
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn integrated metabolomics approach for the research of new cerebrospinal fluid biomarkers of multiple sclerosis†
Damiana
Pieragostino
ab,
Michele
D'Alessandro
ab,
Maria
di Ioia
bc,
Claudia
Rossi
ab,
Mirco
Zucchelli
bd,
Andrea
Urbani
e,
Carmine
Di Ilio
ab,
Alessandra
Lugaresi
bc,
Paolo
Sacchetta
ab and
Piero
Del Boccio
*ab
aDepartment of Medical, Oral and Biotechnological Sciences, University “G. d'Annunzio” of Chieti-Pescara, Chieti, Italy. E-mail: p.delboccio@unich.it; Fax: +39 0871 541598; Tel: +39 0871 541593
bAnalytical Biochemistry and Proteomics Unit, Research Centre on Aging (Ce.S.I), University “G. d'Annunzio” of Chieti-Pescara, Chieti, Italy
cDepartment of Neurosciences and Imaging, University “G. d'Annunzio” of Chieti-Pescara, Chieti, Italy
dSchool of Medicine and Health Sciences, University “G. d'Annunzio” of Chieti-Pescara, Chieti, Italy
eDepartment of Experimental Medicine and Surgery, University of Tor Vergata, Rome, Italy
First published on 6th February 2015
Abstract
Multiple Sclerosis (MuS) is a disease caused due to an autoimmune attack against myelin components in which non proteic mediators may play a role. Recent research in metabolomics and lipidomics has been driven by rapid advances in technologies such as mass spectrometry and computational methods. They can be used to study multifactorial disorders like MuS, highlighting the effects of disease on metabolic profiling, regardless of the multiple trigger factors. We coupled MALDI-TOF-MS untargeted lipidomics and targeted LC-MS/MS analysis of acylcarnitines and aminoacids to compare cerebrospinal fluid metabolites in 13 MuS subjects and in 12 patients with Other Neurological Diseases (OND). After data processing and statistical evaluation, we found 10 metabolites that significantly (p < 0.05) segregate the two clinical groups. The most relevant result was the alteration of phospholipids levels in MuS and the correlation between some of them with clinical data. In particular lysophosphatidylcholines (m/z = 522.3 Da, 524.3 Da) and an unidentified peak at m/z = 523.0 Da correlated to the Link index, lysophosphatidylinositol (m/z = 573.3 Da) correlated to EDSS and phosphatidylinositol (m/z = 969.6 Da) correlated to disease duration. We also found high levels of glutamate in MuS. In conclusion, our integrated mass spectrometry approach showed high potentiality to find metabolic alteration in cerebrospinal fluid. These data, if confirmed in a wider clinical study, could open the door for the discovery of novel candidate biomarkers of MuS.
Introduction
Multiple Sclerosis (MuS) is a chronic demyelinating disease of the Central Nervous System (CNS) characterized by inflammation and axonal degeneration. It is the most common cause of neurologic disability in young adults in the Western world affecting mostly people between 20 and 40 years of age, with a female–male ratio which has increased, in the last decades from 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 3:1.1 In most cases MuS has a relapsing-remitting (RR) course.2
:1 to 3:1.1 In most cases MuS has a relapsing-remitting (RR) course.2
MuS is commonly considered an acquired autoimmune pathology in genetically susceptible individuals when environmental risk factors, such as viral infections, vitamin D deficiency and ultraviolet radiation, occurred.3 While the etiology of MuS is still unclear, a favored hypothesis suggests that one factor contributing to the development of autoreactive T-cells in MuS is a cross-reactive immune response between viral components and CNS antigens (“antigenic mimicry”).4,5 Among possible mediators are proteins, metabolites and lipids thought to be involved in disease mechanisms. Lipids, in particular, represent important targets since they are implicated in many signalling routes and antigen reactions. Recent studies suggest a role for lipids in the autoimmune process and recent evidence has shown lipid metabolism alterations in the CNS of MuS subjects.6,7
Metabolomic and lipidomic approaches revealed to be useful in identifying metabolic alterations in Cerebrospinal Fluid (CSF) in an animal model of MuS and in neurodegenerative diseases in general. In a previous work of Gonzalo et al., through an untargeted approach of metabolomics and lipidomics, several discriminant metabolites were found between MuS and non-MuS patients. Among these differential molecules, the lipid oxidation marker 8-iso-prostaglandine F2α was found to be increased in MuS patients. Also autoantibodies against lipoxidized proteins were increased in MuS, suggesting an enhanced autoimmune response underlying the progression of the disease.8 Moreover, when the limits of metabolomics are exceeded, it will be possible to comprehend the pathogenetic pathways facilitating the development of specific disease treatments. Sphingosine is the molecular backbone of sphingolipids which are the most abundant lipids in the myelin sheath, and Fingolimod, for example, is a sphingosine 1-phosphate receptor (S1PR) modulator, today used as a new oral drug for MuS.9 Furthermore, metabolic homeostasis is deeply modified during pregnancy, which is a typical remission period in MuS course.10,11 Therefore, determining the major epitopes of the different encephalitogenic myelin and neuronal factors implicated in MuS is of major significance not only for devising immuno-specific therapeutic approaches to MuS, but also for understanding the pathophysiology and etiology of the disease. Moreover it is demonstrated that carnitine, involved in fatty acid metabolism, and its derivatives are present in different concentrations in various body fluids and tissues and that they can be implicated in various diseases characterized by upregulated or impaired immune responses.12,13 The therapeutic rationale in MuS derives from the demonstration of a reduction of nitroxidative stress in CSF in active MuS patients treated with acetylcarnitine.14 However a current Cochrane review on the efficacy of carnitine in MuS-related fatigue concludes for an insufficient evidence of a therapeutic advantage of carnitine over placebo or active comparators.15 Recently, a metabolomics study by Noga MJ et al. demonstrated a significant change in amino acid metabolism in CSF during Experimental Allergic Encephalomyelitis (EAE). They found altered levels of metabolites related to pathways including nitric oxide synthesis, altered energy metabolism, polyamine synthesis and levels of endogenous antioxidants.16
On the spur of these recent findings, here we report a metabolomics investigation of CSF including carnitines, amino acids and lipid profiles in MuS subjects and patients with OND by combining untargeted and targeted metabolomics strategies. The aim of our study was to identify new candidate biomarkers for MuS diagnosis, in order to better understand the possible link between metabolic alterations and clinical features of the disease.
Materials and methods
Ethics statement
The study design was made following the guidelines for Good Clinical practice (GCP) of the local Ethics Committee that approved the study (Ethic committee of “G. d'Annunzio” University and ASL N.2 Lanciano-Vasto-Chieti, Italy), and following the ethical standards in conformity with those laid down in the Declaration of Helsinki (World Medical Association, 1997). All patients were informed about the procedures and provided written informed consent to participate in the study. In order to protect human subject identity a number code was employed for specimen identification.Patients
13 patients with Relapsing Remitting MuS, in accordance with the 2010 Polman's criteria,17 were included in this study. Clinical diagnosis was confirmed by cerebral and spinal cord magnetic resonance imaging (MRI) studies and by the presence of oligoclonal bands in CSF. The Expanded Disability Status Scale (EDSS) score was obtained at the time of lumbar puncture. To be enrolled in the study patients should have not been treated with steroids in the month before study entry and should have been never treated with immunomodulatory or immunosuppressive drugs both for MuS and for other diseases. 12 CSF samples of patients with other neurological diseases (OND) were used as the control group. The diagnosis in each of these patients was defined according to individual disease diagnostic criteria. Table S1 in the ESI† summarizes clinical and demographic features of the enrolled patients. The disease duration is defined as the time elapsed (expressed in days) between the first symptom to the day of CSF collection. All patients were selected in order to obtain age, sex and ethnicity (white people) matched cohorts of subjects. Patients with other disease course and comorbidites were excluded. CSF samples were collected at the MuS Center of Chieti (Italy).Sample collection
CSF samples, taken by a routine lumbar puncture at L3/L4 or L4/L5 interspace, were always collected in the morning on the first day of patient observation. Each sample (around 3 mL) was centrifuged at 10000g at 4 °C, for 10 minutes. The supernatant was divided into aliquots and stored at −80 °C. Around 2 mL of CSF from each subject were used for diagnosis. Only 300 microliters of CSF per patient were employed in this study (200 μL for untargeted lipidomics and 10 μL for targeted amino acids and carnitines determination).
