
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents
Ilka
Maschmeyer
*ab,
Alexandra K.
Lorenz
ab,
Katharina
Schimek
a,
Tobias
Hasenberg
ab,
Anja P.
Ramme
ab,
Juliane
Hübner
a,
Marcus
Lindner
a,
Christopher
Drewell
a,
Sophie
Bauer
a,
Alexander
Thomas
a,
Naomia Sisoli
Sambo
a,
Frank
Sonntag
c,
Roland
Lauster
a and
Uwe
Marx
ab
aTechnische Universität Berlin, Institute of Biotechnology, Department Medical Biotechnology, Gustav-Meyer-Allee 25, 13355 Berlin, Germany. E-mail: Ilka.maschmeyer@tissuse.com; Fax: +49 30 314 27914; Tel: +49 30 314 27907
bTissUse GmbH, Markgrafenstrasse 18, 15528 Spreenhagen, Germany
cFraunhofer Institute for Material and Beam Technology, Winterbergstr. 28, 01277 Dresden, Germany
First published on 13th May 2015
Abstract
Systemic absorption and metabolism of drugs in the small intestine, metabolism by the liver as well as excretion by the kidney are key determinants of efficacy and safety for therapeutic candidates. However, these systemic responses of applied substances lack in most in vitro assays. In this study, a microphysiological system maintaining the functionality of four organs over 28 days in co-culture has been established at a minute but standardized microsystem scale. Preformed human intestine and skin models have been integrated into the four-organ-chip on standard cell culture inserts at a size 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold smaller than their human counterpart organs. A 3D-based spheroid, equivalent to ten liver lobules, mimics liver function. Finally, a barrier segregating the media flow through the organs from fluids excreted by the kidney has been generated by a polymeric membrane covered by a monolayer of human proximal tubule epithelial cells. A peristaltic on-chip micropump ensures pulsatile media flow interconnecting the four tissue culture compartments through microfluidic channels. A second microfluidic circuit ensures drainage of the fluid excreted through the kidney epithelial cell layer. This four-organ-chip system assures near to physiological fluid-to-tissue ratios. In-depth metabolic and gene analysis revealed the establishment of reproducible homeostasis among the co-cultures within two to four days, sustainable over at least 28 days independent of the individual human cell line or tissue donor background used for each organ equivalent. Lastly, 3D imaging two-photon microscopy visualised details of spatiotemporal segregation of the two microfluidic flows by proximal tubule epithelia. To our knowledge, this study is the first approach to establish a system for in vitro microfluidic ADME profiling and repeated dose systemic toxicity testing of drug candidates over 28 days.
000-fold smaller than their human counterpart organs. A 3D-based spheroid, equivalent to ten liver lobules, mimics liver function. Finally, a barrier segregating the media flow through the organs from fluids excreted by the kidney has been generated by a polymeric membrane covered by a monolayer of human proximal tubule epithelial cells. A peristaltic on-chip micropump ensures pulsatile media flow interconnecting the four tissue culture compartments through microfluidic channels. A second microfluidic circuit ensures drainage of the fluid excreted through the kidney epithelial cell layer. This four-organ-chip system assures near to physiological fluid-to-tissue ratios. In-depth metabolic and gene analysis revealed the establishment of reproducible homeostasis among the co-cultures within two to four days, sustainable over at least 28 days independent of the individual human cell line or tissue donor background used for each organ equivalent. Lastly, 3D imaging two-photon microscopy visualised details of spatiotemporal segregation of the two microfluidic flows by proximal tubule epithelia. To our knowledge, this study is the first approach to establish a system for in vitro microfluidic ADME profiling and repeated dose systemic toxicity testing of drug candidates over 28 days.
Introduction
The success of organ-on-a-chip microphysiological tissue culture systems has stimulated researchers to challenge a more systemic level of human biology in vitro through so-called “body-on-a-chip” or “human-on-a-chip” systems.1–4 Such systems aim to combine several organ equivalents within a human-like metabolizing environment or aim at in vivo-like pharmacokinetics and pharmacodynamics. The first of these microsystems – the micro cell culture analogue (μCCA) of Shuler and colleagues5 – supports the molecular interplay between individual liver, bone-marrow and tumour cell line culture compartments through a media re-circulation using an external pump and an external common reservoir. Physiological flow velocity and substance residence times can be maintained in the system at an artificially high fluid-to-tissue ratio. The same solution for media re-circulation has been used in the multi-channel 3D microfluidic cell culture system (μFCCS) of Zhang and colleagues6 to combine individual human liver, lung, kidney, and adipose cell culture compartments. By contrast, the total micro bioassay system of Imura and colleagues,7 in its most advanced version, constitutively combines human intestine, liver and breast cancer cell line cultures in a single linear channel applying unidirectional flow without media re-circulation. The advantage of these multi-organ systems is an explicit adjustable fluid flow and a controllable local tissue-to-fluid ratio in the channels. However, these systems suffer from reduced cross-conditioning and reduced molecular crosstalk among tissues, due to a small cell count (mostly below ten thousand per culture compartment) and a significant substance dilution. The effective substance exposure time in most existing microsystems usually ranges from 24 to 72 hours and rarely exceeds seven days.4 We have recently introduced a multi-organ-chip (MOC) platform, enabling the co-culture of two different human 3D organ equivalents, e.g. liver spheroids with skin punch biopsies, neuronal spheroids or intestinal tissues, at homeostasis over weeks for repeated dose substance testing.8–10 An integrated on-chip micropump and interconnecting microchannels support pulsatile medium perfusion at relevant tissue-to-liquid ratios within a surrogate blood circuit of such two-organ-chips. The combination of a human barrier organ equivalent, such as skin, intestine or lung, with a liver model allows the generation of data on substance absorption and distribution between the two organs and its metabolism in human liver tissue.Here we aimed to extend the capability of our MOC platform toward the generation of absorption, distribution, metabolism and excretion (ADME) profiles along the timeline of a repeated dose substance test. Therefore, we manufactured a MOC format able to host four human organ equivalents: i) a reconstructed human 3D small intestine and ii) a skin biopsy opting for oral and dermal substance absorption, iii) a 3D liver equivalent enabling primary substance metabolism and iv) a kidney proximal tubule compartment supporting metabolite excretion. A robust protocol has been established for the co-culture of these four human tissues over at least 28 days. Here we demonstrated remarkably robust homeostasis and functionality of the four organ equivalents over the experimental period of 28 days. Barrier integrity, continuous molecular transport against gradients and metabolic activity could be demonstrated for the four-organ-chip (4OC) co-cultures, thus making it a perfect platform for further in vitro ADME and repeated dose toxicity testing.
Materials and methods
Device design and fabrication
We designed and fabricated a microfluidic MOC device accommodating two microphysiological fluid flow circuits, each operated by a separate peristaltic on-chip micropump and both overlapping in the kidney proximal tubule culture compartment. Fig. 1a illustrates the system at a glance. Micropump software control facilitates both clockwise and anti-clockwise fluid flow. The peristaltic on-chip micropumps on both circuits were modified after Wu and colleagues,11 and each represents three 500 μm-thick polydimethylsiloxane (PDMS) membrane valves in a row. They are consecutively actuated by applying pressure and vacuum. The cover-plates of this 4OC accommodates six air pressure fittings, four inserts forming culture compartments for the four different tissue types and two additional reservoirs for fluids from the excretory organ. The surrogate blood flow circuit comprises a total volume of 830 μl medium, whilst the segregated medium pool of the intestinal lumen has a volumetric capacity of 250 μl and the excretory circuit holds 600 μl medium.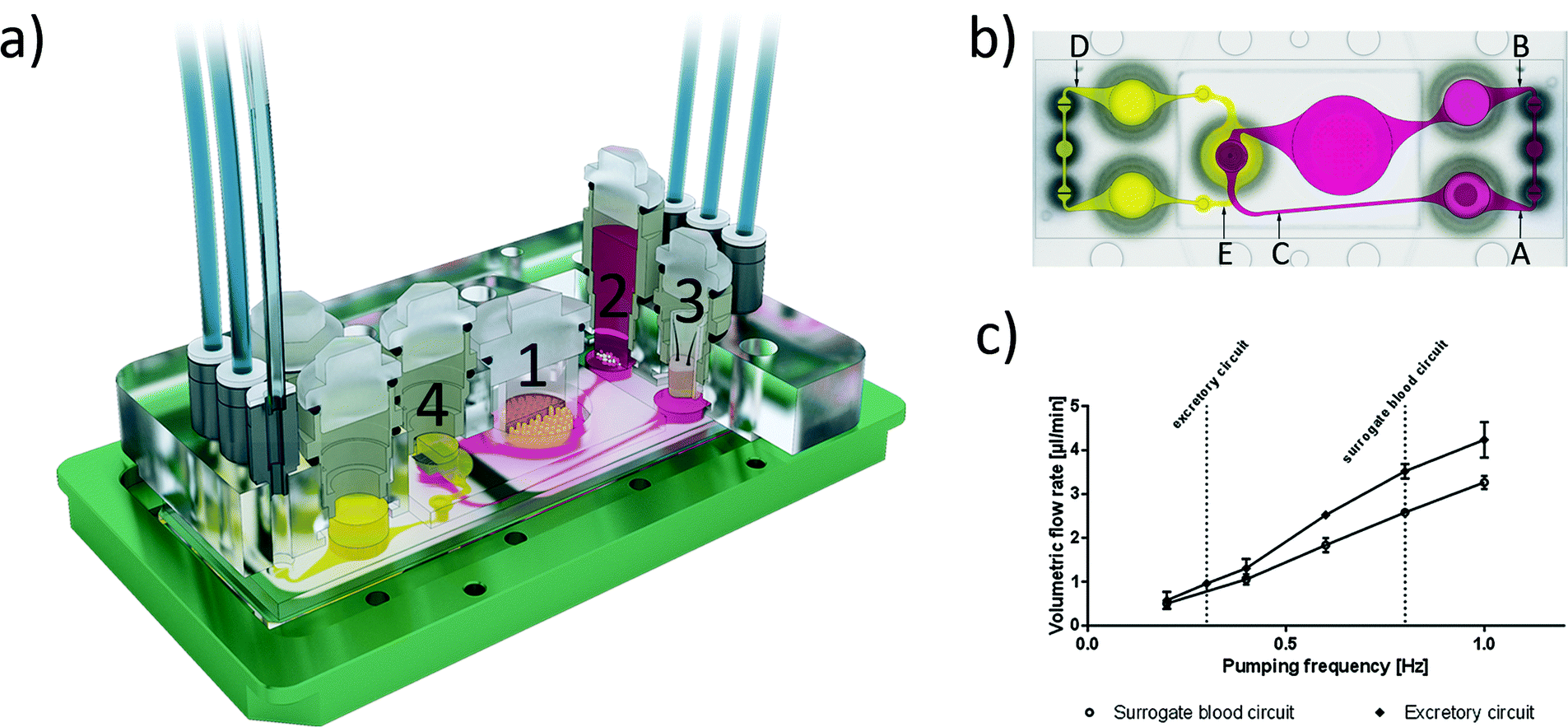 | ||
| Fig. 1 The microfluidic four-organ-chip device at a glance. a) 3D view of the device comprising two polycarbonate cover-plates, the PDMS-glass chip (footprint: 76 mm × 25 mm; height: 3 mm) accommodating a surrogate blood flow circuit (pink) and an excretory flow circuit (yellow). Numbers represent the four tissue culture compartments for intestine (1), liver (2), skin (3), and kidney (4) tissue. A central cross-section of each tissue culture compartment aligned along the interconnecting microchannel is depicted. b) Evaluation of fluid dynamics in the 4OC using μPIV. Top view of the four-organ-chip layout illustrating the positions of three measuring spots (A, B and C) in the surrogate blood circuit and two spots (D, E) in the excretory circuit. c) Average volumetric flow rate plotted against pumping frequency of the surrogate blood flow circuit and the excretory circuit. Co-culture experiments were performed at 0.8 Hz and 0.3 Hz, respectively, indicated by the vertical lines. Error bars are standard error of the mean. | ||
The 4OCs were fabricated by slight modification of a method described previously.8 In brief, two PDMS layers, the upper small and lower large, each 2 mm thick, contain the respective arrangement of channels, micropumps, membranes and openings for culture compartments. The larger layer was permanently bonded to a glass microscope slide with a footprint of 75 × 25 mm (Menzel, Braunschweig, Germany) by low pressure plasma oxidation (Femto; Diener, Ebhausen, Germany), thus forming the respective fluid-tight microfluidics with standard channel heights of 100 μm. Afterwards, a PET membrane (1 μm pores; Polyester Track Etched (PETE) Membrane, GVS, Zola Predosa, Italy) with a diameter of 4 mm was glued to the PDMS layer. Consecutively, the second, smaller PDMS (21.6 × 33.6 mm) layer was permanently bonded to the first layer by low pressure plasma oxidation, forming the connection to the second circuit, separated from the first and lower circuit by the PET membrane. The surrogate blood circuit comprises three culture compartments: i) the intestinal culture compartment is able to hold a cell culture insert with a membrane diameter of 9 mm for the intestinal tissue; ii) the liver spheroid culture compartment, which is directly exposed to the fluid flow; and iii) the skin biopsy culture compartment, where the biopsy tissue is shielded from fluid flow by a standard Transwell® insert (thickness 10 μm, pore size 0.4 μm).
Characterization of fluid dynamics
The microfluidic flow driven by the on-chip micropump was regulated by an external control unit. Among others, the frequency of the actuation, pressure and vacuum above the membrane could be adjusted. Thus, the mechanical forces conveyed by the circulating medium could be adapted to the physiological needs of the respective organ equivalents. We applied micro particle image velocimetry (μPIV) to determine and adjust the flow, described in detail before.12,13 For the flow analysis, a single chip was taken from a large batch of freshly manufactured 4OCs. The fluid flow of the two microfluidic circuits was analysed in selective spots (Fig. 1b). The average flow rate in spots A, B and C was calculated for the surrogate blood circuit and in D and E for the excretory circuit. We used human red blood cells (RBCs) instead of polymeric micro particles. Blood was obtained from donors by venepuncture. After centrifugation at 300g for 5 min, the plasma was discarded and the RBCs were re-suspended in phosphate-buffered saline (PBS). A dilution of 5 × 108 cells per ml was prepared resembling a haematocrit of about 5%. A volume of 600 μl RBC solution was filled into the liver compartment and 300 μl was filled into a reservoir of the excretory circuit. The residual volume of the chip was filled with PBS. A high-speed CMOS-camera (Baumer Optronic HXC40, Radeberg, Germany) coupled to an inverted microscope (Zeiss Axiovert, Jena, Germany) was used to track the movement of the RBCs. The exposure time was set to 4 μs per single image; the frame rate varied, depending on the size of the region of interest (up to 2732 fps). The size of the cells mapped was optimal at a low magnification (2.5×), which resulted in a resolution of 0.23 px μm−1. The displacement of the RBCs in takes of up to 16 s was calculated by the open source MATLAB toolbox PIVlab.14,15 Further calculations were based on the maximum velocity of the laminar plug flow in the channels. Thus, the velocity vectors close to either side of the channel walls were omitted. Specifically, one quarter of the average width was neglected above and below the central velocity field.The spatial and temporal average velocity of the central velocity field (vmax) was determined to calculate the flow rate Q in each spot:
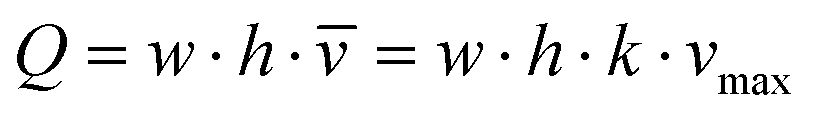 | (1) |
![[v with combining macron]](https://www.rsc.org/images/entities/i_char_0076_0304.gif) .
.
The following pump configuration was used in all experiments: pressure 500 mbar, vacuum −500 mbar, and air flow 0.6 l min−1 at 350 mbar. The frequency was varied from 0.2 to 1 complete pumping cycles per s (Hz).
Human cell and tissue sources
Reconstructed human small intestinal barrier models in cell culture inserts (EpiIntestinal™) of 9 mm size were provided by MatTek Corporation (Ashland, MA, USA). The human HepaRG cell line was obtained from Biopredic International (Rennes, France). Human primary hepatic stellate cells (HHSteC) were purchased from ScienCell Research Laboratories (ScienCell, Carlsbad, CA, USA). Human juvenile prepuce was obtained, with informed consent and ethics approval (Ethic Committee Charité University Medicine, Berlin, Germany), from a paediatric surgery after routine circumcisions, in compliance with the relevant laws. The human proximal tubule cell line RPTEC/TERT-1 was obtained from Evercyte GmbH (Vienna, Austria).Reconstruction of the intestinal barrier model
EpiIntestinal™ models are reconstructions of human organotypic small intestine tissue models, generated from primary human small intestinal epithelial cells that grow in tissue culture inserts. The EpiIntestinal™ models were produced at MatTek Corporation (Ashland, USA) and were released for a two-day overseas shipment at 4 °C after transepithelial electrical resistance (TEER) levels reached a stable range of 50–180 Ω cm2, mimicking the physiologic human small intestine microenvironment.16–18 Upon reception, 350 μl of a specially formulated culture medium was added on the apical side of the EpiIntestinal™ tissue. Then, the cell culture inserts were transferred into the respective compartment of the 4OC systems. Barrier models were positioned about 100 μm above the bottom glass slide of each 4OC circuit to ensure free media passage below the intestinal barrier models.De novo formation of liver equivalents
Cell culture components were purchased from PAN Biotech (Aidenbach, Germany) and cultures were incubated at 37 °C and 5% CO2, unless otherwise stated.The HepaRG cells were maintained, as described by Gripon and colleagues.19 Briefly, cells were cultured in HepaRG medium, consisting of William's Medium E supplemented with 10% human serum, 5 μg ml−1 gentamycin, 5 μg ml−1 human insulin, 2 mM L-glutamine, and 5 × 10−5 M hydrocortisone hemisuccinate (Sigma-Aldrich, St. Louis, USA). Medium was renewed every two or three days. Differentiation was initiated by maintaining the cells in growth medium for two weeks with a regular medium exchange every two or three days in order to reach cellular confluence, according to the manufacturer's instructions. Differentiation medium containing 2% dimethyl sulfoxide (DMSO; Carl Roth GmbH, Karlsruhe, Germany) was then added for a cultivation period of two weeks. When differentiation was completed, the cells were used for further experiments. Human stellate cells were expanded in Stellate Cell Media, provided by ScienCell Research Laboratories (Carlsbad, USA), on poly-L-lysine-coated T75 tissue culture flasks (ScienCell, Carlsbad, USA). Cells were harvested for further use at 80% confluence.
Human liver spheroids were formed combining HepaRG cells and human stellate cells, as described previously,8 using Perfecta3D® 384-Well Hanging Drop Plates (3D Biomatrix, Ann Arbor, USA). Briefly, 20 μl cell suspension containing 4.8 × 104 hepatocytes and 0.2 × 104 HHSteC was pipetted to each access hole. After two days of hanging drop culture, the spheroids were transferred with wide-bore tips to ultra-low attachment 24-well plates (Corning, Amsterdam, The Netherlands). Twenty spheroids were collected together to form a single liver equivalent in the respective culture compartment of the 4OC.
Reconstruction of proximal tubule barrier model
Human proximal tubule cell line RPTEC/TERT-120 was used to reconstruct the proximal tubule barrier and was cultured in a proximal tubule cell medium, which is comprised of a 1 to 1 mixture of DMEM and Ham's F-12 nutrient mix supplemented with 2 mM Glutamin from PAN Biotech (Aidenbach, Germany), 5 μg ml−1 insulin, 5 μg ml−1 transferrin and 5 ng ml−1 sodium selenite (ITS 1×), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin, 10 ng ml−1 epithelial growth factor and 36 ng ml−1 hydrocortisone. Seven days prior to the start of the four-organ culture, 100000 cells were seeded on the PET Membrane separating the two culture circuits in the 4OC. Cells were allowed to adhere to the membrane overnight at static conditions. Subsequently, non-adherent cells were removed and the flow was initiated. The six-day pre-culture allowed the RPTEC/TERT-1 to adjust to the flow and to polarise.
Preparation of the skin biopsy barrier
Prepuce samples with an average size of 2.5 cm2 were stored and transported in 10 ml PBS at 4 °C and prepared for further culture within 4 h following surgery. They were subsequently incubated in 80% ethanol for 30 s. Samples with an average thickness of 2 mm were punched to provide biopsies of 4.5 mm diameter. Samples were loaded with the epidermal-side up into 96-well Transwell® (Corning, Lowell, USA) inserts and were then integrated into the respective 4OC culture compartment.Chip-based co-cultures
The 4OC circuits were loaded with the RPTEC/TERT-1 cells seven days prior to the start of the co-culture experiment, as described above. In order to start the co-culture experiment, all preloaded 4OCs were incorporated with the small intestine barrier inserts, the liver equivalents and the skin biopsy inserts. A frequency of 0.8 Hz in the surrogate blood circuit and 0.3 Hz in the excretory circuit were chosen to ensure pulsatile fluid flow. The height of the tissue and the amount of medium added to the cell culture inserts enabled the tissues to be at the air–liquid interface (skin) or submerged in media (intestine). Three different culture media were used: i) the small intestine culture medium containing 2.5 g l−1 glucose and 10% human serum in the 250 μl lumen on top of the small intestine insert; ii) the liver tissue medium containing 2.0 g l−1 glucose and 10% human serum in the 830 μl volume of the surrogate blood circuit; and iii) the proximal tubule cell medium containing 1.0 g l−1 glucose and no serum in the 600 μl volume of the excretory circuit. Supernatants were collected twice each day from all three media pools separately: 250 μl (100%) from the intestinal lumen, 500 μl (60%) from the surrogate blood circuit and 300 μl (50%) from the excretory circuit for determination of glucose and lactate metabolism. Harvested media volumes were replaced by the respective fresh medium of each medium pool. After 7, 14, 21, and 28 days of culture, tissues were removed from the 4OC and cell behaviour, tissue architecture and transcriptional levels of protein expression were analysed by immunohistochemistry and qRT-PCR techniques. Each experimental set-up was conducted with five replicates.Co-culture analyses
Tissue viability was monitored daily by the measurement of lactate dehydrogenase (LDH) released in the supernatants of the three media pools – intestinal lumen pool (Fig. 1a, brown), surrogate blood circuit pool (Fig. 1a, red) and excretory circuit pool (Fig. 1a, yellow) – segregated from each other by the small intestine and proximal tubule cell barriers. In brief, samples were immediately measured for LDH activity. All absorbance-related measurements were performed in 96-well or 384-well microtitre plates (Corning, Amsterdam, The Netherlands) in a microplate reader (FLUOstar Omega, BMG Labtech, Ortenberg, Germany), if not stated otherwise. The LDH activity of the medium was measured using the Pierce™ LDH Cytotoxicity Assay Kit (Thermo Fisher Scientific Inc. Waltham, USA), according to the manufacturer's instructions, with minor modifications based on our own standard curves LDH Positive Control (Cayman, Ann Arbor, USA): an amount of 12.5 μl of reagent was used and 12.5 μl of the sample was added for each measurement.Metabolic activity of the tissues was monitored daily by the measurement of glucose and lactate concentration in the media supernatants of the three pools. For this, the GLU 142 (Diaglobal, Berlin, Germany) was used with minor modifications: an amount of 99 μl of reagent was used and 1 μl of sample was added. Lactate concentration of the medium was measured using the LAC 142 kit (Diaglobal, Berlin, Germany), with minor modifications based on our own standard curves: an amount of 99 μl of the reagent was mixed with 1 μl of sample and absorbance was measured at 520 nm, using medium as a reference.
Immunohistochemical end-point analyses were performed by staining the liver tissue sections for cytochrome P450 3A4 (CYP3A4, mouse anti-human, Santa Cruz Biotechnology, Heidelberg, Germany). Skin tissues were stained for cytokeratin 10 (Ctk 10, mouse anti-human, Merck-Millipore, Darmstadt, Germany) and cytokeratin 15 (Ctk 15, rabbit anti-human, Abcam, Cambridge, UK), and intestinal tissue for cytokeratin 19 (Ctk 19, rabbit anti-human, DB Biotech, Kosice, Slovakia). Briefly, representative central cryosections of the tissues were fixed in acetone at −20 °C for 10 min and blocked with 10% goat serum in PBS. Tissue sections were then incubated with the respective primary antibody for 2 h and washed with PBS. Goat anti-rabbit IgG Alexa Fluor® 594 and goat anti-mouse FITC antibodies (all purchased from Life Technologies, Darmstadt, Germany) were used for visualization.
Real-time qPCR endpoint analyses were performed to evaluate gene transcription at mRNA level after the MOC cultures were stopped. Tissue equivalents were collected for RNA isolation using the RNeasy Mini Kit (Qiagen, Hilden, Germany). The cDNA was synthesized by reverse transcription of 400 ng total RNA (TaqMan1, Roche Diagnostics, Mannheim, Germany). Real-time qPCR experiments were conducted using the Stratagene system (Agilent Technologies, Böblingen, Germany) and the SensiFast SYBR No-ROX One-Step Kit (Bioline, Luckenwalde, Germany), according to the manufacturer's instructions. The real-time qPCR primers are shown in Table 1.
| Gene | Primer (5′ → 3′) | |
|---|---|---|
| Forward | Reverse | |
| Intestine: | ||
| SGLT1/SLC5A1 | gAgCCCAgCAACTgTCCCAC | CAggCTCCAACACAgACggT |
| Na-K-ATPase | ACCgCCCAgAAATCCCAAAAC | CAgCggTCATCCCAgTCC |
| MDR1 | TggATgTTTCCggTTTggAg | TgTgggCTgCTgATATTTTgg |
| Kidney: | ||
| SGLT2/SLC5A2 | CAACCTCAATgCCCTgCTC | ACTgCCCCTTCCCCTTTTC |
| Claudin 10 | gggCTgTgCTCAATgACTgg | gCCCCgTTgTATgTgTATCTgg |
| TJP3/ZO-3 | gAgAAgCCAgTTTCAAgCgCC | gTCACATCCAggAgCgCATg |
| Liver: | ||
| Albumin | TgCAAggCTgATAAggAg | TTTAgACAgggTgTTggCTTTACAC |
| BSEP/ABCB11 | gCAgACACTggCgTTTgTTg | ATgTTTgAgCggAggAACTgg |
| GSTA2 | CTgAggAACAAgATgCCAAgC | AgCAgAgggAAgCTggAAATAAg |
| CPY3A4 | ggAAgTggACCCAgAAACTgC | TTACggTgCCATCCCTTgAC |
| UGT1A1 | ATgCAAAgCgCATggAgAC | ggTCCTTgTgAAggCTggAg |
| MRP2 | ggggACACTgTTggCTTTgTTC | CCCAgggTgCCTCATTTTCCA |
Immunofluorescence staining of RPTEC/TERT-1 cell monolayers
After culture, the membrane with the RPTEC/TERT-1 monolayer was removed from compartment 4 of the 4OC (Fig. 1a), washed with PBS and fixed with 4% PFA for 10 min in a 48-well plate. After washing, the cells were permeabilised with 0.05% Triton X-100, washed and blocked with 10% serum for 20 min. Then, the membranes were incubated with primary antibody rabbit anti-human NaK-ATPase (Abcam, Cambridge, UK) and mouse anti-human Cytokeratin 8/18 (Santa Cruz Biotechnology, Heidelberg, Germany) for 2 h. Subsequently, the membranes were washed three times and incubated with the secondary antibody goat anti-rabbit Alexa Fluor 594 and goat anti-mouse FITC (both Life Technologies, Darmstadt, Germany) for 45 min. After washing and mounting, 3D stack images of the samples were acquired by two-photon microscopy (TriMScope II; LaVision BioTec, Bielefeld, Germany). The 3D images were reconstructed from the image stack collected, using Imaris software (Bitplane, Zurich, Switzerland).Transepithelial electrical resistance
The barrier function of the small intestinal epithelial tissue was monitored by TEER using the Millicell ERS-2 Volt-Ohm Meter (Millipore Co., Bedford, USA). Millicell inserts were removed from the MOC during measurement and TEER analysis was performed, as described by the manufacturer. The TEER was expressed as Ω cm2 based on the area of Transwell® plate inserts after subtracting the resistance of the supporting filter from the reading and multiplying it by the surface area.Results and discussion
Ensuring good absorption, distribution, metabolism and excretion (ADME) properties are crucial to analyse if a drug reaches its intended target and has a therapeutic effect without causing unacceptable toxicities. However, only few drug companies test for ADME characteristics in the early stages of drug discovery and development.21 Current in vitro models are lacking a systemic approach and therefore fail to predict the interaction of metabolites between organ cultures. To overcome this problem, we combined four human organ equivalents through a microfluidic flow in our 4OC. On top of the surrogate blood flow circuit, a primary human small intestinal model was inserted in a cell culture insert (Fig. 1a) and provided a barrier function between the apical side of the intestine and the surrogate blood circuit, allowing absorption. An on-chip micropump enabled the distribution from the basolateral side of the intestinal model to a liver equivalent, where potential substances could be metabolised. The microfluidic channel further passed a PET membrane, seeded with renal proximal tubule cells. This kidney model separated the surrogate blood circuit from the second, excretory circuit. In this study, we combined this ADME approach with a skin biopsy. The skin biopsy could be either used as an alternative absorption route or to analyse toxicity of the parent drug or its metabolites. However, the skin could also be replaced by any other organ equivalent, like neuronal tissue, lung tissue or others, depending on the toxicity target to be analysed. The 4OC has been designed to circulate 30–40 μl fluid in its channel system to match a surrogate blood volume a 100000-fold smaller than the original volume of an adult human.22,23 Additionally, the culture compartments surrounding the tissues provide a medium volume of about 90–120 μl, corresponding to a 100000-fold reduction to the original interstitial fluid in man. Finally, the four-organ-chip consists of an artificial media reservoir of 650–680 μl above the liver tissue culture compartment. Such a reservoir obviously does not exist in vivo. However, in the 4OC it is necessary to provide sufficient nutrients to the tissues with limited feeding rates. This artificial reservoir is envisioned to be eliminated from the design, as soon as vascularisation of the tissues can be accomplished and the four-organ-chip will be transferred to oral feeding at blood perfusion.
Evaluation of fluid dynamics
Microscopic access to each and every area of the 4OC facilitates in-depth fluid flow analyses at various spots on the chip (Fig. 1b and c). Two robust peristaltic on-chip micropumps, integrated into a surrogate blood flow circuit and an excretory circuit, respectively, are capable of circulating media at sterile conditions over weeks. The flow rate ranged from 0.51 μl min−1 (at 0.2 Hz) to 3.26 μl min−1 (at 1 Hz) in the surrogate blood flow circuit, and from 0.58 μl min−1 to 4.24 μl min−1 in the excretory circuit. The pulse frequency in the surrogate blood flow circuit can be increased up to 2.4 Hz, which corresponds to a high but still physiological heart activity of 144 beats per min in humans. The mean velocity increased almost linearly with the pumping frequency. The pumping frequency used in our experiments (0.8 Hz in the surrogate blood circuit) corresponds to 48 “heart-beats” per min (approximately two thirds of the physiological value of an adult at rest) to avoid untypical high shear stress in the liver compartment. At this frequency, the medium flow rate corresponds to 2.58 μl min−1. Thus, the total volume of 830 μl of the surrogate blood circuit needs 5.3 h for a complete turnover. In the excretory circuit, this takes 10.4 h. The 4OC supports rectified or reversed flow in the two chambers of the common kidney compartment, segregated by a porous membrane. Here we applied reversed flow.We successfully established μPIV using RBCs to characterize fluid flow profiles at different spots on the 4OC circuits. The use of human RBCs was advantageous to synthetic micro-particles used in our earlier fluid dynamic study13 due to their surface potential. It prohibited the occasional adherence of particles to channel walls, cells or cell residues considerably. Since we used RBCs at a density suited for μPIV measurements, no difference was observed to runs with synthetic particles of a similar size (data not shown). Moreover, we envision RBCs as an essential prerequisite for later non-invasive online measurement of fluid flow once synthetic media is substituted by human blood in microphysiological MOC systems.
Systemic tissue viability and barrier organ integrity
We have successfully applied LDH measurement to evaluate the systemic viability of co-cultures in the MOC system in the past.8 Human lactate dehydrogenase is a 140 kD enzyme catalysing the conversion of L-lactate and NAD to pyruvate and NADH in the final step of anaerobic glycolysis in human cells. Here, we evaluated the LDH activity in each of the three media pools separately. We hypothesised that LDH is not able to pass a functional intestine or proximal tubule cell membrane due to its size. Consequently, as long as the barriers are functional, LDH values on the apical site of the epithelia – in the intestinal lumen and the excretory circuit – should be exclusively generated by the physiological turnover of the epithelial cells, and the LDH values in the surrogate blood circuit should be exclusively generated by a cell turnover, appearing in the liver equivalent and in the dermal part of the skin equivalent. The results are summarized in Fig. 2. After a period of 4 days, LDH levels in the 4OC normalized toward a constant steady state in all three media pools, indicating a constant physiological cell turnover within the different organ compartments. The high LDH values in the intestinal lumen and in the surrogate blood circuit at the beginning of the 4OC co-culture are most probably related to increased initial cell death due to shipment conditions for the small intestine and the damaging biopsy procedure for the skin tissues, respectively.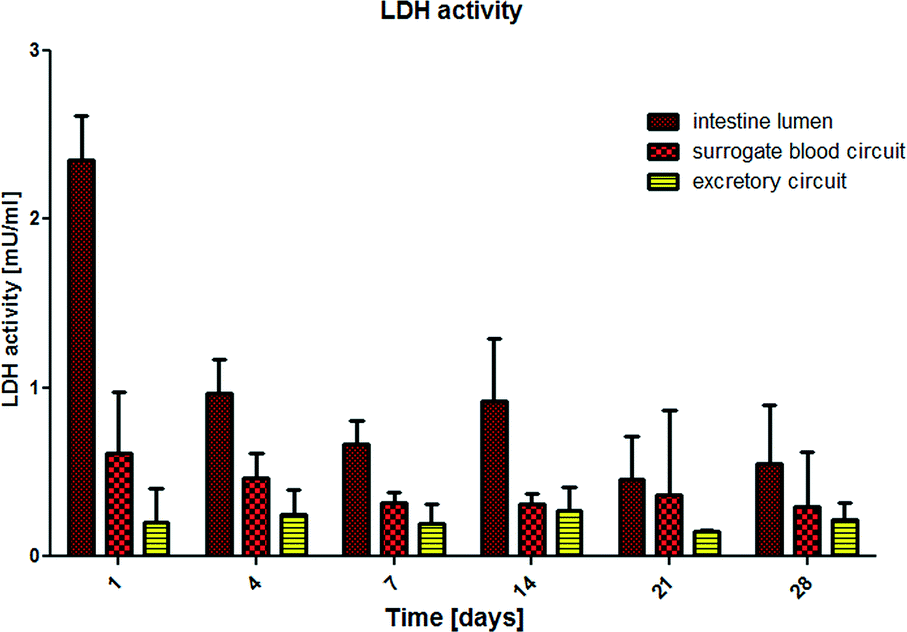 | ||
| Fig. 2 Systemic tissue viability in the 4OC over 28 days of co-culture measured by LDH activity in the intestine lumen, the surrogate blood circuit and the excretory circuit media pools, segregated from each other by a functional small intestine and proximal tubule barrier, respectively. | ||
By comparison, increased LDH values are repeatedly reported in plasma or urine of patients, where they are associated with tumour cell death or renal epithelia cell death, respectively.24,25 It is important to consider that LDH expression in tumour cells is significantly increased in comparison to the respective primary cells, from which the tumour cells originate.26 This is due to the metabolic shift toward anaerobic glycolysis in these immortalized cells. The human HepaRG cell line and the RPTEC/TERT-1 cell line are immortalized cells and, therefore, might express higher LDH levels than primary human hepatocytes and proximal tubule cells at an individual cell level. This might explain the detectable LDH levels in the surrogate blood circuit and the excretory circuit during the entire co-culture period. The higher LDH levels in the intestinal lumen of the 4OC are related to the high turnover of small intestine epithelia, which is in the range of 3–5 days.27 Finally, the differences in LDH activity of the three media pools is a first clear indicator of the biological integrity of the small intestine and proximal tubule cell barriers in the 4OC. A more reliable indicator of the biological integrity and functionality of human intestinal or proximal tubule barriers is the glucose balance generated by these barriers in the context of different high glucose challenges on both sides of each barrier. Glucose concentration measured in the media pools over the 28-day co-culture are summarized in Fig. 3. The amount of glucose stored in the human body is approx. 450 g and 250 g is turned over daily. The normal Western diet provides 180 g glucose per day and the gap between the amount in food and the amount utilised is bridged by stored glucose and gluconeogenesis mainly in the liver and kidneys.28 The brain alone utilises 125 g of the daily glucose turnover. We extrapolated a daily turnover of less than 1.2 mg glucose for the four organ equivalents co-cultured in the 4OC, which were established at a size 100000-fold smaller than their original counterpart organs. We supplemented glucose in excess of close to 2.2 mg every day to the three media pools – 0.62 mg into the intestinal lumen, 1.25 mg into the surrogate blood circuit and 0.3 mg into the excretory circuit – to evaluate the response of the tissues at non-physiologically high amounts of glucose. Homeostatic mechanisms maintain blood glucose within narrow limits of 0.65 g l−1 and 1.1 g l−1 to ensure that an adequate amount of energy is provided to the organs of the body. Glucose is usually absorbed completely in the small intestine. The kidneys assist in keeping blood glucose levels normal by reabsorbing the filtered 180 g of glucose at the glomeruli. Virtually no glucose appears in the urine (<0.5 g per day) because of the presence of glucose transporters in the proximal tubule.28 Here, we report a remarkably stable glucose balance in the three media pools, which has been established in the 4OC system over the entire period of the 28-day co-culture of the four tissues (Fig. 3). The glucose level in the surrogate blood circuit was constantly decreased to values fitting into the narrow range of normal blood glucose. Glucose concentration in the intestinal lumen decreased at least four-fold in comparison to the initial medium content of 2.5 g l−1. Finally, the lowest level of glucose was constantly present in the excretory circuit, although this value never dropped to zero.
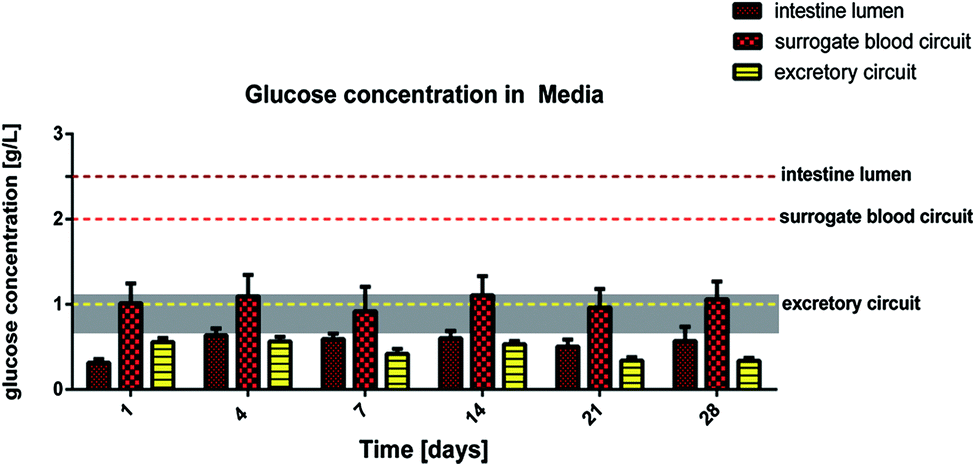 | ||
| Fig. 3 Glucose balance in the three segregated media pools of the 4OC over 28 days. Bars are plotted against the background of the physiological glucose concentration range in human blood of healthy people (grey area). Dotted lines indicate the respective initial glucose level of the cell culture media fed daily into the intestine lumen (brown), the surrogate blood circuit (red) and the excretory circuit (yellow). | ||
The ability of the barrier models to maintain gradients between the three medium compartments was additionally verified by protein concentration measurements (data not shown).
Transepithelial electrical resistance (TEER) measurements were performed for intestinal tissues. The TEER values stayed constant over the culture time and ranged from 75 to 195 Ω cm2, indicating an intact and physiological barrier, comparable to the human intestine, where TEER values range from 50 to 120 Ω cm2.16,17
The distinct glucose and protein concentration gradients between the three media pools and the consistent TEER values verify the functional integrity of the intestinal and the proximal tubule cell barriers throughout the entire co-culture.
Four-organ-co-culture performance
At the end of the 28 day co-culture period, we investigated the tissue architecture of the different organ equivalents using immunohistochemical staining. Fig. 4 illustrates the prime findings of these analyses. The human small intestine barrier exhibited a perfect physiological polarisation, as shown by Ctk 19 expression along the crypt-villus axis (Fig. 4a). In humans, Ctk 19 is present along the entire crypt-villus axis and is mainly expressed in the apical domain of polarised cells. Its downregulation has been shown to decrease the number of microvilli and derange the targeting of apically distributed proteins.29–31 Remarkably, in addition to the microvilli expression, human small intestine epithelia form a villi-like 3D architecture of up to 270 μm height in the 4OC co-culture. Such villi-like structures have rarely been shown to appear to a maximum height of 130 μm in CaCo-2 cell models – the current gold standard for an intestinal barrier in vitro – when shear stress is applied.32 Here, we report such an architectural self-assembly without periodical shear stress applied to the apical site of the intestinal model. The self-assembly capacity of the primary human small intestine epithelia within the right microenvironment might be a possible explanation for this reproducible phenomenon.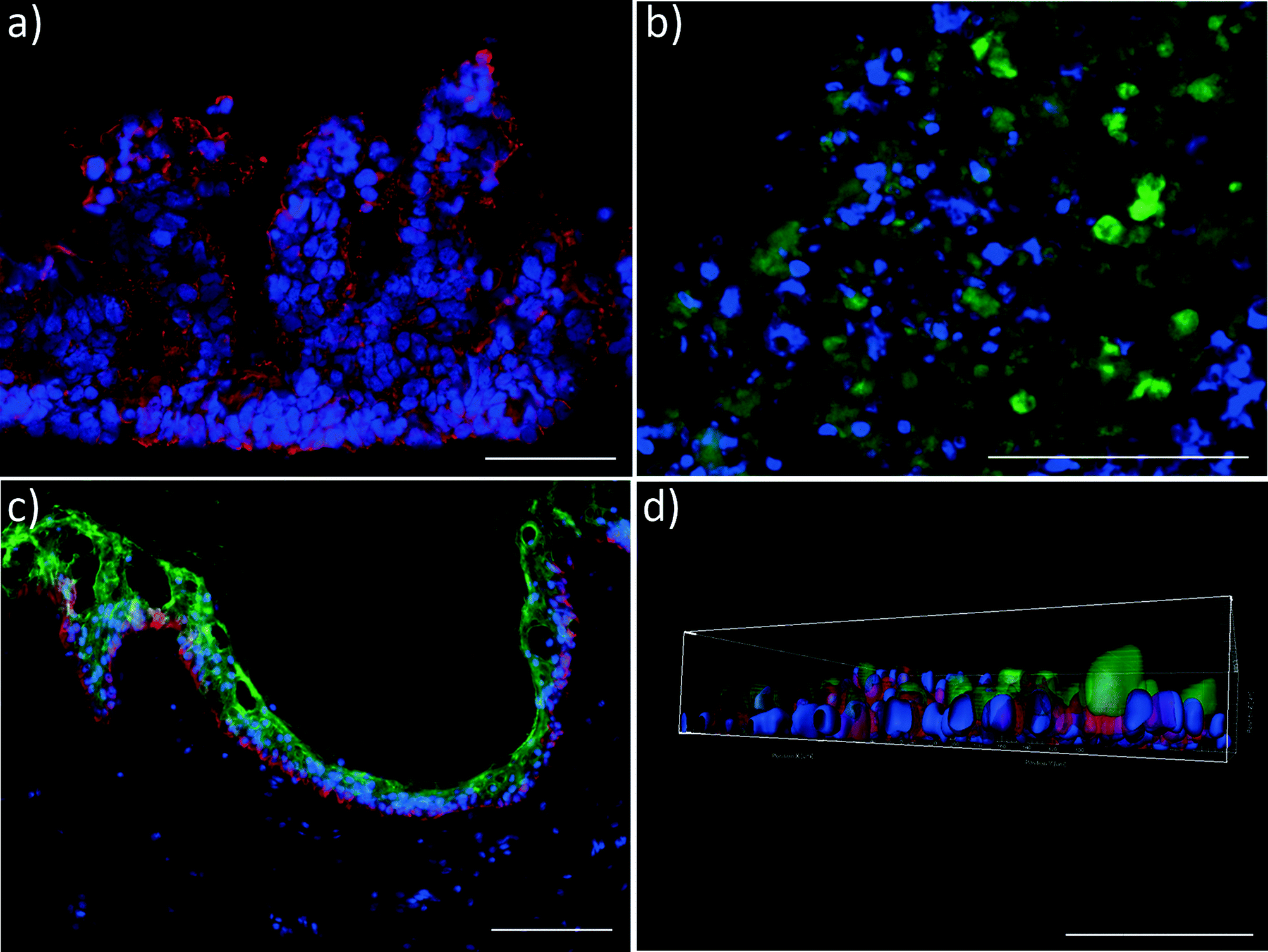 | ||
| Fig. 4 Performance of human tissues in the 4OC after 28 days of co-culture. Staining a) for Ctk 19 (red) expression in small intestinal epithelial tissue, b) CYP3A4 (green) expression in liver aggregates, c) epidermal markers Ctk 10 (green) and Ctk 15 (red) in skin biopsies, d) transporter NaK-ATPase (red) and Ctk 8/18 (green) in proximal tubule epithelial monolayer. (a–d) Nuclei were stained with DAPI (blue). Bars = 100 μm. | ||
Three-dimensional spheroids of hepatocytes co-cultured with human stellate cells, the fibroblasts of the liver, preserve their initial well-defined 3D architecture over the entire co-culture period. Moreover, they still express the key CYP3A4, as exemplarily demonstrated in Fig. 4b.
Skin biopsies reveal a fully functional, proliferative and differentiated epidermis, as staining of epidermal markers Ctk 10 and 15 showed (Fig. 4c). The culture at a physiological air–liquid interface facilitates the maintenance of a stratified stratum corneum, thus providing an in vivo-like architecture for topical substance administration. Various successful MOC experiments with skin biopsies, reconstructed skin and a combination of both have been performed in the past,8,33 verifying the robustness and flexibility of the platform.
Finally, polarisation of the Na-K-ATPase at the basolateral surface of the human proximal tubule cell layer indicates distinct transport function of the sodium-potassium pump during the 28-day co-culture in the 4OC.
In order to further evaluate the functional integrity and capabilities of the established organ equivalents to absorb, distribute, metabolise, reabsorb and excrete molecules, we applied gene expression analysis for selected genes associated with these processes. To investigate the cell barriers of the small intestine (Fig. 5a) and proximal tubule (Fig. 5b) we focused on the expression of the major active sodium-coupled glucose transporters SLC5A1 playing a central role in intestinal glucose absorption,28 and SLC5A2 (SGLT2 gene family), crucial for renal proximal tubule glucose reabsorption34 (Fig. 5a, b). Both transporters are constitutively expressed in their respective tissues over the entire co-culture period. Remarkably, glucose is present in the glomerular filtrate in vivo and is, under usual circumstances, completely reabsorbed by SGLT2 transporters of proximal tubule cells. Recent observations have shown that diabetic patients up-regulate the expression of SGLT2 renal glucose transporters, presumably to compensate for and reduce glycosuria.35 The higher expression of SLC5A2 in the 4OC might be the result of a similar up-regulation due to the exposure of the proximal tubule cells to the high glucose levels in the surrogate blood circuit and the excretory circuit. However, comparing these expressions to conventional culture methods, the expression was 2.5 fold lower in cells cultured under fluid flow (data not shown).
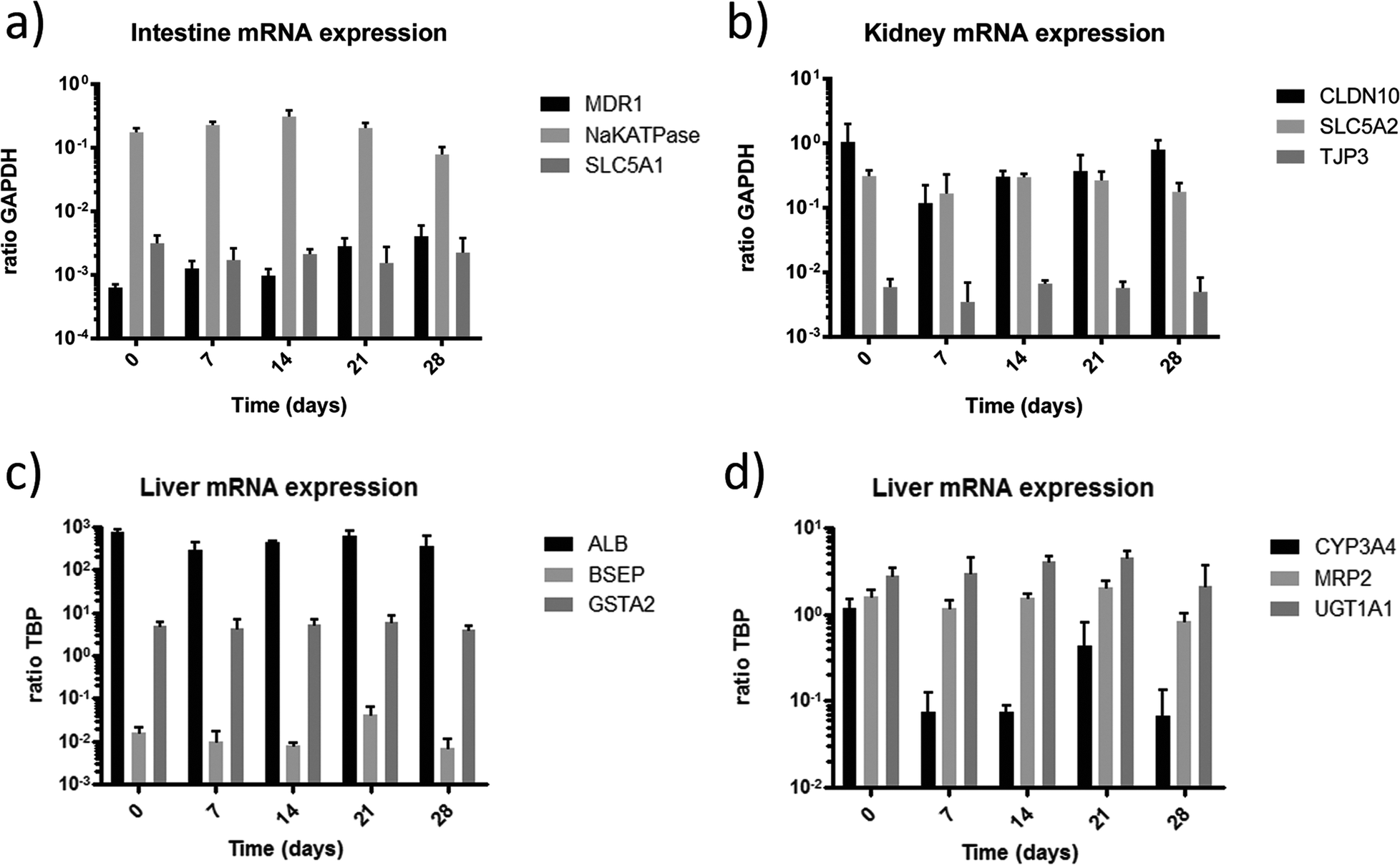 | ||
| Fig. 5 Gene expression in co-cultures of the four-organ-chip over 28 days. a) Absorption potential of the intestine barrier is verified by SLC5A1, Na-K-ATPase and MDR1 expression. b) Reabsorption and consequently excretion capability of the kidney proximal tubules cell barrier is verified by the expression of SLC5A2. The expression of Claudin-10 and TJP3 underpins barrier integrity and cell turnover. c) Physiological function of the liver tissue is verified by stable steady state expression of Albumin, BSEP and GSTA2 at their discrete levels over the entire co-culture period. d) Liver capability to perform phase I, phase II and phase III metabolism is exemplarily shown by expression of CYP3A4, UGT1A1 and MRP2 respectively. | ||
The expression of the sodium–potassium ATPase in the small intestine is a prime prerequisite for the maintenance of physiological ion gradients across the cell membrane and the associated molecular transport capacity. The expression of the sodium–potassium ATPase stayed constant over the culture period. Finally, multidrug resistance transporters, such as MDR1, provide a general protection against hydrophobic xenobiotics in intestine and all other human tissues.36 Over the culture period of 28 days, the mRNA expression of MDR1 in the intestinal tissue increased slightly, but not significantly. The stable expression of these targets demonstrates the homeostasis of the tissue within the 4OC culture.
Tight junctions are membrane proteins that close the intercellular space in epithelial cells. They act as a paracellular barrier regulating the passage of ions, water and various macromolecules. Additionally, they maintain epithelial cell polarity and thus prevent intermixture of membrane components of the apical with those of the lateral side. Claudin-10 is concentrated within the tight junctions of proximal tubule epithelial cells and is responsible for the formation of tight junction strands.37 Tight junction protein 3 (ZO-3) is involved in cell proliferation and epithelial homeostasis through complex formation with cyclin D1.38 The constitutive expression of claudin-10 and TJP3 in the proximal tubule cells indicates physiological function of the tight junctions of this barrier throughout the co-culture period.
Physiological function of the liver tissue in the 4OC co-cultures is verified by the stable steady-state expression of albumin, bile salt export pump (BSEP) and GSTA2 at their discrete levels over the entire co-culture period of 28 days (Fig. 5c). Albumin is constitutively expressed at high levels. Its secretion into the medium circulation of MOCs has been demonstrated before.8 The BSEP or sPgp (sister of P-glycoprotein) is the product of the ABCB11 gene. The activity of this transporter in humans is the major determinant of bile formation and bile flow. This particular protein is responsible for the transport of taurocholate and other cholate conjugates from hepatocytes to the bile.39 Its consistent expression indicates functional bile formation over the 28 days. A major remaining drawback of each and every currently existing liver-on-a-chip model is the lack of bile removal from the liver equivalent through a channel system, segregated from the main medium circuit, as summarized in a respective review.40 Glutathione S-transferase (GSTA2) plays a key role in detoxification of electrophilic compounds by conjugation with glutathione. Therefore, it protects cells from reactive oxygen species and products of peroxidation. Its constant expression serves as a marker of the self-protective capacity of the liver equivalent in the co-culture.
The capability of the liver model to perform phase I, II and III metabolism is exemplarily shown by expression of CYP3A4, UGT1A1 and MRP2, respectively (Fig. 5d). CYP3A4 is an important phase I metabolism enzyme acting by oxidising xenobiotics. A high expression of CYP3A4 can be seen at day 0 before culture. The expression was reduced after 4 days of culture (data not shown) and stayed constant over the following 25 days of culture, at a comparable expression level observed in previous cultures.8,10 The high expression of CYP3A4 at day 0 of culture is related to the pre-culture in 2% DMSO for differentiation of HepaRG prior to the co-culture, as DMSO considerably induces CYP3A4 activity.41 This supports the previous findings, indicating that the system needs 3 to 4 days to adapt to the new microenvironment in order to establish a reproducible homeostasis. A potential substance application should therefore only start after a 4 day adaption phase. Uridine 5′-diphospho-glucuronosyltransferase (UGT1A1) transforms small lipophilic molecules, such as steroids, bilirubin, hormones, and drugs, into water-soluble, excretable metabolites.42 It is a major enzyme of the phase II metabolism in the liver. Multi-drug resistance-associated protein 2 is expressed in the canalicular (apical) part of the hepatocyte and functions in biliary transport (Phase III metabolism). The discrete expression of the three key metabolic proteins, each acting in a different phase of the metabolism, verifies that the liver equivalent has a broad and stable metabolic capacity throughout the entire co-culture period of 28 days. The addition of antibiotics to the culture medium during co-culture exposes a constant stimulation of xenobiotic metabolism in the liver equivalent of the 4OC.
Conclusions
For the first time, we report a microphysiological four-organ-chip system that enables a reproducible 28 day co-culture of four tissues. All tissues maintained a high cell viability and discrete physiological tissue architecture over the entire co-culture period. The human small intestine epithelia formed stable 3D villi-like structures of up to 270 μm height and expressed an apical brush border. Skin biopsies developed a stratified stratum corneum at the air–liquid interface. The human proximal tubule epithelia maintained a functional polarised monolayer barrier. Small intestine and kidney proximal tubule cell barriers were able to keep a continuously stable glucose gradient balance between the intestinal lumen, the surrogate blood circuit and the excretory circuit of the system. Moreover, in-depth metabolic and gene analysis revealed the establishment of a reproducible homeostasis between all four tissues over at least 28 days independent of the individual human cell line or tissue donor background used for each organ equivalent. The four-organ-chip presented here has been designed to support ADME profiling of substances along with repeated dose systemic toxicity testing of drug candidates. We could demonstrate integrity and functionality of the intestine and the kidney biological barriers at a physiologically relevant organ downscale. A separate medium reservoir at the apical surface of the intestinal barrier allows for “oral” administration of a drug compound. Following absorption, a drug will reach the surrogate blood circulation mimicking the plasma compartment. Subsequently, a drug compound will be distributed to the liver equivalent followed by the skin and the kidney equivalent. This arrangement of our four-organ-chip enables physiological absorption, first path metabolism in the liver tissue, secondary metabolism and finally excretion through the kidney model. Permanent access to the different compartments and the organ equivalents themselves enable to evaluate pharmacokinetic and pharmacodynamics parameters, such as effective concentration, maximum tolerable dose, time course and intensity of therapeutic and adverse effects.Acknowledgements
The authors would like to thank Philip Saunders for the outstanding creative assistance and feedback on the manuscript and Peter Mangel for the preeminent graphic visualisation of the 4OC in Fig. 1. EpiIntestinal™ tissues were kindly provided by MatTek cooperation. We thank Dr. Guguen-Guillouzo, Dr. Gripon and Dr. Trepo who made HepaRG cells available. The work has been funded by the German Federal Ministry for Education and Research, GO-Bio grant no. 0315569.References
- M. B. Esch, T. L. King and M. L. Shuler, The role of body-on-a-chip devices in drug and toxicity studies, Annu. Rev. Biomed. Eng., 2011, 13, 55–72, DOI:10.1146/annurev-bioeng-071910-124629.
- D. Huh, G. A. Hamilton and D. E. Ingber, From 3D cell culture to organs-on-chips, Trends Cell Biol., 2011, 21(12), 745–754, DOI:10.1016/j.tcb.2011.09.005.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow, Lab Chip, 2012, 12(12), 2165–2174, 10.1039/c2lc40074j.
- U. Marx, H. Walles and S. Hoffmann, et al., Human-on-a-chip developments: a translational cutting-edge alternative to systemic safety assessment and efficiency evaluation of substances in laboratory animals and man?, ATLA, Altern. Lab. Anim., 2012, 40(5), 235–257 CAS; http://www.ncbi.nlm.nih.gov/pubmed/23215661, Accessed March 23, 2015.
- J. H. Sung, M. B. Esch and J.-M. Prot, et al., Microfabricated mammalian organ systems and their integration into models of whole animals and humans, Lab Chip, 2013 10.1039/c3lc41017j.
- C. Zhang, Z. Zhao, N. A. Abdul Rahim, D. van Noort and H. Yu, Towards a human-on-chip: culturing multiple cell types on a chip with compartmentalized microenvironments, Lab Chip, 2009, 9(22), 3185–3192, 10.1039/b915147h.
- Y. Imura, Y. Asano, K. Sato and E. Yoshimura, A microfluidic system to evaluate intestinal absorption, Anal. Sci., 2009, 25(12), 1403–1407 CrossRef CAS PubMed; http://www.ncbi.nlm.nih.gov/pubmed/20009325, Accessed March 25, 2015.
- I. Wagner, E.-M. Materne and S. Brincker, et al., A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture, Lab Chip, 2013, 13(18), 3538–3547, 10.1039/c3lc50234a.
- E.-M. Materne, A. P. Ramme, A. P. Terrasso, M. Serra, P. M. Alves, C. Brito and U. Marx, A multi-organ chip co-culture of neurospheres and liver equivalents for long-term substance testing, J. Biotechnol., 2015, 205, 36–46 CrossRef CAS PubMed.
- I. Maschmeyer and T. Hasenberg, A. Jaenicke, et al., Chip-based human liver-intestine and liver-skin co-cultures - a first step toward systemic repeated dose substance testing in vitro, Press, 2015 Search PubMed.
- M.-H. Wu, S.-B. Huang, Z. Cui, Z. Cui and G.-B. Lee, A high throughput perfusion-based microbioreactor platform integrated with pneumatic micropumps for three-dimensional cell culture, Biomed. Microdevices, 2008, 10(2), 309–319, DOI:10.1007/s10544-007-9138-3.
- R. Lindken, M. Rossi, S. Grosse and J. Westerweel, Micro-Particle Image Velocimetry (microPIV): recent developments, applications, and guidelines, Lab Chip, 2009, 9(17), 2551–2567, 10.1039/b906558j.
- K. Schimek, M. Busek and S. Brincker, et al., Integrating biological vasculature into a multi-organ-chip microsystem, Lab Chip, 2013, 13(18), 3588–3598 RSC; http://www.ncbi.nlm.nih.gov/pubmed/23743770 .
- W. Thielicke and E. J. Stamhuis, PIVlab – Towards User-friendly, Affordable and Accurate Digital Particle Image Velocimetry in MATLAB, J. Open Res. Softw., 2014, 2(1), e30, DOI:10.5334/jors.bl.
- W. Thielicke, The Flapping Flight of Birds - Analysis and Application, 2014 Search PubMed.
- V. Gupta, N. Doshi and S. Mitragotri, Permeation of insulin, calcitonin and exenatide across Caco-2 monolayers: measurement using a rapid, 3-day system, PLoS One, 2013, 8(2), e57136, DOI:10.1371/journal.pone.0057136.
- P. Artursson, A. L. Ungell and J. E. Löfroth, Selective paracellular permeability in two models of intestinal absorption: cultured monolayers of human intestinal epithelial cells and rat intestinal segments, Pharm. Res., 1993, 10(8), 1123–1129 CrossRef CAS; http://www.ncbi.nlm.nih.gov/pubmed/8415396, Accessed March 25, 2015.
- E. Le Ferrec, C. Chesne and P. Artusson, et al., in vitro models of the intestinal barrier. The report and recommendations of ECVAM Workshop 46. European Centre for the Validation of Alternative methods, ATLA, Altern. Lab. Anim., 2001, vol. 29, 6, pp. 649–668 Search PubMed; http://www.ncbi.nlm.nih.gov/pubmed/11709041, Accessed March 25, 2015.
- P. Gripon, S. Rumin and S. Urban, et al., Infection of a human hepatoma cell line by hepatitis B virus, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(24), 15655–15660, DOI:10.1073/pnas.232137699.
- M. Wieser, G. Stadler and P. Jennings, et al., hTERT alone immortalizes epithelial cells of renal proximal tubules without changing their functional characteristics, Am. J. Physiol., 2008, 295(5), F1365–F1375, DOI:10.1152/ajprenal.90405.2008.
- High-Throughput ADMET Screening: Improving the Efficiency of Drug Discovery, Wiley Handbook of Current and Emerging Drug Therapies, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2006 Search PubMed.
- J. P. Wikswo, E. L. Curtis and Z. E. Eagleton, et al., Scaling and systems biology for integrating multiple organs-on-a-chip, Lab Chip, 2013, 13(18), 3496–3511, 10.1039/c3lc50243k.
- C. Moraes, J. M. Labuz, B. M. Leung, M. Inoue, T.-H. Chun and S. Takayama, On being the right size: scaling effects in designing a human-on-a-chip, Integr. Biol., 2013, 5(9), 1149–1161, 10.1039/c3ib40040a.
- K. Seki, Y. Tsuduki and T. Ioroi, et al., Serum lactate dehydrogenase levels as a predictive marker of oxaliplatin-induced hypersensitivity reactions in Japanese patients with advanced colorectal cancer, Int. J. Med. Sci., 2014, 11(6), 641–645, DOI:10.7150/ijms.7643.
- D. T. Plummer and P. D. Leathwood, Some properties of lactate dehydrogenase found in human urine, Biochem. J., 1967, 103(1), 172–176 CrossRef CAS PubMed; http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1270381&tool=pmcentrez&rendertype=abstract, Accessed March 25, 2015.
- H. Girgis, O. Masui and N. M. White, et al., Lactate dehydrogenase A is a potential prognostic marker in clear cell renal cell carcinoma, Mol. Cancer, 2014, 13, 101, DOI:10.1186/1476-4598-13-101.
- N. Barker, Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration, Nat. Rev. Mol. Cell Biol., 2014, 15(1), 19–33, DOI:10.1038/nrm3721.
- E. M. Wright, B. A. Hirayama and D. F. Loo, Active sugar transport in health and disease, J. Intern. Med., 2007, 261(1), 32–43, DOI:10.1111/j.1365-2796.2006.01746.x.
- A. Habtezion, D. M. Toivola and M. N. Asghar, et al., Absence of keratin 8 confers a paradoxical microflora-dependent resistance to apoptosis in the colon, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(4), 1445–1450, DOI:10.1073/pnas.1010833108.
- M. B. Omary, N.-O. Ku, P. Strnad and S. Hanada, Toward unraveling the complexity of simple epithelial keratins in human disease, J. Clin. Invest., 2009, 119(7), 1794–1805, DOI:10.1172/JCI37762.
- P. C. Sumitran-Holgersson and S. Cytokeratins, in Tools in Oncology, ed. G. Hamilton, InTech, 2012, DOI:10.5772/1888.
- H. J. Kim and D. E. Ingber, Gut-on-a-Chip microenvironment induces human intestinal cells to undergo villus differentiation, Integr. Biol., 2013, 5(9), 1130–1140, 10.1039/c3ib40126j.
- B. Ataç, I. Wagner and R. Horland, et al., Skin and hair on-a-chip: in vitro skin models versus ex vivo tissue maintenance with dynamic perfusion, Lab Chip, 2013, 13(18), 3555–3561, 10.1039/c3lc50227a.
- E. M. Wright and E. Turk, The sodium/glucose cotransport family SLC5, Pflugers Arch., 2004, 447(5), 510–518, DOI:10.1007/s00424-003-1063-6.
- H. Rahmoune, P. W. Thompson, J. M. Ward, C. D. Smith, G. Hong and J. Brown, Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes, Diabetes, 2005, 54(12), 3427–3434 CrossRef CAS PubMed; http://www.ncbi.nlm.nih.gov/pubmed/16306358, Accessed March 25, 2015.
- B. Sarkadi, L. Homolya, G. Szakács and A. Váradi, Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system, Physiol. Rev., 2006, 86(4), 1179–1236, DOI:10.1152/physrev.00037.2005.
- D. Gunzel, M. Stuiver and P. J. Kausalya, et al., Claudin-10 exists in six alternatively spliced isoforms that exhibit distinct localization and function, J. Cell Sci., 2009, 122(10), 1507–1517, DOI:10.1242/jcs.040113.
- C. T. Capaldo, S. Koch, M. Kwon, O. Laur, C. A. Parkos and A. Nusrat, Tight function zonula occludens-3 regulates cyclin D1-dependent cell proliferation, Mol. Biol. Cell, 2011, 22(10), 1677–1685, DOI:10.1091/mbc.E10-08-0677.
- J. C. Chambers, W. Zhang and J. Sehmi, et al., Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma, Nat. Genet., 2011, 43(11), 1131–1138 CrossRef CAS PubMed.
- E.-M. Materne, A. G. Tonevitsky and U. Marx, Chip-based liver equivalents for toxicity testing--organotypicalness versus cost-efficient high throughput, Lab Chip, 2013, 13(18), 3481–3495, 10.1039/c3lc50240f.
- E. LeCluyse, A. Madan, G. Hamilton, K. Carroll, R. DeHaan and A. Parkinson, Expression and regulation of cytochrome P450 enzymes in primary cultures of human hepatocytes, J. Biochem. Mol. Toxicol., 2000, 14(4), 177–188 CrossRef CAS PubMed.
- UGT1A1 UDP glucuronosyltransferase 1 family, polypeptide A1 [Homo sapiens (human)] - Gene - NCBI, http://www.ncbi.nlm.nih.gov/gene/54658, Accessed May 11, 2015 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |