
Alpha-synuclein oligomers and fibrils originate in two distinct conformer pools: a small angle X-ray scattering and ensemble optimisation modelling study†
Cyril C.
Curtain
*ab,
Nigel M.
Kirby
c,
Haydyn D. T.
Mertens‡
c,
Kevin J.
Barnham
ab,
Robert B.
Knott
d,
Colin L.
Masters
b,
Roberto
Cappai
a,
Agata
Rekas
d,
Vijaya B.
Kenche
b and
Timothy
Ryan
b
aDepartment of Pathology and Bio21 Molecular Science and Technology Institute, The University of Melbourne, Victoria 3010, Australia. E-mail: ccurtain@unimelb.edu.au
bThe University of Melbourne, Florey Institute of Neuroscience and Mental Health, Victoria 3010, Australia
cSAXS/WAXS Beamline, The Australian Synchrotron, Clayton, Victoria 3168, Australia
dAustralian Nuclear Science and Technology Organisation (ANSTO), Kirrawee, NSW 2232, Australia
First published on 21st October 2014
Abstract
The 140 residue intrinsically disordered protein α-synuclein (α-syn) self-associates to form fibrils that are the major constituent of the Lewy body intracellular protein inclusions, and neurotoxic oligomers. Both of these macromolecular structures are associated with a number of neurodegenerative diseases, including Parkinson's disease and dementia with Lewy bodies. Using ensemble optimisation modelling (EOM) and small angle X-ray scattering (SAXS) on a size-exclusion column equipped beamline, we studied how the distribution of structural conformers in α-syn may be influenced by the presence of the familial early-onset mutations A30P, E45K and A53T, by substituting the four methionine residues with alanines and by reaction with copper (Cu2+) or an anti-fibril organic platinum (Pt) complex. We found that the WT had two major conformer groups, representing ensembles of compact and extended structures. The population of the extended group was increased in the more rapidly fibril-forming E45K and A53T mutants, while the compact group was enlarged in the oligomer-forming A30P mutant. Addition of Cu2+ resulted in the formation of an ensemble of compact conformers, while the anti-fibril agent and alanine substitution substantially reduced the population of extended conformers. Since our observations with the mutants suggest that fibrils may be drawn from the extended conformer ensemble, we propose that the compact and extended ensembles represent the beginning of oligomer and fibril formation pathways respectively, both of which have been reported to lead to a toxic gain of function. Manipulating these pathways and monitoring the results by EOM and SAXS may be useful in the development of anti-Parkinson's disease therapies.
1. Introduction
Many of the intrinsically disordered proteins (IDPs) that occupy a large fraction of every proteome have essential biological functions. Their structural plasticity may enable IDPs to perform different functions in different environments. Hence, they have been referred to as “molecular chameleons”.1 The disadvantage of this flexibility is that they may also form cross β-spine-structures, creating amyloids and/or cytotoxic oligomers that occur in a number of diseases. One such IDP is α-synuclein (α-syn), whose Lewy body aggregates in the brain are one of the hallmarks of Parkinson's disease (PD).2 PD is a progressive, incurable, complex and disabling neurological condition affecting approximately 1% of the population, mostly over 60 years of age. As is the case with all IDPs, the pathological conformers of α-syn are a difficult drug target because of a lack of defined stable structure that can be solved by X-ray crystallography or NMR. One approach is to understand the environmental conditions that favour formation of toxic oligomers and/or fibrils. A starting point is that IDP structures may not be truly random because side-chain steric interference and long range interactions may frustrate the development of some conformers, while selection of the initial structures from this apparently random pool is poorly understood. Conformational heterogeneity of α-syn has been shown by atomic force microscopy3–5 and electrospray ionisation spectrometry.6 NMR and spin label studies have shown the existence of long-range intra-molecular interactions between the C-terminal tail (residues 120–140) and the central part of α-syn (residues 30–100) and molecular dynamics simulations showed that the native WT is more compact than a random coil.7 Ullman et al.8 used a Bayesian approach combining data from NMR chemical shifts, residual dipolar couplings and small angle X-ray scattering (SAXS) on the monomeric protein with molecular simulations to explain how it can assume different conformations. The existence of a proportion of relatively compact forms is reflected in the findings from SAXS studies that the radius of gyration (Rg) of native α-syn (approx. 40 Å) is larger than the value of a folded globular protein of similar chain length (15 Å), but smaller than that of a fully unfolded random coil with a similar molecular mass (52 Å).9,10 Further, Uversky et al.11,12 pointed out that the uneven distribution of charges along the sequence could contribute to the long range-interactions. There are 8 negative charges in the sequence 120–140 of C-terminus while the central region sequence 30–100 has a net positive charge of 3. In an advance on the charge distribution concept Das and Pappu13 employed an implicit solvation model and underlying force-field paradigm that uses experimentally measured free energies of solvation for model compounds as inputs, coupled with refined parameters for modelling van der Waals interactions based on data for heats of fusion. Their approach allowed IDPs such as α-syn whose sequences included positively and negatively charged residues to be classified into globular conformers with well mixed sequences and those with segregated charges a preference for hairpin conformations arising from long range attractions induced by conformational fluctuations.We have shown previously that SAXS and ensemble optimisation modelling (EOM) of native α-syn can be used to study the distribution of conformers based on their Rg and Pr distribution.14 Oxidation of the protein's four methionine residues markedly reduces the proportion of the most extended members of the ensemble of conformers. Since the native protein readily aggregates to form the fibrils associated with Parkinson's disease Lewy bodies, and the oxidised version is non-fibrillogenic,12 it is plausible that the extended conformer is a key stage on the fibril formation pathway. In this communication we present data from a size exclusion column (SEC) SAXS-EOM study of conformational changes in familial early PD onset mutants of α-syn and the influence of Cu2+ and an inhibitor of fibril formation on conformer distribution.