Lipid extraction procedure and MALDI-TOF-MS analysis
200 μL of CSF per patient were used for lipid extraction. Total lipids were extracted using a modified “Bligh and Dyer” method.18 Briefly, 100 μL of a saturated solution of (NH4)SO4 (Sigma Aldrich) for protein precipitation were added to 2 different aliquots of 100 μL of CSF. After vortexing and centrifugation (15 min at 10000g) 500 μL of MeOH, 250 μL of CHCl3 and 250 μL of H2O were added to the supernatant (180 μL) and vortexed (all solvents were purchased from Sigma Aldrich). After centrifugation for 15 min at 10000g the CHCl3 phases of two aliquots of the same sample were brought together, dried and sealed to be stored at −80 °C. The total lipids extract was re-suspended in 50 μL of MeOH, vortexed, centrifuged for 15 min at 10000g and used for MALDI-TOF-MS analysis. Each CSF sample was fortified with 10 μL of Dimyristoylphosphatidylcholine (DMPC) (Avanti Polar Lipids, inc USA) at 2.5 μg mL−1, used as an internal standard for mass accuracy verification. After spectra acquisition the signals of the DMPC and the signal of an endogenous well characterized signal were used to verify the instrumental mass accuracy (see Fig. S1A for details, ESI†). In order to compare groups, the variability of quantification and the extraction recovery were assessed by using quality controls of CSF fortified with standard DMPC at different concentration levels (2.5, 5, 10 and 25 μL mL−1). As reported in Fig. S1B ESI,† the method showed good linearity response with an RSD% below 20% for each concentration level. The matrix solution was prepared by using a saturated solution of 2,5-dihydroxybenzoic acid (DHB) in acetone–chloroform (9:1) according to Fujiwaki et al.19 Extracted lipids from CSF were mixed in a ratio of 1:1 with the matrix solution and 0.5 μL of the resulting solution were spotted on MTP Ground steel 384 (Bruker Daltonics). All analyses were performed using an Autoflex Speed MALDI-TOF-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) in the mass range 300–3000 Da. Instrument parameters were tuned in order to obtain the highest resolution and sensitivity in the mass range used. All mass spectra were acquired in positive Reflectron mode at a voltage of 19; 16.72 and 8.54 kV for the first and second ion extraction stages and lens, respectively. Every single acquisition run was composed of 500 laser pulses at 1000 Hz. The most abundant lipid signals were characterized by fragmentation experiments by LIFT tandem mass spectrometry analysis. Analyses were performed using the following acquisition settings: ion source 1: 6.0 kV; ion source 2: 5.3 kV; lens: 3.0 kV; reflector 1: 27.0 kV; reflector 2, 11.70 kV; lift 1: 19.0 kV; lift 2: 4.25 kV; pulsed ion extraction 120 ns.
Lipid identification was carried out by database search (mainly by: “Lipid Maps”) using their accurate mass measured and by the mass of characteristic fragments obtained in LIFT experiments. Fig. S2–S4 in ESI† show the fragmentation spectrum of the signals at m/z = 522 Da, 524 Da and 734 Da reported in Table 2 as differential metabolites.
LC-MS/MS analysis of amino acids, free carnitine and acylcarnitines
A targeted metabolic fingerprint strategy using Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS) in multiple reaction monitoring (MRM) mode was applied for the determination of amino acids, free carnitine and acylcarnitines levels in CSF samples. CSF (9.6 μL) was transferred into 1.5 mL tubes (Eppendorf, Hamburg, Germany) and then extracted with a solution containing the stable isotope labeled internal standards for each analyte of interest, according to the principle of isotope dilution internal standardization. The stable isotope labeled internal standards, as well as the extraction solution, were obtained from the NeoBase Non-derivatized MSMS Kit (Perkin Elmer Life and Analytical Sciences, Turku, Finland). The tubes were then capped and vortex mixed. The samples were centrifuged (22582g at 4 °C for 15 minutes), and the supernatant was analyzed by direct infusion mass spectrometry (DIMS) as already reported.20
The NeoBase non-derivatized MSMS Kit is validated for blood spots for the determination of absolute concentrations of amino acids and acylcarnitines. As done in other studies21,22 working with different biological matrices such as plasma and serum, the use of the kit was intended to reveal relevant alterations in amino acids and acylcarnitines profiles in CSF patient samples. Anyway, once accepted the use of the kit for different biological matrices other than for blood spot to determine alterations in the metabolites profile, we decided to extract 9.6 μL of CSF for each sample, after testing the reproducibility at three different volumes of the sample. A quality control (QC) CSF pool was prepared from the CSF patient samples, then extracted and analyzed as described for the CSF samples in the study. A total of 10 QC CSF pool were analyzed during the run. Method accuracy was accessed for each analyte, precision being evaluated as repeatability in terms of coefficient of variation (CV) for the QC samples. The calculated mean CV for the amino acids and acylcarnitines was between 3.9–13% and 6.1–11.5%, respectively.
The DIMS analysis for the evaluation of the metabolic profile in CSF samples was performed using a LC-MS/MS system consisting of an Alliance HT 2795 HPLC Separation Module coupled to a Quattro Ultima Pt ESI tandem quadrupole mass spectrometer (Waters Corporation, Milford, MA, USA). The instrument operated in positive electrospray ionization mode using MassLynx V4.0 Software (Waters) with auto data processing by NeoLynx (Waters Corporation, Milford, MA, USA). Autosampler injections of 30 μL were made into the ion source directly using a narrow peek tube, and the mobile phase was methanol–water 75:25 (v/v) plus 0.01% oxalic acid (Perkin Elmer). The total run time was 1.8 min, injection-to-injection. The mass spectrometer ionization source settings were optimized for maximum ion yields for each analyte. The capillary voltage was 3.25 kV, source temperature was 120 °C, desolvation temperature was 350 °C, and the collision cell gas pressure was 3–3.50 × 10−3 mbar Argon. In ESI† Tables S2–S4 report the detailed list of analytes.
Data processing and statistical methods
All CSF raw MALDI mass spectra acquired were processed using the SpecAlign free software, version 2.4.1, developed by Dr Jason Wong at the University of Oxford (http://powcs.med.unsw.edu.au/research/adult-cancer-program/services-resources/specalign). This procedure allowed peak deconvolution and alignment, denoise and Total Ion Count (TIC) normalization to give a table of mass pairs with associated relative intensities for all the detected MALDI peaks for each CSF sample analyzed. The relative intensities of lipid signals obtained by untargeted MALDI-TOF-MS analysis were subjected to multivariate processing. This data matrix was exported, UV-scaled, mean centered and used for partial least squares discriminant analysis (PLS-DA) using SIMCA-P + 11.0 (Umetrics AB, Umeå, Sweden). The most influential variables responsible for the separation between classes are the variables having the greatest influence in PLS-DA. A parameter named VIP (Variable Importance in the Projection) was employed to reflect the importance of variables in the discriminant analysis. The result of the PLS-DA analysis was a list of all variables (lipids) sorted by their discriminatory power (VIP). We used this result as explorative and selected the major discriminant lipid signals (VIP > 2), obtained by PLS-DA elaboration, for a statistical re-evaluation applying the Student's t-test when normality was accepted, otherwise the Mann–Whitney U-test was applied. From this processing of untargeted data we obtained a list of significant variables listed as potential biomarkers.For targeted analysis, all LC-MS/MS data were subjected to D'Agostino and Pearson omnibus normality test in order to determine the normality of each variable measured in each group. When normality was accepted the Student's t-test was employed, otherwise the Mann–Whitney U-test was used for comparing the groups. All statistical elaborations were performed by using GraphPad Prism (GraphPad Software, Inc. USA). Correlation analysis with clinical parameters to lipids, carnitines and amino acids levels was performed using Statistica 7.0 (StatSoft DemoVersion). GraphPad Prism was employed for ROC curve analysis.
