
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-contact acoustic capture of microparticles from small plasma volumes
Mikael
Evander
*a,
Olof
Gidlöf
b,
Björn
Olde
b,
David
Erlinge
b and
Thomas
Laurell
a
aDepartment of Biomedical Engineering, Lund University, Box 118, 221 00 Lund, Sweden. E-mail: mikael.evander@bme.lth.se
bDepartment of Cardiology, Clinical Sciences, Lund University, Box 118, 221 00 Lund, Sweden
First published on 21st April 2015
Abstract
Microparticles (MP) are small (100–1000 nm) membrane vesicles shed by cells as a response to activation, stress or apoptosis. Platelet-derived MP (PMP) has been shown to reflect the pathophysiological processes of a range of cardiovascular diseases and there is a potential clinical value in using PMPs as biomarkers, as well as a need to better understand the biology of these vesicles. The current method for isolating MP depends on differential centrifugation steps, which require relatively large sample volumes and have been shown to compromise the integrity and composition of the MP population. We present a novel method for rapid, non-contact capture of PMP in minute sample volumes based on a microscale acoustic standing wave technology. Capture of PMPs from plasma is shown by scanning electron microscopy and flow cytometry. Furthermore, the system is characterized with regards to plasma sample concentration and flow rate. Finally, the technique is compared to a standard differential centrifugation protocol using samples from both healthy controls and ST-elevation myocardial infarction (STEMI) patient samples. The acoustic system is shown to offer a quick and automated setup for extracting microparticles from small sample volumes with higher recovery than a standard differential centrifugation protocol.
Introduction
Microparticles (MPs, also known as microvesicles) are a class of extracellular vesicles released from various cell types upon activation, stress or apoptosis.1–3 These 100–1000 nm membrane vesicles are present in the circulation of healthy individuals but their levels are altered as a consequence of various pathophysiological processes.4–7 The biological role of MPs is not fully understood, but recent evidence suggests that they are key mediators of cell–cell communication, transferring mRNA, microRNA and proteins between cells,8–10 regulating processes such as antigen presentation,11 inflammation,12 and hemostasis.13Platelets are the main source of circulating MPs.14 MPs are released from platelets upon activation, either through receptor activation (e.g. by thrombin15), binding of complement proteins16 or shear stress17 and are considered both pro-coagulant18 and pro-inflammatory.19,20 Platelet-derived microparticles (PMP) are identified by the expression of platelet surface markers such as CD41, CD42a/b, CD62p and CD63.21,22 Elevated levels of platelet-derived MPs (PMPs) are associated with many forms of cardiovascular disease, including peripheral arterial disease,22 unstable angina23 and myocardial infarction.22,24,25 Furthermore, PMP levels have been shown to reflect both the myocardium at risk26 and the general level of platelet activation in patients with myocardial infarction.27 Thus, there is both a potential clinical value of using PMPs as biomarkers for various cardiovascular diseases, as well as a biological value in understanding the molecular and physiological functions of PMPs. To this end, the ability to quickly and accurately isolate PMPs, both for quantification and for downstream analyses, such as gene and protein expression analyses, co-culture experiments and other functional studies, is critical.
One concern regarding the analysis of MPs in general is the lack of a consensus protocol for sample preparation.28,29 As demonstrated by Lacroix et al., pre-analytical parameters such as centrifugation speed, agitation of tubes or a delay in sample preparation can have a considerable effect on the outcome of the analysis.29 Another concern is the sample volumes required for standard analysis of MPs. In theory, considering the high concentration of MPs in blood, the sample volume required is in the range of a few microliters. However, the centrifugation steps needed to isolate MPs usually require hundreds of microliters. This is a severe limitation, especially when analyzing biobank samples, where there is often restricted access and only very limited amounts of sample can be obtained.
Most researchers use a series of centrifugation steps to prepare plasma for MP analysis.28 Although a consensus protocol is lacking and the variation between labs is considerable, many researchers seem to favor a two-step centrifugation method of 1500 × g for 15 minutes followed by 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 2 minutes.28 However, the second centrifugation step of 13000 × g for 2 minutes has been shown to both reduce the MP numbers to about 80%30 and create artefactual PMPs.29 Moreover, all centrifugation steps will inevitably create a size bias and a density bias in the microparticle population and a method that could isolate MPs with fewer or no centrifugation steps would potentially increase the accuracy and validity of downstream analysis. Consequently, there is a clear need for an MP isolation method that enables higher recovery, does not negatively affect the MPs integrity and allow limited sample volumes.
000 × g for 2 minutes.28 However, the second centrifugation step of 13000 × g for 2 minutes has been shown to both reduce the MP numbers to about 80%30 and create artefactual PMPs.29 Moreover, all centrifugation steps will inevitably create a size bias and a density bias in the microparticle population and a method that could isolate MPs with fewer or no centrifugation steps would potentially increase the accuracy and validity of downstream analysis. Consequently, there is a clear need for an MP isolation method that enables higher recovery, does not negatively affect the MPs integrity and allow limited sample volumes.
Lab-on-a chip techniques have been suggested as a new approach to vesicle isolation due to the small volumes that can be handled and the excellent fluid control. So far, this approach has primarily been aimed at exosomes, vesicles in the 10–100 nm range, and the most common approach is the use of immunoaffinity capture on chip surfaces.31–35 Immunoaffinity methods have the advantage of being able to selectively capture microparticles by targeting specific antigens expressed on them. However, capturing a variety of vesicles requires a wide range of antibodies and the method is not suitable for recovering vesicles whose surface antigenic profile is unknown. An alternative to a targeted capture through a specific antibody is to attach modified cholesterol to a surface, as presented by Kuhn et al.36 Affinity ligand-based vesicle capture methods, however, still suffer from problems of releasing captured intact vesicles for downstream applications (e.g. co-culture).
Other microfluidic approaches have used filters of different kinds with the inherent problem of clogging the system.37,38 He et al. performed an immunoaffinity assay on magnetic nanoparticles and could therefore detain and concentrate the vesicles in the chip prior to lysis and analysis.39 There have also been efforts to perform a size-selective separation of vesicles using deterministic lateral displacement.40 Here, intact vesicles could be extracted from the system although no enrichment was achieved.
Acoustofluidics have been used in many different aspects of cell handling, primarily separation but also concentration and trapping.41–43 Acoustic standing waves, typically in the MHz range, are used to exert size-dependent forces on cells and particles in a gentle and non-contact way.44 However, influencing objects like bacteria, viruses and vesicles by using acoustic forces is difficult due to their minute size. In acoustic trapping, large seed particles have been used to attract and retain bacteria and nanometer-sized beads against flow by secondary acoustic forces between the seed particles and the bacteria or the nano particles45 and this strategy was recently applied to microparticles as well.45,46 This method opens up the possibility of capturing extracellular vesicles using acoustic forces. The main advantages are non-contact capture without any requirements for labeling and the possibility to recover intact vesicles in sample volumes ranging from 5 to 10 μL.
In this paper, we present a novel method for rapid non-contact capture of platelet-derived microparticles in minute sample volumes based on a microscale acoustic standing wave technology. We demonstrate that by using large seed particles that can easily be retained against flow, acoustic trapping is capable of capturing microparticles on the seed particle cluster from plasma. The system is characterized by fluorescence time-lapse imaging and flow cytometry of the microparticle fraction. Finally, the acoustic technique is compared to a more standardized differential centrifugation protocol using ST-elevation myocardial infarction (STEMI) patient samples from a biobank and healthy controls.
Results and discussion
As a proof-of-principle of the technique and to visualize the trapping of platelet-derived MPs in the cluster of seed particles, cell-free plasma was stained with CD42a-PE and injected into the system under a fluorescence microscope. Images were acquired after injection of 0, 50, 150 and 250 μl of sample and showed a clear accumulation of fluorescent material with increased sample volume (Fig. 1). The average size of the seed cluster used in this study was about 1800 particles and it typically covered a volume of about 20 nl (1 × 1 × 0.02 mm3). | ||
| Fig. 1 Fluorescence time-lapse images showing acoustic capture of washed CD42-stained microparticles. (A) shows a background image containing only 12 μm of plain polystyrene seed particles. (B) was taken after processing 50 μl of microparticle suspension. (C) was taken after a total sample volume of 150 μl and (D) shows the final image after 250 μl sample injection. Microparticle capture can be seen from the increased fluorescence intensity. | ||
To demonstrate that the captured material was actually PMPs, the experiment was repeated with unstained plasma, the cluster was released in PBS, stained and analyzed by flow cytometry. Most of the trapped material (~90%) was contained within the MP gate (Fig. 2A) and approximately 20% of the events stained positive for CD42a (Fig. 2B). Analysis of the cluster after trapping with a scanning electron microscope (SEM) clearly showed the 12 μm seed particles surrounded by an aggregate of microparticle-sized objects, with a size range of 300 nm–1 μm (Fig. 2C). Aggregation of the microparticles is believed to be an effect caused by the fixation agent, glutaraldehyde, which previously has been shown to cause platelet aggregation.47
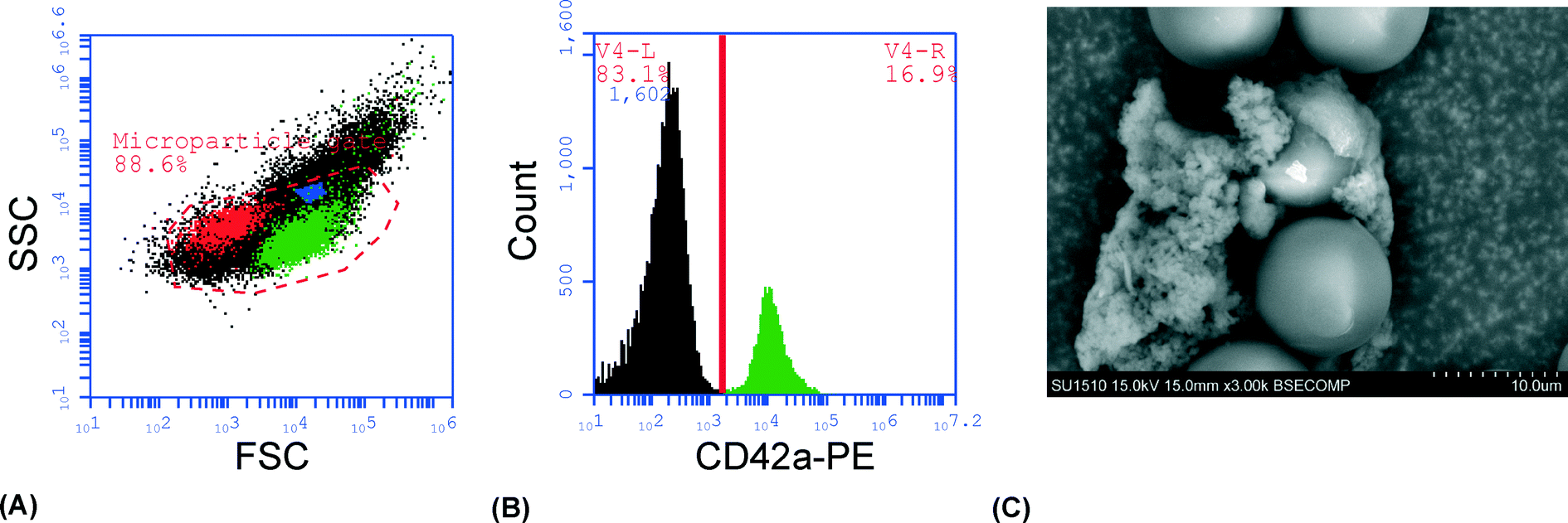 | ||
| Fig. 2 (A) Flow cytometry forward scatter (FSC) vs. side scatter (SSC) plot showing the microparticle gate. The gate was set using 500 and 800 nm beads, shown in red and blue, respectively. CD42a+ microparticles are shown in green. (B) A histogram showing CD42a+ events in the microparticle gate (i.e. platelet-derived microparticles). (C) An SEM-image of the seed cluster with captured microparticles. The 12 μm seed particles can be seen clearly and are surrounded by an aggregate of objects 300 nm–1 μm in size, i.e. matching the expected size range of microparticles. | ||
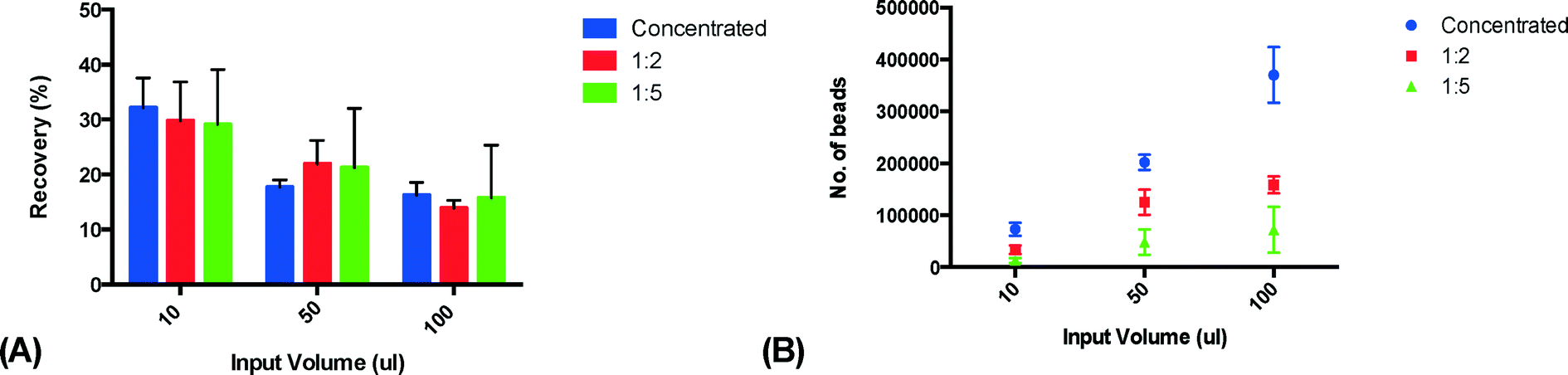
