | alpha-Terpineol | rs2050 | 5 | 39.26 | Aspartic acid | rs7448 | 2 | 63.5 | Taste_sweet |
| rs6687 | 2 | 76.3 | cis-3-Hexenol | rs2050 | 5 | 39.26 | Citric acid | rs7448 | 2 | 63.5 | Taste_tomato |
| rs6687 | 2 | 76.3 | myo-Inositol | rs2050 | 5 | 39.26 | Malic acid | rs7448 | 2 | 63.5 | Taste_unripe |
| rs6687 | 2 | 76.3 | 3-Methylbutanal | rs2050 | 5 | 39.26 | Fructose | rs7448 | 2 | 63.5 | Taste_spicy |
| rs6687 | 2 | 76.3 | Citric acid | rs2050 | 5 | 39.26 | Glucose | rs7448 | 2 | 63.5 | Aftertaste_sweet |
| rs6687 | 2 | 76.3 | Glucose | rs4202 | 6 | 4.03 | Glutamic acid | rs8396 | 3 | 103.79 | Mouthfeel_moist |
| rs6687 | 2 | 76.3 | Fructose | rs4202 | 6 | 4.03 | Sucrose | rs8396 | 3 | 103.79 | Mouthfeel_solid |
| rs6687 | 2 | 76.3 | Sucrose | rs4202 | 6 | 4.03 | Brix | rs8396 | 3 | 103.79 | Aftertaste_rough |
| rs6687 | 2 | 76.3 | Brix | rs3540 | 6 | 14.14 | cis-3-Hexenal | rs7775 | 7 | 56.23 | Scent_smoky |
| rs6691 | 4 | 55.68 | 1-Penten-3-one | rs3540 | 6 | 14.14 | 1-Penten-3-one | rs7775 | 7 | 56.23 | Aftertaste_sweet |
| rs6691 | 4 | 55.68 | beta-Damascenone | rs3540 | 6 | 14.14 | Hexanal | rs8016 | 8 | 11.04 | Scent_smoky |
| rs7153 | 4 | 86.64 | Sucrose | rs3540 | 6 | 14.14 | Benzyl alcohol | rs8591 | 8 | 58.98 | Taste_pungent |
| rs7153 | 4 | 86.64 | Malic acid | rs6254 | 6 | 47.72 | beta-Ionone | rs7089 | 10 | 7.73 | Scent_aromaintensity |
| rs7153 | 4 | 86.64 | Aspartic acid | rs6254 | 6 | 47.72 | 6-Methyl-5-hepten-2-one | rs7089 | 10 | 7.73 | Scent_sweet |
| rs7153 | 4 | 86.64 | Citric acid | rs6254 | 6 | 47.72 | Methyl_salicylate | rs7089 | 10 | 7.73 | Taste_sour |
| rs7153 | 4 | 86.64 | Glutamic acid | rs6254 | 6 | 47.72 | Hexanal | rs7089 | 10 | 7.73 | Aftertaste_sour |
| rs2050 | 5 | 39.26 | Methyl_salicylate | rs8092 | 6 | 56.64 | Citric acid | rs7089 | 10 | 7.73 | Aftertaste_salty |
| rs2050 | 5 | 39.26 | Phenylethyl_alcohol | rs8092 | 6 | 56.64 | Malic acid | rs8434 | 11 | 25.94 | Scent_spicy |
| rs2050 | 5 | 39.26 | cis-3-Hexenal | rs9061 | 9 | 87.48 | Methyl_salicylate | rs8434 | 11 | 25.94 | Taste_unripe |
| rs2050 | 5 | 39.26 | 3-Methylbutanal | rs9061 | 9 | 87.48 | Guaiacol | rs8434 | 11 | 25.94 | Mouthfeel_solid |
| rs2050 | 5 | 39.26 | 1-Penten-3-one | rs9084 | 9 | 94.11 | Citric acid | ||||
| rs2050 | 5 | 39.26 | beta-Ionone | rs9084 | 9 | 94.11 | Malic acid | ||||
| rs2050 | 5 | 39.26 | 3-Methyl-1-butanol | rs6903 | 11 | 86.04 | Phenylethyl_alcohol | ||||
| rs2050 | 5 | 39.26 | 2-Methyl-1-butanol | rs6903 | 11 | 86.04 | Benzeneacetaldehyde | ||||
| rs2050 | 5 | 39.26 | 2-Isobutylthiazole | rs6495 | 12 | 26.59 | beta-Damascenone |
3 Methods
3.1 Outline approach to dependency network reconstruction