Results
Metabolic profiling in CSF by targeted LC-MS/MS analysis
We investigated amino acids, free carnitine and acylcarnitines levels in CSF samples by LC-MS/MS in a targeted metabolomic strategy. Samples were randomized and analyzed by DIMS as described in the Methods section. Table 1 shows CSF relative abundance of short and medium acylcarnitines and amino acids obtained by DIMS analysis in OND and MuS subjects. Long-chain acylcarnitines were not detectable in CSF. As shown in Table 1 only glutamate resulted to be differential between the two clinical groups investigated (p < 0.05, t-test).| Compound (abbreviation name) | MuS | OND | p-value |
|---|---|---|---|
| a Indicates statistical test with a p-value < 0.05. b Isomeric compounds having the same m/z and measured as a single sum value. | |||
| C0 | 4.75(±1.00) | 4.64(±1.71) | 0.85 |
| C2 | 1.50(±0.39) | 1.52(±0.78) | 0.93 |
| C3 | 0.08(±0.02) | 0.10(±0.07) | 0.26 |
| C4 | 0.07(±0.01) | 0.08(±0.03) | 0.77 |
| C5OH/C4DCb | 0.08(±0.01) | 0.09(±0.03) | 0.27 |
| C5DC/C6OHb | 0.17(±0.05) | 0.17(±0.06) | 0.97 |
| C6DC | 0.09(±0.03) | 0.08(±0.03) | 0.55 |
| PRO | 61.33(±13.90) | 55.07(±13.51) | 0.27 |
| VAL | 52.0.6(±6.61) | 50.17(±10.52) | 0.59 |
| LEU/ILE/PRO-OHb | 42.87(±8.24) | 40.41(±12.75) | 0.57 |
| ORN | 25.07(±6.33) | 23.91(±6.92) | 0.67 |
| MET | 10.16(±2.06) | 11.28(±3.97) | 0.38 |
| PHE | 18.08(±4.03) | 16.97(±4.35) | 0.51 |
| ARG | 57.04(±10.65) | 54.27(±12.36) | 0.55 |
| CIT | 24.03(±6.84) | 24.25(±6.69) | 0.94 |
| TYR | 16.86(±4.95) | 17.61(±5.33) | 0.72 |
| GLY | 757.74(±259.78) | 687.75(±243.43) | 0.49 |
| ALA bis | 248.04(±36.44) | 259.50(±64.61) | 0.59 |
| SER | 8.29(±1.94) | 8.47(±2.56) | 0.84 |
| THR | 51.50(±5.90) | 47.97(±6.16) | 0.16 |
| ASN | 3.42(±0.76) | 3.13(±0.89) | 0.38 |
| ASP | 6.50(±1.18) | 5.82(±1.09) | 0.15 |
| LYS/GLNb | 5985.08(±846.54) | 5714.12(±1296.35) | 0.54 |
| GLU | 26.66(±2.37) | 23.58(±4.49) | 0.04a |
| HIS | 57.18(±9.77) | 55.84(±14.48) | 0.79 |
CSF lipidomics by MALDI-TOF-MS
The lipid pool extracted from CSF of each patient was randomized and subjected to MALDI-TOF-MS analysis. Fig. 1 (panel a) shows the average MALDI-TOF mass spectra obtained by the two clinical groups investigated. In panel b the virtual gel showing the lipid profiling of each CSF analyzed is reported. The more populated range of lipid signals in the spectrum was between m/z = 500 Da and 1000 Da. Fragmentation analysis of the main peaks (data not shown) indicated that the most abundant ions contain phosphocoline as the head group at m/z = 184 Da, mainly sphingomyelins (SM) and phosphatidylcholines (PC).23 Once all CSF samples were analyzed, an automatic elaboration of all spectra acquired was performed. After denoising and signals alignment, an average data matrix was obtained containing 642 m/z signals ascribable to lipid molecular species, with an intensity normalized vs. the Total Ion Count (TIC). Data were displayed in a final reference matrix where all molecules studied were reported as variables (642) linked to each observation (patient) in a compatible format for the multivariate analysis with Simca-P software. To explain the maximum separation between defined class samples in the data set, the 642 variables were processed with Partial Least Squares Discriminant Analysis (PLS-DA). In Fig. 2 the PLS-DA score plot of the first three components where the two group of patients were visualized and separated, based on their lipidomics pattern, is reported. However, the model with greater predictive power was constituted of nine components describing a 78% of variation in X (R2X = 78.6%) and 99% of the variation in the response Y (class) (R2Y = 98.9%) with a Q2Y = 0.68. In order to avoid over-parametrization due to the fact that the model was used to many variables with respect to the analyzed cases, we validated this exploratory results by univariate analysis