In order to characterize the system and optimize the acoustic trapping, we assessed the effect of altering key parameters on trapping performance. We hypothesized that the high viscosity of plasma would affect trapping negatively, and therefore compared trapping performance in undiluted plasma with plasma that was diluted 1:2 and 1:5 with PBS, using three different sample volumes (10, 50 and 100 μl). All experiments were performed using a flow rate of 10 μl min−1. Released clusters were stained for CD42a and the amount of captured microparticles was measured using flow cytometry. Undiluted plasma was shown to have the lowest recovery while plasma diluted 1:5 with PBS displayed the highest recovery (Fig. 3A). This is most likely due to the fact that diluting the samples changes the viscosity and density of the media surrounding the PMPs, thus yielding a higher acoustic contrast and therefore increasing the recovery. The result also shows that the recovery decreases with increased sample volumes. We believe this is caused by a further dilution of the plasma in the microfluidic system that is more pronounced for smaller sample volumes due to Taylor dispersion than for larger sample volumes. As the plasma samples are injected through a switch valve upstream of the trapping zone, there will be a total volume of approximately 10–15 μl in the narrow tubing between the injection site and the trapping site. While the sample plug travels through the tubing towards the trapping zone, it will be subjected to a fair amount of dispersion. The dispersion will cause the start and the end of the sample plug to be diluted over a larger volume – leading to a more significant sample dilution for the smaller volumes. Considering the small size of the PMPs and the capacity of the seed particle cluster, it is unlikely that the recovery decrease seen is caused by saturation. However, the total amount of PMP in the sample was comparable between undiluted plasma and plasma diluted 1:2 with PBS as the increased recovery counteracted the decrease in PMP concentration caused by the dilution (Fig. 3B).
 | ||
| Fig. 3 Acoustic capture of microparticles using different plasma concentrations measured using flow cytometry (n = 3). (A) shows that the more diluted plasma leads to higher recovery. A 1:2 dilution was chosen for the following experiment as it had higher recovery than undiluted plasma but with about the same amount of extracted microparticles, as seen in (B). The recovery also decreases as the sample volume increases. This is believed to be caused by dispersion-driven dilution in the microfluidic system. | ||
When diluting the plasma, the viscosity and density of the plasma as well as the concentration of the PMPs are changed. To investigate the effect of plasma density and viscosity further we repeated the experiment using fluorescent beads diluted with PBS. Very little effect of dilution on trapping performance with beads was observed, suggesting that this effect is intrinsic to plasma viscosity and density changes, not the PMP concentration (Fig. 4). For the remainder of the study, a plasma dilution of 1:2 with PBS was used. The intention was to minimize the effect on trapping performance caused by donor dependent variations in plasma viscosity and density.
 | ||
| Fig. 4 Acoustic capture of fluorescent, polystyrene nanoparticles, in different concentrations measured using flow cytometry (n = 3). In (A), the variation in recovery depending on the particle concentration is shown. As in Fig. 3, a slight decrease in recovery with increased sample volumes can be seen, although much less pronounced. Due to a similar recovery for the different concentrations, the actual amount of extracted particles behaves more as expected (B). | ||
The effect of the sample flow rate on trapping performance was tested by injecting the sample with varying flow rates (5, 10 and 25 μl min−1) and volumes (10, 50 and 100 μl). The released sample clusters were analyzed using flow cytometry to determine the recovery and the amount of PMPs in the sample (Fig. 5). No effect of the change in the flow rate could be seen for lower sample volumes, although a slower flow rate slightly improved the trapping performance for the 100 μl sample volume. However, the difference was not sufficient to justify the longer run times resulting from the 5 μl min−1 flow rate and 10 μl min−1 was chosen as the optimal flow rate for the remainder of the study.