Fig. 1 illustrates our integrative method for learning dependency network from the sensory, metabolic and QTL data. First, two GGM approaches, (i) LBNS + StARS and (ii) the PC-skeleton algorithm, were used to obtain the consensus of direct associations among metabolites (Fig. S1B vs. S3B, ESI†) and that among sensory traits (Fig. S2B vs. S3D, ESI†). Second, the lasso + SS was implemented in addition to the above two approaches to get the consensus of dependencies of sensory traits on metabolites (Fig. S4A–C, ESI†). Please note that here brix was taken into account, since it is a major intermediate between metabolites and sensory traits. Specifically, brix was treated as a response of metabolites and a predictor for sensory traits in the lasso + SS. The reason for taking multiple ways to network reconstruction is because the common findings of various methods are considered to be true positive with high-confidence. Third, given (i) the dependencies obtained in the second step and (ii) QTLs previously identified for the metabolites and sensory traits, the directions of associations were inferred by the QPSO algorithm. Last, each metabolite and sensory trait was regressed on its QTLs and estimated parent nodes, respectively. It is worth noting that parent nodes of a metabolite should only be metabolites, while parent nodes of a sensory trait could consist of metabolites and sensory traits. Signs of the fitted coefficients discriminated between positive and negative dependencies. This is particularly helpful to decipher whether the cherry or the round allele contributed to the alteration of a trait. As in Fig. 2–4, positive QTL effects (solid red lines) mean that the round allele increased the level of a trait whereas the cherry allele led to a decrease; conversely, negative QTL effects (dashed red lines) mean that the cherry allele produced an increase while the round allele produced a decrease. The absolute values of the fitted coefficient implied the strength of dependencies, which are depicted by the edge thickness in Fig. 2–4. | ||
| Fig. 1 A schematic diagram of the proposed integrative method for learning dependency networks from the sensory, metabolic and QTL data. | ||
 | ||
| Fig. 2 A dependency network of 29 metabolites and 14 QTLs detected in ripe tomatoes. Red edges connect QTLs to their target traits; blue edges represent the dependencies between metabolites. Line style and thickness are determined by the fitted coefficients of each metabolite being regressed on its QTLs and inferred parent nodes. Specifically, thicker lines indicate stronger dependencies; positive and negative dependencies are distinguished by solid and dashed lines. In particular, a solid red edge indicates the round allele at the QTL increases the trait, while a dashed red edge indicates the cherry allele at the QTL increases the trait. | ||
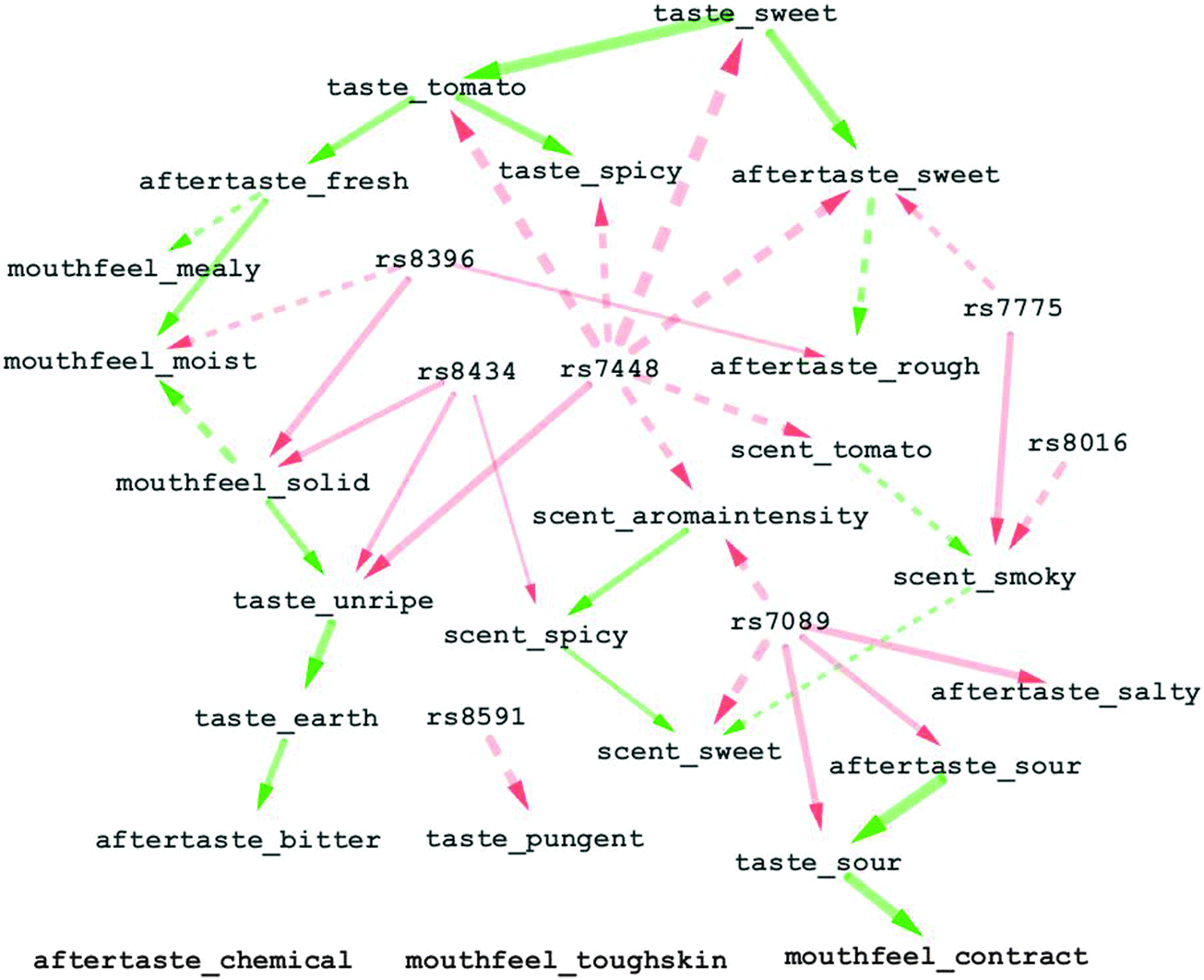 | ||
| Fig. 3 A dependency network of 24 sensory traits and 7 QTLs detected in ripe tomatoes. Red edges connect QTLs to their target traits; green edges represent the dependencies between sensory traits. Line style and thickness are determined by the fitted coefficients of each sensory trait being regressed on its QTLs and inferred parent nodes. Solid and dashed linestyle schemes are identical to those in Fig. 2. | ||
 | ||
| Fig. 4 A dependency network of brix, 29 metabolites, 24 sensory traits and 21 QTLs detected in ripe tomatoes. Black edges represent dependencies of sensory trait on metabolites (via brix). Red, blue and green edges, together with their linestyle and thickness are identical to those in Fig. 2 and 3. | ||
3.2 Gaussian Graphical Models (GGMs)
GGMs are a class of undirected graphs that present only direct associations among multivariate Gaussian random variables. Under the assumption that all involved variables have a multivariate Gaussian distribution, two variables are said to be conditionally independent, i.e. not directly associated, if and only if their partial correlation is zero. Partial correlation measures the degree of correlation between two variables after removing the effects of other variables. It is known that zero entries in the inverse covariance matrix, also known as concentration matrix or precision matrix, correspond to zero partial correlations. In summary, under multivariate normality, non-zero entries of the concentration matrix imply direct associations between pairs of variables, and thereby define the presence of edges in GGM.3.3 Lasso-Based Neighborhood Selection (LBNS) + Stability Approach to Regularization Selection (StARS)
For high-dimensional data with more variables than samples, the concentration matrix cannot be directly estimated from the sample covariance matrix as the latter is non-invertible (singular). In such a case, estimating a sparse concentration matrix is a prerequisite to constructing a GGM. To this end, Meinshausen and Buhlmann proposed the LBNS scheme.11 This scheme first fits a lasso model14 to each variable separately, using all other variables as predictors. It then sets an entry in the concentration matrix, say pij, to be non-zero if the estimated coefficient of variable i on j and/or the estimated coefficient of variable j on i is non-zero.A major challenge when applying lasso-based approaches to graphical modeling is to specify the regularization parameter that controls the sparsity of the resulting graph: larger amounts yield sparser graphs whereas smaller amounts lead to denser graphs. To come up with a general solution that is especially suited to high-dimensional problems, Liu et al. proposed StARS: a stability approach to regularization selection.12 StARS implements subsampling26 to draw a finite number of subsamples (overlapping subsamples are allowed) and constructs a GGM for each subsample. StARS starts with a strong regularization and gradually reduces it until the resulting graphs are simultaneously sparse and replicable across all subsamples. An implementation of LBNS in combination with StARS is available in the R package ‘huge’, which involves a variability threshold with two alternatives 0.1 and 0.05.27 Application of the two thresholds to both metabolic and sensory data suggested that 0.1 would be a better choice in this study (see Section 5.2 for details).
3.4 The PC-skeleton algorithm
The PC algorithm, named after its inventors Peter Spirtes and Clark Glymour, consists of two steps: first, learn an undirected graph from observational data through a series of conditional independence tests; second, orient as many edges as possible according to the estimated conditional independencies and the acyclic constraint. Here we only used the first step, which is referred to as the PC-skeleton algorithm. It starts with a complete graph and removes redundant edges one by one if pairs of corresponding variables are found conditionally independent. For proper implementation of conditional independence tests on different types of data, the PC-skeleton algorithm uses Fisher's z-transformation of the partial correlation for quantitative data and the G2 statistic for categorical data.28 In this study, the significance level of conditional independence tests was set at 0.05. The reason for this will be given in detail in Section 5.2.3.5 The lasso + SS
Though GGMs can effectively reveal direct associations among substances of the same nature, they perform poorly in the identification of associations between substances of different nature. This is mainly because substances of different nature are usually obtained by different measuring techniques and thus have medium to low absolute correlations. This phenomenon was also observed in the present study for associations between metabolic and sensory traits. For this reason, we performed lasso regression14 of each sensory trait on metabolites as a supplement to the implementation of LBNS + StARS and the PC-skeleton algorithm. The proper amount of regularization in the lasso was chosen by SS. More specifically, the lasso was applied to each of a hundred half-size subsamples. The first four predictor metabolites that entered the regularization path for each sensory trait were selected. The final selection retained those predictors that were selected for at least π × 100 percent of the subsamples. π was chosen such that the expected number of false positives, i.e. 42/(p × (2π − 1)), was bounded at 1, where p is the number of metabolites.153.6 The QPSO algorithm
Inferring causal phenotype networks contributes to predicting the effects of external interventions on traits,29 and thereby attracts a surge of research interest.30 Current approaches mainly exploit previously determined QTLs to learn about causal relationships between traits. These methods require at least one unique QTL for each and every trait. This prerequisite, however, is often not met in practice due to various reasons such as limited samples sizes, small QTL effects and high noise levels. To get rid of this unrealistic prerequisite, the QPSO algorithm has been presented very recently.10 This algorithm is applied to a pre-learnt undirected phenotype network, based on which it searches for the optimal causal phenotype network through a heuristic strategy. A major advantage of the QPSO algorithm is that it takes into account the relevant phenotypic interactions in addition to the detected QTLs when orienting an undirected edge between two traits. As a result, it is applicable to general cases where some traits lack unique QTLs, or, come without QTL.4 Results
4.1 A dependency network involving 29 metabolites and 14 QTLs
Fig. 2 presents a dependency network involving 29 metabolites in ripe tomatoes and the most significant 14 QTLs (p-value <0.01) identified by multi-trait mixed model analysis for the metabolites. Except two QTLs, rs6495 and rs8314, which were responsible for beta-damascenone and cis-3-hexenol respectively, all other QTLs were found associated with multiple metabolites. In particular, rs2050 had pleiotropic effects on many metabolites, including eleven volatiles, two sugars and three acids. For two metabolites, eugenol and trans-2-hexenal, no QTL was identified. Another ten metabolites were, respectively, associated with one QTL. Each of the remaining metabolites was associated with two or more QTLs.Fig. 2 indicates a separation between primary and secondary metabolism, i.e., sugars and acids on the left whereas volatiles on the right. Further, (1) sugars and a sugar alcohol, myo-inositol, were grouped together; (2) acids were gathered and linked to sugars; (3) most volatiles interacted, and a few of them were connected with sugars/acids.
Metabolic profiling of ripe tomatoes was carried out at single time points after harvest, that is, it did not produce time series data. The dependency network (Fig. 2) learnt from non-sequential metabolic data cannot be interpreted as a metabolic pathway; instead, it represented directed associations at the level of mean metabolite abundances. These dependencies, though essentially different from pathways, still provide hints on how the effects of allele substitutions propagate through a set of metabolites. For example, genotypic changes at locus rs6691 shall alter the concentration of 1-penten-3-one. This will probably subsequently affect the concentrations of beta-ionone, cis-3-hexenal and aspartic acid. Conversely, variations in the concentration of 1-penten-3-one are unlikely to affect the concentration of trans-2-hexenal, since trans-2-hexenal was found a parent node of 1-penten-3-one in the dependence network.
A better understanding of the dependence structure underlying multiple traits contributes to a better manipulation of those traits. Assume we want to regulate the concentration of beta-ionone, we should control genotypes at loci rs6691 and rs3540 in addition to those at rs2050 and rs6254. The reason is that any allele substitution leading to a change in the concentration of 1-penten-3-one might then alter the concentration of beta-ionone.
4.2 A dependency network involving 24 sensory traits and 7 QTLs
Fig. 3 shows a dependency network involving 24 sensory traits in ripe tomatoes and the most significant 7 QTLs (p-value <0.01) identified by multi-trait mixed model analysis for the sensory traits. Among the 7 QTLs, rs8591 and rs8016 were respectively responsible for one sensory trait; each of the remaining QTLs was associated with multiple sensory traits. From another perspective, 7 sensory traits came without QTLs, while the remaining traits were identified with at least one QTL.Fig. 3 is helpful to predict the simultaneous influence of various allele substitutions on multiple sensory traits. Assume that a genotypic change at locus rs7448 raises the level of scent_tomato. Accordingly there might be a decrease in scent_smoky, and further, an increase in scent_sweet. However, to finely predict one or more phenotypes, a comprehensive consideration of multiple allele substitutions is usually required. For instance, an increase in scent_tomato is not necessarily coupled with a decrease in scent_smoky. This is because apart from QTL rs7448, which had direct negative effect on scent_tomato and, subsequently, indirect positive influence on scent_smoky, scent_smoky was found also being regulated by another two QTLs rs7775 and rs8016. Analogously, scent_sweet was directly or indirectly determined by 5 QTLs, including rs7089, rs8434, rs7448, rs7775 and rs8016.
4.3 A dependency network involving brix, 29 metabolites, 24 sensory traits and 21 QTL
Fig. 4 shows a dependency network involving brix as well as all metabolites, sensory traits and QTLs mentioned above. Brix was found to be dependent on two sugars sucrose and fructose and the sugar alcohol myo-inositol; meanwhile, it was found a main factor influencing taste_sweet. This does not come as a surprise, as brix whilst being a measure of total soluble solids content is most often used to measure sugar content. Indeed, silencing an invertase had a strong influence on brix.31,32 Citric acid was involved in the determination of taste_sour, aftertaste_fresh and taste_tomato. Sucrose, in addition to citric acid, was also a predictor of taste_tomato. Scent_smoky was driven by methyl_salicylate, which was positively affected by guaiacol. This coincides with the previous findings that both methyl_salicylate and guaiacol contribute to the smokey smell of tomatoes,33,34 though a recent study indicates that guaiacol is probably a more important contributor.35In addition to the aforementioned positive directed associations between metabolites and sensory traits, three negative dependencies were respectively found between eugenol and aftertaste_fresh, 2-methyl-1-butanol and aftertaste_chemical, as well as 2-methyl-1-butanol and aftertaste_sweet. The latter two are in agreement with the fact that 2-methyl-1-butanol is often found in fruits (NCBI PubChem) and that it seems to improve or partially impart an Italico-cheese flavor (US 3978242 A), which would not be perceived as a chemical taste but rather associated with natural products.
By taking into account the directed associations from metabolites to sensory traits, we were able to get a more realistic estimation of the dependence structure underlying those sensory traits. An example is that in Fig. 3 aftertaste_sour is present as a parent node of taste_sour, while in Fig. 4 a reversed dependency, which seems more logical, is achieved simply because an additional determinant citric_acid has been introduced to taste_sour.
5 Discussion
5.1 Comparison with known metabolic reactions
As noted above, though the metabolic part of network was learnt from non-time series data and thus cannot necessarily represent a metabolic pathway, it is still to some extent informative about the regulatory mechanisms underlying the metabolites.There was a separation between primary and secondary metabolites. This of course makes sense considering the structural function of primary metabolites and the auxiliary function of secondary metabolites. Interestingly, within the primary metabolites, sucrose was the parent of fructose which in turn was the parent of glucose. This may be due to the enzymatic action of invertase which splits sucrose into glucose and fructose. The direct link between sucrose and glucose was recovered as an indirect one, which is potentially due to the additional action of sucrose synthase utilizing fructose and UDP-glucose.
It is noteworthy that in Fig. 4 fructose and glutamic acid were present as parent nodes of myo-inositol which in turn was the parent of sucrose. Metabolically myo-inositol is synthesized from glucose-6-phosphate viaD-myo-inositol 3-phosphate.36 But since neither glucose-6-phosphate nor D-myo-inositol 3-phosphate were quantified in this study, the network reconstruction and orientation algorithms might have compacted the network. Whilst this leaves the link from glutamic acid unexplained, it seems like a good testable hypothesis for the sugars and the sugar alcohol myo-inositol, which could be further explored.
For glutamic acid a direct and strong influence was observed from aspartic acid. Metabolically this might be explained by the enzymatic action of aspartate aminotransferase that converts glutamic acid and oxaloacetate into 2-oxoglutarate and aspartate. Indeed, aspartate aminotransferase has already been implicated in glutamate content in red tomato fruits.37 Comparatively, the impact of malic acid on aspartic acid seems less obvious. That said, an RNAi approach against PEPCK revealed strongly increased aspartic acid levels coinciding with reduced malate levels. However, silencing of NADP-malic enzyme in the same study showed less aspartic acid and somewhat lower malic acid levels in one transgenic line.38
Turning to volatiles as flavor carrying compounds it is obvious that the most-likely carotenoid derived volatiles beta-damascenone39 and beta-ionone40 were linked because of the common precursor beta-carotene. However, the deduced influence of one on the other might only be explained by hidden variables such as the actual enzyme activities and actual carotenoid concentrations not measured here. Also it is intriguing that 6-methyl-5-hepten-2-one and geranylacetone, both being interconnected, were not linked to the former pair of volatiles despite them also being carotenoid volatiles. The different differential behaviors of these two pairs of volatiles were also observed in earlier studies,39 and it has been reported that 6-methyl-5-hepten-2-one likely stems from lycopene.41 We therefore suspect the difference is attributed to distinct precursors. Apart from these carotenoid derived metabolites, the synthesis of phenylethyl alcohol from benzeneacetaldehyde42 was recovered in our analysis.
Regarding the linked metabolites 3-methyl-1-butanol and 3-methylbutanal, they are most likely leucine derived, whilst the associated 2-methyl-1-butanol likely stems from isoleucine. Also the association between 2-isobutylthiazole and 3-methyl-1-butanol was observed before.19,39 Thus this whole sub-cluster of metabolites is derived from or associated to branched chain amino acids. The current model for the biosynthesis of leucine-derived flavor imparting compounds assumes a decarboxylation to an aldehyde followed by a reduction. The truth, however, is that the alcohols should derive from the aldehydes.
5.2 Choice of methods and parameters
The most straightforward way to construct biological networks is the correlation network (also known as relevance network), which is based on unconditional pairwise correlations. However, though strong correlations are good indicators of dependencies, they cannot distinguish between direct and indirect associations. Thus, correlation networks are typically dense graphs, from which definitive conclusions can hardly be drawn (see examples in Fig. S5A and B, ESI†). To learn less dense but more informative graphs, especially from high-dimensional data with limited sample size, here we used three approaches to graphical modeling: LBNS + StARS, the PC-skeleton algorithm, and the lasso + SS.StARS has been shown to outperform the conventional regularization parameter selection methods, including AIC, BIC and cross-validation, in the reconstruction of high-dimensional graphs.12 In view of this, we exploited StARS to set regularization in LBNS. The R package “huge” implements StARS with two optional variability thresholds: 0.1 and 0.05. We tested both thresholds on the metabolic and sensory data separately, and found that 0.05 led to a bit sparser graph than 0.01 (Fig. S1A vs. S1B, Fig. S2A vs. S2B, ESI†). As we aimed to extract a consensus network, the variability threshold of 0.1 was then used in StARS to ensure that given the same dataset, edges obtained by LBNS can overlap, to a large extent, with those learnt by the PC-skeleton algorithm.
The PC-skeleton algorithm also requires a pre-specified parameter, i.e. the significance level of conditional independence tests. We tested the two most common significance levels, 0.01 and 0.05, on the metabolic and sensory data separately. Results on the same datasets indicated that the significance level of 0.05 recovered a few more edges than the level of 0.01 (Fig. S3A vs. S3B, Fig. S3C vs. S3D, ESI†). Again, to reach as many as possible consensus edges, we took the significance level of 0.05 in this study.
Our strategy, which first overfits an undirected graph by LBNS + StARS and then screens out the unlikely edges by comparison with the outcome of the PC-skeleton algorithm, was also tried on the mixture of metabolic and sensory data. Surprisingly, only a few links between metabolites and sensory traits were discovered by either method (see black edges in Fig. S4A and B, ESI†). After discarding edges unique to one graph, we were left with merely eight common links (see the boldfaced black edges in Fig. S4A or B, ESI†). This implies that the above strategy, when being used to decipher the relationships between substances of different nature, is very likely to produce an underfitted graph. We then tried a third method, i.e. regressing every sensory trait on the metabolites by the lasso + SS, to get the directed graph in Fig. S4C (ESI†). To draw safe conclusions but without losing too much useful information, we extracted those edges that appeared between metabolites and sensory traits at least twice over Fig. S4A–C (ESI†). Finally, 12 edges satisfying this criterion were reported (see black edges in Fig. 4).
5.3 Other aspects
Multi-trait analysis is in general preferred over single-trait analysis for QTL mapping. This is because: (1) multi-trait analysis takes into account the genetic correlations among traits and thus increases the power of detecting QTLs;43 (2) it allows a more straightforward assessment of pleiotropic effects of QTLs.23,24 Nonetheless, the outputs of multi-trait QTL analyses not necessarily fully encompass the results of single-trait analyses. That is, a QTL identified by single-trait analysis can be missed in multi-trait analysis, though this rarely happens. In this study we missed a QTL for scent smoky on chromosome 9, whereas this QTL was clearly identified in another study with the same material.35 We were able to detect the QTL when rerunning a single-trait analysis for scent smoky. A limited multi-trait analysis on scent smoky and some volatiles that are known to be related to scent smoky produced the QTL as well.We have identified a total of 21 QTLs for the 29 metabolites and 24 sensory traits. Most of the QTLs were found to have pleiotropic effects; in particular, a few of them, such as rs2050, rs6687, rs7089 and rs7448, served as hubs in the resulting dependency network (Fig. 4). A particularly noteworthy phenomenon was that a number of directed triangles appeared in Fig. 4, especially around the hubs. One may doubt whether the QTL really affects so many traits? Does its impact on a downstream trait actually pass through the upstream traits? Moreover, will two directly associated traits become independent of each other given their common QTL? A possible solution to these detailed questions is the triad analysis, which aims at identifying causal relationships in configurations consisting of two traits and one QTL.44,45
Though both SS and StARS can choose a proper regularization for high-dimensional sparse linear regression, they are essentially different. Given the same training dataset, StARS tolerates false positives (false edges in the reconstructed graph) but not false negatives (true edges absent in the reconstructed graph) and thus leads to a dense graph with high recall but relatively low precision (in the context of graphical modeling, recall refers to the fraction of true edges that are recovered in the resulting graph; precision refers to the fraction of recovered edges that are actually true); SS, contrariwise, allows false negatives but not false positives and therefore results in a sparse graph with high precision but comparatively low recall.
6. Conclusion
We have investigated the utility of existing methods for GGM reconstruction in combination with the QPSO algorithm for dependency inference between 29 metabolites and 24 sensory traits scored on ripe tomatoes. The resulting network provides hints on how the sensory traits depend upon the metabolites and further upon the detected QTLs. This integrative approach does not require the identification of QTLs for each and every trait studied, and thus has broad applicability across a number of practical settings. Furthermore, it is applicable to a range of population structures, including offspring populations from crosses between inbred parents and outbred parents, association panels and natural population. The novel dependencies emerged in this study form the hypotheses that can be individually tested in future studies.References
- M. Drton and M. D. Perlman, Biometrika, 2004, 91, 591–602 CrossRef PubMed.
- J. Krumsiek, K. Suhre, T. Illig, J. Adamski and F. J. Theis, BMC Syst. Biol., 2011 DOI:10.1186/1752-0509-5-21.
- N. Carreno-Quintero, H. J. Bouwmeester and J. J. B. Keurentjes, Trends Genet., 2013, 29, 41–50 CrossRef CAS PubMed.
- R. H. Li, S. W. Tsaih, K. Shockley, I. M. Stylianou, J. Wergedal, B. Paigen and G. A. Churchill, PLoS Genet., 2006 DOI:10.1371/journal.pgen0020114.
- J. E. Aten, T. F. Fuller, A. J. Lusis and S. Horvath, BMC Syst. Biol., 2008 DOI:10.1186/1752-0509-2-34.
- E. C. Neto, C. T. Ferrara, A. D. Attie and B. S. Yandell, Genetics, 2008, 179, 1089–1100 CrossRef PubMed.
- E. C. Neto, M. P. Keller, A. D. Attie and B. S. Yandell, Ann. Appl. Stat., 2010, 4, 320–339 Search PubMed.
- B. A. Logsdon and J. Mezey, PLoS Comput. Biol., 2010 DOI:10.1371/journal.pcbi.1001014.
- X. D. Cai, J. A. Bazerque and G. B. Giannakis, PLoS Comput. Biol., 2013 DOI:10.1371/journal.pcbi.1003068.
- H. G. Wang and F. A. van Eeuwijk, PLoS One, 2014 DOI:10.1371/journal.pone.0103997.
- N. Meinshausen and P. Buhlmann, Ann. Stat., 2006, 34, 1436–1462 CrossRef PubMed.
- H. Liu, K. Roeder and L. Wasserman, presented in part at the Twenty-Third Annual Conference on Neural Information Processing Systems, 2010.
- P. Spirtes, C. N. Glymour and R. Scheines, Causation, prediction, and search, MIT Press, Cambridge, Mass, 2nd edn, 2000 Search PubMed.
- R. Tibshirani, J. R. Stat. Soc. Ser. B, 1996, 58, 267–288 Search PubMed.
- N. Meinshausen and P. Buhlmann, J. R. Stat. Soc. Ser. B, 2010, 72, 417–473 CrossRef PubMed.
- K. S. Tandon, E. A. Baldwin, J. W. Scott and R. L. Shewfelt, J. Food Sci., 2003, 68, 2366–2371 CrossRef CAS PubMed.
- E. G. Abegaz, K. S. Tandon, J. W. Scott, E. A. Baldwin and R. L. Shewfelt, Postharvest Biol. Technol., 2004, 34, 227–235 CrossRef CAS PubMed.
- P. Carli, S. Arima, V. Fogliano, L. Tardella, L. Frusciante and M. R. Ercolano, J. Exp. Bot., 2009, 60, 3379–3386 CrossRef CAS PubMed.
- Y. Tikunov, A. Lommen, C. H. R. de Vos, H. A. Verhoeven, R. J. Bino, R. D. Hall and A. G. Bovy, Plant Physiol., 2005, 139, 1125–1137 CrossRef CAS PubMed.
- U. Roessner-Tunali, B. Hegemann, A. Lytovchenko, F. Carrari, C. Bruedigam, D. Granot and A. R. Fernie, Plant Physiol., 2003, 133, 84–99 CrossRef CAS PubMed.
- L. W. Sumner, A. Amberg, D. Barrett, M. H. Beale, R. Beger, C. A. Daykin, T. W. M. Fan, O. Fiehn, R. Goodacre, J. L. Griffin, T. Hankemeier, N. Hardy, J. Harnly, R. Higashi, J. Kopka, A. N. Lane, J. C. Lindon, P. Marriott, A. W. Nicholls, M. D. Reily, J. J. Thaden and M. R. Viant, Metabolomics, 2007, 3, 211–221 CrossRef CAS PubMed.
- J. Vrebalov, D. Ruezinsky, V. Padmanabhan, R. White, D. Medrano, R. Drake, W. Schuch and J. Giovannoni, Science, 2002, 296, 343–346 CrossRef CAS PubMed.
- M. Malosetti, J. M. Ribaut, M. Vargas, J. Crossa and F. A. van Eeuwijk, Euphytica, 2008, 161, 241–257 CrossRef.
- N. A. Alimi, M. C. A. M. Bink, J. A. Dieleman, J. J. Magan, A. M. Wubs, A. Palloix and F. A. van Eeuwijk, Theor. Appl. Genet., 2013, 126, 2597–2625 CrossRef CAS PubMed.
- C. J. Jiang and Z. B. Zeng, Genetica, 1997, 101, 47–58 CrossRef CAS.
- D. N. Politis, J. P. Romano and M. Wolf, Subsampling, Springer, 1st edn, 1999 Search PubMed.
- T. Zhao, H. Liu, K. Roeder, J. Lafferty and L. Wasserman, J. Mach. Learn. Res., 2012, 13, 1059–1062 Search PubMed.
- D. Colombo, A. Hauser, M. Kalisch and M. Maechler, Package ‘pcalg’, http://cran.r-project.org/web/packages/pcalg/pcalg.pdf.
- B. D. Valente, G. J. M. Rosa, D. Gianola, X. L. Wu and K. Weigel, Genetics, 2013, 194, 561–572 CrossRef PubMed.
- G. J. M. Rosa, B. D. Valente, G. de los Campos, X. L. Wu, D. Gianola and M. A. Silva, Genet., Sel., Evol., 2011 DOI:10.1186/1297-9686-43-6.
- E. Fridman, T. Pleban and D. Zamir, PNAS, 2000, 97, 4718–4723 CrossRef CAS.
- M. I. Zanor, S. Osorio, A. Nunes-Nesi, F. Carrari, M. Lohse, B. Usadel, C. Kuhn, W. Bleiss, P. Giavalisco, L. Willmitzer, R. Sulpice, Y. H. Zhou and A. R. Fernie, Plant Physiol., 2009, 150, 1204–1218 CrossRef CAS PubMed.
- R. G. Buttery, L. C. Ling and D. M. Light, J. Agric. Food Chem., 1987, 35, 1039–1042 CrossRef CAS.
- R. G. Buttery, G. Takeoka, R. Teranishi and L. C. Ling, J. Agric. Food Chem., 1990, 38, 2050–2053 CrossRef CAS.
- Y. M. Tikunov, J. Molthoff, R. C. H. de Vos, J. Beekwilder, A. van Houwelingen, J. J. J. van der Hooft, M. Nijenhuis-de Vries, C. W. Labrie, W. Verkerke, H. van de Geest, M. V. Zamora, S. Presa, J. L. Rambla, A. Granell, R. D. Hall and A. G. Bovy, Plant Cell, 2013, 25, 3067–3078 CrossRef CAS PubMed.
- C. E. Hegeman, L. L. Good and E. A. Grabau, Plant Physiol., 2001, 125, 1941–1948 CrossRef CAS PubMed.
- S. B. Boggio, J. F. Palatnik, H. W. Heldt and E. M. Valle, Plant Sci., 2000, 159, 125–133 CrossRef CAS.
- S. Osorio, J. G. Vallarino, M. Szecowka, S. Ufaz, V. Tzin, R. Angelovici, G. Galili and A. R. Fernie, Plant Physiol., 2013, 161, 628–643 CrossRef CAS PubMed.
- S. Mathieu, V. D. Cin, Z. J. Fei, H. Li, P. Bliss, M. G. Taylor, H. J. Klee and D. M. Tieman, J. Exp. Bot., 2009, 60, 325–337 CrossRef CAS PubMed.
- S. Baldermann, M. Kato, M. Kurosawa, Y. Kurobayashi, A. Fujita, P. Fleischmann and N. Watanabe, J. Exp. Bot., 2010, 61, 2967–2977 CrossRef CAS PubMed.
- H. Y. Gao, H. L. Zhu, Y. Shao, A. J. Chen, C. W. Lu, B. Z. Zhu and Y. B. Luo, J. Integr. Plant Biol., 2008, 50, 991–996 CrossRef CAS PubMed.
- M. Sakai, H. Hirata, H. Sayama, K. Sekiguchi, H. Itano, T. Asai, H. Dohra, M. Hara and N. Watanabe, Biosci., Biotechnol., Biochem., 2007, 71, 2408–2419 CrossRef CAS PubMed.
- C. J. Jiang and Z. B. Zeng, Genetics, 1995, 140, 1111–1127 CAS.
- Y. Li, B. M. Tesson, G. A. Churchill and R. C. Jansen, Trends Genet., 2010, 26, 493–498 CrossRef CAS PubMed.
- E. E. Schadt, J. Lamb, X. Yang, J. Zhu, S. Edwards, D. GuhaThakurta, S. K. Sieberts, S. Monks, M. Reitman, C. S. Zhang, P. Y. Lum, A. Leonardson, R. Thieringer, J. M. Metzger, L. M. Yang, J. Castle, H. Y. Zhu, S. F. Kash, T. A. Drake, A. Sachs and A. J. Lusis, Nat. Genet., 2005, 37, 710–717 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00477b |
| This journal is © The Royal Society of Chemistry 2015 |