as described below. Simca-P software generates a list of all the variables analysed sorted by a score number named VIP. The VIP score reflects the variable's contribution to classification, and can be used to discover the most relevant differential variables responsible for group separation. We used the results of multivariate analysis as explorative and selected the major discriminant lipid signals for a statistical re-evaluation. The software indicates VIP > 1 significant for discrimination. In this study the differential lipids with a VIP value > 2 (two fold the nominal cut off) were taken into consideration for an independent statistical re-evaluation (Mann–Whitney U test/t-test). Variables with a p < 0.05 obtained from hypothesis testing were taken into consideration as putative biomarkers.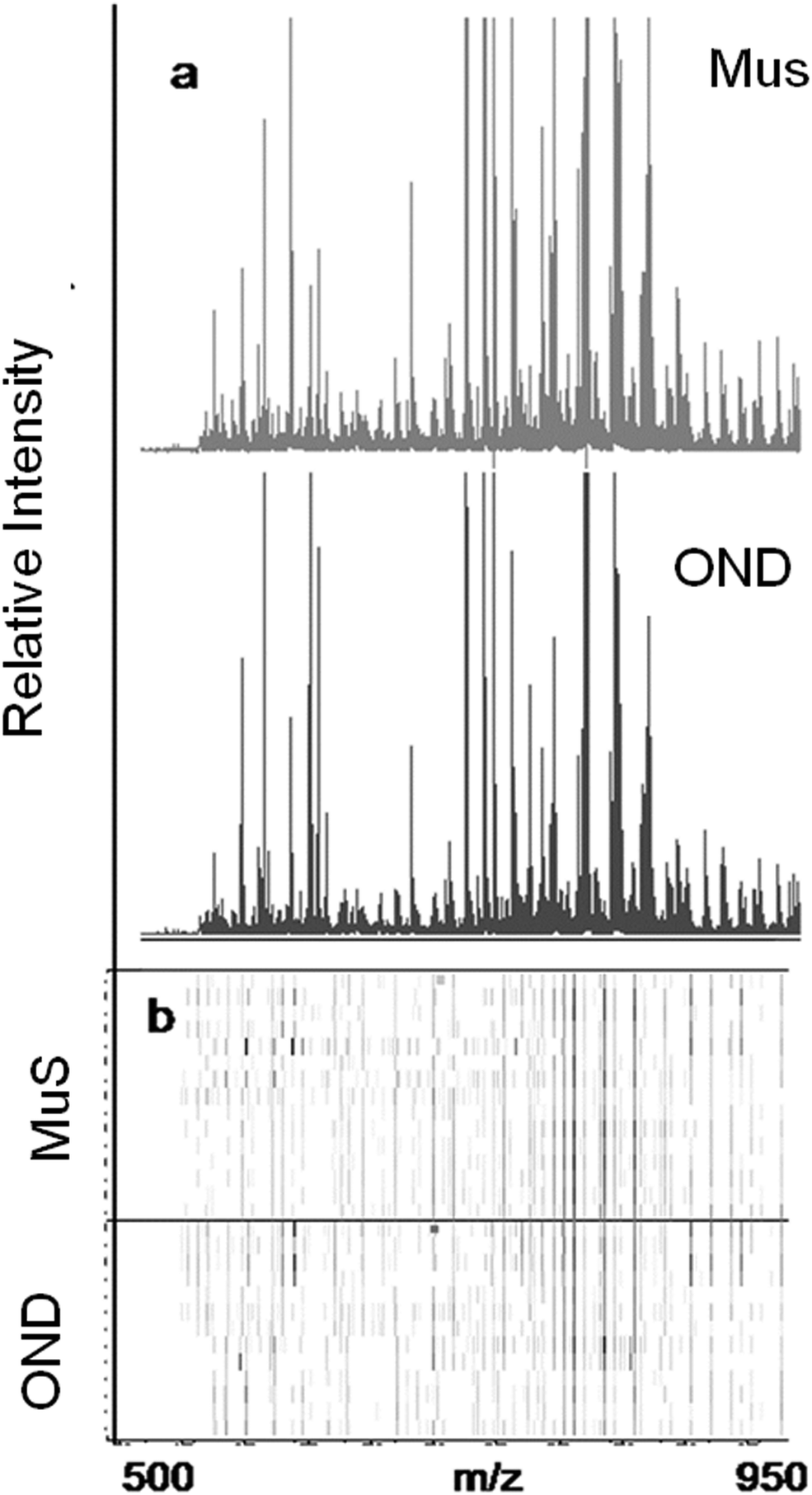 | ||
| Fig. 1 Lipidomic results in CSF by MALDI-TOF-MS. The lipid species show m/z signals between 500 and 1000 m/z. (a) Average MALDI-TOF mass spectrum of CSF obtained from 13 MuS patients (upper spectrum) and from 12 OND patients (bottom spectrum). (b) Virtual gel showing the cases vs. the m/z signals of each CSF sample analyzed. The horizontal line distinguishes the two clinical groups. This image was made using the CinProTools software (Bruker Daltonics, Germany). | ||
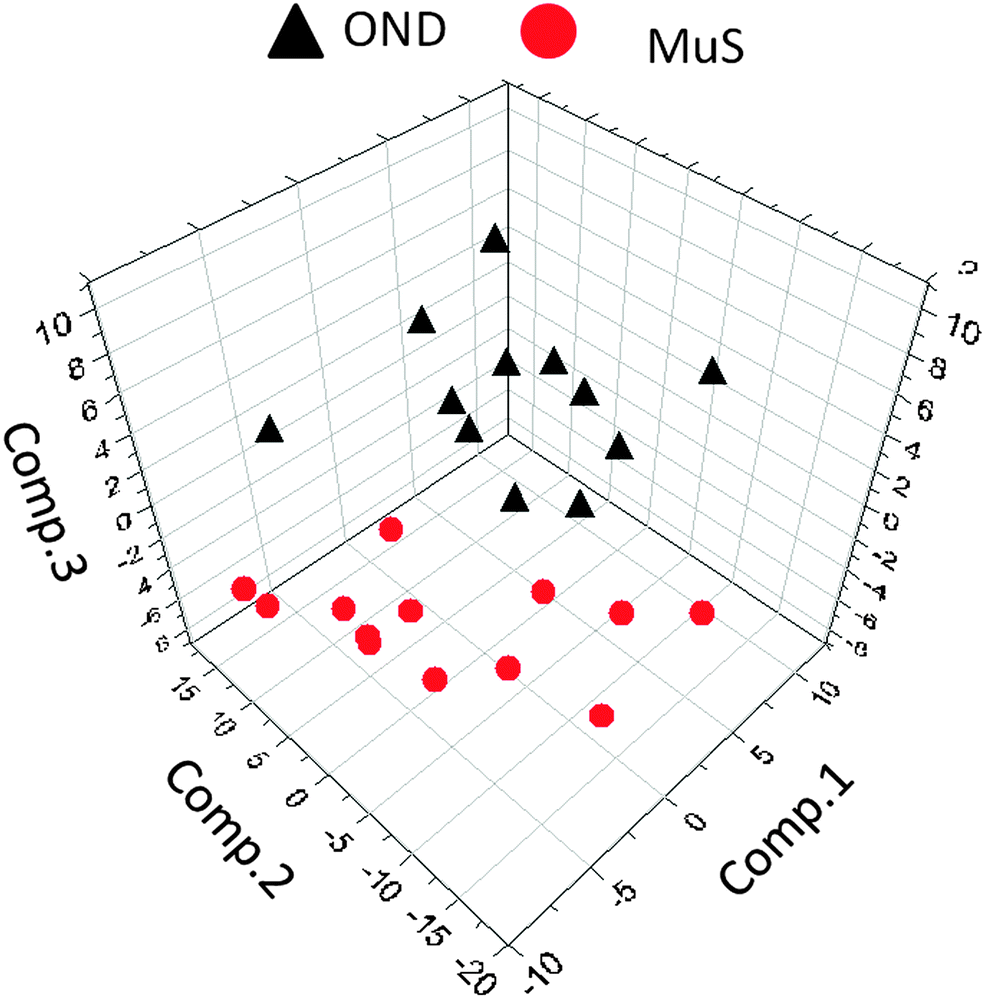 | ||
| Fig. 2 Multivariate analysis of metabolomics data. Partial least squares discriminant analysis (PLS-DA) score plot ([t1]/[t2]/[t3]) of the first three components, from metabolomics data derived from OND subjects (black triangles) and MuS patients (red circles). The most significant variables that drive the separation between the two clinical groups are reported in Table 2. | ||
Potential biomarkers identified
In Table 2 all differential (p < 0.05) metabolites highlighted in this study and in particular nine lipid species and glutamate are reported. Among all the differential lipid signals some of them were tentatively identified through a database search of the accurate mass measured and, when possible, by fragmentation analysis (for mass tolerance in the database search 15 ppm was chosen which is about 2 times higher than the mass accuracy measured for the DMPC standard, considering the large range in ion intensity, during the fast acquisition of the TOF). Fig. 3 shows the distribution of each discriminant metabolite in two groups of patients studied. Three lyso phospholipids at m/z = 522.3 Da (a), 524.3 Da (b) and 573.3 Da (c) identified as two lysophosphatydilcholine (LPC) and lysophosphatidylinositol (LPI), respectively, and the signals at 734.5 Da (d) and 969.6 Da (e) identified as phosphatidylcholine (PC) and phosphatidylinositol (PI) respectively, as well as two unidentified species at m/z = 523.0 Da (f) and 523.9 Da (g), are significantly increased in MuS patients. Otherwise the signals at 673.4 Da (h) identified as phosphatydic acid (PA) and at 727.0 Da (i) (unidentified signal) decreased in MuS patients vs. OND subjects. Considering all amino acids and carnitine species analysed, the glutamate (j) in CSF of MuS patients was significantly elevated vs. OND patients. Table 3 summarizes the discriminatory power of each potential biomarker highlighted, showing a receiver operating characteristic curve (ROC) data based on the relative intensity of each biomarker analyzed. The accuracy of each biomarker in classifying the two groups is outlined by the sensitivity and the specificity in the reclassification of MuS patients. Eight compounds, taken singularly, show significant reclassification and, most of them showed high specificity in classifying the MuS group.| Abbrevation | Observed | Calc. m/z | Molecular formula | Identification tools | p-value | Trend in MuS |
|---|---|---|---|---|---|---|