 | ||
| Fig. 5 Acoustic capture of microparticles in plasma diluted 1:2 with PBS using different sample flow rates measured using flow cytometry (n = 3). (A) shows that there is a slightly improved recovery when processing 50 μl samples and larger at 5 μl min−1. Although small, this increase results in higher absolute PMP counts in these sample (B) but also leads to unnecessarily long processing times. | ||
In order to show the clinical relevance of the technique, we analyzed plasma PMP levels in patients with ST-elevation myocardial infarction (STEMI) and healthy controls, prepared either with acoustic trapping or a standard differential centrifugation protocol.3,29,48 Frozen plasma from six STEMI patients and six age and sex matched controls were thawed and prepared for FACS analysis in parallel. The samples were either processed through acoustic trapping of 25 μl of plasma, injected at 10 μl min−1 and released in 500 μl of PBS, or through centrifugation at 1500 × g for 15 minutes, followed by 13000 × g for 2 minutes. Samples were stained with CD42a-PE and analyzed by FACS. A statistically significant increase in PMPs in STEMI patients compared to healthy controls could be measured using both preparation methods and the results correlated significantly (r2 = 0.38, p = 0.0328) (Fig. 6). MP recovery was slightly better using acoustic trapping (9.8% compared to 6.3%, p = 0.045). The lower general recovery is likely due to the fact that these samples have been stored for a relatively long time at −80 °C, which has previously been shown to affect overall microparticle levels.49 Acoustic trapping required 1:20 of the sample volume used for standard centrifugation (25 μl versus 500 μl) – a critical point when working with biobank samples of limited volume. While the automated version of the acoustic protocol was completed in 7 minutes, the centrifugation protocol was finished after 15 minutes and required manual pipetting steps.
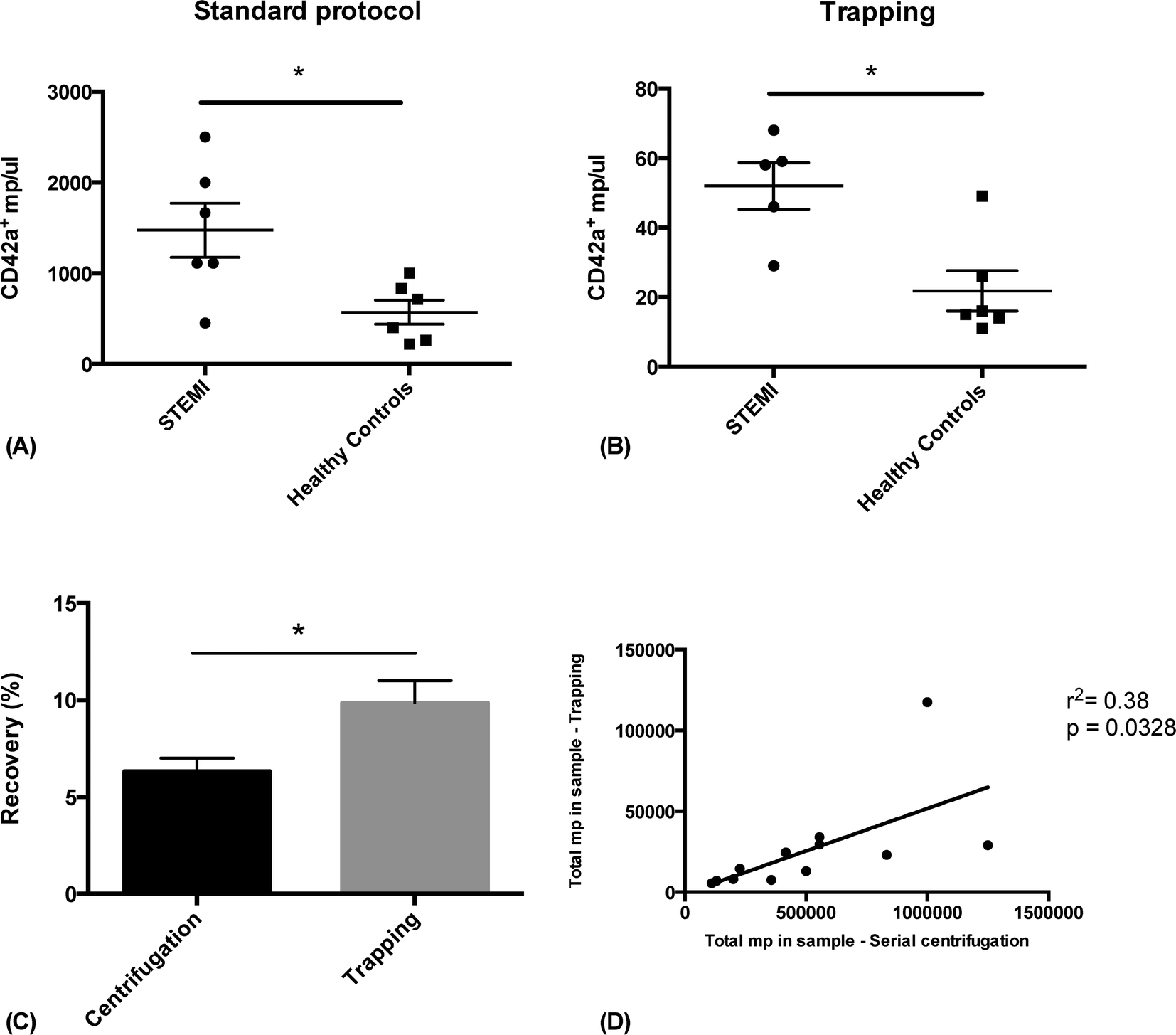 | ||
| Fig. 6 Microparticle extraction from 6 STEMI-patients and 6 healthy controls using differential centrifugation (A) and acoustic trapping (B). Both methods successfully distinguish between the elevated microparticle levels in the STEMI-patients and the lower concentration in the control subjects. One outlier in the STEMI group from the acoustic trapping protocol was identified using Grubb's test and excluded from the analysis. (C) shows the recovery of PMP using either protocol. (D) shows the correlation between trapped PMP for each sample using the two protocols. * indicates p < 0.05 by Student's t-test. | ||
Experimental
Theory
The experimental platform is based on seed particle-enabled acoustic trapping in a rectangular glass capillary.45,50 Ultrasonic standing waves are used to create a local acoustic potential in the fluidic channel. The primary acoustic radiation force can be expressed as the gradient of the acoustic potential, consisting of the square of the time averages of both the pressure and the velocity field.44 A particle that is denser or less compressible than the surrounding media will experience a force toward the minimal acoustic potential – corresponding to the pressure node and the velocity antinode. Most plastic particles and cells fall in this category and will move away from the channel walls into the center of the fluidic channel when using a λ/2 standing wave configuration (see Fig. 7). While in the pressure node, the forces arising from the higher lateral gradient of the velocity field will keep the particles trapped against a fluid flow.51 Secondary acoustic forces, arising from sound waves scattered by the trapped particles (seed particles), create attractive forces between the particles in the pressure node, leading to a tightly packed cluster of particles.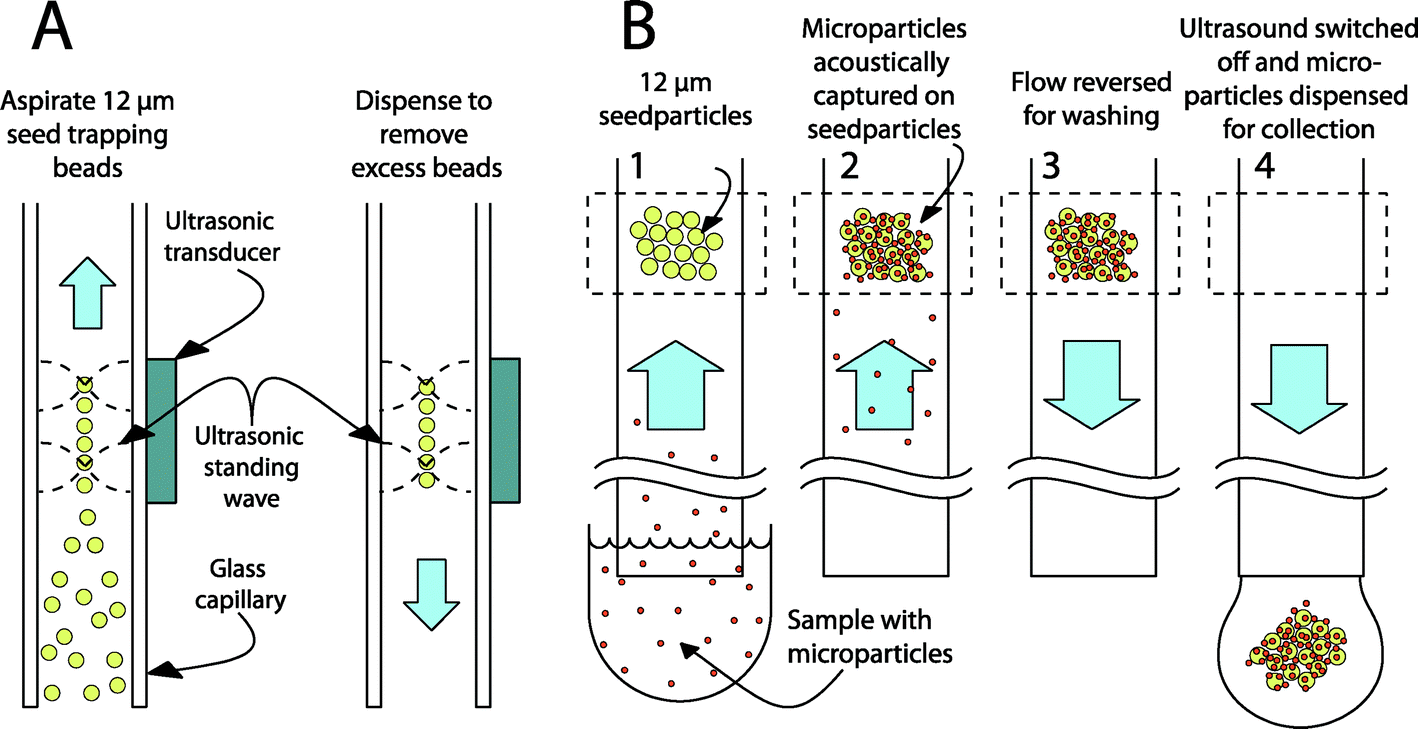 | ||
| Fig. 7 A schematic cross-section image of how an acoustic standing wave will collect microparticles using seed particles. In (A), a piezoelectric transducer is used to create a half wavelength standing wave in the fluidic channel. Seed particles are aspirated and trapped in the acoustic potential minimum in the center of the channel. After washing away any excess particles, a microparticle sample can be aspirated into the channel (B). Microparticles will be attracted to the seed particles by secondary acoustic forces. This will result in a cluster of seed particles and captured microparticles in the standing wave. After completing the sample infusion, the flow is reversed to wash the cluster. Finally, the ultrasound is terminated and the cluster can be released for flow cytometry. | ||
The primary radiation force is not strong enough to affect small particles, as it is strongly size dependent (~r3); the secondary radiation force, however, scales with the distance between the particles (~d−4) making it very strong as particles come near each other.52 This phenomenon is used in seed trapping where large particles, which are easily handled, are initially trapped and can then trap small particles by the secondary forces.
Experimental setup
A rectangular, borosilicate capillary with inner dimensions of 2 × 0.2 mm2 was used as a fluidic channel resulting in a system resonance frequency of around 4 MHz. The capillary is clamped onto a kerfed piezoceramic element and acoustically contacted through a thin film of glycerol.53 The piezoceramic element is surface mounted on a printed circuit board (PCB) to enable actuation through a waveform generator (Fig. 8). | ||
| Fig. 8 A picture of the acoustic trapping system that was used in this study. The piezoelectric transducer is mounted on a PCB with an SMA-contact connected to a waveform generator. The 2 × 0.2 mm2 capillary is clamped to the surface of the transducer and coupled with a thin layer of glycerol. The thin glass walls and small transducer result in a localized standing wave that will trap and hold cells and particles in the center of the capillary above the transducer. | ||
The experimental setup consisted of the acoustic trapping platform, two syringe pumps (neMESYS, Cetoni GmbH, Korbußen, Germany) and an electrically controlled 4-port switch valve (Cheminert C2, Valco Instruments Company Inc., Houston, TX, USA). The piezoelectric transducer was actuated using a waveform generator (Agilent 33220A, Agilent Technologies Inc., Santa Clara, CA, USA) at 10 Vpp. The actuation frequency was determined using a frequency tracking module that automatically finds and holds the optimal resonance frequency in the system, making the system less affected by variations in e.g. plasma constitution and temperature.53 The sample was loaded in a 1 ml disposable syringe in one syringe pump while the other pump handled a 5 ml disposable syringe with phosphate buffered saline (PBS) supplemented with 0.35% bovine serum albumin (BSA). All syringe pumps, the switch valve, the waveform generator and the frequency tracking hardware were controlled through a LabVIEW program.
