
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA miniaturized readout strategy for endogenous histone deacetylase activity†
Jan Oliver
Jost
,
Alfred
Hanswillemenke
and
Dirk
Schwarzer
*
Interfaculty Institute of Biochemistry, University of Tuebingen, Hoppe-Seyler-Str. 4, 72076 Tuebingen, Germany. E-mail: dirk.schwarzer@uni-tuebingen.de; Fax: +49-7071-29-4518; Tel: +49-7071-29-73344
First published on 12th May 2015
Abstract
Histone deacetylases are important drug targets, which are difficult to characterize due to their poor accessibility. We have developed a miniaturized assay for the multi-site readout of deacetylase activity and profiled the substrate selectivity of HDACs for acetylation sites on histone H4 and tumor suppressor protein p53.
Lysine acetylation is a long-known posttranslational modification (PTM) of proteins, which has recently entered the center stage as an important regulator of protein structure and function.1 Initially discovered on histones, many more acetylated proteins were found in eukaryotic and prokaryotic proteomes, rendering lysine acetylation as one of the most abundant PTMs in nature.1 Lysine acetylation is introduced by lysine acetyl-transferases and removed by lysine or histone deacetylases (HDACs).2–5 The latter class of enzymes has gained considerable attention because they have been established as promising drug targets for a wide range of diseases including cancer.6,7 Especially the eleven mammalian HDACs of classes I, II and IV (hereafter referred to as HDACs) are in the focus of biomedical research.6,7 These enzymes require Zn2+ for catalysis and cleave the acetyl groups in a hydrolytic reaction. Despite their medicinal importance, little is known about the substrate selectivity and catalytic properties of these enzymes due to the poor accessibility and activity of recombinant HDACs.8,9 However, compared to most recombinant enzymes, endogenous HDACs produced by mammalian cells appear highly active.8 A wide range of deacetylase-assays was developed, but most of them are incompatible with small amounts of endogenous HDACs which can be isolated from mammalian cells by standard techniques of molecular biology.10–14 For example colorimetric readouts usually require reaction volumes >100 μL.10 In addition several proteins possess acetylation sites in close proximity and only very few assays are capable of a multi-site readout of HDAC activity.15 Based on this we planned to develop a quantitative and miniaturized readout strategy for endogenous HDAC activity, which is compatible with established methods of molecular biology. Furthermore, the detection method should be able to monitor more than one modification site. To this end we resorted to MALDI-TOF-based mass spectrometry (MS). Mass spectrometry requires much less material for analysis than most other detection techniques. However, differences in the ionization yield of educts and deacetylated products resulting from the presence or absence of this charge-altering PTM will likely perturb the MS analysis.16–18 Recent developments in MS-based proteomics have solved this problem by supplying the analysis with isotopic coded references, which can be used for relative and absolute quantification of the MS signals.19,20
We used these findings for establishing the envisioned HDAC assay (Scheme 1). In the simplest setting, MALDI-TOF detects deacetylated products after incubating acetylated peptides with HDACs. At selected intervals, HDAC-inhibitors stop the reactions and added isotopic references enable absolute quantification of the formed products. Multi-site detection requires a further isotopic coding strategy for the acetyl-groups and immunoprecipitation used for isolating native HDACs from cellular lysates should enable the analysis of activity and selectivity of endogenous HDACs.
 | ||
| Scheme 1 Basic concept of the quantitative MALDI-TOF readout of HDAC activity. | ||
At first we established the basic concept with HDAC8, the only human HDAC which can be produced in active form in E. coli.21 Recombinant HDAC8 is able to deacetylate K382ac of tumor suppressor protein p53 (Fig. 1a) and the enzyme was reported to be more active when supplied with Co2+ instead of the canonical Zn2+.22 We synthesized a peptide derived from the C-terminal region of p53 (378–386) covering the acetylation site at K382 (p53K382ac) as well as an isotopic coded reference of the deacetylated product containing homogenously 13C- and 15N-labeled lysine at position 381 (p53K382(iso)) (Table S1, ESI†). Recombinant HDAC8 was incubated with p53K382ac in the presence of either Co2+ or Zn2+, and after stopping the reaction by adding pan-specific HDAC inhibitor trichostatin A (TSA) the mixture was supplied with p53K382(iso) and analyzed by MALDI-TOF. The expected signals of the deacetylated product, isotopic coded reference and starting material were readily detected and the deacetylation rates were calculated from the signal intensities (Fig. S1, ESI†). The analysis showed that recombinant HDAC8 was approximately 10-fold more active in the presence of Co2+ compared to Zn2+ (Fig. 1b). Consequently, we used Co2+ instead of Zn2+ for all further assays with recombinant HDAC8.
 | ||
| Fig. 1 Analyses of HDAC8 activity with the MALDI-TOF readout. (a) Sequence alignment of confirmed and putative substrate sites of HDAC8. Acetylation sites are marked by the black box, important residues are color-coded in grey and the putative consensus sequence is underlined. (b) Metal ion dependency of recombinant HDAC8. The product formation rates were measured in the presence of either Zn2+ or Co2+ at 400 μM p53K382ac. (c) Comparison of HDAC8-catalyzed deacetylation rates measured by MALDI-TOF and HPLC readouts at 100 μM p53K382ac. (d) HDAC8-catalyzed deacetylation of 53K382ac, p53K372ac and p53K320ac. The velocity of product formation is plotted against the respective substrate concentrations. V: velocity of the deacetylation reaction; K: Michaelis constant; V/E: velocity normalized to enzyme concentration at a given substrate concentration; V/K: velocity at substrate saturation divided by Michaelis constant; n.d.: not detectable. | ||
In the following we compared the MALDI-TOF readout with a conventional HPLC-based assay. Nearly identical deacetylation rates were determined with both methods, indicating that the MALDI-TOF readout in combination with isotopic coded references is suitable for analyzing HDAC activity (Fig. 1c).
Encouraged by these findings we investigated the substrate selectivity of HDAC8 in more detail. There are only a few other described substrates of recombinant HDAC8. One of them is the acetylation site at lysine 20 of histone H4.15 Both p53K382ac and H4K20ac share a homologous sequence context (R-H-R/K-Kac-V/L). It is not clear if these sites are somehow connected in a physiological context, but it appears likely that the amino acids flanking the acetylation sites support the substrate recognition by HDAC8 (Fig. 1a).23 Tumor suppressor p53 possesses several acetylation sites and we asked if sites other than K382ac might represent HDAC8 substrates.24 Two sites were of particular interest: p53K373ac and p53K320ac which share limited similarities with the basic N-terminal region of p53K382ac and H4K20ac (Fig. 1a). We synthesized acetylated peptides derived from both sites (p53K373ac and p53K320ac) as well as the isotopic references (p53K373(iso) and p53K320(iso)). The kinetics confirmed that p53K382ac was efficiently deacetylated by HDAC8 (Fig. 1d). The enzyme was also able to deacetylate p53K373ac, but at an approximately 18-fold reduced rate. In contrast, no deacetylation of p53K320ac was observed within the concentration range. These measurements indicate that the substrate selectivity profile of recombinant HDAC8 is rather narrow and that deacetylation rates correlate with the homology to the p53K382ac and H4K20ac sites (Fig. 1a).
Next, we focused on HDAC-substrates with more than one acetylation site. The N-terminal tails of histones represent prime examples of hyper-acetylated peptides. Histone H4 contains acetylated K16 in addition to the above-mentioned K20ac site. However, K16ac of H4 does not belong to the reported substrates of HDAC8.15 We synthesized a bis-acetylated peptide derived from H4 containing K16ac and K20ac (H4K16acK20ac). In order to distinguish both sites, the acetyl group at K20ac was homogenously deuterium- and 13C-coded, while the acetyl group at K16ac remained unlabeled (Fig. 2a). Deacetylation of H4K16acK20ac results in mono-deacetylated and fully deacetylated products and we synthesized the corresponding isotopic coded references H4K16acK20(iso) and H4K16K20(iso) (Fig. S2, ESI†). Recombinant HDAC8 removed K20ac efficiently, while K16ac was deacetylated slowly and only after complete removal of K20ac (Fig. 2b and Fig. S3a, ESI†). Pan-specific HDAC inhibitor TSA blocked the reaction, as well as PCI-34051, which is a synthetic HDAC8-specific inhibitor.
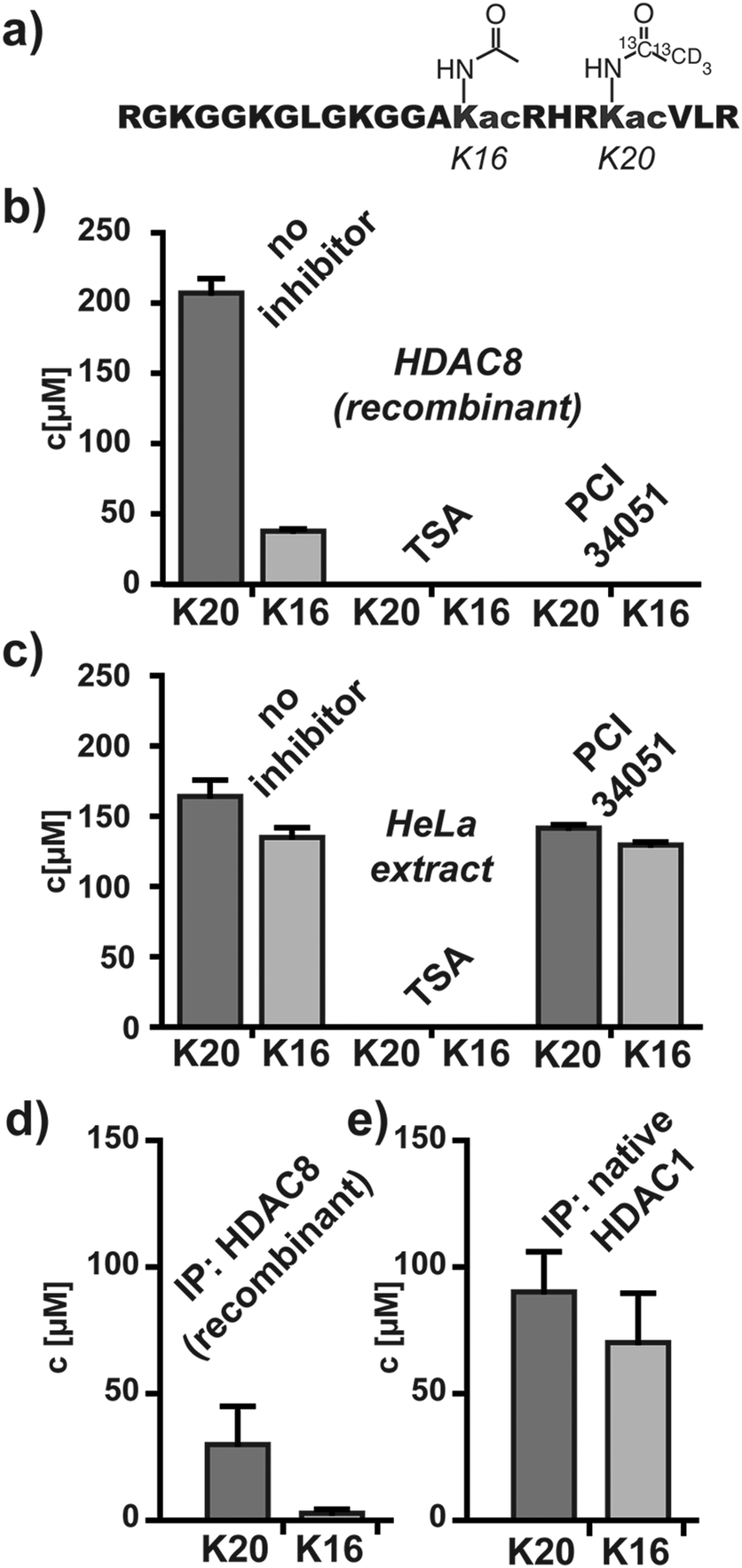 | ||
| Fig. 2 Analyses of deacetylase activities with bisacetylated H4K16acK20ac. (a) Bisacetylated HDAC substrate. Deacetylase activity and inhibition of (b) recombinant HDAC8 and (c) global HDAC activity in HeLa lysates. Deacetylase activity of immunoprecipitated (d) recombinant HDAC8 and (e) endogenous HDAC1. All analyses were performed at a fixed substrate concentration of 200 μM. | ||
Encouraged by these findings we analyzed global HDAC activity in native HeLa extracts. H4K16acK20ac was mixed with the whole-cell HeLa extract and analyzed by MALDI-TOF. We observed efficient and simultaneous deacetylation of K20ac and K16ac. Global deacetylase activity was blocked by TSA but not by PCI-34051 (Fig. 2c and Fig. S3b, ESI†).
After the successful establishment of the miniaturized multi-site readout of HDAC activity we focused on individual endogenous enzymes from HeLa extracts. To this end we established a simple procedure for isolating and assaying HDACs from cellular lysates by immunoprecipitation (Fig. S4, ESI†). At first gel-loading tips were filled with antibody-binding protein A or protein G agarose, which remained in the tips during pipetting steps. In the following anti-HDAC antibodies and subsequently HDACs were immobilized on the resin by pipetting antibody solutions and HeLa lysates. Next, H4K16acK20ac was loaded onto the resin and at selected intervals samples were eluted for MALDI-TOF analysis. We performed this procedure using recombinant HDAC8 and observed the expected preference for K20ac (Fig. 2d). However, when performing the reaction with the deacetylase activities that were immunoprecipitated from HeLa extracts with anti-HDAC1 and selected other anti-HDAC antibodies we observed simultaneous deacetylation of K20ac and K16ac, similar to the activity in whole-cell lysates (Fig. S5, ESI†). The specific activity of endogenous HDACs remains unknown since the amounts and interaction partners of immobilized enzymes are very hard to determine, but the deacetylase activities associated with antibodies against HDACs 1 and 2 were most efficient in deacetylating H4K16acK20ac (Fig. 2e and Fig. S5, ESI†). These findings are consistent with the notion that HDACs 1 and 2 are abundant HDACs and the main mammalian deacetylases of histones in HeLa cells.15,25,26
Finally, we also tested the generality of this approach by analyzing dephosphorylation reactions. Protein phosphatase 1 (PP1) is known to catalyze dephosphorylation of S10ph on histone H3 and we studied this reaction using recombinant PP1α, an H3S10ph substrate and the corresponding H3S10(iso) reference (Fig. S6, ESI†). Kinetics and inhibition studies of PP1α with PP1-inhibitor microcystin-LR were performed and enabled the determination of the kinetic parameters (Fig. S7, ESI†). This finding indicates that the established methodology is broadly applicable for studying PTM-removing enzymes on a small-scale.
In conclusion we have established a miniaturized quantitative method for the simultaneous readout of HDAC activity at two sites. This method appears to be generally applicable and can be combined with immunoprecipitation in order to investigate the substrate selectivity of endogenous HDACs isolated from cellular lysates.
We thank Hubert Kalbacher for support with the MALDI-TOF. This work was supported by the priority program SPP1623 of the Deutsche Forschungsgemeinschaft (DFG) (SCHW 1163/4-1).
Notes and references
- C. Choudhary, B. T. Weinert, Y. Nishida, E. Verdin and M. Mann, Nat. Rev. Mol. Cell Biol., 2014, 15, 536–550 CrossRef CAS PubMed.
- C. D. Allis, S. L. Berger, J. Cote, S. Dent, T. Jenuwien, T. Kouzarides, L. Pillus, D. Reinberg, Y. Shi, R. Shiekhattar, A. Shilatifard, J. Workman and Y. Zhang, Cell, 2007, 131, 633–636 CrossRef CAS PubMed.
- P. A. Cole, Nat. Chem. Biol., 2008, 4, 590–597 CrossRef CAS PubMed.
- M. C. Haigis and D. A. Sinclair, Annu. Rev. Pathol.: Mech. Dis., 2010, 5, 253–295 CrossRef CAS PubMed.
- X. J. Yang and E. Seto, Nat. Rev. Mol. Cell Biol., 2008, 9, 206–218 CrossRef CAS PubMed.
- M. Haberland, R. L. Montgomery and E. N. Olson, Nat. Rev. Genet., 2009, 10, 32–42 CrossRef CAS PubMed.
- S. Minucci and P. G. Pelicci, Nat. Rev. Cancer, 2006, 6, 38–51 CrossRef CAS PubMed.
- W. Fischle, F. Dequiedt, M. J. Hendzel, M. G. Guenther, M. A. Lazar, W. Voelter and E. Verdin, Mol. Cell, 2002, 9, 45–57 CrossRef CAS.
- N. Sengupta and E. Seto, J. Cell. Biochem., 2004, 93, 57–67 CrossRef CAS PubMed.
- A. Dose, J. O. Jost, A. C. Spiess, P. Henklein, M. Beyermann and D. Schwarzer, Chem. Commun., 2012, 48, 9525–9527 RSC.
- A. T. Hauser and M. Jung, Curr. Top. Med. Chem., 2009, 9, 227–234 CrossRef CAS.
- K. Hoffmann, G. Brosch, P. Loidl and M. Jung, Nucleic Acids Res., 1999, 27, 2057–2058 CrossRef CAS PubMed.
- D. Rauh, F. Fischer, M. Gertz, M. Lakshminarasimhan, T. Bergbrede, F. Aladini, C. Kambach, C. F. Becker, J. Zerweck, M. Schutkowski and C. Steegborn, Nat. Commun., 2013, 4, 2327 Search PubMed.
- M. Schutkowski, F. Fischer, C. Roessler and C. Steegborn, Expert Opin. Drug Discovery, 2014, 9, 183–199 CrossRef CAS PubMed.
- A. Dose, S. Liokatis, F. X. Theillet, P. Selenko and D. Schwarzer, ACS Chem. Biol., 2011, 6, 419–424 CrossRef CAS PubMed.
- S. A. Gerber, J. Rush, O. Stemman, M. W. Kirschner and S. P. Gygi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6940–6945 CrossRef CAS PubMed.
- J. Gropengiesser, B. T. Varadarajan, H. Stephanowitz and E. Krause, J. Mass Spectrom., 2009, 44, 821–831 CrossRef CAS PubMed.
- K. Kubota, R. Anjum, Y. Yu, R. C. Kunz, J. N. Andersen, M. Kraus, H. Keilhack, K. Nagashima, S. Krauss, C. Paweletz, R. C. Hendrickson, A. S. Feldman, C. L. Wu, J. Rush, J. Villen and S. P. Gygi, Nat. Biotechnol., 2009, 27, 933–940 CrossRef CAS PubMed.
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS PubMed.
- S. E. Ong, B. Blagoev, I. Kratchmarova, D. B. Kristensen, H. Steen, A. Pandey and M. Mann, Mol. Cell. Proteomics, 2002, 1, 376–386 CAS.
- E. Hu, Z. Chen, T. Fredrickson, Y. Zhu, R. Kirkpatrick, G. F. Zhang, K. Johanson, C. M. Sung, R. Liu and J. Winkler, J. Biol. Chem., 2000, 275, 15254–15264 CrossRef CAS PubMed.
- S. L. Gantt, S. G. Gattis and C. A. Fierke, Biochemistry, 2006, 45, 6170–6178 CrossRef CAS PubMed.
- Z. A. Gurard-Levin, J. Kim and M. Mrksich, ChemBioChem, 2009, 10, 2159–2161 CrossRef CAS PubMed.
- K. H. Vousden and C. Prives, Cell, 2009, 137, 413–431 CrossRef CAS PubMed.
- R. Ferreira, I. Naguibneva, M. Mathieu, S. Ait-Si-Ali, P. Robin, L. L. Pritchard and A. Harel-Bellan, EMBO Rep., 2001, 2, 794–799 CrossRef CAS PubMed.
- A. Kuzmichev, Y. Zhang, H. Erdjument-Bromage, P. Tempst and D. Reinberg, Mol. Cell. Biol., 2002, 22, 835–848 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and further experimental data. See DOI: 10.1039/c5mb00326a |
| This journal is © The Royal Society of Chemistry 2015 |