
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards personalized medicine: chemosensitivity assays of patient lung cancer cell spheroids in a perfused microfluidic platform†
Janine
Ruppen
ab,
Franziska D.
Wildhaber
a,
Christoph
Strub
a,
Sean R. R.
Hall
cd,
Ralph A.
Schmid
cd,
Thomas
Geiser
de and
Olivier T.
Guenat
*ace
aARTORG Lung Regeneration Technologies Lab, University of Bern, Murtenstrasse 50, CH-3010 Bern, Switzerland. E-mail: olivier.guenat@artorg.unibe.ch; Fax: +41 31 632 7576; Tel: +41 31 632 7608
bGraduate School for Cellular and Biomedical Sciences, University of Bern, Switzerland
cDivision of General Thoracic Surgery, University Hospital of Bern, Bern, Switzerland
dDepartment of Clinical Research, University of Bern, Bern, Switzerland
eDivision of Pulmonary Medicine, University Hospital of Bern, Bern, Switzerland
First published on 2nd June 2015
Abstract
Cancer is responsible for millions of deaths worldwide and the variability in disease patterns calls for patient-specific treatment. Therefore, personalized treatment is expected to become a daily routine in prospective clinical tests. In addition to genetic mutation analysis, predictive chemosensitive assays using patient's cells will be carried out as a decision making tool. However, prior to their widespread application in clinics, several challenges linked to the establishment of such assays need to be addressed. To best predict the drug response in a patient, the cellular environment needs to resemble that of the tumor. Furthermore, the formation of homogeneous replicates from a scarce amount of patient's cells is essential to compare the responses under various conditions (compound and concentration). Here, we present a microfluidic device for homogeneous spheroid formation in eight replicates in a perfused microenvironment. Spheroid replicates from either a cell line or primary cells from adenocarcinoma patients were successfully created. To further mimic the tumor microenvironment, spheroid co-culture of primary lung cancer epithelial cells and primary pericytes were tested. A higher chemoresistance in primary co-culture spheroids compared to primary monoculture spheroids was found when both were constantly perfused with cisplatin. This result is thought to be due to the barrier created by the pericytes around the tumor spheroids. Thus, this device can be used for additional chemosensitivity assays (e.g. sequential treatment) of patient material to further approach the personalized oncology field.
1. Introduction
Despite huge efforts by researchers and pharmaceutical companies to find new treatments, cancer remains one of the leading causes of death worldwide. With 1.59 million deaths in 2012,1 lung cancer-related fatalities represent the highest fraction of mortalities in the world among the different cancer types. To reduce this number, a paradigm shift towards a tailored treatment for each patient is emerging.2 Although personalized genomic mutation assays are already performed in clinics to identify specific types of cancers and their corresponding targeted therapies, tumor recurrence and high variability in disease patterns remain an important problem.3 To circumvent these issues, one further envisages testing and analysing the patient's own cells in order to better predict the individual response to a specific therapy regimen.One of the challenges associated with such assays is related to the small amount of patient's material available for testing. Typically, a few micrograms of material can be obtained from biopsies performed using fine needles of which 0.5 to 1 million cells are typically extracted.4 The first fraction of these cells is reserved for histological and genomic assessments, while the second, from which undesired cells are removed, is available for further analysis. Considering the tumor inhomogeneity and the unavoidable loss of cells during the enzymatic digestion procedure, the number of relevant cells available to perform reliable assays is very limited. Another challenge of personalized medicine is in the lack of appropriate in vitro models that have the capability to predict the chemotherapeutic response for each patient. Traditional in vitro models often fail in predicting the in vivo efficacy of specific chemotherapeutic agents5 and are thus starting to be replaced by spheroid models that better reflect the in vivo behavior of cells in tumor tissues.5–7 Besides the three-dimensional cellular assembly, the tumor microenvironment consists of a complex combination of extracellular matrix, stroma cells and interstitial fluids. This complex composition of the tumor microenvironment influences the tumor cell phenotype via mechanical and biochemical factors that ultimately contribute to tumor growth.8,9 Microfluidics, which enables the accurate control of cell culture conditions, can ideally reproduce specific aspects of the tumor microenvironment, such as the continuous transport of nutrients and oxygen as well as the removal of cellular waste products.10,11 In addition, microfluidic systems, in which individual cells can easily and accurately be manipulated,12 make them ideal to handle scarce patient material and are thought to represent the platform of choice for the next generation of in vitro cancer models.13,14
A further challenge in personalized medicine models, besides the use of primary cells15 and the small amount of patient's cells available, represents the reproducibility of in vitro tumors. So far, several groups reported concerning the formation of spheroids on chip using either gravity traps16 or trapping systems based on the creation of small vortexes17 or removable trapping barriers.18 However, the creation of homogeneous spheroid replicates from limited patient material has not yet been addressed. Additionally, a tumor has to be seen as a unique and complex organ that interacts with its microenvironment.19 The tumor microenvironment not only supports the tumor in maintaining proliferation,20 but represents also a barrier for drug delivery.21 Drugs have to overcome several barriers before reaching the tumor where they shall destroy the tumor cells. Barriers for intravenously administered drugs are, for instance the blood vessels, walls through which drugs have to extravasate, or the tumor interstitial site containing extracellular matrix, cancer-associated fibroblasts or pericytes.21 Thus, the tumor microenvironment plays a critical role in tumor development as well as drug administration. Therefore, co-culture systems further strengthen the reliability of in vitro model as in vivo-like systems. In addition, Amann and colleagues22 observed more compact and round microtissue surfaces in co-cultures than in monocultures, showing the importance of co-culture models. Especially pericytes, which play an essential role in the stabilization of microvessels,23 are interesting as they initially accumulate at the interface of tumor and host tissue.24
In this study, we present a microfluidic system that enables the homogeneous distribution of cells and the formation of spheroids in eight microwells using very low number of cells that corresponds to a fraction of those obtained from a tumor biopsy. Cells from a malignant pleural mesothelioma cell line as well as cells obtained from patients with non-small cell lung adenocarcinoma (NSCLC) following lung resection are tested on the chip in terms of distribution homogeneity and spheroid formation. Further, the cell line is used to test cell viability and proliferation on chip to assess the culture conditions. In addition, a chemosensitivity assay with cisplatin is carried out under perfusion using primary human lung adenocarcinoma spheroids. Primary spheroids are cultured either as monoculture with epithelial cells (EpCAM + CD73 + CD90−) only or as co-culture using epithelial cells and pericytes (EpCAM − CD73 + CD90+) from patient-derived primary lung adenocarcinoma.
2. Materials and methods
2.1. Design of the microfluidic device for spheroid formation
The preparation of samples from patient material starts with the enzymatic digestion of tissues obtained from a biopsy or a lung resection. This step is followed by the selection of the cells of interest by fluorescence-activated cell sorting (FACS) and the formation of spheroids using these cells. To minimize the manipulation of the delicate spheroids, we chose to load the suspended FACS-sorted cells directly on the chip, where they aggregate and form spheroids.The first design prerequisite of the microfluidic device is to produce samples with equal number of cells to enable the formation of homogeneous replicates. The second is to do so with a small number of FACS-sorted cells obtained from the available patient material. To address these requirements we were inspired by the dichotomy of the in vivo microvasculature that distribute red blood cells equally to ensure a homogeneous distribution at branch points. A symmetrical tree-like microstructure is designed to distribute the suspended cells evenly in the eight microwells, in which they are trapped by gravity. Per channel, three branches are considered to create a total of eight replicates (ESI† Fig. S1) that enable the secretion of sufficient cytokines or proteases to be detected in the supernatant.11
The dimensions of the microfluidic channels were defined to the minimal lateral resolution of about 100 μm in width that can be achieved by stereolithography, the technique chosen to produce the microfluidic chip. The required lengths of the daughter channels were determined so that the eight microwells would fit within a diameter of 9 mm, which corresponds to the diameter of a well in a 96-well plate (ESI† Fig. S1A). This feature makes the system compatible with modern wide-field high-content imaging systems as well as with standard microplate readers, which will be investigated in further studies.
The trapping of the suspended cells is driven by gravity using a hydrostatic pressure difference between the inlet (loading reservoir) and the outlet (collecting reservoir) of the chip. When the suspended cells are flowing through the 200 μm wide daughter channel 3 and reach the 0.5 mm in diameter microwells, their speed decreases, allowing cell sedimentation at the bottom of the microwells (Fig. 1A).
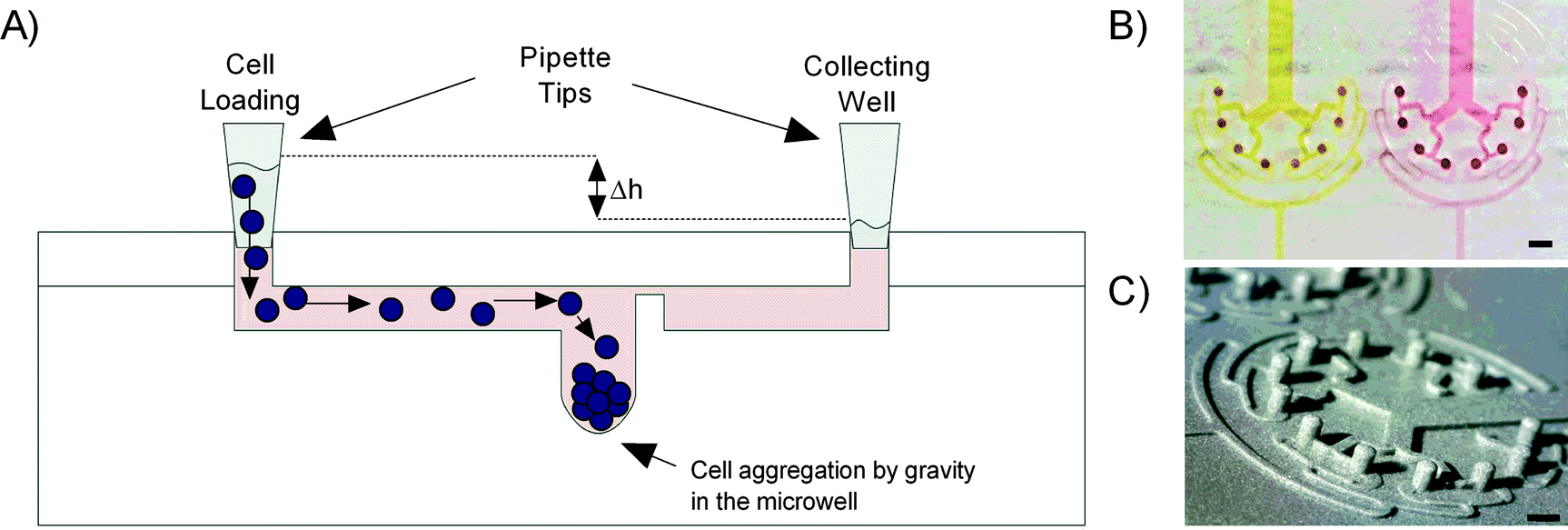 | ||
| Fig. 1 A) Cell loading principle: cells are loaded in the system using hydrostatic pressure and are trapped in the microwells by gravity. B) Top-view picture of two microfluidic channels filled with red and yellow food dyes. C) Image of the epoxy mold with rounded micropillars, representing the negative of the final channels. Scale bar corresponds to 1200 μm. | ||
2.2. Fabrication of the microfluidic device
Soft replicas of polydimethylsiloxane (PDMS, Dow Corning) were cast on an epoxy mold made of Accura Extreme, produced by stereolithography (Proform AG, Switzerland), using standard rapid prototyping protocols.25 Access ports were punched with a 2 mm dermal biopsy punch (Shoney Scientific, India) into a PDMS cover plate. The PDMS cast and cover plate were then cleaned with 100% isopropanol and dried before being exposed to oxygen plasma for 25 s at 650 mTorr (Harrick Plasma, USA) enabling covalent bonding of the two PDMS parts. After bonding, the assembled microfluidic platforms were post-baked overnight at 60 °C in order to strengthen the sealing between the two PDMS parts25 (Fig. 1B). As the process of stereolithography does not allow the production of smooth round-bottom microwells, different processing steps were needed to make round-bottom micropillars. The first PDMS replica was produced using the Accura Extreme mold as cast and cured through baking at 60 °C for 2 hours. Then the PDMS replica was silanized with 1H,1H,2H,2H-perfluoro-octyl-trichlorosilane (Fluorochem, Brunschwig AG) at 80 °C overnight before casting a second PDMS mold out of the silanized PDMS cast. After a curing step of 2 hours at 60 °C in the oven, a drop of PDMS was added on every micropillar of the second PDMS mold by using a needle tip to make round-bottom microwells (ESI† Fig. S2). Then the mold was also cured and silanized. A third PDMS mold was cast out of the silanized second PDMS mold, and also cured and silanized. In the final step, the epoxy casting resin (Weidling C, Weicon) was used to fabricate the final mold out of the third and the last PDMS cast. The mold was cured for 24 h at 40 °C. Finally we checked under a digital microscope (Dino-Lite, IDCP B.V., Netherlands) the formation of the round-bottom microwells (Fig. 1C). After silanization with perfluoro-octyl-trichlorosilane, the epoxy mold was used to fabricate all PDMS casts for the cellular assays as described above. All the experiments were performed using the round-bottom microwells unless otherwise stated.2.3. Cell culture
The microfluidic chip was tested with a mesothelioma cell line (H2052) and with epithelial adenocarcinoma primary cells obtained from patients with lung adenocarcinoma (BE063-T and BE069-T) and squamous carcinoma (BE067-T) as well as primary pericytes from one patient (BE069-T). The protocol for the use of human material for research purposes was approved by the Ethics Committee of the Canton of Bern, CH, and all the patients provided written informed consent for the usage of surgical specimens and materials removed for research purposes. Cells were manipulated in a sterile flow hood and incubated at 37 °C and 5% CO2 unless otherwise stated. Cell culture experiments with the mesothelioma cell line H2052 (ATCC-CRL-5915, ATCC, France) were performed according to a previously described protocol.11 Briefly, cells were maintained in RPMI1640 (Invitrogen) medium containing 10% fetal bovine serum, FBS (Sigma) and 1% of a mixture of penicillin and streptomycin (P/S, Invitrogen). TrypLE™ Express (LubioScience, Invitrogen) was used to harvest cells, which were then counted following Trypan blue staining using a hemocytometer (bright-line Neubauer improved). The appropriate amount of cells was then seeded either on the chip for perfusion experiments or in a 96-well U-bottom plate (BD Falcon) for static experiments in RPMI1640 medium supplemented with 20 ng ml−1 human epidermal growth factor, hEGF (Gibco, Invitrogen), 20 ng ml−1 basic fibroblast growth factor, bFGF (Gibco, Invitrogen), 4 μg ml−1 human insulin (BioReagent, Sigma), 2% serum-free supplement B27 (Gibco, Invitrogen) and 1% P/S.Primary lung epithelial tumor cells (PLETCs) as well as primary pericytes (PCs) were isolated from lung tumor specimens as previously described.26 In brief, PLETCs were prospectively isolated using fluorescence-activated cell sorting (FACS) with an immunophenotypic profile of Lineage-EpCAM + CD73 + CD90−, and were seeded for expansion in a 6-well dish coated with 0.2% gelatin and human collagen IV (Sigma) in CnT-PA growth medium (CellnTec, Switzerland) supplemented with 10 ng ml−1 insulin-like growth factor, IGF-2 (Peprotech) and 10 ng ml−1 heregulin β, HerB (Peprotech). PCs were prospectively isolated with an immunophenotypic profile of Lineage-EpCAM − CD73 + CD90+ and were seeded for expansion in tissue culture plates coated with 0.2% gelatin and human collagen IV in lung pericyte growth medium (L-PC) composed of α-MEM medium (Sigma) supplemented with 20 ng ml−1 hEGF, 20 ng ml−1 bFGF, 4 μg ml−1 human insulin, 1% FBS and a 1% antibiotic–antimycotic solution (Invitrogen). Primary cells were used up to four passages. Following institutional review board approval, the patient, whose lung resection was used in this study, signed the surgical patient consent form of the University Hospital Bern, Switzerland, including the consent for the usage of surgical specimens and materials removed for research purposes.
2.4. Primary pericytes staining
PCs were stained to distinguish them from PLETCs. Depending on the experiment purpose, different stains were used. For short time experiments, a CellTrace™ CFSE cell proliferation kit (Molecular Probes, Life Technologies) was used. Therefore, pericytes were suspended in 10 μM CFSE solution, incubated for 15 minutes, re-suspended in fresh pre-warmed L-PC medium and incubated for another 30 minutes. PCs were then suspended again in fresh L-PC medium and seeded on the chip either mixed with PLETCs or alone. For long time experiments either a CellMask™ orange plasma membrane stain (Molecular Probes, Life Technologies) or a PKH26 red fluorescent cell linker (Sigma) was used. For the CellMask™ orange stain, PCs were suspended in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 dilution of the CellMask™ orange stain, incubated for 10 minutes at room temperature (RT) with occasional mixing and washed once with fresh L-PC medium before use. When using the PKH26 dye, PCs were suspended in 2 × 10−6 M PKH26, incubated for 5 minutes at RT with periodic mixing and followed by adding an equal volume of 1% bovine serum albumin, BSA (Sigma) for 1 minute before adding L-PC medium. Finally cells were washed once with L-PC medium and once with phosphate-buffered saline, PBS (Gibco, Invitrogen) before use.
:1000 dilution of the CellMask™ orange stain, incubated for 10 minutes at room temperature (RT) with occasional mixing and washed once with fresh L-PC medium before use. When using the PKH26 dye, PCs were suspended in 2 × 10−6 M PKH26, incubated for 5 minutes at RT with periodic mixing and followed by adding an equal volume of 1% bovine serum albumin, BSA (Sigma) for 1 minute before adding L-PC medium. Finally cells were washed once with L-PC medium and once with phosphate-buffered saline, PBS (Gibco, Invitrogen) before use.
2.5. Cell loading and spheroid formation
Prior to cell loading, the microchannels were sterilized with ozone (CoolCLAVE personal sterilizer), rinsed with 70% ethanol and degassed in a vacuum pump (KNF Neuberger) before rinsing with sterile deionized water. Then, synperonic F-108 (1% w/v, Fluka, Sigma) was flushed through the microchannels to avoid cell adhesion on the channel walls and incubated for 4 hours. Finally, the chip was rinsed with RPMI1640 medium containing EGF, bFGF, insulin, B27 and P/S for H2052 cell seeding, CnT-PA supplemented with IGF-2 and HerB for PLETCs or a 2:1 mixture of CnT-PA and L-PC media for co-culture cell seeding. A hydrostatic pressure difference, induced by a few millimeter high cell culture medium column containing the suspended cells, was used for cell loading on the chip from the loading reservoir. During cell seeding, fresh medium was regularly added in the inlet and the waste medium was removed from the outlet to maintain the hydrostatic pressure difference. During cell seeding, one minute video sequences were taken at the three branch points by using a digital camera (Moticam 1000, VWR). Video sequences of the cell seeding were used to count the number of cells passing through the right and the left arm at each branch and to determine the homogeneity of the cell distribution in the tree-like structure. After cell seeding, the microfluidic channels were incubated for 48 hours to allow spheroid formation (see video in the ESI†). The medium was exchanged once a day to ensure sufficient nutrient supply to the cells. Furthermore, each microwell was imaged once per day to observe and quantify spheroid formation and size. The formation of selected spheroids on the microfluidic chip was also observed in time-lapse mode (1 picture every 10 minutes for 48 h) using a microscope placed in the incubator (JuLI Smart Fluorescent Cell Analyzer). Table 1 indicates the number of H2052 cells that were loaded on the chip using hydrostatic pressure and the corresponding number of cells in each microwell. After cell seeding, the microfluidic channels were incubated for 11 days and each microwell was imaged once a day to observe and quantify spheroid formation and growth. The medium was exchanged once a day to ensure nutrient supply to cells.
| Number of cells loaded on chip | Theoretical number of cells per well | Diameter of spheroids after 3 days on chip (μm) | Diameter of spheroids after 11 days on chip (μm) |
|---|---|---|---|
| 10000 |
1250 | 324 ± 36 | 357 ± 20 |
| 5000 | 625 | 262 ± 39 | 300 ± 41 |
| 2500 | 312 | 210 ± 20 | 250 ± 37 |
| 1250 | 156 | 186 ± 28 | 175 ± 38 |
Cell seeding for primary co-culture spheroids (PLETCs/PCs spheroids) was done either sequentially or simultaneously. With regard to the sequential seeding, PCs were seeded 48 hours after the PLETCs. During the simultaneous seeding, PLETCs and PCs were mixed in an appropriate ratio, which was obtained by testing different PLETC to PC ratios (2:1, 3:1, 4:1, 5:1, 7:1, 10:1, and 20:1), and then seeded on the chip.
2.6. Proliferation assay on chip
A CellTrace™ CFSE Cell Proliferation Kit (Molecular Probes, Invitrogen) was used to observe cell proliferation during spheroid formation on the chip using dye dilution. Therefore 5 mM CFSE stock solution was diluted with phosphate-buffered saline (PBS, Gibco, Invitrogen) to obtain a 10 μM working concentration. The dye was added to only 1–2% of the cell suspension so that stained cells could easily be distinguished and counted. Cells within the dye solution were then incubated for 15 minutes before being centrifuged for 4 minutes at 200 relative centrifugal force (rcf). Then, the cells were resuspended in fresh pre-warmed medium (RPMI1640) and incubated for another 30 minutes to ensure complete modification of the probe. Finally the cells were washed again with the fresh medium and mixed with the non-stained cells before seeding all cells on the chip. Microscopic pictures were taken once a day using an inverted fluorescence microscope (Leica DMI4000B). In addition, proliferation during spheroid formation was observed by time-lapse microscopy every hour using the JuLI analyzer.2.7. Perfusion and cisplatin treatment on chip
For chemosensitive assays, 5000 cells per channel were seeded and incubated for 48 hours to enable spheroid formation. The ratio between PLETCs and PCs was set to 5:1, meaning 4167 PLETCs and 833 PCs were mixed and seeded per channel. The medium was exchanged once a day to ensure sufficient nutrient supply. After spheroid formation, the inlet channels of the microfluidic chips were connected to syringe pumps (Harvard Apparatus) equipped with 1 ml syringes. For this, polytetrafluoroethylene (PTFE, Milian) tubes, which have an inner diameter (ID) of 0.8 mm, connect the syringes with the inlet of the channels. Each of the tubes is interrupted by an air bubble trap shortly before the channel inlet to avoid the entry of air bubbles into the microfluidic channels. The air bubble trap consists of a polyvinyl chloride (PVC) tubing (ID = 2 mm) enclosing a 1.6 mm PTFE tube (ID = 0.8 mm), which collects the air bubbles. The outlet channels of the chip were connected as well to the PTFE tubes (ID = 0.8 mm) ending in 1.5 ml microtubes (Eppendorf, VWR) that finally collect the supernatant during the whole assay period. Spheroids were perfused for 48 hours under sterile conditions at 37 °C and 5% CO2 with a flow rate of 0.1 μl min−1 and different concentrations of cisplatin (0 μM, 2 μM, 4 μM, 8 μM, 16 μM, 32 μM, 48 μM, 64 μM, 80 μM, 96 μM, 112 μM, 128 μM, and 144 μM). For this purpose, cisplatin (0.5 mg ml−1, Sandoz) was diluted with the appropriate medium. Consequently, RPMI1640 medium supplemented with 20 ng ml−1 hEGF, 20 ng ml−1 bFGF, 4 μg ml−1 human insulin, 2% B27 and 1% P/S was used for experiments with the mesothelioma cell line H2052, whereas PLETCs were perfused with CnT-PA medium supplemented with 10 ng ml−1 IGF-2 and 10 ng ml−1 HerB and co-culture spheroids with a combination of CnT-PA medium supplemented with 10 ng ml−1 IGF-2 and 10 ng ml−1 HerB and L-PC medium in a 2:1 ratio. The final supernatant volume of 288 μl was then analysed for its caspase-3/7 activity. All experiments were done at least in triplicate.
2.8. Caspase-3/7 activity
After cisplatin treatment, supernatants were quickly frozen down in liquid nitrogen and stored at −80 °C until they were used to measure the caspase-3/7 activity. For this purpose, the supernatants were rapidly thawed in a 37 °C water bath. Then, 100 μl of each supernatant was mixed in a 1:1 ratio with the Caspase-Glo®3/7 assay (Promega) in a black 96-well flat-bottom plate and incubated for 60 minutes before luminescence was measured using a plate reader (TECAN Infinite M1000).
2.9. Cell viability staining on chip
A live/dead stain was performed by using calcein and ethidium homodimer 1 (EthD1, Viability Cytotoxicity Kit, Invitrogen). In addition, Hoechst 33342 (Molecular Probes, Invitrogen) was used to stain the cell nuclei. For this, 0.5 μM calcein was mixed with 1 μM EthD1 and 1 μg ml−1 Hoechst 33342 in RPMI1640 medium containing EGF, bFGF, insulin, B27 and P/S. The combined reagents were added to the spheroids and incubated for 3 hours. Following this, the spheroids were washed with RPMI1640 medium containing EGF, bFGF, insulin, B27 and P/S and observed under the fluorescence microscope.2.10. Microscopy and image analysis
Microscopic pictures were taken using an inverted Leica fluorescence microscope (Leica DMI4000B) with a CCD camera (Leica DFC360 FX) or with a laser scanning microscope (Zeiss LSM710) for fluorescence-labelled cells. The JuLI analyzer was used for time serial images for longer duration and the digital Moticam camera was used to take daily bright-field images as well as video sequences. The images were processed using the Leica AF image analysis software and were further treated and visually analyzed using the Fiji image analysis software based on ImageJ (http://rsbweb.nih.gov/ij/).2.11. Statistical analysis
Prism6 (GraphPad Software, Inc., La Jolla, CA) was used for statistical analysis. The statistical significance was set at a value of 0.05.3. Results and discussion
3.1. Cell loading and distribution in the microfluidic branch
In contrast to other microfluidic systems aimed at creating spheroids on chip,16,27–29 the aim of the present system is to load a defined number of cells on the chip to form spheroids with a minimum cell loss. A known amount of cells (MPM H2052 have an approximate size of 20 μm) is pipetted at the inlet of the channel and transported by hydrostatic pressure in the microfluidic network. At the first branch, the cells are distributed equally in the two daughter branches. 51.8 ± 4.5% of the cells are transported in the left branch, while 48.2 ± 4.5% in the right branch (ESI† Fig. S3). At the second branch, the cells split equally again (51.4 ± 3.9% (left branch) and 48.6 ± 3.9% (right branch)), as well as at the last intersection, 52.8 ± 7.2% are counted in the left and 47.2 ± 7.2% in the right branch. The standard errors are within a few percentages and are thus acceptable. The distribution of the cells as a function of the position of the microwells in the microvasculature is homogeneous with a slightly higher number of cells in the first and last wells. This difference is however not significant in comparison with the number of cells trapped in the other microwells (ESI† Fig. S4). Overall, a homogeneous distribution of the cells is obtained with this device even when a very low number of cells are seeded. Almost all loaded cells are trapped in the microwells, with no remaining cells observed either in the inlet of the device, or after the trapping section. Indeed, about 150 cells are found in each microwell for the smallest cell density loaded. These results demonstrate that the efficiency of the device in terms of cell loss is excellent.3.2. Spheroid formation with the H2052 cell line
During the first three days after seeding, the diameters of the cellular aggregates significantly decrease regardless of the number of cells seeded (Fig. 2). During this time, the cell–cell contacts become stronger and tight junctions are created, which results in a compact cellular structure (Fig. 2B). After this initial formation phase of the spheroids, the spheroid diameters start to increase as a consequence of cellular proliferation. This tendency is observed until the end of the culture at day 11 after cell seeding. Between day 3 and day 11 in the culture, the diameters of the spheroids increase by 19% in the channels seeded with 2500 cells, and by 14% in the channels seeded with 5000 cells and by 10% in the channels seeded with 10000 cells (Fig. 2A). This increase in spheroid size is statistically significant for 2500 cells seeded between day 3 and day 10 as well as for 5000 cells seeded between day 3 and days 9, 10 and 11. The smaller spheroids formed with only 156 cells changed minimally over time. The lowest diameter is reached merely after 7 days and increases by only 2% until day 11 in the culture. A minimal amount of cells seems to be needed to favor cellular proliferation. In the present case, the critical limit is situated between 150 to 250 MPM H2052 cells. This may be due to insufficient production of extracellular matrix from the cells. Similar results were observed in a standard 96-well plate (results not shown), which suggests that this limit is not due to the microfluidic confinement.
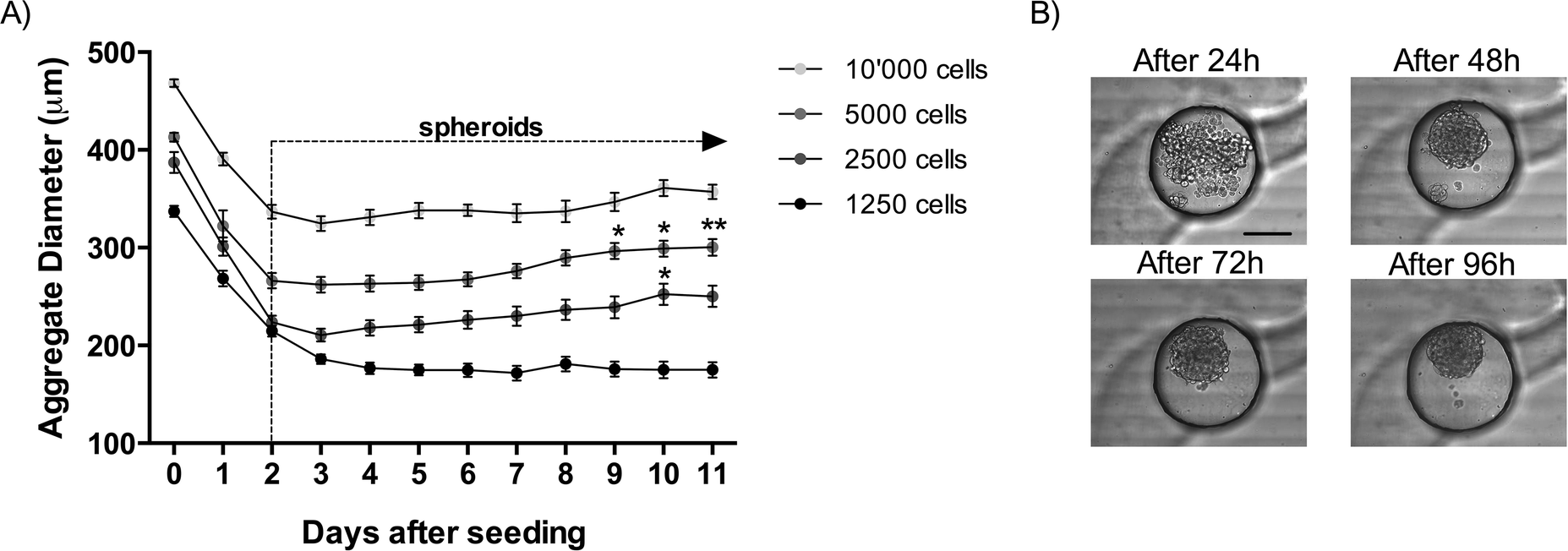 | ||
| Fig. 2 A) Graph showing the cellular aggregate diameter over a period of 11 days as a function of the number of cells seeded. The data are presented as mean ± SEM. Statistical significance was calculated compared to day 3 after seeding. N ≥ 8. B) Bright-field images of spheroid formation on chip with round-bottom microwells. The images were taken 24, 48, 72 and 96 hours after cell seeding. Scale bar corresponds to 250 μm. N ≥ 8. | ||
3.3. Difference between flat-bottom and round-bottom wells
Stereolithography, the technique used to create the PDMS mold, does not enable the creation of smooth spherical micropillars to be used for the molding of round-bottom microwells, due to its resolution limits. Rounded micropillars are thus produced by manually adding uncured PDMS droplets on the stereolithographic mold. This process appears to be reproducible (Fig. 1C). For 10000 cells loaded per channel, an important difference in the number of spheroids formed in each microwell is observed between flat-bottom and round-bottom microwells (Fig. 3A and B). In the flat-bottom microwells, after 24 hours in culture, only 14% of the wells contains a single spheroid, whereas 23.4% contains two spheroids, 35.9% contains three spheroids and 26.6% of the microwells even with four spheroids (Fig. 3A). In sharp contrast, 75% of the round-bottom receptacles contain only one spheroid after one day in culture. In addition, the presence of multiple spheroids was only observed in 20.3% of the round-bottom microwells (two spheroids) and in 3.2% of the microwells (three or four spheroids) (Fig. 3B). We observe that the number of spheroids per cavity decreases with time. After 3 days in culture, a single spheroid is formed in 42.2% of the flat-bottom microwells, and in 78.1% of the round-bottom microwells, respectively (Fig. 3C). Our results demonstrate the importance of the microwell shape and of the cell culture time to obtain similar replicates. Fig. 3D shows representative images of round and flat-bottom wells. As a result, all subsequent tests are carried out with the round-bottom microwells.
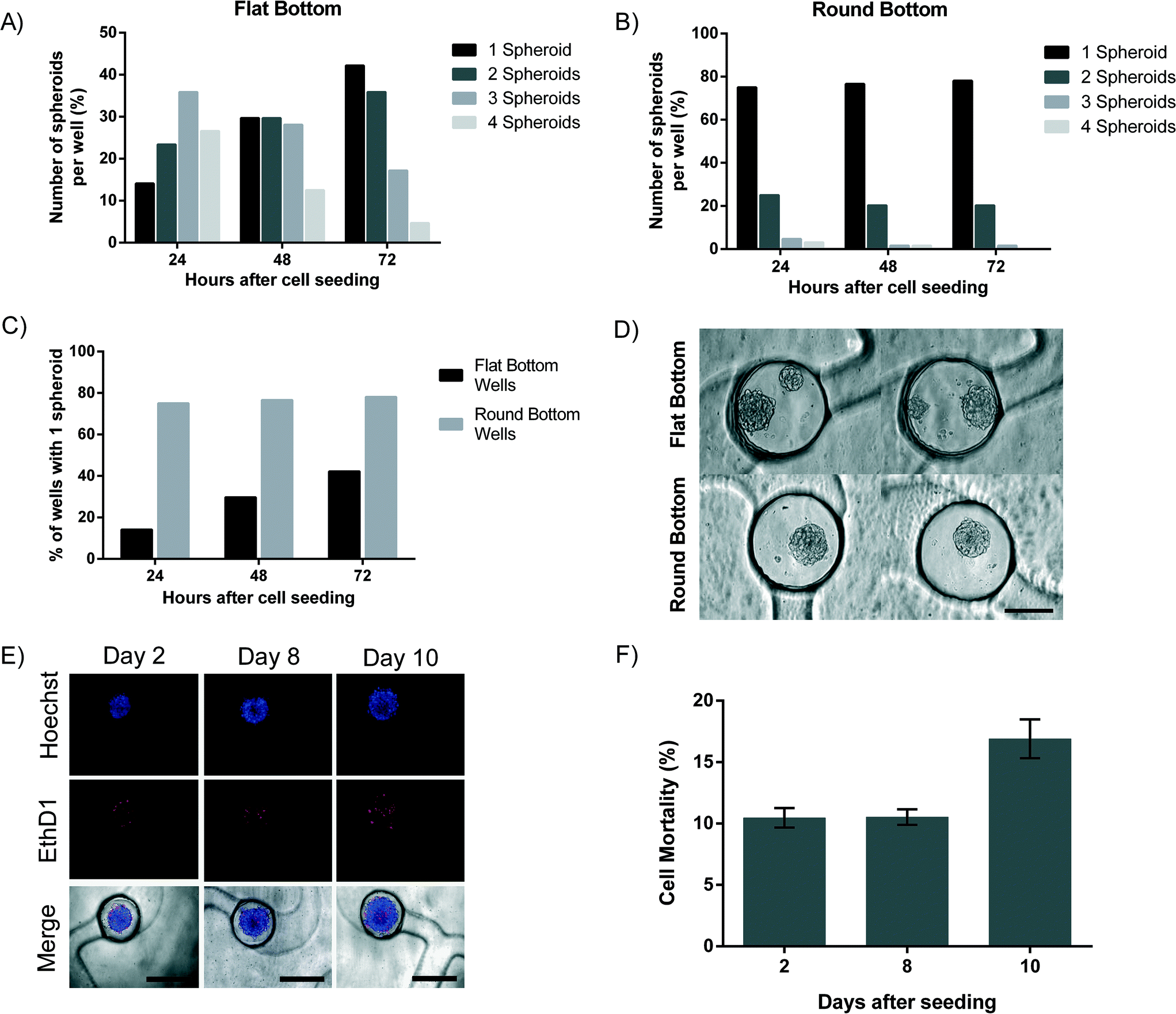 | ||
| Fig. 3 Experiments with MPM H2052 cell line. Percentage of microwells containing either one spheroid (dark), two spheroids (dark grey), three spheroids (grey) or four spheroids (light grey) in flat-bottom microwells A) or round-bottom microwells B). C) Comparison between percentage of microwells with a flat or round-bottom microwells containing one spheroid. D) Representative images of spheroids formed in flat-bottom microwells and round-bottom microwells after three days in culture. Scale bar corresponds to 250 μm. For all experiments N = 64. E) Images showing spheroids stained with Hoechst (blue) and Ethidium homodimer-1 (EthD1, red) indicating cell death. Scale bar corresponds to 500 μm. F) Graph representing cell mortality in spheroids after 2 days (N = 16), 8 days (N = 16) and 10 days (N = 8) on chip. The data are represented as mean ± SEM. | ||
3.4. Cell proliferation and viability in the H2052 cell line
Cell viability and proliferation in the spheroids were investigated for a period of at least 8 days (Fig. 3E and F). Cell death was assessed by the loss of plasma membrane integrity using ethidium homodimer-1 and found to be 10.5 ± 3% after 2 days. Cell mortality remains stable over a period of 8 days with a cell mortality of 10.5 ± 2.9%. These results fit with our previous findings where a cell mortality of 10% was found.11 After 10 days on the chip, cell mortality increases slightly to 16.9 ± 4.9% (Fig. 3F). This can be explained by the formation of a necrotic core typical to spheroids that are larger than 300 μm.7 Cell proliferation was assessed on spheroids with only 150 cells, which enables the imaging of cell division within the spheroids. CFSE covalently binds to intracellular molecules and therefore the fluorescent CFSE can be retained within the cell for a long time. In our case, CFSE was used to track cell proliferation within the chip over a period of 8 days (ESI† Fig. S5). We chose to stain only few cells with CFSE to ease the observation of proliferation in the spheroids. Cells proliferate slower in 3D cultures compared to a standard monolayer cell culture, as reported earlier by our group using MPM cells11 and by additional groups using other cells.30–343.5. Human lung primary cell spheroids: mono- and co-culture
As the ultimate aim of this platform is to perform personalized chemosensitivity assay, primary tumor cells from patients with NSCLC are loaded on the chip. 10000 PLETCs obtained from lung tumor resection of one adenocarcinoma patient (BE063-T) and one squamous carcinoma patient (BE067-T) were loaded and cultured in the round-bottom microwell platform. Cellular distribution in the eight microwells was homogeneous, which resulted in spheroid diameters with a standard error of 4.6 μm after three days. Surprisingly, in contrast to the 75% of single MPM H2052 spheroids obtained per well, about 90% of the microwells contain a single spheroid after 24 hours (Fig. 4A). In addition, no wells contain more than two spheroids. Thus, intercellular adhesion and formation of the cellular aggregates may increase with primary cells. The percentage of wells containing only one spheroid remains stable over a period of three days (Fig. 4A). The diameter of the cellular aggregates decreases from 220 ± 45 μm after 24 hours on the chip to 176 ± 35 μm after three days (Fig. 4B), which corresponds to the formation of a compact spheroid. The cells from both patients formed uniformly sized spheroids. In total, 32 wells were observed and analyzed per patient. However, a difference was observed between spheroids from the two patients. When using the cells from patient BE063, a primary adenocarcinoma, the number of wells containing more than one spheroid increases from 2 wells in the first day to 5 wells after three days. In contrast, the number of wells containing multiple spheroids decreases with the cells from patient BE067, a primary squamous carcinoma, from 1 well at day one to 0 wells after three days.
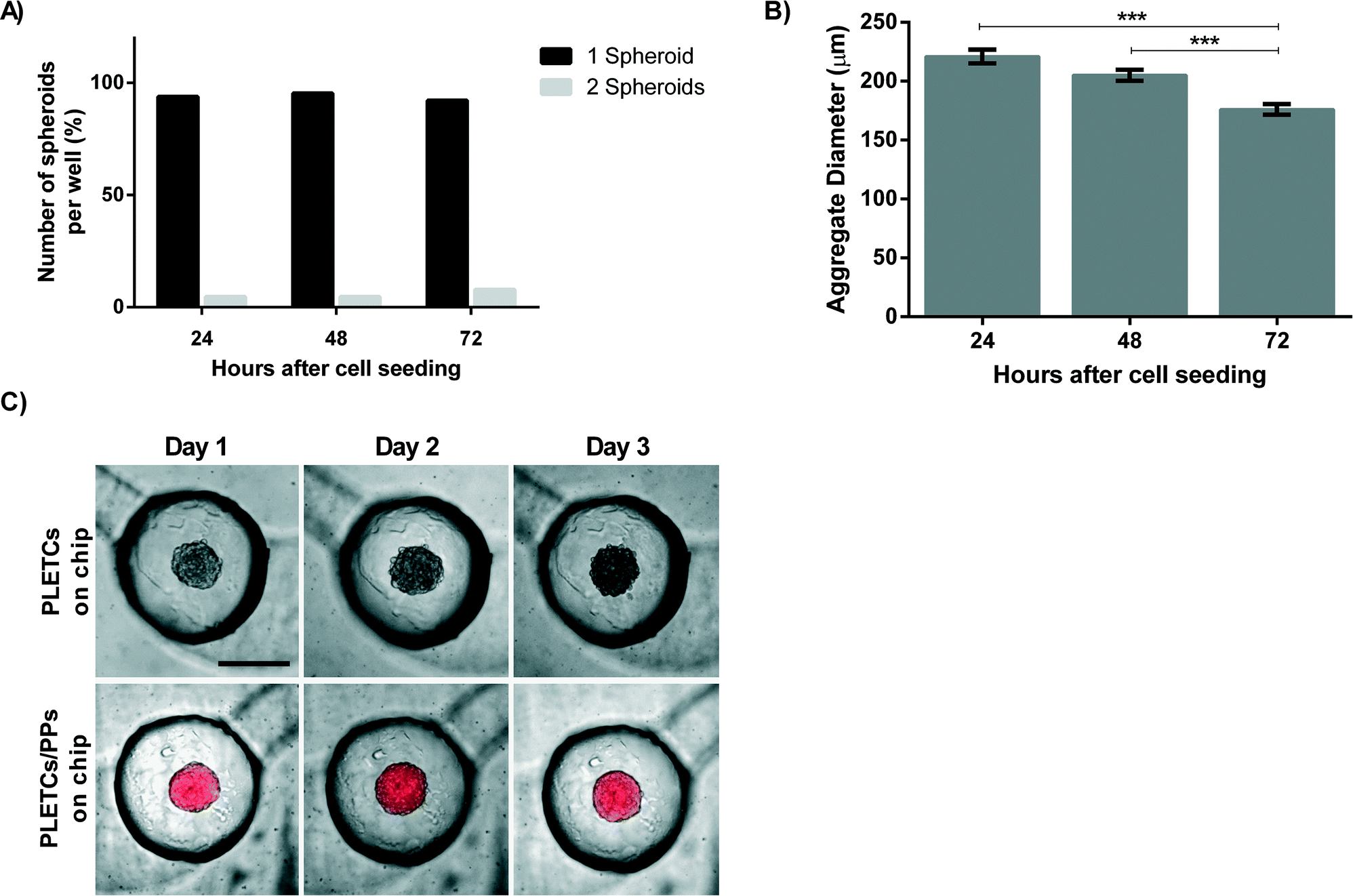 | ||
| Fig. 4 Experiments with primary lung tumor cells. Experiments in A) and B) were performed with primary cells from two patients (BE063-T (adenocarcinoma) and BE067-T (squamous carcinoma)), whereas experiments in C) were done with patient BE069-T (adenocarcinoma). A) Number of microwells containing one or two spheroids. B) Diameter of primary cells spheroids. The data are presented as mean ± SEM. (N ≥ 59). C) Images taken daily during 3 days of mono- or co-culture spheroids cultured on chip. Pericytes were stained in red with PKH26. Scale bar corresponds to 250 μm. | ||
The complexity of the tumor microenvironment imposes several barriers that limit drug diffusion to the cancerous cells. Extracellular matrix, smooth muscle cells, pericytes, cancer-associated fibroblasts and others are creating a protective barrier from the drug around the tumor.21 In an attempt to further reproduce a part of this barrier, primary pericytes (PCs) are co-cultured with primary lung adenocarcinoma epithelial cells (PLETCs). PCs as well as PLETCs that are tested on the chip were collected from the same patient (BE069-T). Co-seeding and sequential seeding strategies are tested to create co-cultured spheroids. As sequential seeding often results in the formation of two distinct spheroids, one with PCs and the second with PLETCs (data not shown), the decision was taken to continue with the co-seeding strategy, where homogeneous spheroids formed after 24 hours. The optimal ratio between PLETCs and PCs was found to be 5:1. At lower ratios no spheroid formation was observed, but instead an accumulation of loose cells. At higher ratios, homogeneous spheroids formed (ESI† Fig. S6).
Fluorescence microscopy images strikingly illustrate the difference in the spheroids obtained from the monoculture or from the co-culture of primary cells. The co-culture spheroids are covered with pericytes, which seem to constrain the spheroid diameter, whereas the spheroids without pericytes are slightly larger (Fig. 4C). However, no difference was observed in terms of spheroid formation between monoculture and co-culture spheroids.
3.6. Cisplatin assay
Cisplatin, a platinum-containing anti-cancer drug that causes crosslinking of DNA and thus triggers apoptosis, is commonly used to treat patients with lung adenocarcinoma. The same drug is used here. In our earlier work we presented a difference in chemosensitivity of cisplatin under static and perfusion conditions in H2052 cells.11 The quantification of the chemoresistance was performed by analyzing the perfused supernatant collected after 48 hours of cisplatin exposure. In the present study, the objective is to investigate the chemoresistive effect of the pericytes co-cultured with lung cancer epithelial cells. For this purpose, PLETCs and PLETC/PC spheroids with similar number of cells, estimated at about 600, were perfused with different concentrations of cisplatin (Fig. 5). Interestingly, PLETC/PC spheroids in our perfused system show a maximum number of apoptotic cells at a cisplatin concentration of 80 μM, whereas in monoculture spheroids the maximum is already reached at 16 μM of cisplatin. The decrease in apoptotic signal after this apoptosis peak is explained by the induction of necrotic cell death, which is not measured by the caspase-3/7 assay, due to cisplatin toxicity. Hence in our microfluidic chip PLETC spheroids are more sensitive to cisplatin than the PLETC/PC spheroids under perfusion. Thus, primary pericytes seem to play a protective role for primary lung cancer epithelial cells.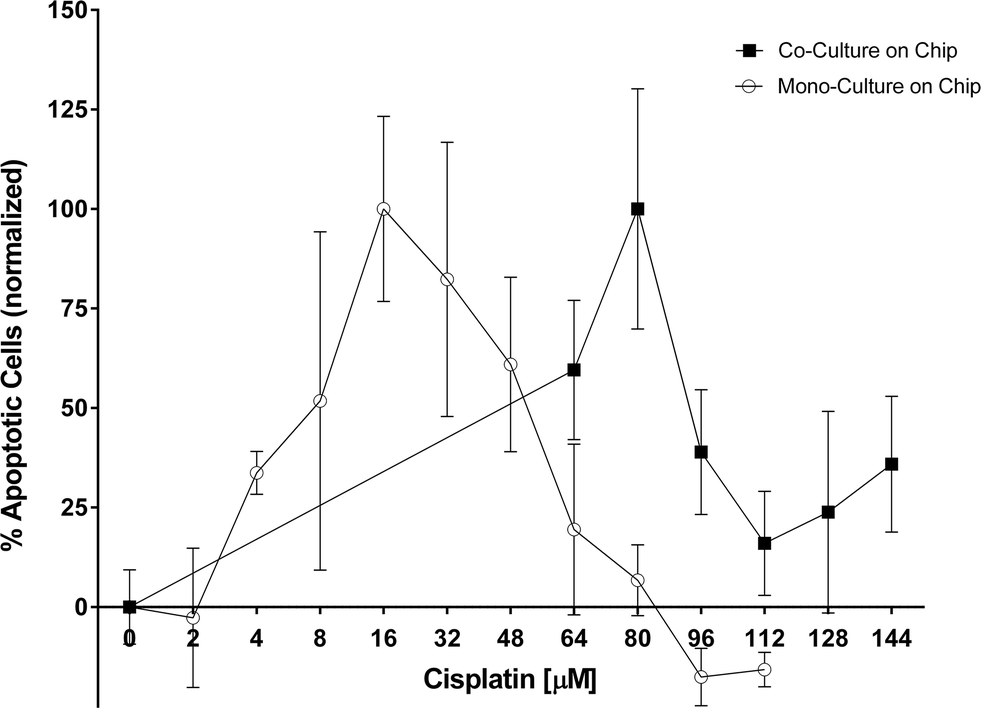 | ||
| Fig. 5 Graph showing the response to cisplatin of primary mono and co-culture spheroids of one patient under perfusion. A maximum of apoptotic cells was measured at 16 mM for monoculture spheroids and at 80 mM for co-culture spheroids on chip. The data are presented as mean ± SEM and were normalized to control. N ≥ 3, except for 2 mM cisplatin where only 2 samples were tested. | ||
4. Conclusions
Microfluidic devices aimed for personalized chemotherapy are widely seen as having the potential to become important tools for the prediction of a patient's response to chemotherapeutic treatment.13 The scarce material obtained from patient's tumors need to be used carefully and as efficiently as possible to extract the maximum amount of information (genomic, proteomic, histology and cell-based assays). Thus, physicians are assisted in the decision-making process aimed at determining the most appropriate chemotherapy. Given that the number of available cells is very limited for a cell-based assay, one of the important parameters of a microfluidic platform for personalized chemotherapy is to provide reliable results despite this constraint. Thus, homogeneous spheroid formation from patient's material in an in vivo-like environment is necessary to obtain reliable results. Therefore, the first objective of this study was to design a microfluidic device that enables the homogeneous distribution of a given number of cells leading to reproducible spheroid formation across test samples. In the second step, the drug response of primary cells in a perfused microenvironment was tested.This study demonstrates the first step in the direction of personalized oncology application with the formation of spheroids of equal sizes from a very limited number of cells and drug perfusion on human primary cells. This microfluidic device is able to form single, uniformly sized spheroids from either cell line or human primary cells. As little as 1250 cells per channel, translating into around 156 cells per spheroid, were loaded on the platform and formed spheroids. Importantly, the system's efficiency is high with almost no cell loss in the microfluidic network. Further, the cell viability and proliferation of the confined spheroids was revealed to be robust, which was demonstrated by the constant cell viability over 8 days. Moreover the optimal ratio between the primary epithelial lung tumor cells (PLETCs) and the primary pericytes (PCs) in the presented microfluidic system was found to be 5:1. The most important finding is that pericytes have a protective effect on the lung cancer epithelial cells from the damaging effects of a chemotherapeutical drug, leading to a higher chemoresistance of PLETC/PC spheroids compared to the PLETC spheroids.
These results demonstrate that we could reproduce at least partly the barrier induced by the tumor microenvironment that protects the tumor from drug exposure. To mimic this microenvironment even further, other constituents of the tumor, such as the endothelial microvasculature, are needed. Microfluidic systems like this one will also allow reproduction of combined or sequential chemotherapies that are often used in clinics. These developments will bring such microfluidic chips closer to being a potential tool to be used in personalized oncology.
Acknowledgements
The images were acquired on equipment supported by the Microscopy Imaging Center of the University of Bern. The authors are grateful to the University of Bern Research Foundation for funding part of the equipment used in this project. In addition, we would like to thank the Thoracic Surgery Group of the Department of Clinical Research, especially Thomas Marti and Renwang Peng for helpful discussion during the project.References
- World Health Organisation, Media Center – Cancer – Key Facts, http://www.who.int/mediacentre/factsheets/, 2014, Accessed: 2014-08-15 Search PubMed.
- L. Chin, J. N. Andersen and P. A. Futreal, Nat. Med., 2011, 17, 297–303 CrossRef CAS PubMed.
- L. Cheng, R. E. Alexander, G. T. Maclennan, O. W. Cummings, R. Montironi, A. Lopez-Beltran, H. M. Cramer, D. D. Davidson and S. Zhang, Mod. Pathol., 2012, 25, 347–369 CrossRef CAS PubMed.
- L. Welker, R. Akkan, O. Holz, H. Schultz and H. Magnussen, Diagn. Pathol., 2007, 2, 31 CrossRef PubMed.
- D. V. LaBarbera, B. G. Reid and B. H. Yoo, Expert Opin. Drug Discovery, 2012, 7, 819–830 CrossRef CAS PubMed.
- K. M. Yamada and E. Cukierman, Cell, 2007, 130, 601–610 CrossRef CAS PubMed.
- F. Hirschhaeuser, H. Menne, C. Dittfeld, J. West, W. Mueller-Klieser and L. A. Kunz-Schughart, J. Biotechnol., 2010, 148, 3–15 CrossRef CAS PubMed.
- A. C. Shieh, Ann. Biomed. Eng., 2011, 39, 1379–1389 CrossRef PubMed.
- A. C. Shieh and M. A. Swartz, Phys. Biol., 2011, 8, 015012 CrossRef PubMed.
- T. Das, L. Meunier, L. Barbe, D. Provencher, O. Guenat, T. Gervais and A.-M. Mes-Masson, Biomicrofluidics, 2013, 7, 011805 CrossRef PubMed.
- J. Ruppen, L. Cortes-Dericks, E. Marconi, G. Karoubi, R. A. Schmid, R. Peng, T. M. Marti and O. T. Guenat, Lab Chip, 2014, 14, 1198–1205 RSC.
- R. N. Zare and S. Kim, Annu. Rev. Biomed. Eng., 2010, 12, 187–201 CrossRef CAS PubMed.
- D. Wlodkowic and J. M. Cooper, Curr. Opin. Chem. Biol., 2010, 14, 556–567 CrossRef CAS PubMed.
- K. E. Sung and D. J. Beebe, Adv. Drug Delivery Rev., 2014, 79–80, 68–78 CrossRef CAS PubMed.
- I. A. Cree, S. Glaysher and A. L. Harvey, Curr. Opin. Pharmacol., 2010, 10, 375–379 CrossRef CAS PubMed.
- K. Lee, C. Kim, J. Young Yang, H. Lee, B. Ahn, L. Xu, J. Yoon Kang and K. W. Oh, Biomicrofluidics, 2012, 6, 14114–141147 Search PubMed.
- A. Hsiao, Y.-S. Torisawa, Y. Tung, S. Sud, R. Taichman, K. Pienta and S. Takayama, Biomaterials, 2009, 30, 3020–3027 CrossRef CAS PubMed.
- H.-J. Jin, Y.-H. Cho, J.-M. Gu, J. Kim and Y.-S. Oh, Lab Chip, 2010, 115–119 Search PubMed.
- M. J. Bissell and D. Radisky, Nat. Rev. Cancer, 2001, 1, 46–54 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- S. K. Sriraman, B. Aryasomayajula and V. P. Torchilin, Tissue Barriers, 2014, 2, e29528 CrossRef PubMed.
- A. Amann, M. Zwierzina, G. Gamerith, M. Bitsche, J. M. Huber, G. F. Vogel, M. Blumer, S. Koeck, E. J. Pechriggl, J. M. Kelm, W. Hilbe and H. Zwierzina, PLoS One, 2014, 9, e92511 Search PubMed.
- D. E. Sims, Tissue Cell, 1986, 18, 153–174 CrossRef CAS.
- Y. Minami, T. Sasaki, J.-I. Kawabe and Y. Ohsaki, in Research Directions in Tumor Angiogenesis, InTech, 2013, ch. 4, pp. 73–88 Search PubMed.
- J. C. McDonald and G. M. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS PubMed.
- A. O. Stucki, J. D. Stucki, S. R. R. Hall, D. M. Felder, Y. Mermoud, R. A. Schmid, T. Geiser and O. T. Guenat, Lab Chip, 2015, 15, 1302–1310 RSC.
- K. Kwapiszewska, A. Michalczuk, M. Rybka, R. Kwapiszewski and Z. Brzozka, Lab Chip, 2014, 14, 2096–2104 RSC.
- Y.-S. Torisawa, B. Mosadegh, G. D. Luker, M. Morell, K. S. O'Shea and S. Takayama, Integr. Biol., 2009, 1, 649–654 RSC.
- B. Patra, Y.-H. Chen, C.-C. Peng, S.-C. Lin, C.-H. Lee and Y.-C. Tung, Biomicrofluidics, 2013, 7, 54114 CrossRef PubMed.
- A. Takagi, M. Watanabe, Y. Ishii, J. Morita, Y. Hirokawa, T. Matsuzaki and T. Shiraishi, Anticancer Res., 2007, 27, 45–53 CAS.
- R. Edmondson, J. J. Broglie, A. F. Adcock and L. Yang, Assay Drug Dev. Technol., 2014, 12, 207–218 CrossRef CAS PubMed.
- K. Chitcholtan, P. H. Sykes and J. J. Evans, J. Transl. Med., 2012, 10, 38 CrossRef CAS PubMed.
- B. Fallica, J. S. Maffei, S. Villa, G. Makin and M. Zaman, PLoS One, 2012, 7, e48024 CAS.
- A. C. Luca, S. Mersch, R. Deenen, S. Schmidt, I. Messner, K.-L. Schäfer, S. E. Baldus, W. Huckenbeck, R. P. Piekorz, W. T. Knoefel, A. Krieg and N. H. Stoecklein, PLoS One, 2013, 8, e59689 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Video showing spheroid formation. See DOI: 10.1039/c5lc00454c |
| This journal is © The Royal Society of Chemistry 2015 |