| a Variables derived from lipidomics analysis, six of them were tentatively identified by database search and fragmentation analysis. b Glutamate was analyzed by LC-MS/MS. c Our data do not provide information about the backbone structure of lipids, thus the ID is not univocal when the compound can have more combinations of instauration positions and fatty acid substitutions. In these cases the ID returned from the database was selected from a list of probable species with identical MW. Lipid Maps: on-line database resource at http://www.lipidmaps.org/tools for lipid identification. MS/MS: fragmentation analysis by tandem MS. Spectra are shown in ESI Fig. S2–S4. Trend in MuS: abundance of each analyte in MuS CSF compared to the abundance in OND CSF. nd: not determined. | ||||||
| LPC(18:1(9Z)/0:0)a | 522.361 | 522.3554 | C26H53NO7P | Lipid maps; MS/MS | 0.015 | Up |
| Unidentifieda | 523.040 | nd | nd | 0.004 | Up | |
| Unidentifieda | 523.993 | nd | nd | 0.017 | Up | |
| LPC(18:0/0:0)a | 524.382 | 524.3711 | C26H54NO7P | Lipid maps; MS/MS | 0.03 | Up |
| LPI(16:0/0:0)a | 573.311 | 573.3035 | C25H50O12P | Lipid maps | 0.009 | Up |
| PAa,c | 673.475 | 673.4803 | C37H70O8P | Lipid maps | 0.034 | Down |
| Unidentifieda | 727.021 | nd | nd | 0.022 | Down | |
| PCa,c | 734.559 | 734.5695 | C40H81NO8P | Lipid maps; MS/MS | 0.037 | Up |
| PIa,c | 969.651 | 969.6427 | C53H94O13P | Lipid maps | 0.039 | Up |
| Glutamateb | — | — | — | — | 0.04 | Up |
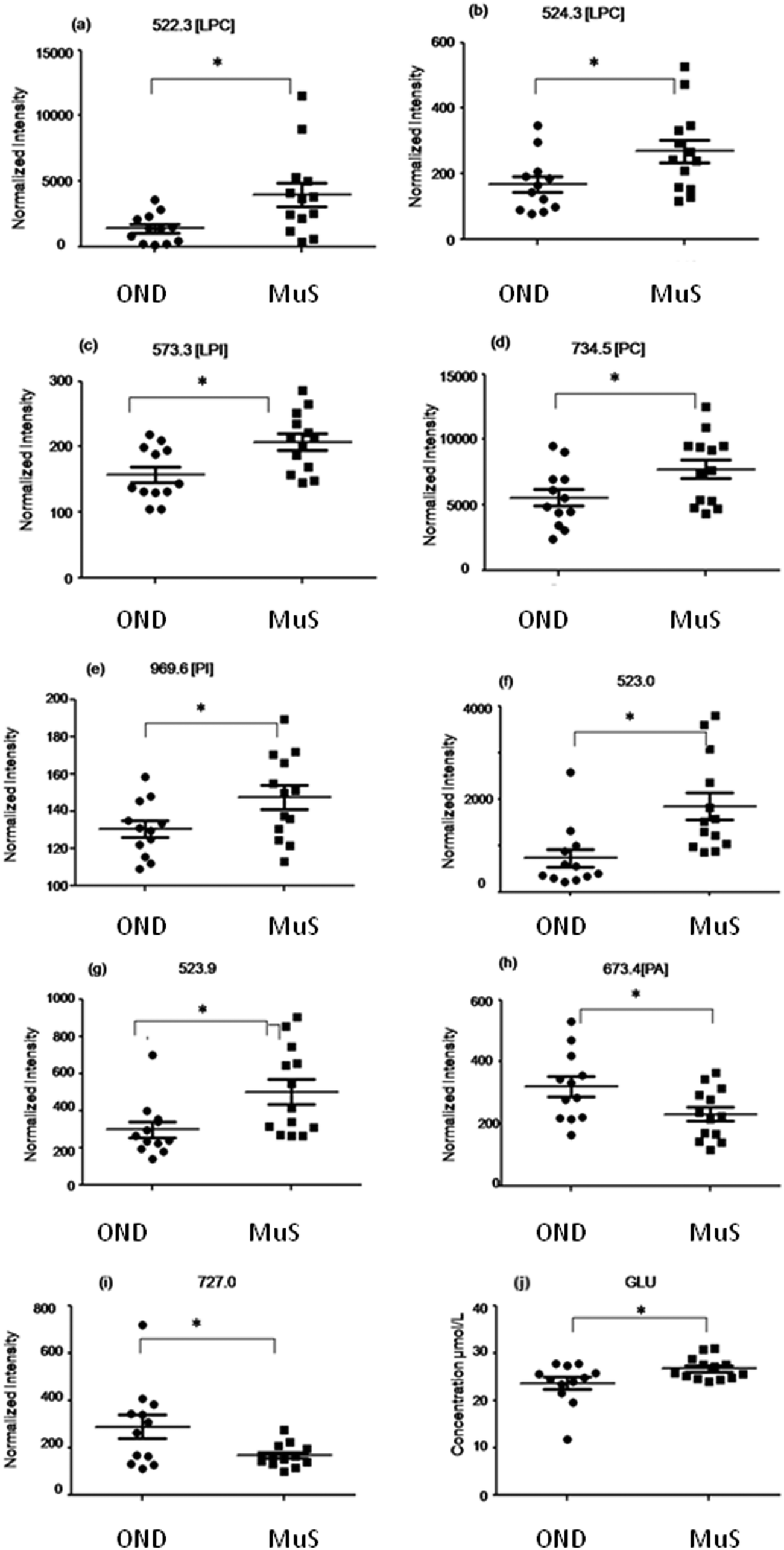 | ||
| Fig. 3 Distribution of each discriminant metabolite in two clinical groups studied. The compounds reported in panels (a–g) are significantly increased in MuS patients. The signals at m/z = 673.4 (h) and m/z = 727.0 (i) decreased in MuS patients vs. OND subjects. The level of glutamate (j) in CSF of MuS patients significantly elevated vs. OND patients. * indicates univariate test with a p-value < 0.05. | ||
| Biomarker | Cutoff (relative intensity) | Specificitya (%) | Sensitivitya (%) | AUC | p-value |
|---|---|---|---|---|---|
| a Sensitivity and specificity values were chosen (along with their 95% confidence interval) by selecting a possible cutoff between MuS and non-MuS. The cutoff was selected considering the better compromise of sensitivity and specificity with a major likelihood ratio returned by the software (Graphpad Prism). The likelihood ratio equals sensitivity/(1.0-specificity). | |||||
| LPC(18:1(9Z)/0:0) | >2391 | 83.33 | 69.23 | 0.80 | 0.010 |
| Unidentified (m/z = 523.040) | >1007 | 83.33 | 76.92 | 0.87 | 0.001 |
| Unidentified (m/z = 523.993) | >404.8 | 91.67 | 53.85 | 0.79 | 0.012 |
| LPC(18:0/0:0) | >208.4 | 83.33 | 69.23 | 0.76 | 0.029 |
| LPI(16:0/0:0) | >199.7 | 83.33 | 61.54 | 0.81 | 0.007 |
| PA | <256.2 | 66.67 | 61.54 | 0.72 | 0.064 |
| Unidentified (m/z = 727.021) | <242.7 | 58.33 | 92.31 | 0.72 | 0.057 |
| PC | >1441 | 83.33 | 61.54 | 0.74 | 0.044 |
| PI | >135.4 | 75.00 | 69.23 | 0.73 | 0.050 |
| Glutamate | >25.72 | 75.00 | 53.85 | 0.71 | 0.081 |
Correlation with clinical parameters
To study the potential of the detected metabolites to identify ongoing disease activity we evaluated the relationship of the signal intensity values of the discriminatory compounds with clinical parameters shown in Table S1 of the ESI.† The intensity of two LPC species at m/z = 522.3 Da, 524.3 Da and an unidentified signal at m/z = 523.0 Da (elevated in MuS patients) correlated positively with the Link index as shown in Fig. 4(a–c). The intensity of the signal at m/z = 969.6 Da (elevated in MuS) identified as PI, correlated positively with disease duration (Fig. 4d), while the lyso isoform (LPI) at m/z = 573.3 Da (elevated in MuS) showed a negative correlation with the EDSS (Fig. 4e). Other variables studied did not show significant correlations with the clinical parameters considered.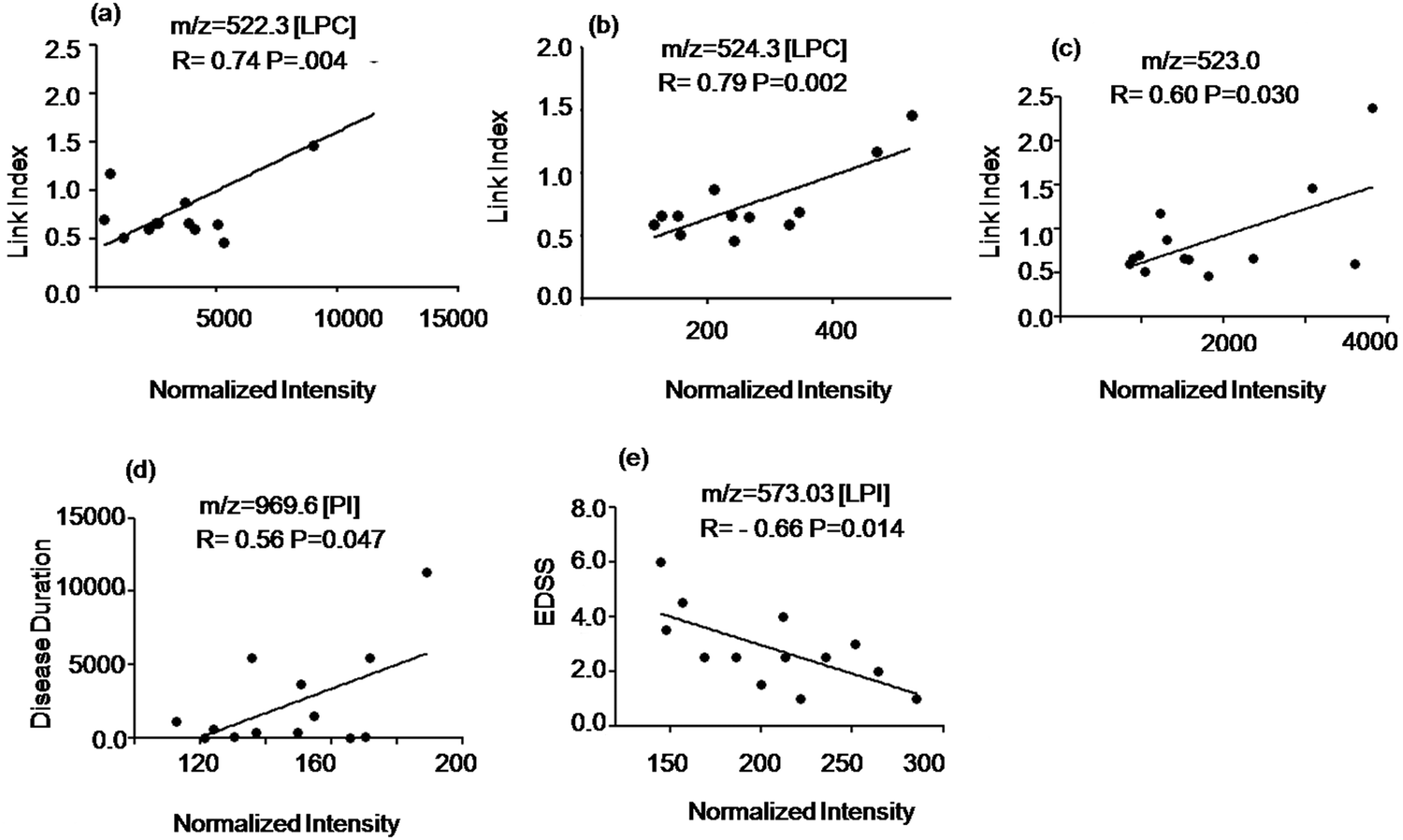 | ||