2. Experimental
2.1 Materials
Wild-type (WT) α-syn and early onset PD familial mutants, A30P, E46K, A53T, were over-expressed in Escherichia coli BL21 (DE3) that had been transformed with pRSETB plasmids containing individual cDNA sequences. The sequence of α-syn with its 4 methionine residues substituted with alanine (M1A/M5A/M116A/M127A), herein referred to as 4M4A, was cloned into pRK172 plasmid and over-expressed in the same way. The proteins were isolated and purified as described previously.14,15 The purity of the protein was determined by SDS-PAGE. The protein concentration was determined using the 280 nm extinction coefficient of 5120 M−1 cm−1. Each protein was dissolved to a concentration of 10 mg mL−1 in 6 M guanidine hydrochloride (GuHCl) before the SAXS experiments.Copper/glycine (CuGly) 10 mM was prepared as described previously16 and the synthesis and characterisation of the anti-fibril agent ([PtII(N,N-dimethyl-2-[2-(quinolin-8-yl)-1H-benzimidazol-1-yl]ethanamine) Cl2] referred to as VK7) has also been described previously.17 All buffers and other reagents were of analytical grade or the highest possible purity.
2.2 Methods
SAXS measurements were made at 27 °C on the SAXS/WAXS beam line of the Australian Synchrotron (Clayton, Victoria, Australia).18 Using the high-intensity undulator source on this instrument, an energy resolution of 10−4 was obtained from a cryo-cooled Si(111) double-crystal monochromator, and the beam size (fwhm focused at the sample) was 250 × 100 μm with a total photon current of about 2 × 1012 photons per s. Scattering patterns were recorded with an X-ray energy of 12 keV using a Pilatus 1 M detector with an active area of 170 × 170 mm and a sample to detector distance of 3300 mm. This corresponded to a q range from 0.005 to 0.25 Å−1 (where q = 4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ/λ, 2θ is the X-ray scattering angle and λ the X-ray wavelength).
sinθ/λ, 2θ is the X-ray scattering angle and λ the X-ray wavelength).
The protein samples were subjected to in-line size-exclusion chromatography (SEC) using an automated HPLC apparatus (Shimadzu) fitted with a 10 mm by 300 mm Superdex 75 size exclusion column (GE Healthcare) with a bed volume of 22 mL equilibrated with 10 mM pH 7.4 phosphate buffer at a flow rate of 0.6 mL min−1. 100 μL of each sample at 10 mg mL−1 was injected from a 96 well plate auto-changer and the column's eluent was directed through a UV protein absorption detector set at 280 nm with a 10 mm path length to a 1.5 mm quartz capillary where it was exposed to the X-ray beam. At least 700 detector images of sequential 2 s exposures (2.1 s repeat time) were collected, corresponding to a total elution volume of 14.5 mL. When there was no further signal post the elution of the main α-syn absorbance peak, SAXS data collection was halted. After each sample the column and capillary were flushed with 100 μL of 6 M guanidine hydrochloride solution to remove any aggregated protein and the column was allowed to re-equilibrate in the phosphate buffer for a further 45 mL. Absolute intensities were calibrated using water in the 1.5 mm ID quartz capillary.
A small amount of aggregation, virtually undetectable by its absorption at 280 nm, occurred in the SEC column causing an increase in scattering that impacted on the quality of the scattering profiles represented by the leading edge of the SEC protein peak. This effect is illustrated in the Fig. S1 (ESI†). The aggregate formation could be triggered by contact with the column packing or could represent the beginnings of the aggregate nucleation that also seems to be a characteristic of other amyloid forming IDP's.19 We detected the influence of aggregates by checking the ratio of the value of the forward scattering intensity I0 against the protein concentration (c) determined from the absorbance at 280 nm acquired from the UV detector on the SEC using the absorption coefficient of 5120 M−1 cm−1 (Table 1). This was linear over a range of concentrations for all samples (Fig. S2, ESI†). We also estimated the linearity of the Guinier plot, which would be sensitive to the presence of oligomers and aggregates. However, this linearity only holds over a restricted region of the scattering profile where qRg < 1.0, or 1.3 for well-folded proteins. For IDPs with their multiple sizes, this region can be restricted to qRg < 0.8 or less,20 limiting the accuracy of the measurement because of the small number of useable points in the SAXS profile. We adopted qRg = 0.8 as a cut-off for deciding whether to include a given profile in the number to be averaged for each sample.
| Sample | Low range peak (%) | High range peak (%) | ½ height peak width low range Rg (Å) | ½ height peak width high range Rg (Å) | Low range max cut-off Rg (Å) | High range min cut-off Rg (Å) | High range max cut-off Rg (Å) |
|---|---|---|---|---|---|---|---|
| WT | 60 | 40 | 8 | 8 | 42 | 49 | 69 |
| A30P | 100 | 0 | 5 | — | 38 | — | — |
| E46K | 55 | 45 | 7 | 6 | 36 | 44 | 60 |
| A53T | 40 | 60 | 8 | 8 | 36 | 46 | 60 |
| WT + Cu2+ | 90 | 10 | 8 | — | 36 | — | — |
| A30P + Cu2+ | 100 | 0 | 9 | — | 37 | — | — |
| A53T + Cu2+ | 80 | 20 | 10 | 20 | 40 | 25 | 60 |
| 4M4A | 70 | 30 | 10 | 8 | 44 | 42 | 61 |
| 4M4A + Cu2+ | 100 | 0 | 6 | — | 33 | — | — |
| WT + VK7 | 70 | 30 | 9 | 13 | 37 | 35 | 62 |
All two-dimensional scattering patterns obtained were reduced to one-dimensional profiles of intensity I versus q using the ScatterBrain software package on line. The 280 nm absorbance was also displayed on a spread-sheet in real time so that the protein peak could be matched to the scattering profiles. Radius of gyration (Rg) calculation, Kratky and Guinier analyses were made using the ATSAS suite of programs,21–25 2.5.0–2 release available from http://www.embl-Hamburg.de/ExternalInfo/Research/Sax/index.html. The Kratky analysis was used to confirm that all of the scattering profiles used indicated essentially unstructured protein. The MM of each sample was determined from I0 calculated on the absolute scale by employing the method of Orthaber et al.,26 where the relative I0 of the protein is divided by the experimental scattering of water and then multiplying by the absolute scattering of water. The MM = [NAI0/c]/ΔρM2 where NA is Avogadro's number, I0/c is the absolute scale forward scattering normalised by concentration and ΔρM2 is the scattering contrast per mass.
EOM (Advanced EOM 2.0 (ref. 27 and 28) available in the ATSAS suite) was used for the analysis of the flexibility and size distribution of possible structures of the conformers of the protein contributing to the experimental scattering pattern. Both “Native” and “random” options were used for each run. The outputs were very similar and the “random” option data are presented here. This approach is based on the generation of a large pool (typically 10000) of theoretical structures derived with side-chain interaction constraints from the primary sequence of the protein. The theoretical X-ray scattering profiles calculated from these structures are then matched for fit using a genetic algorithm against the experimental scattering profile to create an ensemble of best fit structures. The structure-related outputs of the EOM analysis are the Rg and Dmax (the maximum distance between two Cα atoms within a conformer). A wide distribution of Rg and Dmax indicates a random distribution of structures that could fit the experimental SAXS profile, and a multi-modal distribution indicates the existence of a number of dominant conformers.29,30
3. Results
3.1 The Rg and Dmax distributions of WT and the pathological mutants
The Rg distributions of the ensembles determined by EOM are given in Fig. 1 for the WT (A) and the familial mutants A30P (B), E46K (C) and A53T (D). The WT, E46K and A53T mutants gave bimodal distributions. Deconvolution by fitting Gaussian distributions to the data, showed that a Rg range of 36–42 Å covered maximum cut-offs of the compact distribution and 44–49 Å for the extended forms (Table 1). The deconvolutions are shown in Fig. S3 (ESI†). The proportion of higher Rg's from integrating the peaks was 40% for the WT, 60% for the A53T and 45% for the E46K (Table 1). On the other hand, the Rg distribution of the A30P mutant was restricted to lower values. Its peak width at half-height of 5 Å and the cut-off at 38 Å was slightly narrower than with the WT and the E46K and A53T mutants (Table 1).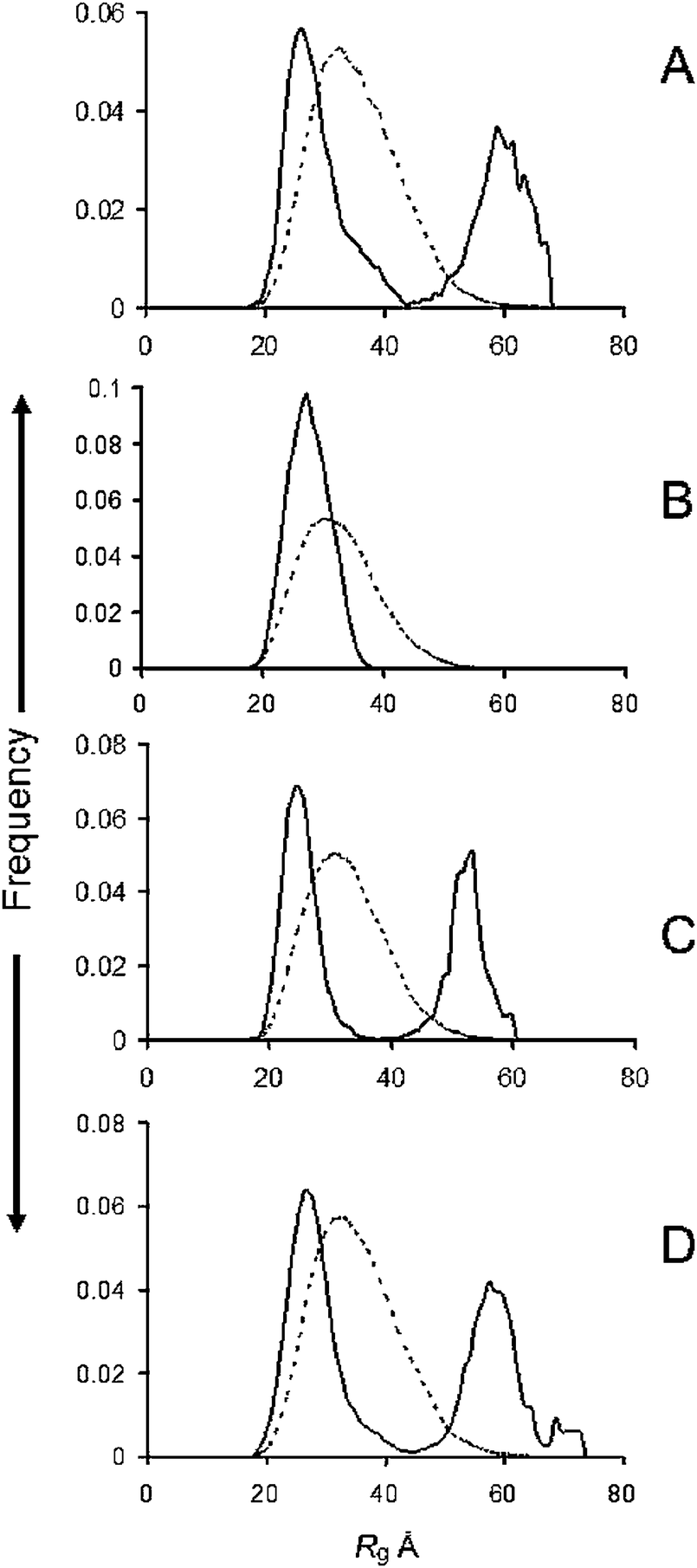 | ||
| Fig. 1 R g distributions from ensemble optimisation modelling of (A) WT, (B) A30P; (C) E46K; (D) A53T familial mutants of α-syn. Solid lines, ensemble; dotted lines, pool. | ||
The fitted SAXS profiles, Dmax distribution, SEC Abs280, Kratky and Guinier plots are available in the Fig. S4 (ESI†). The Dmax distributions closely followed those of the Rg's and SEC analysis showed that the samples were monodisperse. The values of average Rg, average ensemble Dmax, and χ for each profile fit are available in Table S1 (ESI†). All the profiles gave a reasonably good fit with an even distribution of errors. The average Rg value for the WT ensemble of 44.34 Å is close to those reported previously9,10 and gives some comfort as to the reproducibility of the scattering data.
The Rg values calculated from a Guinier plot of the experimental scattering profiles, the absolute scale I0, the protein concentration determined from the 280 nm absorbance and the derived molecular masses (MM) are given in Table 2. The latter values are within the accepted 10% error.26 The I0 and corresponding concentrations are averages of 5 readings taken around the UV absorption maximum, although more values were taken from each side of it to confirm that I0/c remained reasonably constant. These are shown as scatter plots in Fig. S2 (ESI†).
| Sample | R g (Å) | I 0 (corrected26) | c (mM) | I 0/c | MM (kD) |
|---|---|---|---|---|---|
| WT | 42.64 ± 0.97 | 137.84 | 0.2 | 685 | 15.8 |
| A30P | 43.64 ± 0.91 | 125.46 | 0.18 | 689 | 16.0 |
| E46K | 27.57 ± 0.40 | 130.57 | 0.19 | 687 | 15.9 |
| A53T | 27.23 ± 0.86 | 148.24 | 0.22 | 674 | 15.8 |
| WT + Cu2+ | 26.49 ± 0.61 | 132.48 | 0.23 | 669 | 15.6 |
| A30P + Cu2+ | 31.37 ± 0.35 | 134.44 | 0.20 | 673 | 15.7 |
| A53T + Cu2+ | 31.87 ± 0.22 | 172.64 | 0.26 | 664 | 15.4 |
| 4M4A | 30.99 ± 0.03 | 155.25 | 0.23 | 675 | 15.7 |
| 4M4A + Cu2+ | 21.11 ± 1.0 | 174.02 | 0.26 | 673 | 15.7 |
| WT + VK7 | 29.16 ± 0.24 | 94.08 | 0.14 | 672 | 15.6 |
3.2 The effect of CuGly on Rg and Dmax distributions
In this experiment equimolar amounts of CuGly were added to each of the protein solutions before they were injected into the column. The Rg distributions determined by EOM are given in Fig. 2 for the WT (A) and the A30P (B) and A53T (C) mutants after passage through the SEC column. We were unable to obtain satisfactory data for the E46K mutants in the presence of CuGly because of extensive aggregation occurring in the column. It can be seen that the Rg distributions of the ensembles were all narrowed and shifted to the lower end of the range covered by the ensemble, with the cut-offs in the range 35–40 Å (Table 1). The plot of I0/c, as determined above was linear for each set of profiles (Fig. S2, ESI†). The Guinier plot Rg from the experimental scattering profile, the absolute scale I0 and its corresponding protein concentrations and calculated MM are given in Table 2. The fitted SAXS profiles, Dmax, Guinier plot and the Abs280 plots from the SEC are available in Fig. S5 (ESI†), and the values of average Rg, average ensemble Dmax, and χ for each profile fit are available in Table S1 (ESI†).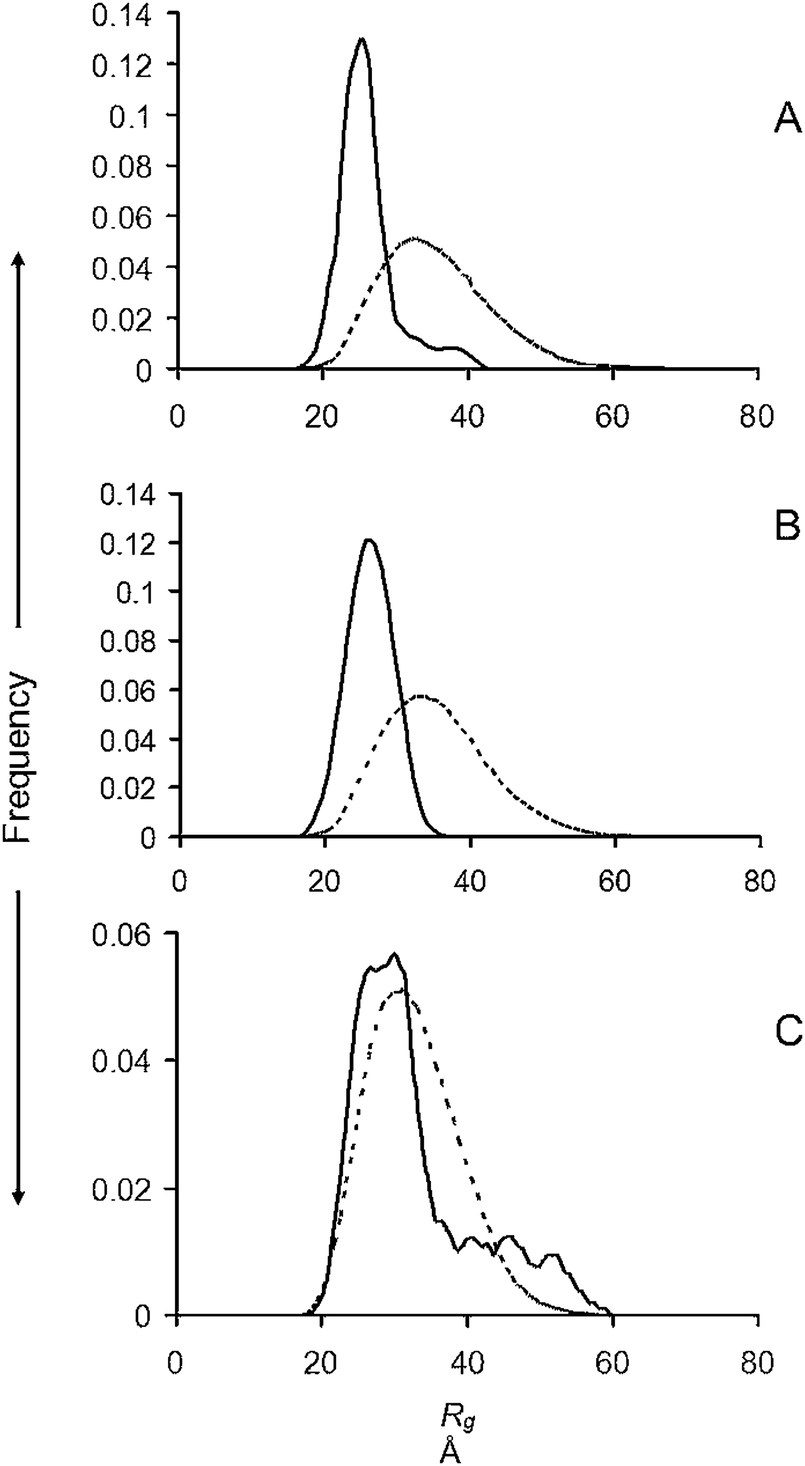 | ||
| Fig. 2 R g distributions from ensemble optimisation modelling after the addition of 1 M/M CuGly to (A) WT and (B) A30P; (C) A53T familial mutants of α-syn. Solid lines, ensemble; dotted lines, pool. | ||
3.3 The effect of the methionine substitutions and the VK7 compound on Rg and Dmax distributions
Substituting the 4 methionine residues of α-syn with alanine residues 4M4A, thereby simulating oxidation of the methionine residues, caused a marked reduction in the proportion of higher Rg's in the ensemble compared with WT α-syn (Fig. 3A and Table 1). A further reduction was observed upon addition of 1 M/M CuGly, which gave a monomodal narrow distribution of low-Rg conformers (Fig. 3B). A significant reduction was also observed with 1 M/M of the fibril inhibitor Pt compound added to WT α-syn (Fig. 3C). The Guinier plot Rg from the experimental scattering profile, the absolute scale I0 and its corresponding protein concentrations and calculated MM are given in Table 2.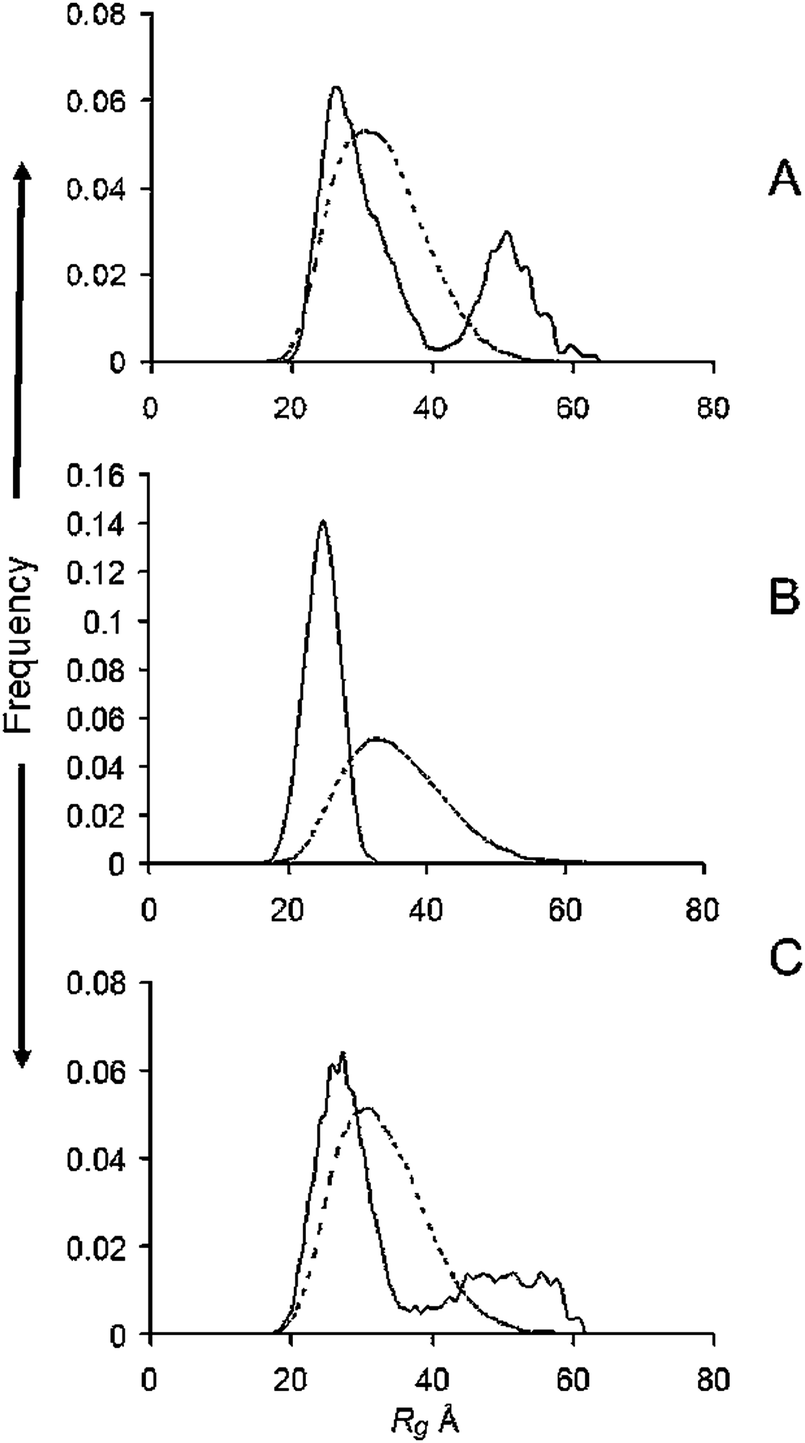 | ||
| Fig. 3 R g distributions from ensemble optimisation modelling of (A) the 4M to 4A substituted α-syn; (B) after the addition of 1 M/M CuGly to the substituted α-syn; (C) after adding the antifibril agent VK7 at 1 M/M to WT α-syn. Solid lines, ensemble; dotted lines, pool. | ||
The fitted SAXS profiles, Dmax, Guinier plot and the Abs280 plots from the SEC are available in Fig. S6 (ESI†), and the values of average Rg, average ensemble Dmax, and χ for each profile fit are available in Table S1 (ESI†). The plot of I0/c, was linear for each set of profiles (Fig. S2, ESI†).
4. Discussion
Various techniques have been used to probe the highly dynamic conformational space of α-syn. Atomic force microscopy-based single-molecule force spectroscopy (SMFS), which exploits protein molecules' resistance to mechanical pulling, showed three major distinct conformations occurring within monomeric alpha-synuclein.3 These were random coil, mechanically weak, and β-like structures. The latter were proposed to be directly related to the aggregation of WT α-syn and the pathological mutations A30P, A53T and E46K, and other factors that increase the protein's fibrillisation or oligomerisation propensity, such as high ionic strength and the presence of Cu2+ shifted the conformational equilibrium towards the β-like structures. Another SMFS study focussed on the role of different segments of the sequence of WT and pathological mutants on dimer formation.4 Electrospray ionisation studies5 have shown that four different conformations of α-syn co-exist in solution under equilibrium, significantly differing from each other by their degree of compactness. A highly structured compact conformation persisted over the pH range 2.5–10 and dimers were observed over the whole range. In our previously reported SAXS-EOM data for WT α-syn a tri-modal distribution of Rg and Dmax was observed with less sharp separation of the peaks,14 and there has been a recent report of SAXS-EOM measurements, in combination with NMR measurements, of WT α-syn showing that the protein adopts a single broad distribution of Rg in 10 mM phosphate buffer.31The reported variations in conformer distribution make it clear that sample preparation conditions and measurement techniques influence the result. A recent advance on SAXS/WAXS beamlines has been to install SEC in line with the capillary in the X-ray beam,32,33 permitting precise control of conditions including matching concentration of the subtraction buffer, an important factor in the study of IDPs whose long range interactions may be disturbed or masked by slight changes in buffer conditions. In the present study SAXS profiles were measured 900 s after the protein left the GuHCl chaotrope behind in the SEC. This approach enabled the early establishment of intra-molecular contacts creating the conformer distribution. Deconvolution showed that with the WT there was a sufficient distinction between the maximum cut-off Rg value of the low range peaks and the minimum value of the high range to justify the conclusion that this early distribution was bi-modal. It could be argued that the broader distributions seen earlier have undergone further conformer development occurring during sample preparation. The increase in the proportion of extended conformers in the ensembles of the rapid fibrillisation-prone E46K and A53T familial PD mutants should be viewed in the light of earlier data suggesting that these conformers represent the pool from which fibrils are derived.3 Although the mutation at residue 46 would alter the balance of charges and could favour the extended conformer13 it is less clear that a decrease in hydrophobicity could lead to a similar effect. However, the A30P mutant, which is also a pathogenic mutation in α-syn, displays no extended conformers and a lower Rg distribution cut-off. A comparison of the oligomerisation and fibrillisation propensities of the A30P and A53T showed that the former tended to form oligomers, particularly when mixed with the WT.34,35 Bertoncini et al.36 showed that residual dipolar couplings were both considerably reduced at both the A30P and A53T mutation sites and, taking into account site-directed spin labelling and paramagnetic broadening data, concluded that both could more easily overcome the barrier to oligomerisation. However, they suggested that the presence of Pro at position 30 could hinder the incorporation of the N-terminus into the β-sheet structure needed to form fibrils. The A30P mutant has been found to bind less thioflavin T during fibrillisation experiments than the WT.37,38 These results suggest that the extended conformer is the basis for fibril formation, as those mutations that have less of it have significantly reduced propensity to aggregate into amyloid.
While pathogenic mutations do cause a significant proportion of clinical cases of PD, the disease is more often associated with a sporadic presentation. This may be due to external factors, which can also affect α-syn folding, such as metal ions, small compounds and other biological macromolecules. One such factor is copper (Cu2+). Cu2+ dysregulation has been proposed as one of the factors in the development of PD (reviewed ref. 39), and Brown et al.40 showed that the addition of Cu2+ favoured the formation of stellate-shaped toxic oligomers, adding to other findings that the presence of Cu2+ would favour oligomer formation under various conditions.41,42 These findings suggested that Cu2+ may have a significant effect on the initial distribution of conformers, possibly by coordinating with His 50 and other residues, altering the balance of charges.13 Indeed, the addition of Cu2+ to the WT, A30P and A53T mutants and the Met to Ala-substituted protein produced a narrow distribution, suggesting that these very compact conformers constitute the ensemble from which the Cu2+-induced toxic oligomers40 might be derived. The narrower Dmax and Rg distributions of the A30P mutant, even in the absence of Cu2+, support the suggestion that the compact conformers are at the commencement of the toxic oligomer pathway. Whether this compact conformer is initiated by a mutation favouring a compact distribution, by the presence of Cu2+ or by some means yet to be described is unfortunately unclear at this time.
Since Cu2+ has such a significant effect on the conformer distribution, which may lead to deleterious effects, there is hope that other factors, such as small molecules, may possibly influence the starting conformation in a beneficial fashion. To test this hypothesis, data was acquired for α-syn in the presence of a platinum based agent, VK7. This compound has previously been observed to be non-toxic and to reduce the amyloid plaque burden in human Aβ transgenic mice17 and has recently been observed to inhibit α-syn fibrillisation (Kench, unpublished). The Rg and Dmax distribution induced by the Pt compound shows significant reduction in the proportion of elongated structures, indicating induction of a compact form. However, this distribution is significantly broader then the narrow range of ensemble parameters induced by Cu2+, suggesting that a wider range of conformations are present in the sample. These results suggest that the compact region of the distribution may represent the starting pool for both toxic and non-toxic oligomers.
Given the observations described here, altering the balance of long-range interactions that promote the creation of the compact and extended formations in favour of those promoting the conformations with Rg values in the range of 35–45 Å appears to represent a potential therapeutic approach to the misfolding of α-syn, and may form the basis of a targeted compound screen. In addition, the study of other IDPs, such as β2-microglobulin,43,44 amylin44,45 and transthyretin mutants involved in familial amyloidotic neuropathy46 where alteration of long range interactions could have pathological consequences may also benefit from the SEC-EOM-SAXS approach.
Acknowledgements
The authors wish to acknowledge the support of the Australian Synchrotron and the Victorian Government Operational Infrastructure Support Program. TMR is supported by the Australian Alzheimer's disease foundation. CLM and KJB are consultants for Prana Biotechnology Pty Ltd. CLM, KJB and RC are the principal investigators on an Australian National Health and Medical Research Council program grant.References
- V. N. Uversky, J. Biomol. Struct. Dyn., 2003, 21, 211–234 CAS.
- N. Abbas, C. B. Lücking, S. Ricard, A. Dürr, V. Bonifati, G. De Michele, S. Bouley, J. R. Vaughan, T. Gasser, R. Marconi, E. Broussolle, C. Brefel-Courbon, B. S. Harhangi, B. A. Oostra, E. Fabrizio, G. A. Böhme, L. Pradier, N. W. Wood, A. Filla, G. Meco, P. Denefle, Y. Agid and A. Brice, French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease, Hum. Mol. Genet., 1999, 8, 567–574 CrossRef CAS PubMed.
- M. Brucale, M. Sandal, S. Di Maio, A. Rampioni, I. Tessari, L. Tosatto, M. Bisaglia, L. Bubacco and B. Samori, ChemBioChem, 2009, 10, 176–183 CrossRef CAS PubMed.
- M. Sandal, F. Valle, I. Tessari, S. Mammi, E. Bergantino, F. Musiani, M. Brucale, L. Bubacco and B. Samori, PLoS Biol., 2008, 6, 99–108 CAS.
- A. V. Krasnoslobodtsev, I. L. Volkov, J. M. Asiago, J. Hindupur, J. C. Rochet and Y. L. Lyubchenko, Biochemistry, 2013, 52, 7377–7386 CrossRef CAS PubMed.
- A. K. Frimpong, R. R. Abzalimov, V. N. Uversky and I. A. Kaltashov, Proteins, 2010, 78, 714–722 CAS.
- M. M. Dedmon, K. Lindorff-Larsen, J. Christodoulou, M. Vendruscolo and C. M. Dobson, J. Am. Chem. Soc., 2005, 127, 476–477 CrossRef CAS PubMed.
- O. Ullman, C. K. Fisher and C. M. Stultz, J. Am. Chem. Soc., 2011, 133, 19536–19546 CrossRef CAS PubMed.
- M. Tashiro, M. Kojima, H. Kihara, K. Kasai, T. Kamiyoshihara, K. Ueda and S. Shimotakahara, Biochem. Biophys. Res. Commun., 2008, 369, 910–914 CrossRef CAS PubMed.
- A. Rekas, R. B. Knott, A. Sokolova, K. J. Barnham, K. A. Perez, C. L. Masters, S. C. Drew, R. Cappai, C. C. Curtain and C. L. Pham, Eur. Biophys. J., 2010, 39, 1407–1419 CrossRef CAS PubMed.
- V. N. Uversky, E. M. Cooper, K. S. Bower, J. Li and A. L. Fink, FEBS Lett., 2002, 515, 99–103 CrossRef CAS.
- W. Zhou, C. Long, S. H. Reaney, D. A. Di Monte, A. L. Fink and V. N. Uversky, Biochim. Biophys. Acta, 2010, 1802, 322–330 CrossRef CAS PubMed.
- R. K. Das and R. V. Pappu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13392–13397 CrossRef CAS PubMed.
- C. L. Pham, N. Kirby, K. Wood, T. Ryan, B. Roberts, A. Sokolova, K. J. Barnham, C. L. Masters, R. B. Knott, R. Cappai, C. C. Curtain and A. Rekas, Proteins, 2014, 82, 10–21 CrossRef CAS PubMed.
- R. Cappai, S. L. Leck, D. J. Tew, N. A. Williamson, D. P. Smith, D. Galatis, R. A. Sharples, C. C. Curtain, F. E. Ali, R. A. Cherny, J. G. Culvenor, S. P. Bottomley, C. L. Masters, K. J. Barnham and A. F. Hill, FASEB J., 2005, 19, 1377–1379 CAS.
- V. B. Kenche, I. Zawisza, C. L. Masters, W. Bal, K. J. Barnham and S. C. Drew, Inorg. Chem., 2013, 52, 4303–4318 CrossRef CAS PubMed.
- V. B. Kenche, L. W. Hung, K. Perez, I. Volitakes, G. Ciccotosto, J. Kwok, N. Critch, N. Sherratt, M. Cortes, V. Lal, C. L. Masters, K. Murakami, R. Cappai, P. A. Adlard and K. J. Barnham, Angew. Chem., Int. Ed., 2013, 52, 3374–3378 CrossRef CAS PubMed.
- N. M. Kirby, S. T. Mudie, A. Hawley, D. J. Cookson, H. D. Mertens, N. Cowieson and V. Samardzic-Boban, J. Appl. Crystallogr., 2013, 46, 1670–1680 CrossRef CAS.
- A. Nayak, A. K. Dutta and G. Belfort, Biochem. Biophys. Res. Commun., 2008, 369, 303–307 CrossRef CAS PubMed.
- D. A. Jacques, J. M. Guss, D. I. Svergun and J. Trewhella, Acta Crystallogr., 2012, 68, 620–626 CrossRef CAS PubMed.
- P. V. Konarev, M. V. Petoukhov, V. V. Volkov and D. I. Svergun, J. Appl. Crystallogr., 2006, 39, 277–286 CrossRef CAS.
- P. V. Konarev, V. V. Volkov, A. V. Sokolova, M. H. Koch and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 1277–1282 CrossRef CAS.
- P. V. Konarev, M. V. Pethoukov, V. V. Volkov and D. I. Svergun, J. Appl. Crystallogr., 2001, 34, 527–532 CrossRef CAS.
- D. I. Svergun, Biophys. J., 1999, 76, 2879–2886 CrossRef CAS.
- D. I. Svergun, A. Becirevic, H. Schrempf, M. H. Koch and G. Gruber, Biochemistry, 2000, 39, 10677–10683 CrossRef CAS PubMed.
- D. Orthaber, A. Bergmann and O. Glatter, J. Appl. Crystallogr., 2000, 33, 218–225 CrossRef CAS.
- P. Bernado, E. Mylonas, M. V. Petoukhov, M. Blackledge and D. I. Svergun, J. Am. Chem. Soc., 2007, 129, 5656–5664 CrossRef CAS PubMed.
- M. V. Pethoukov, D. Franke, A. V. Shkumatov, G. Tria, A. G. Kikheny, M. Gajda, C. Gorba, H. D. T. Mertens, P. V. Konarev and D. I. Svergun, J. Appl. Crystallogr., 2012, 45, 342–350 Search PubMed.
- M. V. Pethoukov and D. I. Svergun, Int. J. Biochem. Cell Biol., 2012, 45, 429–437 CrossRef PubMed.
- P. Bernado and D. I. Svergun, Mol. BioSyst., 2012, 8, 151–167 RSC.
- M. Schwalbe, V. Ozenne, S. Bibow, M. Jaremko, L. Jaremko, M. Gajda, M. R. Jensen, J. Biernat, S. Becker, E. Mandelkow, M. Zweckstetter and M. Blackledge, Structure, 2014, 22, 238–249 CrossRef CAS PubMed.
- N. J. Gunn, M. A. Gorman, R. C. Dobson, M. W. Parker and T. D. Mulhern, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2011, 67, 336–339 CrossRef CAS PubMed.
- Y. Watanabe and Y. Inoko, Anal. Bioanal. Chem., 2011, 399, 1449–1453 CrossRef CAS PubMed.
- K. A. Conway, J. D. Harper and P. T. Lansbury, Nat. Med., 1998, 4, 1318–1320 CrossRef CAS PubMed.
- K. A. Conway, S. J. Lee, J. C. Rochet, T. T. Ding, R. E. Williamson and P. T. Lansbury Jr., Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 571–576 CrossRef CAS.
- C. W. Bertoncini, Y. S. Jung, C. O. Fernandez, W. Hoyer, C. Griesinger, T. M. Jovin and M. Zweckstetter, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1430–1435 CrossRef CAS PubMed.
- A. Rekas, C. G. Adda, J. Andrew Aquilina, K. J. Barnham, M. Sunde, D. Galatis, N. A. Williamson, C. L. Masters, R. F. Anders, C. V. Robinson, R. Cappai and J. A. Carver, J. Mol. Biol., 2004, 340, 1167–1183 CrossRef CAS PubMed.
- S. B. Nielsen, F. Macchi, S. Raccosta, A. E. Langkilde, L. Giehm, A. Kyrsting, A. S. Svane, M. Manno, G. Christiansen, N. C. Nielsen, L. Oddershede, B. Vestergaard and D. E. Otzen, PLoS One, 2013, 8, e67713 CAS.
- R. M. Rasia, C. W. Bertoncini, D. Marsh, W. Hoyer, D. Cherny, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4294–4299 CrossRef CAS PubMed.
- D. R. Brown, FEBS J., 2007, 274, 3766–3774 CrossRef CAS PubMed.
- S. R. Paik, H. J. Shin, J. H. Lee, C. S. Chang and J. Kim, Biochem. J., 1999, 340, 821–828 CrossRef CAS.
- J. A. Wright, X. Wang and D. R. Brown, FASEB J., 2009, 23, 2384–2393 CrossRef CAS PubMed.
- V. J. McParland, N. M. Kad, A. P. Kalverda, A. Brown, P. Kirwin-Jones, M. G. Hunter, M. Sunde and S. E. Radford, Biochemistry, 2000, 39, 8735–8746 CrossRef CAS PubMed.
- E. Chatani, H. Yagi, H. Naiki and Y. Goto, J. Biol. Chem., 2012, 287, 22827–22837 CrossRef CAS PubMed.
- N. Reixach, S. Deechongkit, X. Jiang, J. W. Kelly and J. N. Buxbaum, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2817–2822 CrossRef CAS PubMed.
- T. R. Foss, R. L. Wiseman and J. W. Kelly, Biochemistry, 2005, 44, 15525–15533 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mb00356j |
| ‡ Present address: Biological Small Angle Scattering Group European Molecular Biology Laboratory c/o DESY Notkestrasse 85, Geb. 25a22603 Hamburg, Germany. |
| This journal is © The Royal Society of Chemistry 2015 |