Work flow for characterization and optimization of acoustic trapping
For optimization and characterization of the system, either plasma from a healthy volunteer or 520 nm fluorescent polystyrene particles (FS03F/5069, Bangs Laboratories, Inc., Fishers, IN, USA) were used. Plasma was obtained by centrifugation of heparinized blood at 1600 × g for 15 minutes followed by addition of 50 mM EDTA. PBS buffer was used as a diluent. Polystyrene particles were suspended in PBS supplemented with 0.35% bovine serum albumin (BSA) at a concentration of 50 particles per microliter.After activation of the ultrasound, 20 μl of a suspension containing seeding particles was aspirated from the outlet. Trapped seeding particles were washed with 20 μl of PBS buffer to remove any excess particles. The average size of the seed cluster used in this study was about 1800 particles and it typically covered a volume of about 20 nl (1 × 1 × 0.02 mm3). The valve was switched, connecting the sample syringe to the trapping capillary, and infusion of plasma or polystyrene particle suspension was initiated. After completing the sample infusion, the valve was switched again and the trapping capillary was washed with 40 μl of PBS buffer. Finally, the ultrasound was deactivated and the cluster flushed into a FACS tube in 500 μl of PBS buffer for analysis.
The analysis of trapped material was conducted by flow cytometry. CD42a was used as a marker for PMPs throughout the study due to its excellent consistency in PMP staining and a good separation of negative and positive populations. Plasma samples were stained with a phycoerythrin-conjugated anti-CD42a antibody (558819, BD) for 20 minutes protected from light and analyzed on a BD Accuri C6 flow cytometer. The threshold settings were set to 4000 on FSC and 1000 on SSC, and the microparticle gate was set using 500 nm and 800 nm beads (Fig. 2A). For PMP quantification, the number of CD42a+ events in the FL-2 channel was determined. Samples containing polystyrene beads were analyzed in the FL-1 channel. The sample volume was set to 200 μl and a minimum of 10000 events were analyzed in each sample. To calculate the recovery of trapped PMPs, the number of PMPs in the plasma before acoustic trapping was determined. Recovery was calculated by dividing the number of trapped PMPs with the number of PMPs in the plasma before trapping.
Validation of the technique
To validate the system and compare the method with a standard microparticle preparation protocol using differential centrifugation, clinical samples from a biobank was used. Plasma from six patients with ST-elevation myocardial infarction were obtained from the LundHeartGene biobank, Skåne University Hospital, Lund, Sweden. Written informed signed consent had previously been given. STEMI diagnosis was based on patient history and ECG-criteria. Plasma had been prepared as described above and stored at −80 °C. As controls, six healthy, age and sex matched volunteers were recruited and plasma was obtained and stored as described above. Before analysis, samples were thawed at room temperature. For acoustic trapping, a 25 μl aliquot was diluted 1:2 with PBS and run in the system at 10 μl min−1, washed and released in 500 μl of PBS with 0.35% BSA. In contrast to the method described above, samples were aspirated from the end of the capillary rather than injected through a syringe. This method enabled the analysis of small sample volumes that would have been problematic to load in a syringe. However, since the trapping site is roughly centered on the capillary (see Fig. 8), there is a volume of about 10–15 μl at the end of the capillary from which no PMPs will be captured. The 25 μl that was withdrawn then correlates to injecting 10 μl as was done with the other samples during characterization. The volume preceding the trapping site can be minimized by utilizing a shorter capillary.
For comparison, a well established differential centrifugation protocol was used.28,48 The protocol consists of two centrifugation steps, 1600 × g for 15 minutes followed by 13000 × g for 2 minutes. The decision to use this protocol as a reference was based on several criteria. First, it is one of the most commonly used protocols (see e.g. the summary by Yuana et al.54). Second, seeing as platelet contamination is considered one of the major pre-analytical problems in processing plasma for MP measurement;54 the use of a second centrifugation step of 13000 × g, which has previously been shown to eliminate 99% of platelets from platelet poor plasma,55 was considered important for validity of MP quantification after sample preparation. Third, the use of MP isolation with high speed centrifugation (17000–20000 × g) as used by many researchers (see e.g. Berckmans et al.56 and van der Zee et al.22) has been shown to result in a significant loss of MPs as well as an alteration in MP integrity,57 and was therefore disregarded in this study. 500 μl of platelet poor plasma from patients or controls was subjected to the centrifugation protocol. The number and recovery of CD42a+ MPs obtained with acoustic trapping and differential centrifugation was determined by flow cytometry as described above.
SEM analysis
A sample run was performed according to the processing flow described in the sample processing subsection using 100 μl of cell-free plasma. After the final rinse of the cluster, a 2.5% glutaraldehyde solution was withdrawn in the capillary to fix the MPMs. After a 5 min incubation, the fixing solution was removed by washing with PBS for 10 minutes. Microparticles were dehydrated using an ethanol dilution series at 10 minutes each at 30%, 50%, 75%, 95% and 99.7%. Finally, the cluster was released onto a conductive carbon adhesive film and analyzed at 100 mbar using a Hitachi SU1510 Variable Pressure SEM.Statistical analysis
All experiments were done in triplicates and statistical analysis was carried out in GraphPad Prism version 6.0c (GraphPad, La Jolla, CA, USA). The data represent the mean ± SEM and differences between groups were analyzed by Student's t-test. Correlation was analyzed using linear regression. Statistical significance was considered when p < 0.05. Outlier detection was performed with Grubb's test with a significance level of 0.05.Future developments
The possibility of releasing intact microparticles enables a wide variation of analysis methods, e.g. flow cytometry, qRT-PCR, mass spectrometry, western blotting, as well as functional assays and co-culture experiments. The miniaturized flow-through format also enables integration with other miniaturized techniques, e.g. integrating a plasmapheresis system upstream58 to extract microparticles directly from whole blood. It would also be possible to develop a system with multiple acoustic traps where one trap would hold a cell population that upon stimulation releases vesicles that subsequently are captured by a second acoustic trap and then further analyzed downstream. This would enable a targeted analysis of vesicles from a specific cell group in a well-defined microenvironment.Conclusion
We have shown that acoustic trapping can be used for capture of platelet-derived microparticles from human plasma samples. The technique offers a quick and automated setup for isolating microparticles from small sample volumes with higher recovery than a standard differential centrifugation protocol.Acknowledgements
This work was funded by the Knut and Alice Wallenberg Foundation and the Swedish Foundation for Strategic Research. The authors also would like to thank Martin Bengtsson for help with the SEM analysis, Siv Svensson for sample collection and all the volunteers who helped us by providing blood samples.References
- E. Biró, J. W. N. Akkerman, F. J. Hoek, G. Gorter, L. M. Pronk, A. Sturk and R. Nieuwland, J. Thromb. Haemostasis, 2005, 3, 2754–2763 CrossRef PubMed
.
- A.-C. Vion, B. Ramkhelawon, X. Loyer, G. Chironi, C. Devue, G. Loirand, A. Tedgui, S. Lehoux and C. M. Boulanger, Circ. Res., 2013, 112, 1323–1333 CrossRef CAS PubMed
- F. Dignat-George and C. M. Boulanger, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 27–33 CrossRef CAS PubMed
- S. Mezouar, D. Mege, R. Darbousset, D. Farge, P. Debourdeau, F. Dignat-George, L. Panicot-Dubois and C. Dubois, Semin. Oncol., 2014, 41, 346–358 CrossRef CAS PubMed
- J.-M. Sinning, J. Losch, K. Walenta, M. Böhm, G. Nickenig and N. Werner, Eur. Heart J., 2011, 32, 2034–2041 CrossRef CAS PubMed
- T. Nozaki, S. Sugiyama, K. Sugamura, K. Ohba, Y. Matsuzawa, M. Konishi, J. Matsubara, E. Akiyama, H. Sumida, K. Matsui, H. Jinnouchi and H. Ogawa, Eur. J. Heart Failure, 2010, 12, 1223–1228 CrossRef PubMed
- T. Helbing, C. Olivier, C. Bode, M. Moser and P. Diehl, World J Cardiol., 2014, 6, 1135–1139 CrossRef PubMed
- M. Mack, A. Kleinschmidt, H. Bruhl, C. Klier, P. J. Nelson, J. Cihak, J. Plachy, M. Stangassinger, V. Erfle and D. Schlondorff, Nat. Med., 2000, 6, 769–775 CrossRef CAS PubMed
- H. Valadi, K. Ekstrom, A. Bossios, M. Sjostrand, J. J. Lee and J. O. Lotvall, Nat. Cell Biol., 2007, 9, 654–659 CrossRef CAS PubMed
- O. Gidlof, M. van der Brug, J. Ohman, P. Gilje, B. Olde, C. Wahlestedt and D. Erlinge, Blood, 2013, 121(19), 3908–3917 CrossRef PubMed
- M. Mittelbrunn, C. Gutierrez-Vazquez, C. Villarroya-Beltri, S. Gonzalez, F. Sanchez-Cabo, M. A. Gonzalez, A. Bernad and F. Sanchez-Madrid, Nat. Commun., 2011, 2, 282 CrossRef PubMed
- A. Zernecke, K. Bidzhekov, H. Noels, E. Shagdarsuren, L. Gan, B. Denecke, M. Hristov, T. Koppel, M. N. Jahantigh, E. Lutgens, S. Wang, E. N. Olson, A. Schober and C. Weber, Sci. Signaling, 2009, 2, ra81 CrossRef PubMed
- P. L. Gross, B. C. Furie, G. Merrill-Skoloff, J. Chou and B. Furie, J. Leukocyte Biol., 2005, 78, 1318–1326 CrossRef CAS PubMed
- S. F. Mause and C. Weber, Circ. Res., 2010, 107, 1047–1057 CrossRef CAS PubMed
- A. P. Bode and D. H. Hickerson, Platelets, 2000, 11, 259–271 CrossRef CAS PubMed
- P. J. Sims, E. M. Faioni, T. Wiedmer and S. J. Shattil, J. Biol. Chem., 1988, 263, 18205–18212 CAS
- Y. Miyazaki, S. Nomura, T. Miyake, H. Kagawa, C. Kitada, H. Taniguchi, Y. Komiyama, Y. Fujimura, Y. Ikeda and S. Fukuhara, Blood, 1996, 88, 3456–3464 CAS
- A. P. Owens 3rd and N. Mackman, Circ. Res., 2011, 108, 1284–1297 CrossRef PubMed
- E. Boilard, P. A. Nigrovic and K. Larabee, Science, 2010, 45 Search PubMed
- G. T. Brown and T. M. McIntyre, J. Immunol., 2011, 186, 5489–5496 CrossRef CAS PubMed
- F. Mobarrez, J. Antovic, N. Egberg, M. Hansson, G. Jörneskog, K. Hultenby and H. Wallén, Thromb. Res., 2010, 125, e110–116 CrossRef CAS PubMed
- P. M. van der Zee, E. Biró, Y. Ko, R. J. de Winter, C. E. Hack, A. Sturk and R. Nieuwland, Clin. Chem., 2006, 52, 657–664 CAS
- C. Vidal, C. Spaulding and F. Picard, J. Thromb. Haemostasis, 2001, 784–790 CAS
- O. Morel, P. Ohlmann, E. Epailly, B. Bakouboula, F. Zobairi, L. Jesel, N. Meyer, M.-P. Chenard, J.-M. Freyssinet, P. Bareiss, J.-P. Mazzucotelli and F. Toti, J Heart Lung Transplant., 2008, 27, 38–45 CrossRef PubMed
- A. E. Michelsen, E. Brodin, F. Brosstad and J.-B. Hansen, Scand. J. Clin. Lab. Invest., 2008, 68, 386–392 CrossRef CAS PubMed
- C. Jung, P. Sörensson, N. Saleh, H. Arheden, L. Rydén and J. Pernow, Atherosclerosis, 2012, 221, 226–231 CrossRef CAS PubMed
- E. Stępień, E. Stankiewicz, J. Zalewski, J. Godlewski, K. Zmudka and I. Wybrańska, Arch. Med. Res., 2012, 43, 31–35 CrossRef PubMed
- W. Jy and L. L. Horstman, J. Thromb. Haemostasis, 2004, 1842–1843 CrossRef CAS PubMed
- R. Lacroix, C. Judicone, P. Poncelet, S. Robert, L. Arnaud, J. Sampol and F. Dignat-George, J. Thromb. Haemostasis, 2012, 10, 437–446 CrossRef CAS PubMed
- L. L. Horstman, W. Jy and J. J. Jimenez, Keio J. Med., 2004, 210–230 CrossRef CAS
- B. A. Ashcroft, J. de Sonneville, Y. Yuana, S. Osanto, R. Bertina, M. E. Kuil and T. H. Oosterkamp, Biomed. Microdevices, 2012, 14, 641–649 CrossRef CAS PubMed
- C. Chen, B.-R. Lin, H.-K. Wang, S.-T. Fan, M.-Y. Hsu and C.-M. Cheng, Microfluid. Nanofluid., 2014, 16, 849–856 CrossRef CAS
- C. Chen, J. Skog, C. H. Hsu, R. T. Lessard, L. Balaj, T. Wurdinger, B. S. Carter, X. O. Breakefield, M. Toner and D. Irimia, Lab Chip, 2010, 10, 505–511 RSC
- H. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18, 1835–1840 CrossRef CAS PubMed
- S. S. Kanwar, C. J. Dunlay, D. M. Simeone and S. Nagrath, Lab Chip, 2014, 14, 1891–1900 RSC
- P. Kuhn, K. Eyer, T. Robinson, F. I. Schmidt, J. Mercer and P. S. Dittrich, Integr. Biol., 2012, 4, 1550–1555 RSC
- R. T. Davies, J. Kim, S. C. Jang, E. J. Choi, Y. S. Gho and J. Park, Lab Chip, 2012, 12, 5202–5210 RSC
- Z. Wang, H. J. Wu, D. Fine, J. Schmulen, Y. Hu, B. Godin, J. X. Zhang and X. Liu, Lab Chip, 2013, 13, 2879–2882 RSC
- M. He, J. Crow, M. Roth, Y. Zeng and A. K. Godwin, Lab Chip, 2014, 14, 3773–3780 RSC
- S. M. Santana, M. A. Antonyak, R. A. Cerione and B. J. Kirby, Biomed. Microdevices, 2014, 16, 869–877 CrossRef CAS PubMed
- A. Lenshof, C. Magnusson and T. Laurell, Lab Chip, 2012, 12, 1210–1223 RSC
- M. Nordin and T. Laurell, Lab Chip, 2012, 12, 4610–4616 RSC
- M. Evander and J. Nilsson, Lab Chip, 2012, 12, 4667–4676 RSC
- H. Bruus, Lab Chip, 2012, 12, 1014–1021 RSC
- B. Hammarstrom, T. Laurell and J. Nilsson, Lab Chip, 2012, 12, 4296–4304 RSC
- M. Evander, O. Gidlöf, D. Erlinge and T. Laurell, presented in part at the Micro Total Analysis Systems 2014, San Antonio, TX, USA, 2014 Search PubMed.
- J. A. Zeller, R. E. Dayhoff, K. Eurenius, W. Russell and R. S. Ledley, Clin. Lab. Haematol., 1984, 6, 145–155 CrossRef CAS PubMed
- J. M. Freyssinet, J. Thromb. Haemostasis, 2003, 1, 1655–1662 CrossRef CAS
- E. Dey-Hazra, B. Hertel, T. Kirsch, A. Woywodt, S. Lovric, H. Haller, M. Haubitz and U. Erdbruegger, Vasc. Health Risk Manage., 2010, 6, 1125–1133 Search PubMed
- B. Hammarstrom, M. Evander, H. Barbeau, M. Bruzelius, J. Larsson, T. Laurell and J. Nilsson, Lab Chip, 2010, 10, 2251–2257 RSC
- P. Glynne-Jones, C. E. M. Demore, Y. Congwei, Q. Yongqiang, S. Cochran and M. Hill, IEEE Trans. Ultrason., Ferroelect., Freq. Control, 2012, 59, 1258–1266 Search PubMed
- M. Groschl, Acustica, 1998, 84, 432–447 Search PubMed
- B. Hammarstrom, M. Evander, J. Wahlstrom and J. Nilsson, Lab Chip, 2014, 14, 1005–1013 RSC
- Y. Yuana, R. M. Bertina and S. Osanto, Thromb. Haemostasis, 2011, 105, 396–408 CrossRef CAS PubMed
- W. L. Chandler, Blood Coagulation Fibrinolysis, 2013, 24, 125–132 Search PubMed
- R. J. Berckmans, R. Nieuwland, A. N. Boing, F. P. Romijn, C. E. Hack and A. Sturk, Thromb. Haemostasis, 2001, 85, 639–646 CAS
- S. Robert, P. Poncelet, R. Lacroix, L. Arnaud, L. Giraudo, A. Hauchard, J. Sampol and F. Dignat-George, J. Thromb. Haemostasis, 2009, 7, 190–197 CrossRef CAS PubMed
- A. Lenshof, A. Ahmad-Tajudin, K. Järås, A.-M. Swärd-Nilsson, L. Åberg, G. Marko-Varga, J. Malm, H. Lilja and T. Laurell, Anal. Chem., 2009, 81, 6030–6037 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |