
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Production of indole antibiotics induced by exogenous gene derived from sponge metagenomes†
Yuya
Takeshige
,
Yoko
Egami
,
Toshiyuki
Wakimoto
* and
Ikuro
Abe
*
Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: abei@mol.f.u-tokyo.ac.jp; Fax: +81 3 5841 4744; Tel: +81 3 5841 4741
First published on 25th March 2015
Abstract
Sponge metagenomes are accessible genetic sources containing genes and gene clusters responsible for the biosynthesis of sponge-derived bioactive natural products. In this study, we obtained the clone pDC112, producing turbomycin A and 2,2-di(3-indolyl)-3-indolone, based on the functional screening of the metagenome library derived from the marine sponge Discodermia calyx. The subcloning experiment identified ORF 25, which is homologous to inosine 5′-monophosphate dehydrogenase and required for the production of 2,2-di(3-indolyl)-3-indolone in Escherichia coli.
Introduction
Marine sponges are prolific sources of bioactive molecules as well as highly complex consortia, including significantly large populations of symbiotic bacteria.1 These symbiotic bacteria often cannot be cultured under normal laboratory conditions.2 To access the genes and gene clusters responsible for the biosynthesis of sponge-derived bioactive natural products, the sponge metagenome is a unique and promising genetic resource.3 There are two possible methods to mine the interesting genes related to secondary metabolites: homology-based screening and function-based screening. Homology-based screening of the metagenome library, which relies on the conserved sequence motifs of biosynthetic genes, offers the opportunity to obtain the genes responsible for natural product production by the symbiotic bacteria.4 Indeed, we recently reported the biosynthetic gene cluster of a cytotoxic compound, calyculin A, which is composed of NRPS-PKS hybrid genes encoded by an uncultured sponge symbiont, Candidatus ‘Entotheonella sp.’.5Alternatively, function-based screening, which depends on a phenotypical alteration by the heterologous expression of the inserted genes in the host, has resulted in the isolation of small molecules, such as terragines,6 isocyanides,7 porphyrins,8,9 and siderophores,10 from metagenomic libraries originating from soil or marine environments. Once a positive clone is found by the function-based screening, such as antibacterial activity, it potentially harbors all three factors, including inserted genes, functional enzymes, and bioactive compounds.11 Moreover, a random screening method regardless of sequence homology is suitable for the discovery of novel enzymes responsible for the production of functional small molecules. These aspects provide an advantage over the homology-based screening. Herein, we performed a function-based screening of a sponge metagenome library, to identify an antibacterial clone and the gene involved in the production of the antibiotics.12
Results and discussion
We performed antibacterial screening with a marine sponge Discodermia calyx metagenome library, by means of an overlay assay with Bacillus cereus as the test bacterial strain.13 Almost 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 colonies were screened, and as a result, a red-pigmented clone (pDC112) exhibited an inhibition halo around the colony. In our previous studies on pDC112, we isolated porphyrin derivatives as pigments without antibacterial properties.9 Thus, we considered that pDC112 would be likely to produce other antibiotics, either directly or indirectly encoded by an exogenous gene derived from the metagenome library. In this study, the culture solution of pDC112 was further subjected to antibacterial activity-guided fractionation.
000 colonies were screened, and as a result, a red-pigmented clone (pDC112) exhibited an inhibition halo around the colony. In our previous studies on pDC112, we isolated porphyrin derivatives as pigments without antibacterial properties.9 Thus, we considered that pDC112 would be likely to produce other antibiotics, either directly or indirectly encoded by an exogenous gene derived from the metagenome library. In this study, the culture solution of pDC112 was further subjected to antibacterial activity-guided fractionation.
After cultivation at 30 °C for 2 days, the culture solution (9 L) was centrifuged, and the supernatant was fractionated by Diaion HP-20, ODS, silica gel column chromatography and ODS-HPLC, monitored by the antibacterial activity, to yield the antibacterial compounds 1 and 2.
Compound 1 was isolated as a red-colored compound, with a molecular formula of C25H18N3 determined by HR-ESI-MS (positive) at m/z 360.1496 (calcd for C25H18N3, 360.1501). One amino proton at δH 7.96 (3H, brs, NH), two doublets at 7.62 (3H, d, J = 8 Hz), 7.36 (3H, d, J = 8 Hz), and two triplets at δH 7.19 (3H, t, J = 8 Hz) and 7.09 (3H, t, J = 8 Hz) in the 1H NMR spectrum revealed the presence of indole rings (Table S1, ESI†). Based on the molecular weight, this compound is composed of an indole trimer, and its NMR data were coincident with those of turbomycin A (1, Fig. 1).14 Reportedly, 1 has been isolated as an antibacterial compound from several microbial species, such as Vibrio parahaemolyticus and Saccharomyces cerevisiae.15
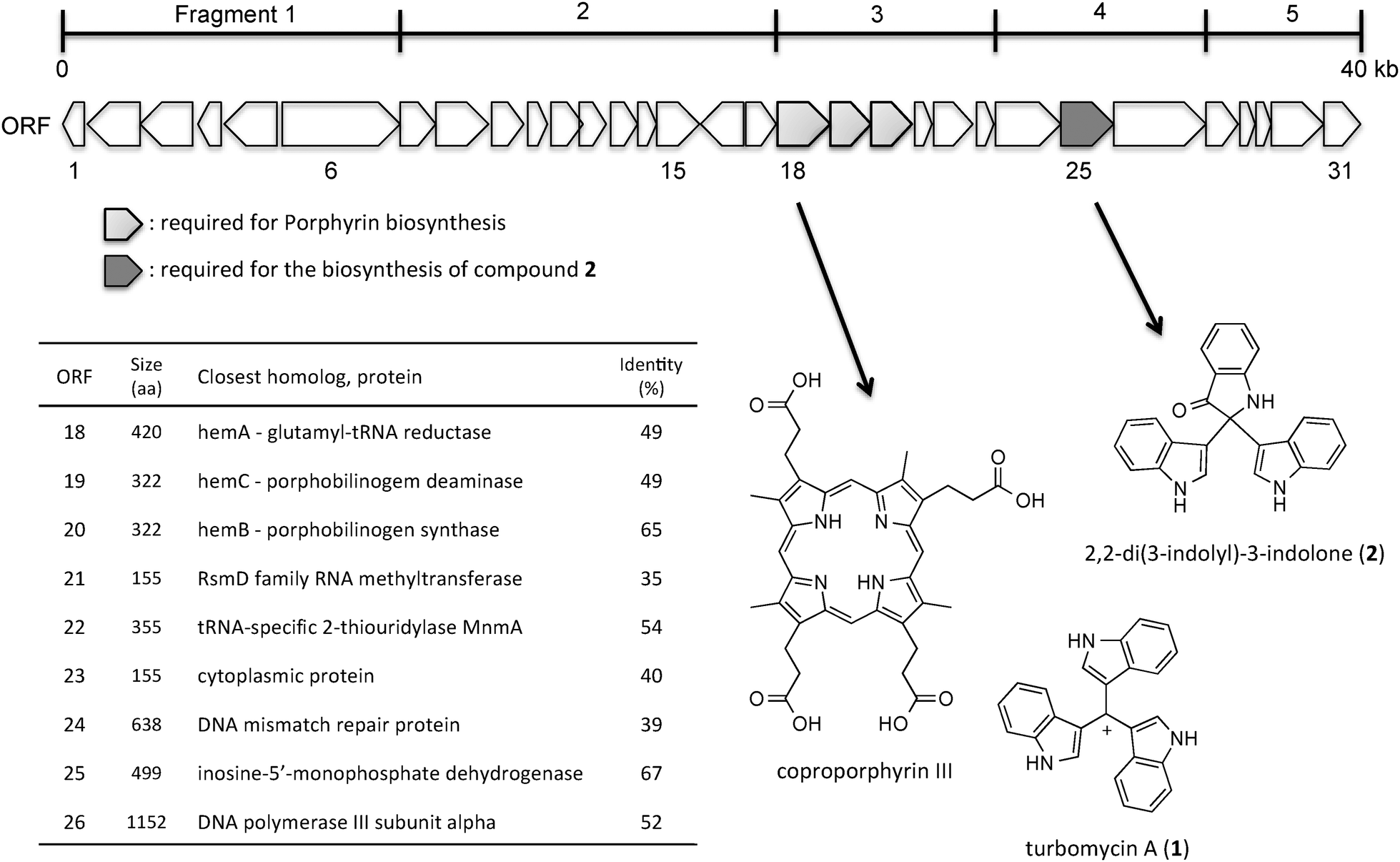 | ||
| Fig. 1 Open reading frames in the DNA insert of pDC112. | ||
Compound 2 was also obtained as a red amorphous powder, with a molecular formula of C24H17N3O determined by HR-ESI-MS (positive) at m/z 364.1441 (calcd for C24H18N3O, 364.1444). The interpretation of the 1H-NMR and COSY data revealed the presence of two 3-substituted indole rings, according to the proton signals at δH 8.10 (2H, brs, NH), 7.36 (2H, d, J = 8 Hz), 7.62 (2H, d, J = 8 Hz), 7.09 (2H, t, J = 8 Hz), 7.19 (2H, t, J = 8 Hz) and 6.94 (2H, s). In addition, the proton signals at δH 8.09 (1H, s, NH), 6.91 (1H, d, J = 8 Hz), 7.73 (1H, d, J = 8 Hz), 6.89 (1H, t, J = 8 Hz) and 7.51 (1H, t, J = 8 Hz) are reminiscent of the other indole ring system (Table S2, ESI†). The HMBC and HMQC correlations allowed us to connect these moieties, and as a result, compound 2 was determined to be 2,2-di(3-indolyl)-3-indolone, which was originally isolated from a Vibrio sp. separated from the marine sponge Hyrtios altum (Fig. 1).16 In line with the previous reports,17,18 compounds 1 and 2 showed antibacterial activity against Bacillus cereus, with 7 mm diameter growth inhibition at 25 μg per paper disk and 5 mm diameter growth inhibition at 100 μg per paper disk, respectively.
Next, to identify the gene responsible for the production of these two antibacterial compounds, we subcloned the insert DNA, which was almost in 40 kb length with 31 putative ORFs (Table S3, ESI†). The insert DNA was divided into five fragments, and each fragment was amplified and transformed into Escherichia coli EPI300. In agreement with our previous report,9 the culture solution of the transformant harboring fragment 3, encoding the enzyme homologous to glutamyl-tRNA reductase, became red due to the enhancement of porphyrin biosynthesis (Fig. 1).19 However, 2,2-di(3-indolyl)-3-indolone (2) was detected only in the culture solution of E. coli carrying fragment 4, by the LC-MS analysis. Encouraged by these results, we focused on each ORF encoded in fragment 4, which was subcloned further and transformed again into E. coli, and the culture solutions of the transformants were subjected to the LC-MS analysis. The results revealed that only ORF 25 is required for the production of compound 2 (Fig. S1, ESI†). ORF 25 shared homology with the inosine 5′-monophosphate dehydrogenase (IMPD) derived from the sulfur-reducing Gram-negative bacterium Deferribacter desulfuricans, with 67% identity.
However, 1 was not detected in any of the transformants prepared during our subcloning experiment. Gillespie and co-workers previously reported the isolation of turbomycins A and B from a soil metagenome library.18 They demonstrated that the 4-hydroxyphenylpyruvate dioxygenase (4HPPD), which catalyzes the conversion of 4-hydroxyphenylpyruvate into homogentisate,20 is required for the production of turbomycins. The proposed biosynthesis of turbomycins involves both the normal E. coli genes for indole production and the heterologously introduced 4HPPD gene, and is catalyzed by the predominant melanin complex, which is generated through the spontaneous oxidation and polymerization of homogentisic acid (HGA). Although there is no ORF similar to 4HPPD in the insert gene of pDC112, the involvement of as-yet unidentified genes in the production of 1 presently cannot be ruled out.
IMPD is an enzyme originally identified as participating in the primary metabolism that catalyzes the rate-limiting reaction of de novo GTP biosynthesis. This enzyme catalyzes the oxidation step from inosine 5′-monophosphate (IMP) to xanthosine 5’-monophosphate (XMP), in an NAD+-dependent manner.21 The catalytic mechanism proceeds through a covalent adduct between the 2-position of the IMP purine ring and the sulfur of an active site cysteine. Hydride transfer from the covalent enzyme-IMP species to NAD+ yields a thioimidate, which is subsequently hydrolyzed.22
Regarding the biosynthesis of 2, two possible biosynthetic pathways can be considered, including the direct or indirect involvement of the IMPD-catalyzed reaction. In terms of the latter possibility, it was assumed that the subcellular ratio of IMP or XMP somehow affects the endogenous pathway for indole production and may enhance the production level of 2. To address this possibility, the gene encoding IMPD from E. coli was transformed and overexpressed in E. coli EPI 300. However, 2 could not be detected, suggesting that the ratio between IMP and XMP is not associated with the pathway to produce 2. Otherwise, the IMPD homolog would directly participate in tryptophan metabolism in the E. coli system.
Next, we set out to examine the in vitro reaction with the IMPD homolog. First, the IMPD homolog was expressed in E. coli BLR or Rosetta, but accumulated only as an insoluble inclusion body. Therefore, we chose the methylotrophic yeast Pichia pastoris as the host, and the expressed enzyme was obtained in the soluble form and purified by Ni-chelate affinity column chromatography. With the soluble enzyme in hand, in vitro experiments were performed with cofactors such as NAD+ or NADP+, and putative substrates including indole, tryptophan, isatin and 3-hydroxy indole. The combinations of these substrates were also used for the enzyme reaction. However, no enzyme activity could be detected under any conditions.
Many Gram-positive and Gram-negative bacteria, including E. coli, produce indole as a signal molecule, which influences numerous aspects of bacterial physiology, such as plasmid stability, biofilm formation, virulence, and stress responses.23,24 Considering tryptophan metabolism in E. coli, tryptophanase and monooxygenase are responsible for the transformation from tryptophan to 3-oxyindole. If the IMPD homolog accepts 3-oxyindole as a substrate, then it would be converted into isatin. Isatin is very reactive, and two indole molecules would attack the amide carbonyl of the isatin to produce 2 (Scheme 1). Reportedly, the related compound, trisindolin, which is presumably generated by the nucleophilic attack on the keto carbonyl group, was also biosynthesized in E. coli heterologously expressing an oxygenase gene derived from a Rhodococcus strain.25
 | ||
| Scheme 1 Proposed biosynthesis for compound 2. | ||
Conclusions
In summary, we obtained the red-pigmented clone pDC112, producing antibacterial substances, by the functional screening of the metagenome library generated from the marine sponge D. calyx. The antibacterial compounds were identified as turbomycin A and 2,2-di(3-indolyl)-3-indolone, and the latter compound was originally isolated from a sponge-derived Vibrio sp. The subcloning experiment revealed that only ORF 25 in the DNA insert of pDC112 is essential for the biosynthesis of 2 in E. coli. The closest homolog of ORF 25 is the inosine 5′-monophosphate dehydrogenase of the Gram-negative bacterium, D. desulfuricans. To the best of our knowledge, this is the first report of a gene responsible for the biosynthesis of an antibacterial 2,2-di (3-indolyl)-3-indolone.Experimental section
General experimental procedures
1H and 13C NMR spectra were recorded on a JEOL ECX-500 spectrometer in CDCl3. 1H and 13C NMR chemical shifts were reported in parts per million and referenced to solvent peaks: δH = 7.26 and δC = 77.2 ppm. LC-MS and HRMS data were obtained from an Agilent 1100 series HPLC-micro TOF mass spectrometer (Bruker Daltonics), using Electrospray Ionization with a COSMOSIL 5C18 MS-II column (2.0 i.d. × 75 mm).Antibacterial screening
Almost 100 colonies on each plate were grown on LB agar medium containing chloramphenicol (15 μg mL−1 final concentration), at 30 °C for 2 days. Subsequently, 0.5% LB soft agar medium containing a Bacillus cereus culture solution was poured onto each agar plate, which was cultivated for 12 hours at 37 °C. After the cultivation, the inhibition halo around each colony was monitored. When a positive clone was detected, it was picked up, inoculated into LB medium containing chloramphenicol, and maintained as a glycerol stock at −80 °C.Production and isolation of anti bacterial compounds
Each antibacterial clone was cultured in LB medium (9 L) supplemented with chloramphenicol (12.5 μg mL−1 as final concentration), with shaking at 30 °C and 120 rpm for 3 days. After centrifugation at 7000 rpm for 10 min, the supernatant was subjected to solid phase extraction with Diaion HP-20 resin. The resin was washed with water and eluted with MeOH. The resulting extract was concentrated by rotary evaporation and fractionated by C18 column chromatography, with a stepwise gradient system from water to methanol. The antibacterial activity was detected in the 100% MeOH fraction. The active fraction was subsequently chromatographed on silica gel, with mixed solvent systems of CHCl3 and MeOH. The fractions that eluted with CHCl3:MeOH = 8:2 were further purified by reversed phase HPLC, using a COSMOSIL 5C18 MS-II column (10 i. d. × 250 mm), to yield turbomycin A (1) and 2,2-di(3-indolyl)-3-indolone (2) as red powders (3.4 mg and 1.5 mg, respectively).
Antibacterial activity
Screening plates containing Bacillus cereus were prepared with LB agar medium. The isolated compounds were dissolved in MeOH to a concentration of 1 mg mL−1, and 10, 25, 50, and 100 μL were applied to 0.7 mm paper disks (ADVANTEC). The disks were placed on the prepared plates and incubated at 30 °C for 12 hours. Inhibition was scored visually, and zones of inhibition were reported as the diameter of the clear zone in millimeters. The assay was conducted in duplicate trials.Sub cloning
Fosmid (15 ng) was used as the template for PCR, and fragments 1–5 were amplified using the primer pairs listed in Table S4 (ESI†). All PCRs were conducted on a 50 μl scale, with reaction solutions containing 1.75 mM MgCl2, 1 μM of each primer, 0.3 mM dNTPs and 1.25 U of KAPA Taq Extra DNA polymerase (Nippon Genetics). The PCR cycling conditions were as follows: initial denaturation (95 °C for 5 min), with 30 cycles of denaturation (95 °C for 30 s), annealing (55 °C for 30 s) and extension (72 °C for 10 min), and a final extension step (72 °C for 10 min). PCR products were ligated into the fosmid vector (Epicentre), and sequenced with the M13 universal and reverse primers using an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). The plasmid vectors were transformed into E. coli EPI300, and the samples for LC-MS were prepared as follows. The strain bearing each subclone was cultivated in 500 mL of LB medium, for 3 days with shaking at 30 °C, 120 rpm. The culture solution was centrifuged at 7000 rpm for 10 min. Diaion HP-20 resin was added to the supernatant, and the mixture was stirred for 3 hours. The resin was washed with water, and then eluted with MeOH. The concentrated extract was fractionated by C18 column chromatography, with a stepwise gradient system from water to methanol. The 100% MeOH fraction was subjected to the LC-MS analysis.Over expression with IMPD derived from E. coli
The genomic DNA of the E. coli BL21 strain was extracted with a QIAGEN genomic DNA extraction kit. The DNA thus obtained was used as the template for PCR on a 50 μl scale, in a reaction solution containing 1.75 mM MgCl2, 1 μM of each primer, 0.3 mM dNTPs and 1.25 U of PrimeSTAR® HS DNA Polymerase (Takara). The PCR cycling conditions were as follows: initial denaturation (95 °C for 5 min), with 30 cycles of denaturation (95 °C for 30 s), annealing (55 °C for 30 s) and extension (72 °C for 90 s). The primers are listed in Table S5 (ESI†). The PCR products were ligated into the fosmid vector (Epicentre), and were sequenced with the M13 universal and reverse primers using an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). The constructed plasmid was transformed into E. coli EPI300, and the sample for LC-MS was prepared as described above.Enzyme expression with Pichia pastoris
To amplify the ORF 25 region, 15 ng of plasmid was used in a 50 μl PCR solution, containing 1.75 mM MgCl2, 1 μM of each primer, 0.3 mM dNTPs and 1.25 U of KAPA Taq Extra DNA polymerase (Nippon Genetics). The PCR cycling conditions were the same as those used for the subcloning experiment. The primers are listed in Table S6 (ESI†). The PCR product was ligated into the pPICZ_N vector, and the constructed plasmid was then transformed into DH5α. After cultivation with LB + Zeocin (100 μg mL−1) medium, transformation was achieved. Subsequently, electroporation was conducted according to the Easy Select TM Pichia Expression Kit manual (Invitrogen). Purification by Ni-chelate column chromatography was performed with wash and elution buffers, as follows (wash buffer: 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 5% glycerol, 10 mM imidazole; elution buffer: 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 5% glycerol, 300 mM imidazole).Acknowledgements
This work was partly supported by a Grant-in-Aids from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. Y. T. is a recipient of the JSPS Fellowship for Young Scientists.Notes and references
- S. E. Brantley, T. F. Molinski, C. M. Preston and E. F. DeLong, Tetrahedron, 1995, 51, 7667–7672 CrossRef CAS.
- R. I. Amann, W. Ludwig and K.-H. Schleifer, Microbiol. Rev., 1995, 59, 143–169 CAS.
- A. Schirmer, R. Gadkari, C. D. Reeves, F. Ibrahim, E. F. DeLong and C. R. Hutchinson, Appl. Environ. Microbiol., 2005, 71, 4840–4849 CrossRef CAS PubMed.
- J. Piel, D. Hui, G. Wen, D. Butzke, M. Platzer, N. Fusetani and S. Matsunaga, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16222–16227 CrossRef CAS PubMed.
- T. Wakimoto, Y. Egami, Y. Nakashima, Y. Wakimoto, T. Mori, T. Awakawa, T. Ito, H. Kenmoku, Y. Asakawa, J. Piel and I. Abe, Nat. Chem. Biol., 2014, 10, 648–655 CrossRef CAS PubMed.
- G. Y. Wang, E. Graziani, B. Waters, W. Pan, X. Li, J. McDermott, G. Meurer, G. Saxena, R. J. Anderson and J. Davies, Org. Lett., 2000, 2, 2401–2404 CrossRef CAS PubMed.
- S. F. Brady and J. Clady, Angew. Chem., Int. Ed., 2005, 44, 7063–7065 CrossRef CAS PubMed.
- J.-S. Kim, H. K. Lim, M. H. Lee, J.-H. Park, E. C. Hwang, B. J. Moon and S.-W. Lee, FEMS Microbiol. Lett., 2009, 295, 42–49 CrossRef CAS PubMed.
- R. He, T. Wakimoto, Y. Takeshige, Y. Egami, H. Kenmoku, T. Ito, B. Wang, Y. Asakawa and I. Abe, Mol. BioSyst., 2012, 8, 2334–2338 RSC.
- M. J. Fujita, N. Kimura, H. Yokose and M. Otsuka, Mol. BioSyst., 2012, 8, 482–485 RSC.
- H. A. Iqbal, J. W. Craig and S. F. Brady, FEMS Microbiol. Lett., 2014, 354, 19–26 CrossRef CAS PubMed.
- M. J. Fujita, N. Kimura, A. Sakai, Y. Ichikawa, T. Hanyu and M. Otsuka, Biosci., Biotechnol., Biochem., 2012, 75, 2283–2287 CrossRef PubMed.
- H. K. Lim, E. J. Chung, J.-C. Kim, G. J. Choi, K. S. Jang, Y. R. Chung, K. Y. Cho and S.-W. Lee, Appl. Environ. Microbiol., 2005, 71, 7768–7777 CrossRef CAS PubMed.
- R. Veluri, I. Oka, I. W. Döbler and H. Laatsch, J. Nat. Prod., 2003, 66, 1520–1523 CrossRef CAS PubMed.
- H. Budzikiwicz and H. Eckau, Tetrahedron Lett., 1972, 36, 3807–3810 CrossRef.
- M. Kobayashi, S. Aoki, K. Matsunami, M. Kurosu and I. Kitagawa, Chem. Pharm. Bull., 1994, 42, 2449–2451 CrossRef CAS.
- S.-X. Cai, D.-H. Li, T.-J. Zhu, F.-P. Wang, X. Xiao and Q.-Q. Gu, Helv. Chim. Acta, 2010, 93, 791–795 CrossRef CAS.
- D. E. Gillespie, S. F. Brady, A. D. Bettermann, N. P. Cianciotto, M. R. Liles, M. R. Rondon, J. Clardy, R. M. Goodman and J. Handelsman, Appl. Environ. Microbiol., 2002, 68, 4301–4306 CrossRef CAS.
- D. V. Wettstein, S. Gough and C. G. Kannangara, Plant Cell, 1995, 7, 1039–1057 CrossRef PubMed.
- G. R. Moran, Arch. Biochem. Biophys., 2005, 433, 117–128 CrossRef CAS PubMed.
- M. D. Sintchak and E. Nimmesgern, Immunopharmacology, 2000, 47, 163–184 CrossRef CAS.
- G. C. Patton, P. Stenmark, D. R. Gollapalli, R. Sevastik, P. Kursula, S. Flodin, H. Schuler, C. T. Swales, H. Eklund, F. Himo, P. Nordlund and L. Hedstrom, Nat. Chem. Biol., 2011, 7, 950–958 CrossRef CAS PubMed.
- J.-H. Lee and J. Lee, FEMS Microbiol. Rev., 2010, 34, 426–444 CAS.
- S. P. Fernandez, C. Chimerel, U. F. Keyser and D. K. Summers, J. Bacteriol., 2011, 193, 1793–1798 CrossRef PubMed.
- M. Yoo, S.-U. Choi, K. Y. Choi, G. H. Yon, J.-C. Chae, D. Kim, G. J. Zylstra and E. Kim, Biochem. Biophys. Res. Commun., 2008, 376, 96–99 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: LC-MS, NMR, and sequence data. See DOI: 10.1039/c5mb00131e |
| This journal is © The Royal Society of Chemistry 2015 |