| Fig. 4 Correlation of Link Index, disease duration and EDSS with specific peak intensities in CSF. (a–c) Correlation of m/z at 522.3, 524.3 and 523.0 with Link Index. (d) Correlation of the signal at m/z = 969.6 with disease duration. (e) Correlation of the signal at m/z = 573.3 with EDSS values. Spearman correlation coefficients (r) are indicated in the figure. | ||
Discussion
In this study we reported a targeted LC-MS/MS of carnitines and amino acids and MALDI-TOF-MS untargeted lipidomics profiling in order to improve knowledge of metabolic pathways modulation in the CSF of MuS patients. Therefore we looked at a possible correlation of the metabolomics data with clinical parameters to study their relationship with disease pathophysiology. By targeted analysis we studied 36 acyl carnitines and 19 amino acids species, while by untargeted analysis we obtained hundreds of peaks associated to lipid species. The choice of high throughput screening by MALDI-TOF-MS technology was driven to obtain high sensitivity to analyze CSF lipids and to be able to carry out identification of unknown compounds, as well as to apply a robust methodology easy to transfer into clinical practice. Our main objective is to identify new candidate biomarkers for MuS diagnosis, in order to better understand the possible link between metabolic alterations and clinical features of the disease. The potential biomarkers, in the lipid screening, were tentatively identified by accurate mass and, when possible, by fragmentation experiments. Lipids containing phosphocholines were assigned in a better fashion due to their major susceptibility to ionization of the quaternary nitrogen in the head group, giving a typical m/z at the fragment (m/z = 184 Da). Ten metabolites that significantly (p < 0.05) segregate the two clinical groups analyzed were obtained. The most relevant result of this approach was the detection of an altered level of specific phospholipids in MuS when compared to OND subjects. In particular we found a significantly increased level of LPC (18:1/0:0), LPC (18:0/0:0) and LPI (16:0/0:0) in MuS patients and other m/z signals (523.9 Da and 523.04 Da) unidentified but probably associated to similar compounds in CSF. Moreover we identified the signal at m/z = 734.5 Da as a PC species that was observed to be higher in MuS than OND patients. LPC (18:1/0:0), LPC (18:0/0:0) and the unidentified lipid at m/z = 523.04 Da are well correlated (p < 0.05) to the Link Index (also known as “IgG Index”) a parameter that indicates high levels of intrathecal IgG synthesis. The intrathecal IgG synthesis is a common event in MuS. Interestingly, our previously published data demonstrated a significant decrease of this specific LPC species (LPC-18:1/0:0 and LPC-18:0/0:0) in serum of MuS patients with respect to healthy control subjects,24 while similar levels were found between MuS and OND. Here we obtained overexpression of these metabolites in CSF of MuS, a cerebral compartment physiologically independent from the serum, suggestive for a possible function of these lipids as candidate biomarkers, reflecting intrathecal synthesis and CNS inflammation.25We considered a single OND control group even if disease parameters are quite variables in the considered clinical cohort. However, aware of the limitation of such choice, we believe our findings are strengthened from the heterogeneity of the OND group highlighting a specific trend in MuS. Moreover, we investigated the lipid composition between gender in the MuS and OND groups, showing no significant differences between males and females, demonstrating that there is no gender influence on the lipid profile in CSF (Fig. S5, ESI†). Unfortunately, we do not have information about the body mass index of patients and this can be considered a biasing factor, even if the CSF should not be strongly influenced by this index like the serum. However, we attempted to insert a homogeneous group of patients.
PC is the major phospholipid species of eukaryotic membranes and removal of one of the fatty acids results in LPC usually through the enzymatic action of a phospholipase A2 (PLA2). Several studies suggested altered levels of PCs in neurodegenerative diseases concluding that secretory PLA2 activity in the CSF might serve as a valuable biomarker of neuroinflammation as demonstrated in Alzheimer’s disease.26,27 In EAE, the blockade of cytosolic PLA(2)α was highly efficacious in ameliorating the disease course probably reducing T cell proliferation, proinflammatory cytokine production, preventing activation of CNS-resident microglia and increasing oligodendrocyte survival. The authors, administrating a cPLA(2)α inhibitor in a relapsing-remitting model of EAE, completely protected mice from subsequent relapses.28 The therapeutic effect of Fingolimod is probably also due to inhibition of cPLA(2)α activity, as previously demonstrated, directly in CNS.29 Here we can speculate that the pathological overstimulation of PLA2 determines cutting of PC from the membranes, resulting in accumulation of LPC species into the damaged tissue, confirmed by high levels of circulating LPCs in CSF of MuS patients.
Moreover LPC is recognized as an important factor underlying signal transduction and plays a functional role in various diseases by LPC specific G-protein-coupled receptors.30 LPC is released into the brain under pathological conditions linked to high levels of pro-inflammatory cytokines Interleukin 1b (IL-1b).31 LysoPC released from apoptotic cells could also act as a chemotactic factor for monocytic cells and primary macrophages.32 It was demonstrated both in vivo and in vitro that LPC induces deramification of murine microglia. In particular LPC (16:0) and (18:0) are able to induce IL-1b release, an important pro-inflammatory cytokine, from microglial cells through activation of the P2X7 receptor.33 This is consistent with the timing of CSF withdrawal in MuS patients during a relapse. Another interesting result of our study was the identification of LPI (16:0/0:0) species and high molecular weight PI that are elevated in MuS subjects vs. OND patients, even if in this case the identification was obtained without fragmentation data due to the low intensity of these signals in the spectra. The levels of LPI in MuS negatively correlated to the EDSS score indicating that an increased level of this metabolite in CSF may be associated to a protective role against progression and severity of symptoms of the disease. On the other hand we found a positive correlation of PI levels in CSF with disease duration; this result may also reflect a role of the metabolite in broadening of neurodegeneration due to inflammation.34 These data may seem contradictory, since the EDSS value, usually, is proportional to disease duration, but they might reflect different roles of these metabolites. However, it is arduous to discuss a possible involvement of LPI and its high expression in the CSF of MuS patients considering that our preliminary evidence pertains to a limited group of patients. However we may speculate that the role of such metabolite may be compared with other LPIs for biological activities. Consistently with this observation there is fascinating new evidence indicating the involvement of LPIs in neuroprotection, mediating modulation of microglia function through G protein-coupled receptor 55 (GPR55).35 Actually, the physiological roles of GPR55 and its possible involvement in the pathophysiology are emerging. Recent studies established LPI as an activator of GPR55 and its implication in pain transmission, where cannabinoids are potent inhibitors of such machine.36 In summary, LPI is developing as a key modulator of cell proliferation, migration, and function, and holds important pathophysiological implications due to its high levels in diseased tissues.37
Regarding all aminoacids and acyl-carnitines quantified in CSF, glutamate levels seem to be slightly increased in MuS during the acute phase of inflammation, although it is known that glutamate is increased also in other neurological diseases, thus it cannot be considered a specific marker of MuS. However, this result is in agreement with recent findings that demonstrated the role of glutamate, the first excitatory neurotransmitter of CNS, in MuS and EAE.38,39 It was already reported that glutamate levels were elevated in CSF of MuS patients and these levels correlated to disease severity.40 Interestingly, this result was in agreement with high levels of LPC in CSF considering that glutamate release, calcium influx, and activation of cellular PLA2 were reported as important steps initiating membrane breakdown.41 These considerations could indicate a potential use of glutamate as a biomarker of MuS severity even in patients without lesions in NAWM. Moreover according to Tejani et al. carnitine levels do not seem to be influenced by the disease.15 In conclusion, even though OND patients show metabolic patterns similar to MuS subjects, some of these features are distinctive and can be considered specific for MuS. In Table 3 the discriminatory power of each potential biomarker is summarized. Eight compounds, taken singularly, show significant reclassification and, most of them showed high specificity in classifying the MuS group. Even if the discriminatory power of each compound is not excellent, taken together, the pattern can represent a specific cerebral metabolic alteration in MuS disease. The limit of this study is the lack of external validation of the results, consisting in a computing prediction for an independent set of test observations. However, a confirmation of these preliminary results in a more wider clinical study could lead to a better understanding of the metabolic (dys)homeostasis in the pathogenesis of MuS, providing the opportunity for new functional biomarkers and new promising targets for therapeutic interventions.
Acknowledgements
This study was supported by the Italian Ministry of Health, Grant number: GR-2010-2307655. Maria di Ioia is supported by a FISM-Fondazione Italiana Sclerosi Multipla-training fellowship Cod. 2012/B/2.References
- H. Tremlett, Y. Zhao, P. Rieckmann and M. Hutchinson, Neurology, 2010, 74, 2004–2015 CrossRef PubMed.
- F. D. Lublin and S. C. Reingold, Neurology, 1996, 46, 907–911 CrossRef CAS.
- R. Milo and E. Kahana, Autoimmun. Rev., 2010, 9, A387–A394 CrossRef CAS PubMed.
- M. Sospedra and R. Martin, Annu. Rev. Immunol., 2005, 23, 683–747 CrossRef CAS PubMed.
- R. Hohlfeld and H. Wekerle, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(suppl 2), 14599–14606 CrossRef CAS PubMed.
- D. Wheeler, V. V. Bandaru, P. A. Calabresi, A. Nath and N. J. Haughey, Brain, 2008, 131, 3092–3102 CrossRef PubMed.
- F. J. Quintana, M. F. Farez, V. Viglietta, A. H. Iglesias, Y. Merbl, G. Izquierdo, M. Lucas, A. S. Basso, S. J. Khoury, C. F. Lucchinetti, I. R. Cohen and H. L. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18889–18894 CrossRef CAS PubMed.
- H. Gonzalo, L. Brieva, F. Tatzber, M. Jove, D. Cacabelos, A. Cassanye, L. Lanau-Angulo, J. Boada, J. C. Serrano, C. Gonzalez, L. Hernandez, S. Peralta, R. Pamplona and M. Portero-Otin, J. Neurochem., 2012, 123, 622–634 CrossRef CAS PubMed.
- J. A. Cohen and J. Chun, Ann. Neurol., 2011, 69, 759–777 CrossRef CAS PubMed.
- C. L. Jensen, Am. J. Clin. Nutr., 2006, 83, 1452S–1457S CAS.
- A. Tsui and M. A. Lee, Curr. Opin. Obstet. Gynecol., 2011, 23, 435–439 CrossRef PubMed.
- G. Famularo, C. De Simone, V. Trinchieri and L. Mosca, Ann. N. Y. Acad. Sci., 2004, 1033, 132–138 CrossRef CAS PubMed.
- C. Lebrun, H. Alchaar, M. Candito, V. Bourg and M. Chatel, Mult. Scler., 2006, 12, 321–324 CrossRef CAS.
- V. Calabrese, G. Scapagnini, A. Ravagna, R. Bella, D. A. Butterfield, M. Calvani, G. Pennisi and A. M. Giuffrida Stella, Neurochem. Res., 2003, 28, 1321–1328 CrossRef CAS.
- A. M. Tejani, M. Wasdell, R. Spiwak, G. Rowell and S. Nathwani, Cochrane Database Syst. Rev., 2012, 5, CD007280 Search PubMed.
- M. J. Noga, A. Dane, S. Shi, A. Attali, H. van Aken, E. Suidgeest, T. Tuinstra, B. Muilwijk, L. Coulier, T. Luider, T. H. Reijmers, R. J. Vreeken and T. Hankemeier, Metabolomics, 2012, 8, 253–263 CrossRef CAS PubMed.
- C. H. Polman, S. C. Reingold, B. Banwell, M. Clanet, J. A. Cohen, M. Filippi, K. Fujihara, E. Havrdova, M. Hutchinson, L. Kappos, F. D. Lublin, X. Montalban, P. O'Connor, M. Sandberg-Wollheim, A. J. Thompson, E. Waubant, B. Weinshenker and J. S. Wolinsky, Ann. Neurol., 2011, 69, 292–302 CrossRef PubMed.
- E. G. Bligh and W. J. Dyer, Can. J. Biochem. Physiol., 1959, 37, 911–917 CrossRef CAS.
- T. Fujiwaki, M. Tasaka, N. Takahashi, H. Kobayashi, Y. Murakami, T. Shimada and S. Yamaguchi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 832, 97–102 CrossRef CAS PubMed.
- V. Sirolli, C. Rossi, A. Di Castelnuovo, P. Felaco, L. Amoroso, M. Zucchelli, D. Ciavardelli, C. Di Ilio, P. Sacchetta, S. Bernardini, A. Arduini, M. Bonomini and A. Urbani, Blood Transfus., 2012, 10 Suppl 2, s78–s88, DOI:10.2450/2012.012S.
- L. Di Liberato, A. Arduini, C. Rossi, A. Di Castelnuovo, C. Posari, P. Sacchetta, A. Urbani and M. Bonomini, J. Nephrol., 2014, 27, 699–706 CrossRef PubMed.
- S. Rizza, M. Copetti, C. Rossi, M. A. Cianfarani, M. Zucchelli, A. Luzi, C. Pecchioli, O. Porzio, G. Di Cola, A. Urbani, F. Pellegrini and M. Federici, Atherosclerosis, 2014, 232, 260–264 CrossRef CAS PubMed.
- C. Lohmann, E. Schachmann, T. Dandekar, C. Villmann and C. M. Becker, J. Neurochem., 2010, 114, 1119–1134 Search PubMed.
- P. Del Boccio, D. Pieragostino, M. Di Ioia, F. Petrucci, A. Lugaresi, G. De Luca, D. Gambi, M. Onofrj, C. Di Ilio, P. Sacchetta and A. Urbani, J. Proteomics, 2011, 74, 2826–2836 CrossRef CAS PubMed.
- K. M. Brennan, F. Galban-Horcajo, S. Rinaldi, C. P. O'Leary, C. S. Goodyear, G. Kalna, A. Arthur, C. Elliot, S. Barnett, C. Linington, J. L. Bennett, G. P. Owens and H. J. Willison, J. Neuroimmunol., 2011, 238, 87–95 CrossRef CAS PubMed.
- S. Chalbot, H. Zetterberg, K. Blennow, T. Fladby, I. Grundke-Iqbal and K. Iqbal, Clin. Chem., 2009, 55, 2171–2179 CAS.
- A. Walter, U. Korth, M. Hilgert, J. Hartmann, O. Weichel, M. Hilgert, K. Fassbender, A. Schmitt and J. Klein, Neurobiol. Aging, 2004, 25, 1299–1303 CrossRef CAS PubMed.
- P. Thakker, S. Marusic, N. L. Stedman, K. L. Lee, J. C. McKew, A. Wood, S. J. Goldman, M. W. Leach, M. Collins, V. K. Kuchroo, S. F. Wolf, J. D. Clark and M. Hassan-Zahraee, J. Immunol., 2011, 187, 1986–1997 CrossRef CAS PubMed.
- S. G. Payne, C. A. Oskeritzian, R. Griffiths, P. Subramanian, S. E. Barbour, C. E. Chalfant, S. Milstien and S. Spiegel, Blood, 2007, 109, 1077–1085 CrossRef CAS PubMed.
- Y. Asaoka, M. Oka, K. Yoshida, Y. Sasaki and Y. Nishizuka, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 6447–6451 CrossRef CAS.
- J. H. Kabarowski, Y. Xu and O. N. Witte, Biochem. Pharmacol., 2002, 64, 161–167 CrossRef CAS.
- K. Lauber, E. Bohn, S. M. Krober, Y. J. Xiao, S. G. Blumenthal, R. K. Lindemann, P. Marini, C. Wiedig, A. Zobywalski, S. Baksh, Y. Xu, I. B. Autenrieth, K. Schulze-Osthoff, C. Belka, G. Stuhler and S. Wesselborg, Cell, 2003, 113, 717–730 CrossRef CAS.
- T. Schilling, F. Lehmann, B. Ruckert and C. Eder, J. Physiol., 2004, 557, 105–120 CrossRef CAS PubMed.
- J. M. Frischer, S. Bramow, A. Dal-Bianco, C. F. Lucchinetti, H. Rauschka, M. Schmidbauer, H. Laursen, P. S. Sorensen and H. Lassmann, Brain, 2009, 132, 1175–1189 CrossRef PubMed.
- S. Kallendrusch, S. Kremzow, M. Nowicki, U. Grabiec, R. Winkelmann, A. Benz, R. Kraft, I. Bechmann, F. Dehghani and M. Koch, Glia, 2013, 61, 1822–1831 Search PubMed.
- S. Anavi-Goffer, G. Baillie, A. J. Irving, J. Gertsch, I. R. Greig, R. G. Pertwee and R. A. Ross, J. Biol. Chem., 2012, 287, 91–104 CrossRef CAS PubMed.
- V. Gangadharan, D. Selvaraj, M. Kurejova, C. Njoo, S. Gritsch, D. Skoricova, H. Horstmann, S. Offermanns, A. J. Brown, T. Kuner, A. Tappe-Theodor and R. Kuner, Pain, 2013, 154, 2801–2812 CrossRef CAS PubMed.
- C. Bolton and C. Paul, Mediators Inflammation, 2006, 2006, 93684 Search PubMed.
- G. Mandolesi, A. Musella, A. Gentile, G. Grasselli, N. Haji, H. Sepman, D. Fresegna, S. Bullitta, F. De Vito, G. Musumeci, C. Di Sanza, P. Strata and D. Centonze, J. Neurosci., 2013, 33, 12105–12121 CrossRef CAS PubMed.
- A. Tisell, O. D. Leinhard, J. B. Warntjes, A. Aalto, O. Smedby, A. M. Landtblom and P. Lundberg, PLoS One, 2013, 8, e61817 CAS.
- J. Klein, J. Neural Transm., 2000, 107, 1027–1063 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mb00700j |
| This journal is © The Royal Society of Chemistry 2015 |