
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStability analysis of chemically modified mRNA using micropattern-based single-cell arrays†
Mehrije
Ferizi‡
a,
Carolin
Leonhardt‡
b,
Christian
Meggle
b,
Manish K.
Aneja
c,
Carsten
Rudolph
c,
Christian
Plank
ac and
Joachim O.
Rädler
*b
aInstitute of Molecular Immunology - Experimental Oncology, Technische Universität München, Munich, 81675 Germany
bFaculty of Physics and Center for NanoScience, Ludwig Maximilian University, Munich, 80539 Germany. E-mail: raedler@lmu.de; Fax: +49 89 2180 3182; Tel: +49 89 2180 2438
cEthris GmbH, Planegg, 82152 Germany
First published on 16th July 2015
Abstract
The measurement of mRNA turnover in living cells plays an important role in the search for stable mRNA constructs for RNA-based therapies. Here we show that automated time-lapse microscopy combined with micropatterned arrays allows for efficient high-throughput monitoring of fluorescent reporter protein expression at the single-cell level. The fluorescence time courses after mRNA transfection yield the distribution of individual mRNA expression and degradation rates within a population. We compare mRNA constructs with combinations of 5′ and 3′ UTR sequences and find a systematic broadening and shift towards longer functional half-lives for UTR stabilized mRNA. At the same time the life time distribution of the destabilized EGFP reporter protein was found to be constant and narrowly distributed. Using mathematical modeling, we show that mRNA functional life-time predicts the time-integrated protein level, i.e. the area under the curve (AUC) of mRNA translation. Our approach paves the way for quantitative assessment of hitherto unexplored mRNA functional life time heterogeneity, possibly predicated on multiple mRNA secondary structures and its dependence on UTR sequences.
Introduction
In recent years, messenger RNA (mRNA) has become increasingly relevant as a new drug entity. As opposed to DNA-based gene therapeutics, mRNA does not need to be transported into the nucleus but is directly translated into protein in the cytoplasm.1,2 This makes mRNA safer in avoiding potential insertional mutagenesis, an unlikely but existent risk of DNA gene medicines. As a consequence, mRNA therapeutics are emerging as promising alternatives for gene and protein replacement therapies in a broad variety of medical indications.1–4 However, the strong immunogenicity as well as the limited stability of conventional mRNA has to be overcome to further establish its clinical applicability. In particular, mRNA stability is an essential parameter for envisaged medical applications because it determines, for example, dosing and the dosing intervals of mRNA drugs.Several strategies have proven successful both at increasing the stability and reducing the immunogenic response triggered by mRNA administered to cells or organisms. Amongst these is the inclusion of chemically modified nucleotides.5 Kormann et al. have shown that the replacement of only 25% of uridine and cytidine residues by 2-thiouridine and 5-methyl-cytidine suffices to increase mRNA stability as well as to reduce the activation of innate immunity triggered by externally administered mRNA in vitro.3
Also, untranslated regions (UTRs) in mRNAs have been reported to play a pivotal role in regulating both mRNA stability and mRNA translation. UTRs are known to influence translational initiation, elongation, and termination, as well as mRNA stabilization and intracellular localization through their interaction with RNA binding proteins.6,7 Depending on the specific motives within the UTR, it can either enhance or decrease mRNA turnover.8–11 Recently, data on mRNA half-lives and the corresponding UTR sequences have been published.12,13
One mRNA with a particularly long half-life is the one transcribed from the human cytochrome b-245 alpha polypeptide (CYBA) gene. The CYBA gene comprises specific 5′ and 3′ UTRs. In general, 5′ UTR motives such as upstream open reading frames (uORFs) or internal ribosomal entry sites (IRES) are known to be involved in gene regulation, particularly in translational initiation.14 The 3′ UTRs can comprise even more regulatory functions than the 5′ UTRs, some of them even hindering mRNA translation.15 While no regulatory motives are known for the CYBA 5′ UTR unit, the CYBA 3′ UTR contains two of them. Firstly, the polyadenylation signal (PAS), which interacts with the cytoplasmic polyadenylation element binding protein (CPEB), as well as with the cleavage and polyadenylation signaling factor (CPSF).11 CPEB is responsible for the prolongation of the poly-A tail in the cytoplasm, whereas CPSF primes the pre-mRNA through cleavage at a specific site for the upcoming addition of poly-A.11,15 As a second regulatory motif, the CYBA 3′ UTR contains the insulin 3′ UTR stability element (INS_SCE). The INS_SCE sequence has been shown to bind to the polypyrimidine tract binding protein (PTB) under reducing conditions, increasing the mRNA half-life of insulin.16 UTRs of CYBA are shown in Table S2.† Another important feature influencing mRNA translation efficiency is the poly A-tail, which is located on the 3′ end. It has been shown that a prolongation of the poly A-tail to 120 nucleotides has beneficial effects on protein expression, assumingly because of the protective effect of longer poly A-tails against mRNA degradation.17 In contrast to long poly A-tails, mRNAs with poly A-tails shorter than 50 nucleotides are claimed not to be translated at all.11,18 Hence, in mRNA therapy, recombinant mRNA constructs are advantageously furnished with a poly A-tail of 120 nucleotides. Degradation of most mRNAs in eukaryotic cells begins with 3′ to 5′ exonucleolytic deadenylation, resulting in removal of most of the poly A-tail. Subsequently, two major pathways that are responsible for the degradation of the rest of the mRNA body are known to come into play. On the one hand, the 5′ end is decapped by the Dcp1/Dcp2 complex, followed by 5′–3′ exonucleolytic degradation that is catalyzed by Xrn1p. On the other hand, the exosome enables 3′–5′ exoribonucleolytic degradation with the 5′ cap being retained.19 Moreover, it is known that the 5′ cap interaction with the 3′ poly A-tail results in circular forms of the mRNA. It is assumed that the circular shape of the mRNA increases the initiation rate of ribosomes and also protects mRNA against degradation.20
We were interested in whether the reported long half-life of natural CYBA mRNA can be conferred to a foreign mRNA by virtue of flanking its coding sequence with combinations of CYBA 5′- and 3′-UTRs. So far, mRNA stability and turnover have been measured by a variety of approaches including metabolic labelling, mass spectrometry, quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR), microarrays, or fluorescence labelling techniques for imaging mRNA inside cells.17,21–26 Most of these are ensemble measurements and data represent mean of all cells without taking into account the fluctuations in the expression amounts (high vs. low) or timing of expression (early or late). Hence, population measurements exhibit an averaged response and are of limited use for computational analysis of the underlying biochemical network. In contrast, single-cell distribution functions provide additional information about the heterogeneity in transfection experiments.
Recently, we have shown that single-cell analysis of mRNA transfection time-lapse movies is capable of assessing individual expression time-courses, yielding mRNA decay rates at the single-cell level.27 Furthermore, we have reported on the use of regular micropatterns to position cells on a regular grid of adhesion sites.28 Hence, we were interested in applying this single-cell micro-array technology to screen and compare different mRNA constructs in a more rapid and standardized format than single-cell studies in diluted culture can do. To address this question, we have chosen the coding sequence of destabilized enhanced green fluorescence protein (d2EGFP) to artificially shorten the life cycle of the reporter protein inside the cell.29 The combinations included insertion of the respective CYBA UTRs at 5′ or 3′ ends, respectively, at both 5′- and 3′ ends, at the 5′ end combined with two repeats of the 3′ UTR at the 3′ end, or two repeats of 3′ UTR without 5′ UTR. All of these compared to a control construct without UTRs. Protein and mRNA functional life times and the expression rates from each of the compared transcripts were assessed using both the single-cell approach and population based methods like flow cytometry, fluorescence microscopy and qRT-PCR. As expression of d2EGFP is a direct readout of the functionality of d2EGFP stable non-immunogenic mRNA (SNIM® RNA), expression kinetics were used to determine the “functional” half-life of d2EGFP SNIM® RNA.
Results
Fluorescence microscopy and analysis via flow cytometry (FC)
To evaluate the effect of different UTR combinations on transgene expression kinetics, two different cells lines were transfected using Lipofectamine™ 2000 with different d2EGFP mRNA constructs containing a 5′ UTR alone, a 3′ UTR, 5′ + 3′ UTR, two copies of 3′ UTR and 5′ + 2 × 3′ UTR. A schematic representation of the building blocks of all constructs can be seen in Fig. 1A.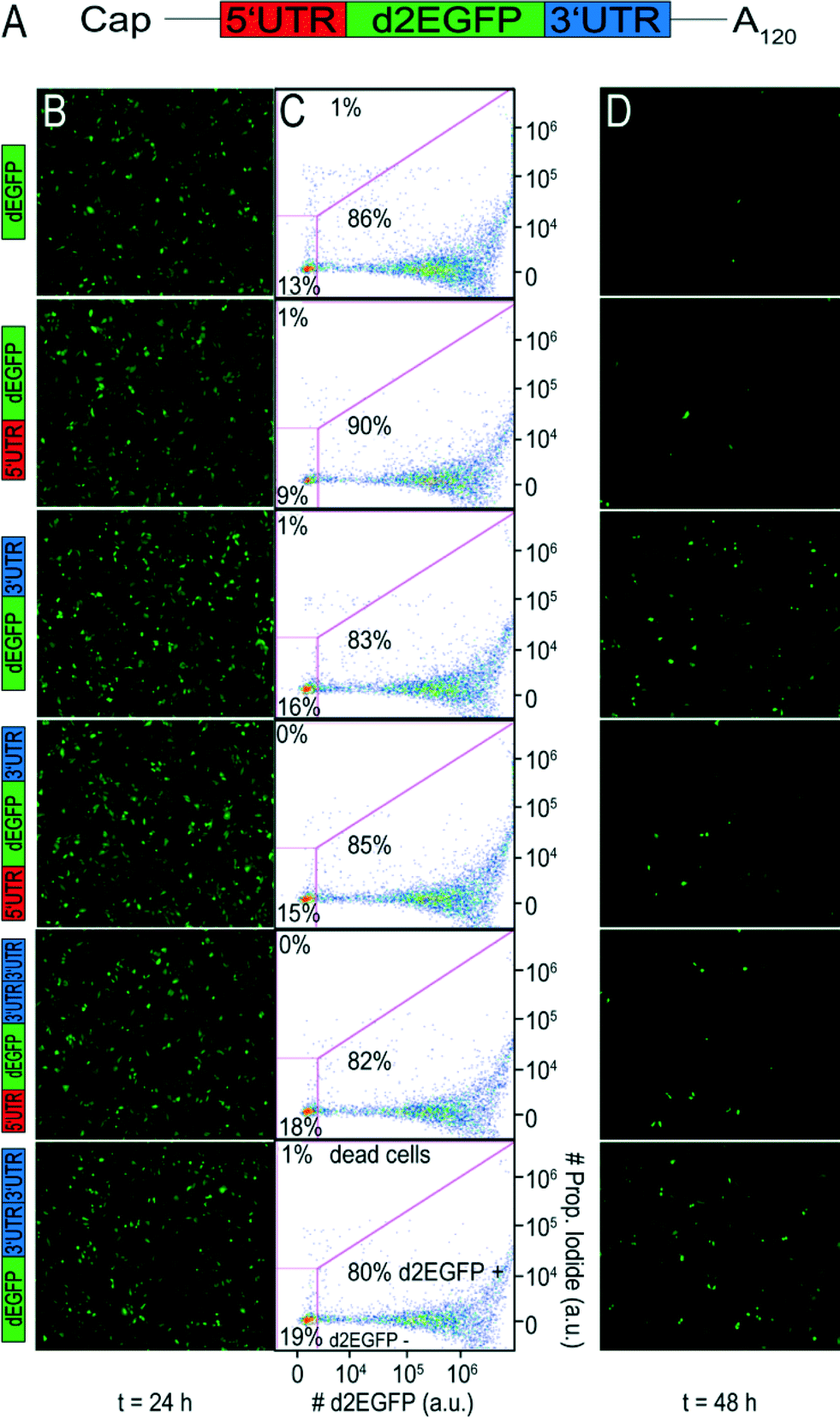 | ||
| Fig. 1 Fluorescence microscopy and flow cytometry data of A549 cells. (A) Schematic illustration of therapeutic mRNA, consisting of a 5′ cap, a 5′ UTR, an encoding region, a 3′ UTR and a poly-A tail. (B) Fluorescence microscopy pictures taken with 4× magnification (JULY™) at 24 h post-transfection. All constructs showed improved protein expression levels as compared to the control. (C) The percentage of d2EGFP positive cells as determined by FC is similar for all constructs. Propidium iodide was used to detect dead cells. The applied gates ensured exclusion of dead cells and untransfected cells. (D) At 48 h post transfection, sustained protein expression was higher for the stabilized constructs as compared to the control. | ||
At different time points through three days post-transfection, d2EGFP expression was quantified using FC. An exemplary dot plot for t = 24 h, illustrating d2EGFP expression levels of live A549 cells, is shown in Fig. 1C (see Fig. S4B† for corresponding Huh7 data). In addition, we imaged the cells using fluorescence microscopy (see Fig. 1B and D and Fig. S4A and C†). Comparable transfection efficiencies for all mRNA constructs were confirmed 24 hours post transfection (Fig. 1B and S5A†). Thereby, we can rule out differential transfer efficiencies to be a causal factor for the observed differences in expression kinetics. Based on fluorescence microscopy images, we detected a drastic reduction of d2EGFP expression for all constructs at 48 h post-transfection (see Fig. 1B and D, S4A and C†). However, higher d2EGFP expression levels with respect to the control were found for all UTR-stabilized mRNAs. More specifically, mRNA constructs containing 3′ UTRs seemed to enhance expression more than constructs without 3′ UTRs. We observed this for A549 and Huh7 cells (see Fig. 1 and S4,† respectively). At time points later than 48 h, this effect was even more pronounced (data not shown). In Fig. 2A and B, the time-courses of the mean fluorescence intensities (MFI) as determined by FC are shown for all constructs in both cell types.
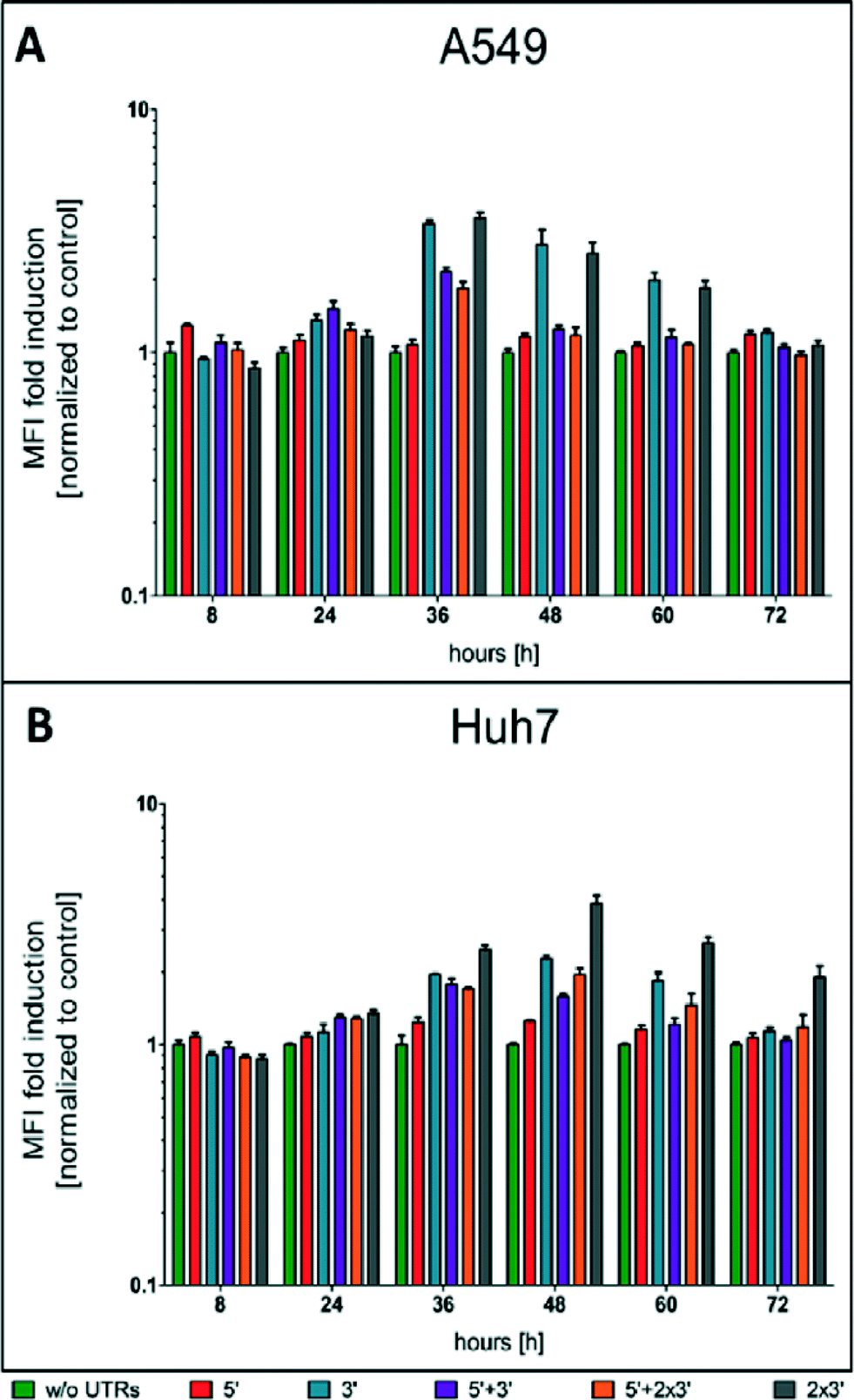 | ||
| Fig. 2 Time-courses of protein expression as determined by FC for A549 cells (A) and Huh7 cells (B). Mean fluorescence intensities normalized to the control are plotted versus time in a log-linear plot. With increasing time post transfection, the elevated protein expression levels of the stabilized constructs become more and more evident. The green, red and blue bars correspond to the control/5′ UTR/3′ UTR constructs, respectively. The purple, orange and grey bars correspond to the constructs that are shown on the bottom of the figure. | ||
Also here, all UTR-containing mRNA constructs showed higher MFI values than the control construct in both cell lines at all time points. Taken together, our fluorescence microscopy and FC data suggest that mRNA molecules furnished with CYBA UTRs show persistent d2EGFP expression for more than 24 hours.
Quantitative real-time PCR
We conducted qRT-PCR measurements as an additional approach to determine the physical mRNA half-life of the different constructs. Binding of our selected primers to d2EGFP occurred 600 nt downstream of the start codon. Hence, measurements of physical mRNA half-life comprise both intact mRNAs and those which have either been decapped but not yet degraded or both decapped and degraded up to base 599. It also includes mRNA that has been removed from the translational pool and stored in P-bodies.30–33 Only intact mRNAs yield d2EGFP protein, while decapped and/or partially degraded transcripts and those in P-bodies do not lead to any expression. Determination of physical mRNA half-lives did not reveal any significant life time prolongation by the UTRs compared to the control in the A549 and Huh7 cells (see ESI† S5A and B, respectively). Interestingly, we observed a decrease in mRNA physical half-lives for 5′, 3′, 5′ + 2 × 3′ and 2 × 3′ UTR constructs in both cell lines.Single-cell expression arrays
We fabricated microstructured, cell-adhesive substrates as shown in Fig. 3A and B as a platform for single-cell time-lapse microscopy.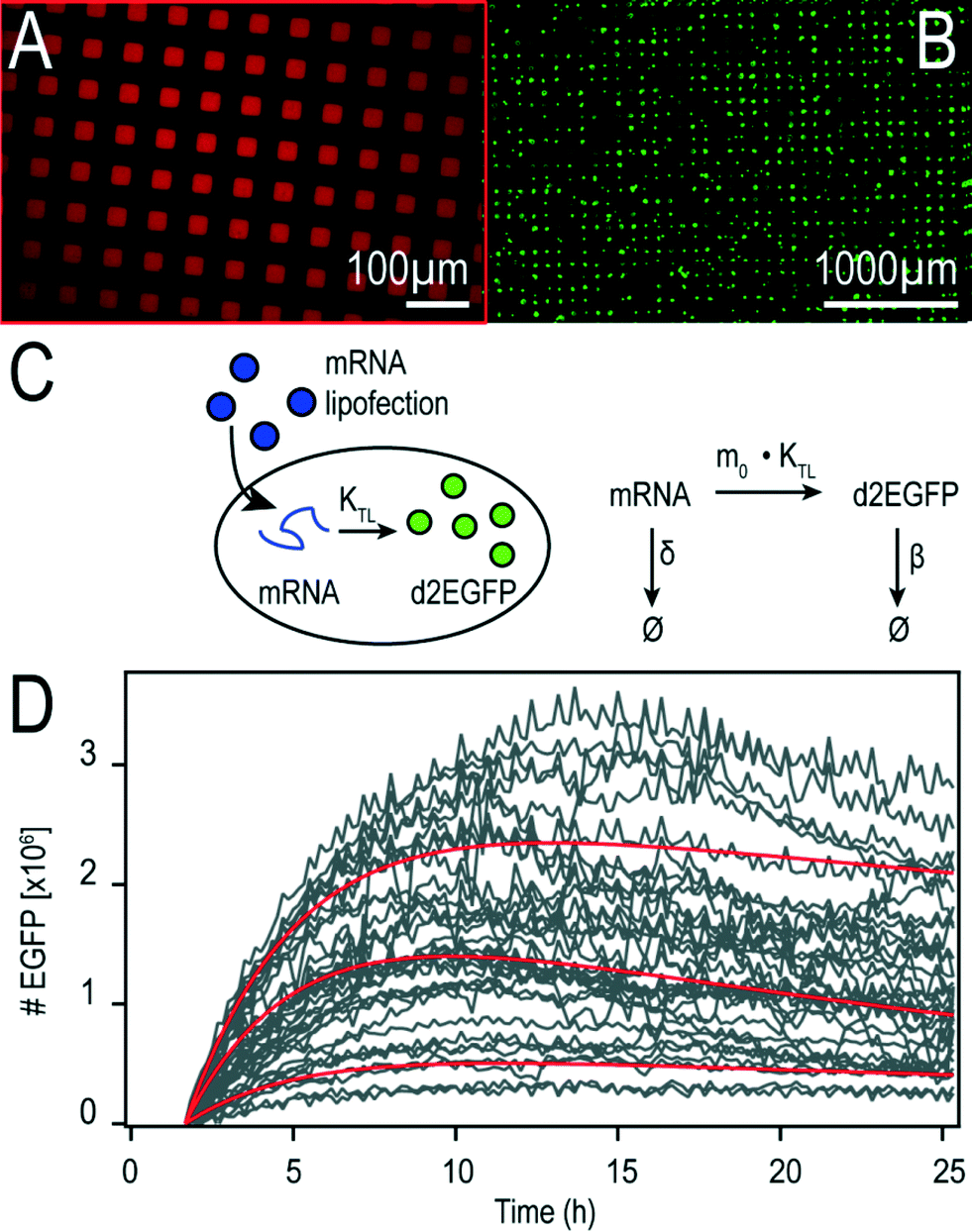 | ||
| Fig. 3 Microstructured multi-channel slides for parallel single-cell assays to test differently stabilized mRNA constructs. (A) Cell-adhesive, microstructured protein patterns with cell-repellent PEG areas in between allow ordered cell arrangement. Fluorescently labeled fibronectin was used to visualize the micropattern. (B) Fluorescent A549 cells adhering to fibronectin patterns inside a microchannel (three hours after seeding). (C) Schematic drawing of mRNA lipofection (on the left) and reaction scheme underlying our analytical solution (on the right). (D) Exemplary time-courses of mRNA-mediated d2EGFP expression in A549 cells. Red lines are representative fits to the theoretical translation model. | ||
The rectangular squares are functionalized with the extracellular matrix protein fibronectin, while the surrounding dark area is passivated with cell repellent PLL-g-PEG. Cells were seeded at an appropriately dilute cell density such that after about three hours cells adhered to the rectangular squares. This cellular self-organization process has been studied in detail before.28 The size of the squares was 30 μm for optimal filling with single cells. The distance between the squares was just big enough (60 μm) to minimize bridging effects of cells adhering to more than one square at the same time. Time-lapse fluorescence microscopy and automated image analysis of the fluorescence signal per square yields hundreds of individual time-courses. A typical set of background corrected raw data is shown in Fig. 3D. The red lines represent exemplary fits to the mathematical expression for mRNA translation (see also Materials and Methods section). Data were analyzed as described recently27 by fitting each time-course with the analytical solution for mRNA-induced protein expression,
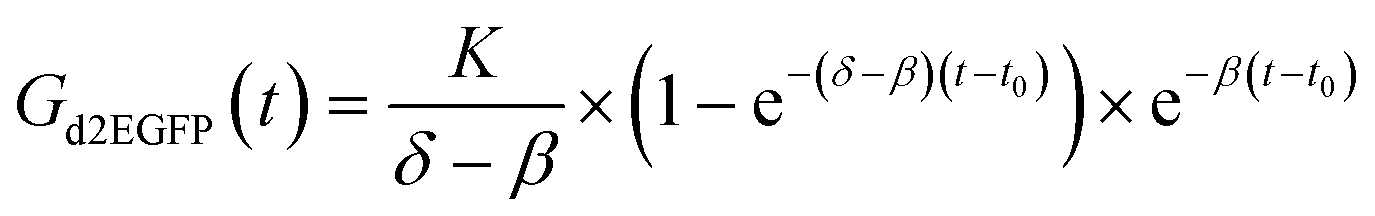 | (1) |
In vitro transfection on cell arrays
In a typical experiment, cells were allowed to adhere to the micropatterns for three hours before transfection. Each of the six microchannels was filled with a different lipoplex solution, containing one of the constructs of interest. In initial experiments, we compared two different, commercially available transfection reagents (namely Lipofectamine™ 2000 and DOGTOR). We found higher transfection efficiencies for Lipofectamine™ 2000 than for DOGTOR (see Fig. S1†). Because we additionally obtained high cell viability rates of above 80% with Lipofectamine™ 2000 (data not shown), all further transfection experiments were conducted using Lipofectamine™ 2000. As mRNA-mediated protein expression starts shortly after transfection, incubation time was kept to a minimum. Accordingly, the ratio between mRNA dosage and incubation time was adjusted to achieve high transfection efficiencies (see also Fig. S1†) and negligible toxic effects caused by over-expression of the reporter protein. At an mRNA dose of 5 pg per cell, an incubation time of one hour was found to be optimal.Expression rates
All results for the two cell types are based on four independent measurements under the same experimental conditions. Time-lapse data of about thousand A549 cells and thousand Huh7 cells have been analyzed. The distributions of the obtained expression rates K are shown in Fig. 4A and the corresponding mean values can be seen in Fig. 4D.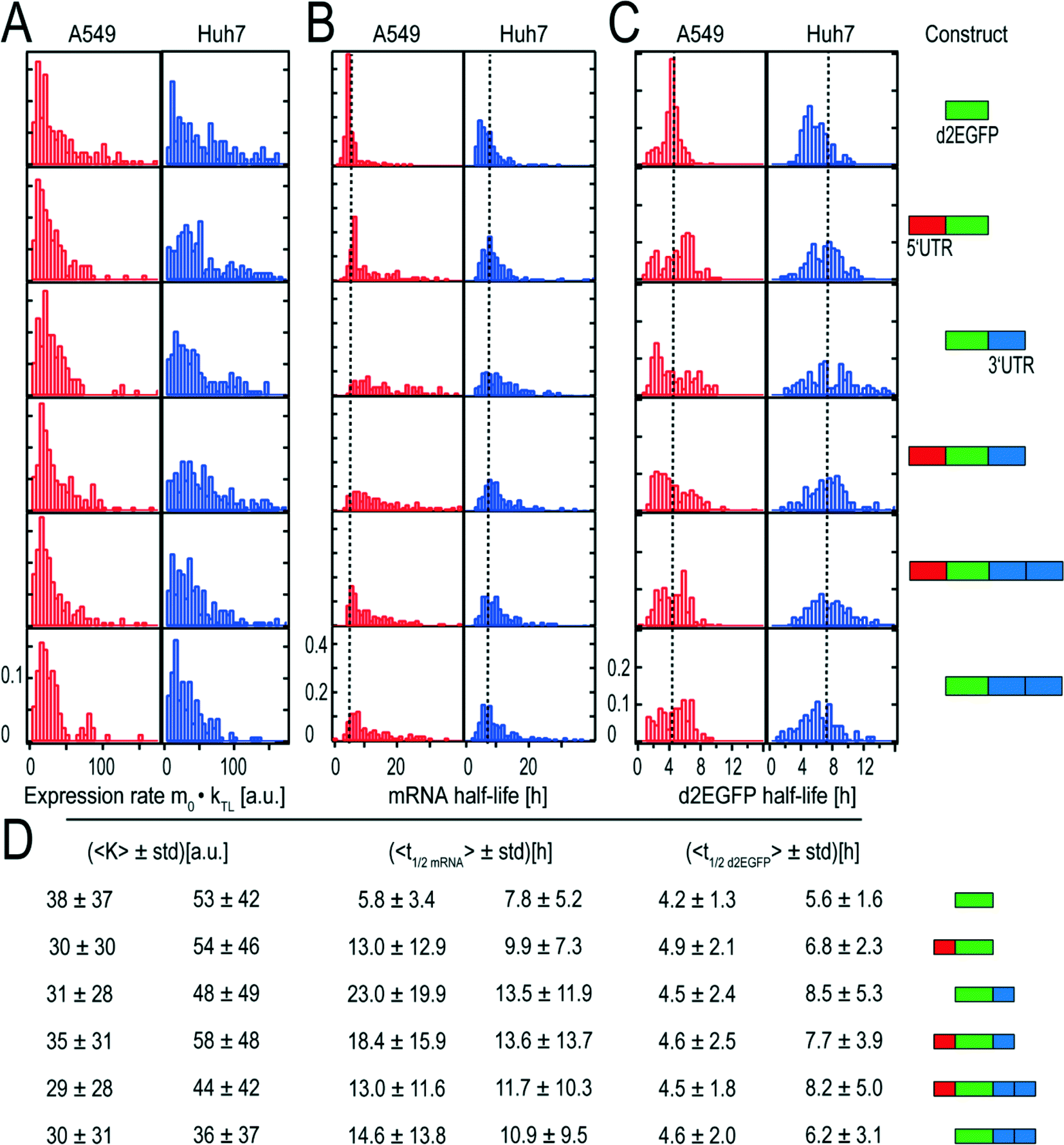 | ||
| Fig. 4 Distributions of expression rates K, mRNA functional life times, and d2EGFP life times and corresponding mean values with schematic representations of the constructs. (A) Distributions of expression rate K, which is the product of the initial number of mRNA molecules and the translation rates. The fact that the distributions are similarly shaped indicates that the transfection kinetics and the translation rates are very similar. (B) The distributions of the mRNA functional half-lives show great variations in their broadness. As a guide to the eye, dotted lines indicate the mean half-life of the control. (C) Distributions of d2EGFP half-lives. As expected, the distributions of the different constructs are similarly shaped and show comparable mean values. As a guide to the eye, the overall mean half-life of d2EGFP based on all measured half-lives is shown as a dotted line. (D) Mean values and the corresponding standard deviations (std) of the fitted rates. Although the control construct yields high mean K values in both cell types, the short mRNA functional half-life of this construct leads to small AUC values as compared to the stabilized constructs. This can be seen in Fig. 6. Schematic representations of the constructs can be seen on the right hand side. All constructs have the same 5′ cap and a poly A-tail. Data from 895 single A549 and 1355 Huh7 cells were analysed. | ||
Both the mean expression rates and the shape of their distributions were found to be rather similar for the different constructs.
mRNA functional half-lives
We converted the fitted mRNA-degradation rates δ into mRNA functional half-lives according to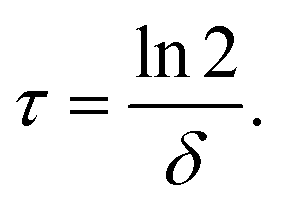 | (2) |
Fig. 4B shows the functional half-life distributions of the examined mRNA constructs in A549 and Huh7 cells, respectively. Here, it becomes evident that for constructs with UTRs, both mean functional half-life and broadness of the underlying distribution increase as compared to the reference construct without UTRs.
An overview of all determined functional half-lives is given in Fig. 4D. Both for A549 and for Huh7 cells, we found longer functional half-lives for mRNAs stabilized by UTR elements compared to the control construct (5.8 hours for A549 cells and to 7.8 hours for Huh7 cells) that does not contain any stabilizing UTR. The functional life time prolonging effect was more pronounced in A549 cells.
Protein half-lives
The distributions of protein (d2EGFP) degradation life times are presented in Fig. 4C. As expected, the half-lives of the expressed protein do not vary for the different mRNA constructs. The determined mean life times range from 4.2 to 4.9 hours for A549 cells and from 5.6 to 8.5 hours for Huh7 cells as shown in Fig. 4D. The coefficients of variation are about 0.29 (A549) and 0.45 (Huh7) and hence are significantly smaller than the coefficient of variation of up to 0.6 that we found for the distribution on mRNA functional life times. As a control, we also measured the half-lives in an alternative approach, where translation was inhibited by addition of cycloheximide at a given time point, t0, after transfection (see Fig. S3†). In this case, protein expression is induced for a while and then stopped. The exponential decay in fluorescence after inhibition yields protein life times. These half-lives were found to be smaller by a factor of about two, compared to the above experiments without inhibition. In both experiments, however, the relative ratios of the protein life times in Huh7 cells as compared to those in A549 cells is the same.Master curves of protein expression
The features of mRNA induced protein expression become evident in the so-called master curve of protein expression as depicted in Fig. 5A (A549) and B (Huh7).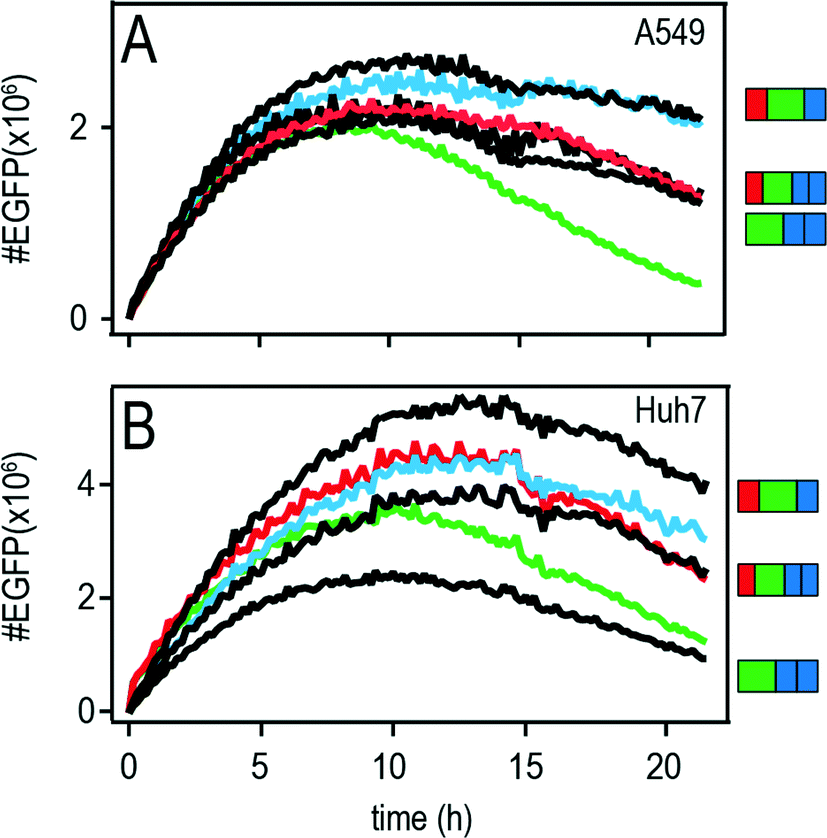 | ||
| Fig. 5 Master curves of the different constructs. Population averages of A549 (A) and Huh7 (B) cells with the onset-time shifted to zero. The green, red, and blue curves correspond to the control/5′ UTR/3′ UTR constructs, respectively. The black curves correspond to the constructs on the right hand side. | ||
The master curve is the population average of the onset-time corrected single-cell traces, i.e. all onset-times were shifted to time point zero. Fluorescence intensities were converted into actual numbers of d2EGFP as described before in reference.27 The superior properties of the 3′ and the 5′ + 3′ UTR mRNA constructs are illustrated in the master curve plot. These constructs showed the shallowest decrease in protein expression with time and hence the longest functional half-lives in addition with higher protein expression values as compared to the other constructs.
Area under the curve (AUC)
In pharmacokinetics, the concentration-time curve of a drug in the blood circulation is known as the “area under the curve” (AUC) which is a measure of the bio-availability of a drug. Here, we use the term for the cumulative time-dose of the protein that is encoded by the mRNA. In this respect, the AUC simultaneously quantifies the translational efficiency and the stability and hence the overall efficacy of a chosen mRNA construct. Given the biochemical rate model (see Fig. 3A), the AUC can be explicitly calculated (see also Fig. S2†):| AUC = 0.48 × m0 × kTL × τmRNA × τd2EGFP | (3) |
Hence an optimal therapeutic mRNA construct should desirably have both long mRNA function half-life, τmRNA, as well as a protein half-life, τd2EGFP, and high translational efficiency, kTL. In addition, the transfer efficiency which determines the initial amount of therapeutic mRNA, m0, is directly proportional to the AUC. An illustrative explanation for the theoretical time-course of protein expression and the calculated AUC can be seen in Fig. 6A.
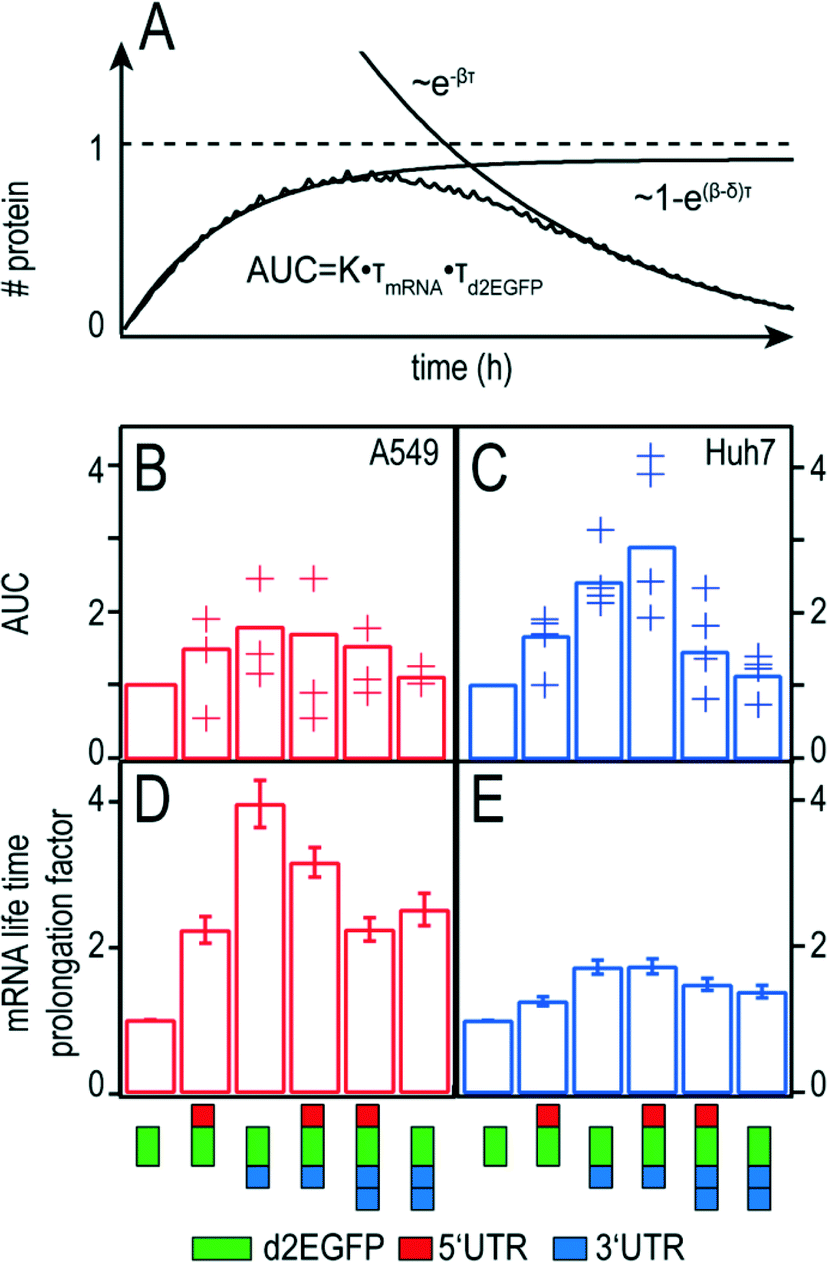 | ||
| Fig. 6 AUC and mRNA functional life time prolongation factors of the different constructs. (A) Schematic representation of the AUC to illustrate the interplay between mRNA translation and degradation of mRNA and protein. (B) and (C) AUC of the different constructs as analysed for t → ∞. Crosses show relative AUCs of different experiments, the bars correspond to the mean of all single-cell AUCs. (D) and (E) mRNA functional life time prolongation factors. All modifications result in prolonged mRNA functional life times as compared to the control. Similar trends are observed in A549 (D) and Huh7 (E) cells. Error bars in (D) and (E) indicate standard deviation. | ||
If there was no protein degradation (β = 0), the amount of protein inside a cell would run into a steady state level as a consequence of a balanced flux of mRNA translation and mRNA degradation. In this case, the expression dynamics follows 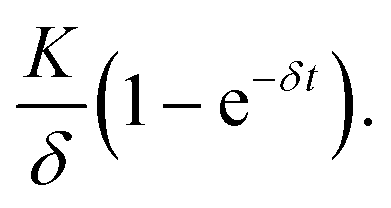 The same would be true in an analogous manner for the case where δ was equal to zero. The superposition of this with the permanent, exponential decay of the d2EGFP protein (following e−βt) results in the characteristic shape of the AUC as shown in Fig. 6A. Fig. 6B and C show the overall mean relative AUCs as well as the “per-experiment” relative AUCs normalized to the mean AUC of the control, the latter being the AUC of protein expression after transfection with the control construct. In both cell types, we find the highest relative AUCs for the 3′ UTR- and the 5′ + 3′ UTR-stabilized construct. This is consistent with the observed long functional half-lives for these constructs, because they contribute to the AUC as seen in eqn (3). The detailed, single-cell AUC distributions can be found in Fig. S2 of the ESI.†
The same would be true in an analogous manner for the case where δ was equal to zero. The superposition of this with the permanent, exponential decay of the d2EGFP protein (following e−βt) results in the characteristic shape of the AUC as shown in Fig. 6A. Fig. 6B and C show the overall mean relative AUCs as well as the “per-experiment” relative AUCs normalized to the mean AUC of the control, the latter being the AUC of protein expression after transfection with the control construct. In both cell types, we find the highest relative AUCs for the 3′ UTR- and the 5′ + 3′ UTR-stabilized construct. This is consistent with the observed long functional half-lives for these constructs, because they contribute to the AUC as seen in eqn (3). The detailed, single-cell AUC distributions can be found in Fig. S2 of the ESI.†
Functional life time prolongation factor
The functional life time prolongation factors for A549 and Huh7 cells are shown in Fig. 6D and E, respectively. As expected, all UTR-flanked constructs yield functional life time prolongation factors higher than one, meaning that the insertion of UTRs at either end causes mRNA stabilization. However, the 3′ UTR mRNA construct shows longer mRNA functional life times than the 2 × 3′ UTR construct. Similarly, the 5′ + 3′ UTR construct is more persistent than the 5′ + 2 × 3′ construct. These results hold true for both cell types. Interestingly, the stabilizing effects are significantly more pronounced in A549 cells than in Huh7 cells in all cases.Discussion
Determination of mRNA functional stability and its expression are two major factors to be considered when it comes to developing new mRNA therapeutics. Here, we used different combinations of UTRs, a 5′ UTR, 3′ UTR, a 5′ + 3′ UTR, 5′ + 2 × 3′ UTR, and two copies 3′ UTR to improve mRNA in terms of functional stability and its expression. The AUC of the d2EGFP time-course is also evaluated, because the total protein expression is relevant for a sustained therapeutic effect. In order to get detailed time-resolved data and monitor protein expression dynamics at the single-cell level, we used microstructured single-cell arrays for parallel, quantitative measurements of mRNA stability and translational efficiency. The regular arrangement of cells guaranteed reproducible microenvironments and enabled fast and automated image-analysis, which are prerequisites for comparative, high-throughput single-cell studies. The approach allows the determination of distribution functions for (i) protein half-life, (ii) expression rates, and (iii) functional mRNA half-life.In both A549 and Huh7 cells, mean protein half-lives of d2EGFP were narrowly distributed and independent of the UTR sequence. The calculated half-life values of 4.5 hours for A549 cells and 7.4 hours for Huh7 cells could be attributed to cell type specific differences between the compared cell lines. Such cell specific differences in d2EGFP half-life have been published previously. A study in NIH3T3 cells using a similar imaging cytometry approach recorded a half-life of 2.8 h within a measurement window of 12 hours.34 An even shorter half-life of less than two hours has been reported for CHO cells by Li et al.35 Here, protein degradation was measured by Western blotting and flow cytometry for three hours only.
To validate our findings from single-cell data analysis, we additionally determined d2EGFP life times in direct measurements using cycloheximide (see Fig. S3†). We found shorter life times as compared to the values observed from single-cell data analysis. This might be due to the fact that in single-cell data analysis, a constant initial number of mRNA molecules was assumed as part of the combined expression rate K = m0 × kTL (see eqn (1)). However, regardless of the fact that cells have been washed after one hour incubation time, it is still likely that the number of mRNA molecules is not constant from the start of observation. As a consequence, mRNA molecules that are available for translation later on, leading to protein expression, might result in longer half-life values obtained from single-cell expression time-course fitting. When we compare the mean half-life determined for A549 cells with the mean half-life determined for Huh7 cells, we find the same ratio of roughly 1.64 for both measurement methods. Also, even a possible systematic over-estimation of mRNA and protein half-lives does not change the qualitative order of the mRNA performance as determined by us.
The expression rate depends on the initial number of mRNA molecules, m0, as well as on the translation rate KTL. Note that the number of successfully delivered mRNA molecules varies due to the intrinsic stochasticity of the delivery process. The mean number of mRNA molecules, however, is expected to be the same, since the transfection protocol has scrupulously been kept up in all experiments. In contrast, the translational activity (KTL) of the various UTR constructs might vary. Still, the fact that the distributions as well as the mean values of the expression rate K are rather similar for all constructs (see Fig. 4A and D) indicates that the translation rate is merely influenced by the inserted UTRs.
The parameter of highest interest is the mRNA functional half-life. Here, we compared functional mRNA half-life to physical mRNA half-life. Our results with single-cell transfection studies suggest that any insertion of 5′ or 3′ UTRs into the mRNA sequence increases its functional half-life which was observed for all modifications tested in this study (see Fig. 4 and 6) as measured by fluorescence microscopic imaging and FC (see Fig. 1 and 2). In contrast to the functional mRNA half-life, the physical mRNA half-life determined by qRT-PCR showed a decrease in mRNA stability for 5′, 3′, 5′ + 2 × 3′ and 2 × 3′ UTR in both cell lines (see Fig. S5A and B†).
One major difference is that the physical half-life as measured by qPCR does not reflect the translational (“functional”) productivity of a given mRNA construct. qPCR as used here merely quantifies the physical presence of an mRNA fragment and is inappropriate to predict the utility of an mRNA construct in a therapeutic application. Similar findings have been reported by Gallie et al.36 In contrast, the functional half-life as measured here is a function of the translational capacity of an mRNA, determines the amount of total protein produced (Area Under the Curve) and predicts the utility of an mRNA construct for a therapeutic application. For a therapeutic mRNA, it can be imperative that the molecule is functional for as long or as short as possible and yields a maximum or a minimum of protein. Furthermore, the heterogenic distribution of the functional half-lives points out the importance of single-cell measurement techniques, because these effects are obscured in ensemble measurements (see Fig. 2, 4 and S5A† and B). An interesting conclusion from the observed discrepancy of physical and functional half-lives is that obviously cells make more productive use of moderate amounts of mRNA than of the initial high amounts present at the early time points after transfection. In fact, the impact of the UTRs examined here on mRNA productivity become increasingly evident at the later time points after transfection.
Interestingly, a positive effect on protein expression was observed for 5′ UTR alone, although so far, no known motif in the CYBA 5′ UTR has been discovered. For the first time, we could show that CYBA UTRs at either end suffice to increase both peak and persistence of protein expression in both cell lines. These findings are consistent with publications claiming individual or synergistic behaviour of 5′ UTRs and 3′ UTRs.15 In contrast to Holtkamp et al.,17 no additional increase in protein expression or mRNA functional stability could be observed with two sequential copies of the 3′ UTR as compared to one single 3′ UTR (see Fig. 4). Conversely, it even resulted in shorter functional life times both for 5′ + 3′ versus 5′ + 2 × 3′ UTR insertion and for 3′ versus 2 × 3′ UTR insertion. This might be due to the fact that a different type of cells (namely dendritic cells) was used in the study by Holtkamp et al.17 Similar cell type-specific effects have been reported for hepatocytes, too.37 Another contributing factor affecting mRNA functional stability might be the secondary structure of the different mRNAs. Such effects of mRNA secondary structure in regulating gene expression have been reported before.38,39
Important structural characteristics together with their minimum free energy for the mRNA constructs used in the current study are summarized in Table S1.† The persistent protein expression of the 5′ + 3′ UTR stabilized construct could be due to binding of the 5′ to the 3′ end, which facilitates circularization of the mRNA.20 Because we could not find any stable secondary structures within the 5′ UTR, we assume that this feature enables an early expression onset.40 In contrast, we identified secondary structures within the 3′ UTRs. These might protect the mRNA from the 3′–5′ degradation pathway. Two 3′ UTRs showed even more secondary structures (two hairpins) with the best minimum free energy, indicating more persistent expression. Taken together, these findings could be the explanation for the inferior onset expression of the 2 × 3′ UTR compared to the 5′ UTR and the persistent expression at later time points of mRNA constructs containing 3′ UTRs.
In accordance with protein half-lives, longer functional half-life values were obtained for mRNAs stabilized with UTRs. This was observed in both cell lines with cell-specific differences most likely affecting the absolute values. In A549 cells, mRNA functional half-lives for the constructs with UTRs ranged from 13.0 h to 23.0 h as compared to 5.8 h for the control. In Huh7 cells, functional half-lives from 9.9 h to 13.6 h were measured for UTR-containing constructs, as opposed to a functional half-life of 7.8 h for the control mRNA. The functional half-life of the 3′ UTR-flanked mRNA in A549 cells is in good agreement with mRNA life times of similarly stabilized mRNAs that were reported previously.17,27 The fact that functional stability and decay kinetics of mRNA and protein differ in different cell types is most likely due to differences in the complex networks of interactions between mRNA and proteins which are very likely to be cell-type dependent.
Taken together, our results in both A549 and Huh7 cells, independent of the analysis method (FC or single-cell analysis), suggest that sustained, high levels of protein expression can be induced by CYBA UTR flanked mRNA. The choice of UTR combination depends on the need of the experiment of application. Where persistent protein expression with reduced mRNA decay is desired, mRNA flanked with a 3′ UTR alone might serve the purpose. However, the combination of 5′ + 3′ UTR results in additional desirable features of early onset, high peak and cumulative protein expression.
Our study demonstrates that micro-array based single-cell analysis of mRNA-induced protein expression is a means to characterize and improve kinetic properties of mRNA constructs. Using automated time-lapse microscopy and automated image analysis, we were able to assess the intracellular bioavailability of different mRNA constructs at the single-cell level in a highly parallel format. The micro-arrays provide a standardized platform to transfect arrayed cells at high number density on a chip and with defined boundary conditions such as the cell area. This allows to acquire single-cell time traces from equally treated cells and hence a more reliable basis to identify sequences yielding sustained protein expression. We found prolonged persistence of protein expression for constructs stabilized by UTR insertions using a single-cell model and FC analysis in two cell types. This finding is desired in case of developing mRNA therapeutics. Messenger RNA constructs with persistent protein expression over a period of time (AUC) is desirable and allows proper reduced dosing into a patient with a final therapeutic outcome. We hope that our study helps to further refine strategies for the manipulation of mRNA stability for future pharmaceutical applications.
Experimental
Plasmid vectors
Destabilized Enhanced Green Fluorescent Protein (d2EGFP) was excised from pd2EGFP-N1 (Clontech) and cloned in pVAXA120 (ref. 3) to generate pVAXA120-d2EGFP. Based on previously published data with respect to mRNA stability, preselected 5′ and 3′ UTR sequences of CYBA gene were synthesized by Eurofins MWG (Germany) and cloned upstream (5′ UTR) and/or downstream (3′ UTR or 2 × 3′ UTR) of d2EGFP in pVAXA120-d2EGFP, thereby generating the constructs with respective UTR combinations.mRNA production
To generate in vitro transcribed mRNA (IVT mRNA), plasmids were linearized downstream of the poly-A tail by NotI digestion and purified by chloroform extraction and ethanol precipitation. Purified linear plasmids were used as template for in vitro transcription using RiboMax Large Scale RNA production System-T7 (Promega, Germany). Anti-Reverse Cap Analog (ARCA) was added to the reaction mix to generate 5′ capped mRNA. Additionally for the production of SNIM® RNAs, chemically modified nucleotides namely methyl-CTP and thio-UTP (Jena Bioscience, Germany) were added to a final concentration of ATP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CTP:UTP:methyl-CTP:thio-UTP:GTP of 7.57 mM:5.68 mM:5.68 mM:1.89 mM:1.89 mM:1.21 mM. The complete IVT mix was incubated at 37 °C for 2 hours followed by a DNA disgestion with DNaseI for 20 minutes at 37 °C. RNA was precipitated with ammonium acetate (final concentration 2.5 M) and washed with 70% EtOH. The washing step was performed twice. Finally, the RNA pellet was re-suspended in RNAse-free water. All mRNAs were verified on 1% agarose gels. A schematic representation of an exemplary mRNA construct can be seen in Fig. 1A. The exact sequences of the UTRs are given in the ESI† (Table S1).
:CTP:UTP:methyl-CTP:thio-UTP:GTP of 7.57 mM:5.68 mM:5.68 mM:1.89 mM:1.89 mM:1.21 mM. The complete IVT mix was incubated at 37 °C for 2 hours followed by a DNA disgestion with DNaseI for 20 minutes at 37 °C. RNA was precipitated with ammonium acetate (final concentration 2.5 M) and washed with 70% EtOH. The washing step was performed twice. Finally, the RNA pellet was re-suspended in RNAse-free water. All mRNAs were verified on 1% agarose gels. A schematic representation of an exemplary mRNA construct can be seen in Fig. 1A. The exact sequences of the UTRs are given in the ESI† (Table S1).
Flow cytometry (FC)
The experimental set-up looks as follows: 20000 cells in 150 μl medium were seeded per well in 96-well plates and transfected 24 hours post-seeding. Cells were transfected at a dose of 5 pg mRNA per cell using the commercial transfection reagent Lipofectamine™ 2000. Complexes were prepared at a ratio of 2.5 μl Lipofectamine™ 2000 per 1 μg mRNA. For the formation of lipoplexes, Lipofectamine™ 2000 and mRNA were diluted separately in OptiMEM transfection medium in a total volume of 50 μl, each. These mixtures were incubated at room temperature for 5 minutes. The mRNA solution was then mixed with the Lipofectamine™ 2000 solution, followed by another 20 minutes of incubation at room temperature. After incubation, 900 μl of OptiMEM were added to the lipoplex solution. Finally, 50 μl of the complex solution were added to the cells and incubated for 1 hour. For every mRNA construct, biological triplicates were prepared. After incubation, the lipoplex-solution was discarded and fresh 150 μl medium was added to each well. d2EGFP expression was measured after 8, 24, 36, 48, 60 and 72 hours using FC. Fluorescence microscopy images were taken at each of these time points. For FC measurements, the cell culture medium was discarded and the cells were washed with 1× DPBS (Gibco Life Technology). Subsequently, 20 μl of TrypLE Express (Gibco Life Technology) were added per well and incubated for 5min at 37 °C. The reaction was neutralized by adding 80 μl 1 × PBS, supplemented with 2% FBS. Cells were mixed by pipetting and were transferred into a 96 well plate appropriate for flow cytometric measurements. Finally, 5 μl of Propidium iodide (final concentration 1 μg ml−1) were added per well and measured with Attune Auto Sampler (Applied Biosystems). Please note that fluorescence images were taken prior to FC analysis with a JULY™ microscope.
Quantitative real-time PCR
A qRT-PCR analysis was used to determine the d2EGFP mRNA amount at time intervals of 4, 8, 24, 36, 48, 60 and 72 hours in A549 and Huh7 cells. Additionally, the mRNA expression kinetic itself was used to calculate the mRNA half-life of each UTR. Here, the cells were transfected similarly to the protocol described above (see FC). A cell density of 200000 cells per well was found to be sufficient for RNA isolation. RNA isolation was performed according to the manufacturer's protocol using NucleoSpin RNA (Macherey Nagel). The isolated total RNA was examined in RNA concentration and quality by spectrophotometric measurements and gel analysis. Further, 0.5 μg of the total RNA of each UTR constructs and the control were used for cDNA synthesis using Oligo(dT)s from First Strand cDNA Synthesis Kit (Thermo Scientific). Equivalent amounts of cDNA (diluted 1:50) were tested with 125 nM of each d2EGFP-Primer (forward Primer: 5′-CAA CCA CTA CCT GAG CAC CC-3′; reverse Primer:5′-GTC CAT GCC GAG AGT GAT CC-3′) using SsoAdvanced™ Universal SYBR® Green Supermix (BioRad). As a standard for the absolute quantification, pure d2EGFP mRNA produced by IVT was used for synthesis of cDNA. Absolute mRNA quantification was performed on a Lightcycler 96 device (Roche).
Surface patterning and sample preparation
Microstructured surfaces were produced by selective oxygen plasma treatment (Femto Diener, 40 W for 3 min) on a top as substrate (ibidi GmbH) with subsequent passivation. Selectivity was achieved using a polydimethylsiloxane (PDMS) stamp (cast from a master produced by photolithography) as a mask. The parts exposed to plasma were passivated by incubation for 30 min with PLL(20k)-g(3.5)-PEG(2k) at a concentration of 1 mg ml−1 in aqueous buffer (10 mM HEPES pH 7.4 and 150 mM NaCl). Thereafter, the samples were rinsed with PBS and the PDMS stamps were removed. The foils were then fixed to adhesive six-channel slides (sticky μ-slide VI). Each channel was filled with a solution of 50 μg ml−1 fibronectin in PBS for one hour to render the remaining sectors cell-adhesive. Probes were thoroughly rinsed with PBS three times. The samples were stored in cell medium at room temperature before cell seeding. For this study, square adhesion sites of 30 μm × 30 μm were used because this size turned out to be reasonable for single-cell adhesion of A549 as well as Huh7 cells. Cells were seeded at a density of 10000 cells per channel so that roughly one cell could adhere on each cell-adhesive island. To obtain fluorescent micropatterns as shown in Fig. 3A, a mixture of 20 μg ml−1 fibronectin and 30 μg ml−1 fibrinogen conjugated with Alexa Fluor 488 was used.
Materials
FBS, Leibovitz's L-15 Medium (Gibco), Lipofectamine™ 2000, and OptiMEM (Gibco) were purchased from Invitrogen, Germany. Sterile PBS was prepared in-house. Ham's F-12K, DMEM, and Trypsin-EDTA were purchased from c.c.pro GmbH, Germany. Channel slides were purchased from ibidi, Germany. Fibronectin was purchased from Yo Proteins, Sweden. PLL-g-PEG was purchased from SuSoS AG, Switzerland. Alexa Fluor 488 was purchased from Life Technologies, Germany. The plasmid pd2EGFP-N1 was purchased from BD Biosciences Clontech, Germany.Cell culture
A human alveolar adenocarcinoma cell line (A549, ATCC CCL-185) was grown in Ham's F12K medium supplemented with 10% FBS. A human hepatoma epithelial cell line (Huh7, JCRB0403, JCRB Cell Bank, Japan) was cultured in DMEM medium, supplemented with 10% fetal bovine serum. All cell lines were grown in a humidified atmosphere at 5% CO2 level.In vitro transfection
Three hours prior to transfection, 10000 cells per channel were seeded in a 6-channel slide. Cells were transfected at a dose of 5 pg mRNA per cell using the commercial transfection reagent Lipofectamine™ 2000 at a ratio of 2.5 μl Lipofectamine™ 2000 per 1 μg mRNA. The complex formation was prepared as follows: Lipofectamine™ 2000 and mRNA were separately diluted in OptiMEM transfection medium to add up to a total volume of 45 μl, each. These mixtures were incubated at room temperature for 5 minutes. The Lipofectamine™ 2000 solution was then mixed with the mRNA solution, followed by another 20 minutes of incubation at room temperature. Please note that the microchannels were never empty during all subsequent rinsing steps: immediately before transfection, the cells were washed with PBS. Finally, the lipoplex solutions containing different mRNAs constructs were filled into the six channels. All five different mRNA constructs plus the reference construct could thus be measured under the same experimental conditions. The cells were incubated in a total transfection volume of 90 μl at 37 °C (5% CO2 level) for one hour. The transfection medium was thereafter removed and the cells were washed with PBS. Subsequently, the cells were re-incubated with Leibovitz's L-15 Medium containing 10% FBS. A drop of anti-evaporation oil (ibidi GmbH, Germany) was added on top of each medium reservoir before microscopic monitoring of d2EGFP expression.
Data acquisition and quantitative image analysis
Live-cell imaging was performed on a motorized inverted microscope (Nikon, Eclipse Ti-E) equipped with an objective lens (CFI PlanFluor DL-10×, Phase1, N.A. 0.30; Nikon) and with a temperature-controlled mounting frame for the microscope stage. We used an ibidi heating system (Ibidi GmbH, Germany) with a temperature controller to stabilize the temperature of the samples at 37 °C (±2 °C) throughout the measurements. To acquire cell images, we used a cooled CCD camera (CLARA-E, Andor). A mercury light source (C-HGFIE Intensilight, Nikon) was used for illumination and a filter cube with the filter set 41024 (Chroma Technology Corp., BP450-490, FT510, LP510-565) was used for d2EGFP detection. An illumination shutter control was used to prevent bleaching. Images were taken at 10 fold magnification with a constant exposure time of 600 ms at 10 minute-intervals for at least 25 hours post-transfection. Fluorescence images were consolidated into single-image sequence files. Quantitative analysis of characteristic parameters of single-cell expression kinetics allows the comparison of various vector performances in terms of expression efficiency and stability. Image analysis consisted of several steps and was done using in-house-developed software based on ImageJ. First, a rectangular grid was overlaid with the original time-lapse movie and adjusted to the size and orientation of the underlying cell-pattern. Next, the software automatically detected d2EGFP-expressing cells by reading out the fluorescence intensities of all squares. Unoccupied squares were used for background correction. The software calculates the cells' fluorescence over the entire sequence and connects corresponding intensities to time courses of the fluorescence per cell. Finally, single-cell fluorescence intensities per square were extracted.
Data were then analyzed as described recently by fitting each time-course with the analytical solution for mRNA-induced protein expression (see eqn (1)) using IgorPro software, which is the solution to the differential equations for mRNA and d2EGFP,
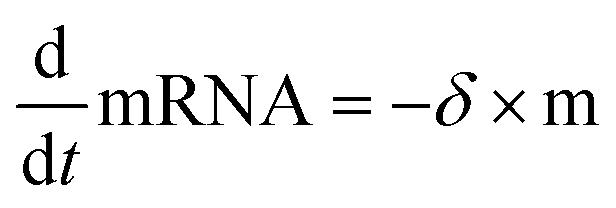 | (4) |
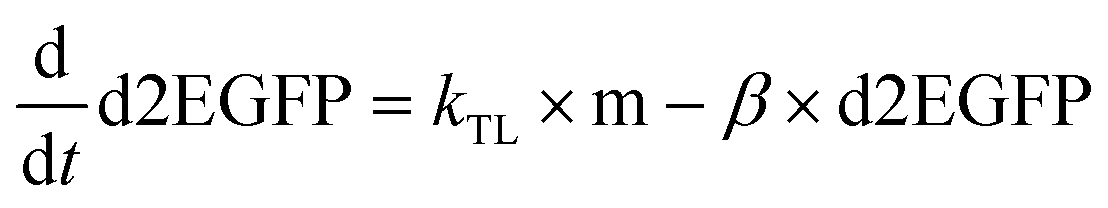 | (5) |
Conclusions
Single-cell micro-arrays present themselves as a highly parallel and standardized tool for rapid transfection and quantitation of mRNA expression kinetics in a much shorter time frame compared to the conventional, population-based molecular biological measurements. The resulting data at the single-cell level are in good correlation with those measured with quantitative real-time and FC analysis. As such, this work represents an advance in automated assessment and predictive modeling of mRNA structures with respect to their expression kinetics and functional stability at the single-cell level.Conflict of interest statement
CP and CR are founders and shareholders of Ethris GmbH, a company which develops mRNA therapeutics. MKA is an employee of Ethris GmbH.Acknowledgements
We thank Max Albert for wafer fabrication, Gerlinde Schwake for helpful discussions and Thomas S. Ligon for proof-reading of the manuscript. Financial support by the German Federal Ministry of Education and Research Go-Bio grant 0315986, by the Deutsche Forschungsgemeinschaft (DFG) via project B1 within the SFB 1032, the Excellence Cluster ‘Nanosystems Initiative Munich (NIM)’, and FP7 EU grants NanoTransKinetics is gratefully acknowledged. Financial support by the Elitenetwork of Bavaria is gratefully acknowledged by CL.Notes and references
- G. Tavernier, O. Andries, J. Demeester, N. N. Sanders, S. C. De Smedt and J. Rejman, J. Controlled Release, 2011, 150, 238–247 CrossRef CAS PubMed.
- A. Yamamoto, M. Kormann, J. Rosenecker and C. Rudolph, Eur. J. Pharm. Biopharm., 2009, 71, 484–489 CrossRef CAS PubMed.
- M. S. D. Kormann, G. Hasenpusch, M. K. Aneja, G. Nica, A. W. Flemmer, S. Herber-Jonat, M. Huppmann, L. E. Mays, M. Illenyi, A. Schams, M. Griese, I. Bittmann, R. Handgretinger, D. Hartl, J. Rosenecker and C. Rudolph, Nat. Biotechnol., 2011, 29, 154–157 CrossRef CAS PubMed.
- M. Esteller, Nat. Rev. Genet., 2011, 12, 861–874 CrossRef CAS PubMed.
- K. Kariko and D. Weissman, Curr. Opin. Drug Discovery Dev., 2007, 10, 523 CAS.
- G. Pesole, G. Grillo, A. Larizza and S. Liuni, Briefings Bioinf., 2000, 1, 236–249 CrossRef CAS.
- T. V. Pestova, J. R. Lorsch and C. U. Hellen, Translational Control in Biology and Medicine, 3rd edition of Cold Spring Harbor Monograph Archive, 2007, vol. 48, pp. 87–128 Search PubMed.
- L. Barrett, S. Fletcher and S. Wilton, Cell. Mol. Life Sci., 2012, 69, 3613–3634 CrossRef CAS PubMed.
- F. Mignone, G. Grillo, F. Licciulli, M. Iacono, S. Liuni, P. J. Kersey, J. Duarte, C. Saccone and G. Pesole, Nucleic Acids Res., 2005, 33, D141–D146 CrossRef CAS PubMed.
- M. J. Moore, Science, 2005, 309, 1514–1518 CrossRef CAS PubMed.
- X. Pichon, L. A. Wilson, M. Stoneley, A. Bastide, H. A. King, J. Somers and A. E. Willis, Curr. Protein Pept. Sci., 2012, 13, 294–304 CrossRef CAS PubMed.
- P. A. C. 't Hoen, M. Hirsch, E. J. D. Meijer, R. X. D. Menezes, G. J. van Ommen and J. T. D. Dunnen, Nucleic Acids Res., 2011, 39, 556–566 CrossRef PubMed.
- L. V. Sharova, A. A. Sharov, T. Nedorezov, Y. Piao, N. Shaik and M. S. Ko, DNA Res., 2009, 16, 45–58 CrossRef CAS PubMed.
- G. Pesole, F. Mignone, C. Gissi, G. Grillo, F. Licciulli and S. Liuni, Gene, 2001, 276, 73–81 CrossRef CAS PubMed.
- F. Gebauer and M. W. Hentze, Nat. Rev. Mol. Cell Biol., 2004, 5, 827–835 CrossRef CAS PubMed.
- L. Tillmar, C. Carlsson and N. Welsh, J. Biol. Chem., 2002, 277, 1099–1106 CrossRef CAS PubMed.
- S. Holtkamp, S. Kreiter, A. Selmi, P. Simon, M. Koslowski, C. Huber, O. Tureci and U. Sahin, Blood, 2006, 108, 4009–4017 CrossRef CAS PubMed.
- C. H. D. Moor, H. Meijer and S. Lissenden, Semin. Cell Dev. Biol., 2005, 16, 49–58 CrossRef PubMed.
- N. L. Garneau, J. Wilusz and C. J. Wilusz, Nat. Rev. Mol. Cell Biol., 2007, 8, 113–126 CrossRef CAS PubMed.
- E. Szostak and F. Gebauer, Briefings Funct. Genomics, 2013, 12(1), 58–65 CrossRef CAS PubMed.
- S. Tyagi, Nat. Methods, 2009, 6, 331–338 CrossRef CAS PubMed.
- H. Y. Park, H. Lim, Y. J. Yoon, A. Follenzi, C. Nwokafor, M. Lopez-Jones, X. Meng and R. H. Singer, Science, 2014, 343, 422–424 CrossRef CAS PubMed.
- C. Miller, B. Schwalb, K. Maier, D. Schulz, S. Dümcke, B. Zacher, A. Mayer, J. Sydow, L. Marcinowski, L. Dölken, D. E. Martin, A. Tresch and P. Cramer, Mol. Syst. Biol., 2011, 7 Search PubMed.
- T. Nolan, R. E. Hands and S. A. Bustin, Nat. Protoc., 2006, 1, 1559–1582 CrossRef CAS PubMed.
- M. Rabani, J. Z. Levin, L. Fan, X. Adiconis, R. Raychowdhury, M. Garber, A. Gnirke, C. Nusbaum, N. Hacohen, N. Friedman, I. Amit and A. Regev, Nat. Biotechnol., 2011, 29, 436–442 CrossRef CAS PubMed.
- B. Schwanhausser, D. Busse, N. Li, G. Dittmar, J. Schuchhardt, J. Wolf, W. Chen and M. Selbach, Nature, 2011, 473, 337–342 CrossRef PubMed.
- C. Leonhardt, G. Schwake, T. R. Stögbauer, S. Rappl, J.-T. Kuhr, T. S. Ligon and J. O. Rädler, Nanomed.: Nanotechnol., Biol. Med., 2014, 10, 679–688 CrossRef CAS PubMed.
- P. J. F. Rottgermann, A. P. Alberola and J. O. Radler, Soft Matter, 2014, 10, 2397–2404 RSC.
- P. Corish and C. Tyler-Smith, Protein Eng., 1999, 12, 1035–1040 CrossRef CAS PubMed.
- J. J. Rossi, Nat. Cell Biol., 2005, 7, 643–644 CrossRef CAS PubMed.
- U. Sheth and R. Parker, Cell, 2006, 125, 1095–1109 CrossRef CAS PubMed.
- A. Jakymiw, K. M. Pauley, S. Li, K. Ikeda, S. Lian, T. Eystathioy, M. Satoh, M. J. Fritzler and E. K. L. Chan, J. Cell Sci., 2007, 120, 1317–1323 CrossRef CAS PubMed.
- R. Parker and U. Sheth, Mol. Cell, 2007, 25, 635–646 CrossRef CAS PubMed.
- M. Halter, A. Tona, K. Bhadriraju, A. L. Plant and J. T. Elliott, Cytometry, Part A, 2007, 71A, 827–834 CrossRef CAS PubMed.
- X. Li, X. Zhao, Y. Fang, X. Jiang, T. Duong, C. Fan, C.-C. Huang and S. R. Kain, J. Biol. Chem., 1998, 273, 34970–34975 CrossRef CAS PubMed.
- D. R. Gallie, Genes Dev., 1991, 5, 2108–2116 CrossRef CAS PubMed.
- B. Kren and C. Steer, FASEB J., 1996, 10, 559–573 CAS.
- J.-M. Chen, C. Férec and D. Cooper, Hum. Genet., 2006, 120, 301–333 CrossRef CAS PubMed.
- P. Gaspar, G. Moura, M. A. Santos and J. L. Oliveira, Nucleic Acids Res., 2013, 41, e73–e73 CrossRef CAS PubMed.
- J. R. Babendure, J. L. Babendure, J.-H. Ding and R. Y. Tsien, RNA, 2006, 12, 851–861 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1, S2; Figure S1–S5. See DOI: 10.1039/c5lc00749f |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2015 |