
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A single inlet two-stage acoustophoresis chip enabling tumor cell enrichment from white blood cells†
Maria
Antfolk
*a,
Christian
Antfolk
a,
Hans
Lilja
bcd,
Thomas
Laurell
a and
Per
Augustsson
*ae
aDepartment of Biomedical Engineering, Lund University, Box 118, SE-221 00 Lund, Sweden. E-mail: maria.antfolk@bme.lth.se; per.augustsson@bme.lth.se
bDepartments of Laboratory Medicine, Surgery, and Medicine, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, New York, NY 10065, USA
cNuffield Department of Surgical Sciences, University of Oxford, Oxford, OX3 7DQ, UK
dDepartment of Translational Medicine, Lund University, SE-205 02 Malmö, Sweden
eDepartment of Electrical Engineering and Computer Science, Massachusetts Institute of Technology, Cambridge, MA, USA
First published on 23rd March 2015
Abstract
Metastatic disease is responsible for most cancer deaths, and hematogenous spread through circulating tumor cells (CTC) is a prerequisite for tumor dissemination. CTCs may undergo epithelial–mesenchymal transition where many epithelial cell characteristics are lost. Therefore, CTC isolation systems relying on epithelial cell markers are at risk of losing important subpopulations of cells. Here, a simple acoustophoresis-based cell separation instrument is presented. Cells are uniquely separated while maintained in their initial suspending medium, thus eliminating the need for a secondary cell-free medium to hydrodynamically pre-position them before the separation. When characterizing the system using polystyrene particles, 99.6 ± 0.2% of 7 μm diameter particles were collected through one outlet while 98.8 ± 0.5% of 5 μm particles were recovered through a second outlet. Prostate cancer cells (DU145) spiked into blood were enriched from white blood cells at a sample flow rate of 100 μL min−1 providing 86.5 ± 6.7% recovery of the cancer cells with 1.1 ± 0.2% contamination of white blood cells. By increasing the acoustic intensity a recovery of 94.8 ± 2.8% of cancer cells was achieved with 2.2 ± 0.6% contamination of white blood cells. The single inlet approach makes this instrument insensitive to acoustic impedance mismatch; a phenomenon reported to importantly affect accuracy in multi-laminar flow stream acoustophoresis. It also offers a possibility of concentrating the recovered cells in the chip, as opposed to systems relying on hydrodynamic pre-positioning which commonly dilute the target cells.
Introduction
The incidence rate of cancer in the world is increasing, largely due to an aging population and changes in lifestyle factors.1 Early diagnosis can improve outcomes, but metastatic spread of the cancer to secondary tissues still contributes the majority of cancer deaths.2 Metastases are formed when cells are disseminated from the primary tumor into the blood circulation (where they are referred to as circulating tumor cells [CTC]), until they reach remote organs and tissues where they may establish secondary tumors.3 To improve survival, it is critical to monitor the cancer's propensity to metastasize; CTC enumeration in blood is prognostic for survival.4 Several techniques to enumerate and detect CTCs, including the FDA-approved system CellSearch®, are based on the use of immunolabels for specific epithelial cell markers such as EpCAM, or cytokeratins.5 However, epithelial cell markers are frequently lost in the epithelial–mesenchymal transition, which the cells undergo to escape the primary epithelial tumor and become CTCs.6,7 Therefore, the effectiveness of using only epithelial cell markers to isolate a purified CTC population prior to the enumeration process is questionable since subpopulations of CTCs may remain undetected.Microfluidic technology offers a large number of cell separation principles, all relying on the deterministic behavior of laminar flow.8–11 Since the dimensions of microchannels match the length scales of cells, microfluidics has the potential to contribute to cell separation by the ability to accurately control the position of the cells within the channels.12 Microfluidic systems also offer potential for lower sample and reagent consumption.13 To date, many microfluidic separators process cells by moving them from one laminar flow stream into a second with cell-free medium, as originally presented by Giddings.14 This can be beneficial, for example when processing crude samples that need to be washed.15–18 Multiple laminar flow streams are also used in cell separation to hydrodynamically pre-position the cells in the channel to increase resolution19,20 or as part of the separation mechanism itself.21,22 However, in many applications the inclusion of several laminar flow streams complicates the fluidic system of the chip, involving extra inlets, outlets and pumps, and an increased need for flow control. Furthermore, hydrodynamic pre-positioning of cells leads to high flow velocities in the separation channel, which is often a major limiting factor in terms of throughput and detector accuracy. Assuming that the separation channel is run at its limiting flow velocity, the sample volume throughput can be increased by replacing the hydrodynamic pre-positioning with an external field acting directly on the cells.
Acoustophoresis has been shown to be a robust, accurate and high-throughput method for performing unit operations on cells in suspension.23 Furthermore, it is a gentle cell handling method that does not compromise cell viability or function, and allows for culturing and phenotypic characterization of the extracted cells.24,25 In acoustophoresis-based cell separation, the sample is commonly laminated to the channel sides by a central stream of cell-free medium and the cells are then acoustically pushed into this cell-free medium. Cells are separated based on their acoustophoretic mobility, resulting in a cell-specific lateral displacement while flowing through the channel.26–31
However, the use of multiple inlet streams in acoustophoresis becomes complicated by the need to match the acoustic impedances of the fluids. The fluid with the highest acoustic impedance must be located where the acoustic standing wave pressure node is positioned. If not, the liquids themselves may relocate while flowing through the channel.32 This relocation hampers the separation capabilities of the device, thus the acoustic impedance of the cell-free central laminar flow stream must be matched relative to the sample to be processed.
An optimal microfluidic system for isolation of CTCs should offer unbiased, label-free separation, simplicity in the fluidic setup and no need for matching the acoustic properties of liquids. Furthermore, it should perform high-throughput separation that can process clinically relevant sample volumes typically within an hour, yielding high recovery and purity of the collected sample. To meet this need, an acoustophoresis-based cell or particle sorter is now presented that is capable of separating cancer cells from white blood cells from a single inlet laminar flow stream. The separation is enabled through acoustic pre-alignment of the cells or particles in two dimensions33,34 into well-defined positions and flow velocities before separation.
Separating or concentrating cells or particles using two-dimensional acoustic pre-alignment has previously been shown to be superior to separating without acoustic pre-alignment.27,35,36 Here, instead of using a separate cell-free laminar flow stream for hydrodynamic pre-positioning of cells, ultrasound is used to acoustically pre-align the cells prior to separation while they remain in their initial suspending medium. This simplifies the fluidic setup, and also paves the way for an increased sample throughput since the sample input flow rate equals the total system flow rate during separation. This study demonstrates how both cancer cells and particles can be separated in this system.
Materials and methods
Device design
The chip was fabricated in <100> silicon using standard photolithography and anisotropic wet etching in KOH (0.4 g mL−1 H2O, 80 °C). Holes for the inlet and outlets were drilled in the silicon using a diamond drill (Tools Sverige AB, Lund, Sweden) before sealing the chip by anodic bonding to a borosilicate lid. The microfluidic chip has a single inlet for cell suspension and two outlets for the separated cells (Fig. 1A). The first part of the chip is a 23 mm long cell pre-alignment channel, etched to a width of 310 μm and a depth of 150 μm. The second part of the chip, the cell separation channel, is 22 mm long and etched to a width of 375 μm and a height of 150 μm. At the end of the channel, the flow is split in a trifurcation outlet where the central branch exits through the central outlet while the two side branches are recombined to a single side outlet. A photograph of the chip has been included as ESI† S1. | ||
| Fig. 1 Illustration of the chip and particle trajectories from (A) the top and (B) the side. Cells or particles are infused at (1) and are acoustically pre-aligned in two dimensions to two positions along the width (y-axis) of the chip and are at the same time levitated to mid-height (z-axis) of the channel (purple lines). The pre-aligned cells then enter the wider separation channel at (a–a), where the larger, denser, or less compressible cells are focused faster towards the center of the channel (red line). Thus, these cells or particles are separated from smaller, less dense, or more compressible cells (blue line) and the two different fractions can be collected in the two outlets (2) and (3). (C) The cross sections at (a–a) and (b–b) in A, where the grey arrows indicate the necessary sideways shift of a cell to exit through the central outlet and the green arrows indicate the cell–cell distance at (b–b). The dashed black lines indicate the center outlet flow stream, which can be tuned by adjusting the relative flow rates in the center outlet (2) with respect to the flow rate in (3). | ||
Underneath the pre-alignment and separation channels, piezoceramic transducers (PZ26, Ferroperm Piezoceramics, Kvistgaard, Denmark) were bonded to the back of the chip by cyanoacrylate glue (Loctite Super glue, Henkel Norden AB, Stockholm, Sweden). The pre-alignment channel was actuated at a frequency of 4.530 MHz and the separation channel was actuated at 2.001 MHz. To drive the ultrasound actuation, a dual-channel function generator (AFG 3022B, Tektronix UK Ltd., Bracknell, UK) was used and the signals were amplified using an in-house built power amplifier based on an LT1012 power amplifier (Linear Technology Corp., Milpitas, CA, USA) and a commercial amplifier (AG Series Amplifier, T&C Power Conversion Inc., Rochester, NY, USA). The applied voltage amplitudes over the piezoceramic transducers were monitored using an oscilloscope (TDS 2120, Tektronix UK Ltd.).
A constant temperature of 37 °C was maintained throughout all experiments through a feedback control loop using a Peltier-controller (TC2812, Cooltronic GmbH, Beinwil am See, Switzerland). A Peltier element (Farnell, London, UK) was glued underneath the 2 MHz actuator and a Pt1000 resistance temperature detector (Farnell) was glued to the chip surface.
Driving the flow
The flows in the inlet and outlets were controlled by glass syringes (Hamilton Bonaduz AG, Bonaduz, Switzerland) mounted on syringe pumps (Nemesys, Cetoni GmbH, Korbussen, Germany). The inlet flow rate was set to 100 μL min−1, while the center outlet flow rate was set to 25 μL min−1 or 10 μL min−1 and the side outlet flow rate was correspondingly set to 75 μL min−1 or 90 μL min−1, throughout the whole experiment. Samples were collected using two 2-position, 6-port valves connected in series with the center and side outlets. 100 μL of sample was collected in the loops of the valves.Microparticles
The system was characterized using polystyrene particles of diameters 7 (7.11) μm and 5 (4.99) μm (Sigma-Aldrich, Buchs, Switzerland). The particles were suspended in PBS, with 0.002% Triton-X100 (Sigma-Aldrich, Switzerland) added to avoid aggregation, at a particle concentration on the order of 105 mL−1.Prostate cancer cells
Prostate cancer cell line DU145 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and grown according to their recommendations. Briefly, RPMI-1640 medium (Sigma-Aldrich, Switzerland) was supplemented with 10% fetal bovine serum (Sigma-Aldrich, Switzerland), 55 IU mL−1 penicillin and 55 μg mL−1 streptomycin (Sigma-Aldrich, Switzerland). The cells were cultured at 37 °C in a humid atmosphere containing 5% CO2. Before the experiments, 5 × 105 cells were detached using trypsin/EDTA, washed and resuspended in 80 μL FACS buffer (PBS supplemented with 1% BSA and 2 mM EDTA [pH 7.4]) to which was added 20 μL of direct conjugated EpCAM-PE antibody (BD Bioscience, San Jose, CA, USA) and incubated on ice for 25 minutes. The cells were then fixated using 2% PFA and incubated on ice for 10 minutes. Finally, the cells were resuspended in 50 μL FACS buffer and stored on ice until spiked in the white blood cell sample prior to experiments. The final concentration of the cancer cells was 5 × 104 cells per mL.Blood samples
Blood was obtained, with informed consent, from healthy volunteers at the Lund University Hospital (Lund, Sweden) using vacutainer tubes (BD Bioscience) containing EDTA as an anticoagulant. Aliquots of 200 μL whole blood were incubated with 20 μL of direct conjugated CD45-APC (BD Bioscience) antibody for 25 minutes at room temperature. The red blood cells were then lysed using 2 mL BD FACS lysis buffer (BD Bioscience), diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 in MilliQ H2O, and incubated for 15 minutes at room temperature. The lysis was followed by fixation by incubating the cells in 2% PFA for 10 minutes on ice. Finally, the sample was resuspended in FACS buffer and stored on ice. The samples were diluted 10 times in FACS buffer and spiked with the cancer cells just prior to the acoustophoresis experiments.
:10 in MilliQ H2O, and incubated for 15 minutes at room temperature. The lysis was followed by fixation by incubating the cells in 2% PFA for 10 minutes on ice. Finally, the sample was resuspended in FACS buffer and stored on ice. The samples were diluted 10 times in FACS buffer and spiked with the cancer cells just prior to the acoustophoresis experiments.
Sample analysis
The samples collected from the center and side outlets during each run through the acoustophoresis chip were stored on ice until analysis with FACS Canto or FACS Canto II (BD Bioscience). White blood cells were characterized as CD45-positive and EpCAM-negative, and the cancer cells were characterized as CD45-negative and EpCAM-positive. To calculate the separation efficiency, the number of cells collected in the central outlet was compared to the total number of collected cells from the central and the side outlets during each run.Flow and separation simulations
Matlab2014a (MathWorks, Natick, MA, USA) was used to calculate the flow profile and particle trajectories as they were subjected to an acoustic field.Results and discussion
In this paper an acoustophoresis system is presented that is able to separate 5 versus 7 μm particles or cancer cells from white blood cells from a single inlet laminar flow stream. Our main objective was to investigate how the reduced complexity of the acoustofluidic set-up affected the separation performance compared to previously described systems for acoustic separation, which use multiple inlet hydrodynamic pre-positioning.Operating principle
The chip utilizes an acoustic pre-alignment channel of width w = 300 μm to position the cells or particles to be separated to two points in the plane transverse to the flow. The acoustic field of the pre-alignment channel has two pressure minima located at distances 1/4w away from each side-wall (Fig. 1A) and which are elevated to mid-height above the channel floor (Fig. 1B). The pre-alignment of the cells is vital for the operation of this system, as only a modest acoustic separation result can be achieved if the cells are randomly distributed in the transverse cross-section upon entering the separation channel. This initial acoustic pre-alignment of the sample eliminates the need for the otherwise essential central inlet cell-free liquid used to hydrodynamically pre-position the cells towards the channel walls prior to the separation step. We have provided a schematic functional comparison between this single-inlet acoustophoresis system and systems using hydrodynamic pre-positioning in the ESI† S2.After acoustic pre-alignment the sample enters the separation channel where particles are focused towards the channel center in an acoustic field having a single centrally located pressure node. Particles or cells that are large, have high density or are of low compressibility move faster in the acoustic field than particles that are small, light and compressible. By correct matching of the flow rate and the acoustic amplitude, the particles of high mobility can be collected in the central outlet of the separation channel (outlet 2 in Fig. 1A & B) while slow-moving particles are collected in the combined side outlet (outlet 3 in Fig. 1A & B).
The acoustic pre-alignment of particles in two dimensions assures that all particles experience identical initial flow conditions, which leads to deterministic separation that is undistorted by the flow velocity distribution in the channel. That is, a particle's sideways deflection in the acoustic field will truly reflect its acoustofluidic mobility, which depends on particle, size morphology, density and compressibility as well as the viscosity, density and compressibility of the suspending liquid.
Without acoustic pre-alignment, the retention time of a particle in the acoustic field depends strongly on its position in the width and height of the channel, due to the flow velocity profile in the channel (see ESI† S3). For instance, in a system without acoustic pre-alignment, a particle of low acoustic mobility that flows slowly near the bottom of the channel experiences the acoustic field for a longer time than a particle of high acoustic mobility flowing in a high flow rate region. Since the sideways deflection is then a function of both the acoustic mobility of the cell and the retention time in the field, the two different particles could end up in the same outlet.27
Numerical separation optimization
To find the optimal separation conditions, particle trajectories were computed taking into account both the flow velocity distribution in the microchannel and the acoustic radiation force acting transversely to the flow. Such trajectories were first described by Mandralis and Feke37 for a parallel-plate channel geometry. A comprehensive theoretical framework governing the motion of a particle in an acoustic field inside a channel with the dimensions used in this work can be found in Barnkob et al.,38 for a no-flow condition.The microchannel flow is governed by the Hagen–Poiseuille equation, for which there exists no exact analytical solution for a rectangular geometry, so the positions of the particles were computed numerically in short time-steps. The fundamental assumptions were that the velocity of a particle in the direction of the channel is always the same as the flow velocity at that point, and that the particle velocity orthogonal to the flow is only governed by the acoustic radiation force. A code example for execution in MATLAB® is shown in the supplementary files exampletrajectory.m, poiseuille.m, and acoustopath.m.
Fig. 2A shows the trajectories of a 5 and a 7 μm particle for identical starting positions after acoustic pre-alignment. For a given outlet flow rate configuration, the optimal separation can be assumed to occur when the two particles are located on opposite sides and at an equal distance from the virtual interface between the central and the side outlet flow streams. For each point along the length of the channel in Fig. 2A, the mean transverse position of each of the two particles was calculated. The central outlet volume flow rate, corresponding to these mean positions, was then derived by integrating the flow velocity profile from each position to the center of the channel.
 | ||
| Fig. 2 (A) Simulated trajectories of 5 μm (blue) and 7 μm (red) polystyrene microparticles starting from an initial position of ideal acoustic pre-alignment in width and height. Arrows indicate the point of maximal separation (d), the dashed black line indicates the channel center, and the green line indicates the interface between the side and central outlet flow streams. (B) Plot showing the maximum achievable particle–particle distance versus the relative center outlet flow rate when separating pre-aligned 5 and 7 μm particles. | ||
Fig. 2B shows the result of a simulation of the separation distance of 5 μm and 7 μm particles at the end of the separation channel versus the relative central outlet volume flow rate. The longest particle–particle distance after separation will be reached when the center outlet flow rate is set to 24.2% of the total flow. The particle–particle distance after separation is in this case 26.3 μm. The reason that the separation optimum does not correspond to the longest sideways deflection is explained by considering the acoustophoretic velocity with which the particles travel towards the channel center. The acoustic radiation force on a particle varies sinusoidally over the width w of the channel and produces maximum velocity for a particle at the symmetrically equivalent positions 1/4w and 3/4w.39 After passing this position (1/4w or 3/4w), the velocity gradually decreases until it reaches zero at the channel center. As the larger particle reaches the central region of the channel, its velocity will at some point become lower than that of a smaller trailing particle, thus reducing the inter–particle distance. Hence, a maximum inter–particle distance can be found with respect to exposure time in the acoustophoresis zone.
Characterization
5 and 7 μm polystyrene particles were used because they have been observed to move at rates similar to those of white blood cells and cancer cells, respectively, when influenced by an acoustic field.27 Therefore, to determine the system separation characteristics, we used equal number concentrations of the two sizes of particles. The outlet flows were configured according to the optimal settings from the simulations withdrawing 25% of the total flow from the central outlet. The separation efficiency of 5 and 7 μm polystyrene particles was investigated by gradually increasing the applied voltage to the transducer.Increasing the sound intensity leads to a higher acoustic migration velocity, which has been shown to increase linearly with the square of the applied transducer voltage.38,40 Since the acoustic migration velocity is also proportional to the square of the particle diameter39,40 the 7 μm particles can be expected to move towards the channel center with approximately twice the velocity as that of the 5 μm particles.
Fig. 3A shows the separation efficiency as the proportion of particles collected in the center outlet (outlet 2 in Fig. 1) compared to the total number of collected particles of that type in the center and side outlets combined (outlets 2 and 3, respectively, in Fig. 1). The larger 7 μm particles have a lower transition voltage, above which they exit through the central outlet, than do the 5 μm particles. At voltage amplitudes (peak to peak) between about 240 V2 and 275 V2, the vast majority of the 7 μm particles could be collected in the center outlet while the majority of the 5 μm particles were collected through the side outlet. At 262 V2, 99.6 ± 0.2% of the 7 μm particles were collected through the center outlet while 98.8 ± 0.5% of the 5 μm particles were collected through the side outlet. The initial purity of 50% for each particle size leads to enrichment factors of 102, reflecting this system's deterministic separation capability. In acoustophoresis, this high enrichment factor in combination with the 2 μm particle size difference has previously only been achieved using a combination of multiple-inlet hydrodynamic pre-positioning and acoustic pre-alignment.27
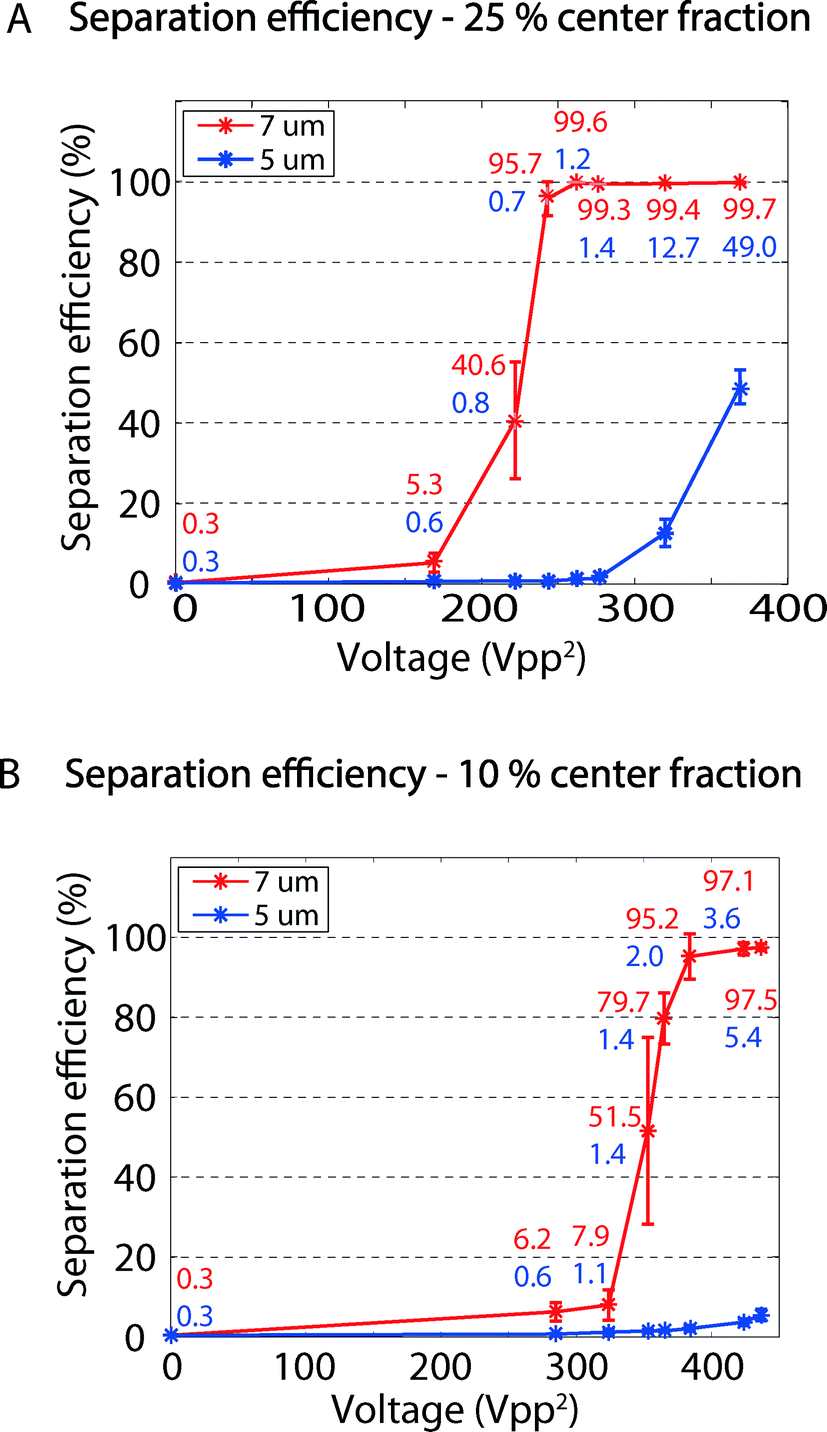 | ||
| Fig. 3 (A) Separation efficiency of 5 μm versus 7 μm polystyrene particles when extracting 25% of the total flow in the center outlet. The total sample flow rate was 100 μL min−1 and the particles were collected through the center outlet with a flow rate of 25 μL min−1. (B) Separation efficiency of 5 μm and 7 μm polystyrene particles when taking out 10% of the total fluid flow in the center outlet. The total sample flow rate was 100 μL min−1 and the particles were collected through the center outlet with a flow rate of 10 μL min−1. The error bars represents the standard deviation for n = 3. | ||
According to the simulations, a lower central outlet flow rate, i.e., a larger sideways shift for the particles, should not further improve the separation efficiency since the particle–particle distance after separation will not be greater. To investigate this, the separation experiments of 5 μm and 7 μm particles were repeated. The center outlet flow rate was now set to 10 μL min−1 and the side outlet flow rate was set to 90 μL min−1, and thus a total sample flow rate of 100 μL min−1 was maintained.
Fig. 3B shows the proportion of particles recovered in the center outlet for the narrow central outlet stream. The transition voltage of the 7 μm particles is now higher and the best separation was achieved for voltage amplitudes ranging from 380 V2 to 420 V2. At 380 V2, 95.2 ± 5.7% of the 7 μm particles are collected in the center outlet while 98 ± 0.7% of the 5 μm particles exit through the side outlet. The higher transition voltage can be explained by the longer sideways shift that the particles have to make to exit the central outlet when only 10% instead of 25% of the total flow rate is extracted through the center outlet. As anticipated, the longer separation distance did not further improve the separation efficiency.
A system dependent on both hydrodynamic pre-positioning and acoustic pre-alignment has the apparent advantage that the cell-free liquid places the pre-positioned cells closer to the channel walls, which theoretically increases the resolution. However, the experimental separation efficiency presented in this paper is comparable to that reported by Augustsson et al.,27 using combined acoustic pre-alignment and hydrodynamic pre-positioning. This suggests that the shorter sideways shift is compensated for by the more stable flow system, something that is more easily attainable with fewer inlets and outlets.
Enrichment of tumor cells spiked into white blood cells
The system was further evaluated for its ability to enrich tumor cells from white blood cells to assess whether this system may be useful to isolate CTCs from patient samples. In this in vitro model prostate cancer cells (cell line, DU145) were spiked at final concentrations of 5 × 104 mL−1 into white blood cell fractions from red blood cell-lysed whole blood. Although the levels of CTCs anticipated in patient samples, as measured by epithelial cell maker-affinity based capture, are commonly two to three magnitudes lower (1–1000 cells per mL) the higher cell concentrations used herein enables the use of conventional flow cytometry to determine tumor cell recovery and purity. Also, from a mechanistic perspective, tumor cell recovery as reported herein is not compromised by 10–100 fold lower concentrations of cancer cells. The total sample flow rate was set to 100 μL min−1 and the center outlet flow rate was set to 25 μL min−1 as determined from simulations and experiments using particles. To save sample and reagents, the level of white blood cells in the samples was one tenth of that of whole blood; however, it has previously been shown that there was no difference in the performance of the acoustophoretic separation using a ten-fold dilution of white blood cells as compared with undiluted samples.27Fig. 4 shows the results from the cell separation experiment. The transition voltage for cancer cells to exit through the center outlet is lower than for the white blood cells, which are smaller. The two cell populations displayed partially overlapping acoustophoretic mobility and could thus not be perfectly separated by this system. Even so, by proper adjustment of the transducer voltage, high recovery of cancer cells could be accomplished while discriminating them from the white blood cells. At 240 V2, 86.5 ± 6.7% of the cancer cells were collected in the center outlet with a contamination of only 1.1 ± 0.2% white blood cells. At 280 V2, there was two-fold higher white blood cell contamination (2.2 ± 0.6%), but 94.8 ± 2.8% of the cancer cells could be recovered in the center outlet. These separation levels are comparable to previous results using acoustophoresis together with hydrodynamic pre-positioning.27 The current system, however, provides a simpler microfluidic setup as well as faster sample processing flow rate of 6 mL h−1, even though the flow rate has not been fully optimized. The simpler fluidic setup, not involving a second liquid flow, leads to a concentration of the cells instead of a dilution as will most often happen when hydrodynamic pre-positioning is used. When separating rare cells such as circulating tumor cells the sample will most likely need to be concentrated before analysis. The possibility to concentrate the sample directly on the chip instead of diluting it is thus a further advantage.
 | ||
| Fig. 4 Separation efficiency of prostate cancer cell line, DU145, and white blood cells (WBC) when extracting 25% of the total flow in the center outlet. The total sample flow rate was 100 μL min−1 and the cells were collected through the center outlet with a flow rate of 25 μL min−1. The error bars represents the standard deviation for n = 3. | ||
The acoustophoretic velocity of a cell scales with the size to the power of two.41 The leukocyte size distribution, as previously measured by impedance cytometry (Coulter counter), range from 7–14 μm while the cancer cells range from 15–25 μm (data not shown), and are thus not overlapping in size. Given the strong size dependence and the distinct size difference of the populations, the separation is likely predominantly based on size.
The experiments and simulations presented here indicate that this method in its current manifestation holds promise, in terms of throughput and accuracy, for further development toward isolation of CTC from patient blood samples. Given the relative rareness of CTCs in patient blood, a 100- to 1000-fold reduction of white blood cells will not allow for direct label-free enumeration of CTCs but the method can be an important unit in a sequence of isolation steps. Further refinements to increase the purity of the isolated cells relative to the white blood cells would be of value to expand its applicability.
Based on the findings, two measures may be taken to further improve the accuracy and throughput to shorten the sample-to-answer time and to make the separation truly deterministic. First, the acoustic pre-alignment channel can be elongated at the expense of the separation channel. In the separation channel, the cells of higher acoustic migration rate must be deflected sideways only a short distance while in the pre-alignment channel all cells must be transferred from their initial random positions in the channel cross-section to the two pre-alignment locations. Second, the separation channel can be widened to improve the separation performance. By doing this, the sideways displacement of the cells increases, leading to a longer absolute distance between separated cells at the outlet. Simulations show that increasing the width of the separation channel to 750 μm, and actuating at the corresponding frequency of 1 MHz, leads to a doubled distance between the separated particles at the outlet (see ESI† S4). This increased distance is anticipated to improve overall separation performance and reduce the sensitivity to phenomena such as flow fluctuations or long-term drift.
Conclusions
This paper presents a simple microfluidic cell sorter for continuous-flow, unbiased, label-free separation of cancer cells from white blood cells based on acoustophoresis. Even though the lateral displacement of a particle in the acoustic field is less than 50 μm, the platform can separate cells and particles with high precision. The single inlet approach leads to simple and robust flow conditions for acoustic pre-alignment and separation of cells.An advantage of this system is that separation is carried out directly in the particles' suspending medium and thus does not require matching of the acoustic properties of the sample relative to a system using multiple laminar flow streams.
This system also paves the way for increased sample throughput, currently enabling clinical sample processing up to 6 mL h−1, since the sample inflow rate equals the total flow rate of the system. This is in contrast to devices relying on hydrodynamic pre-positioning of cells where the volume flow of cell-free medium adds to the net flow velocity of the particles in the separation channel, limiting the sample throughput.
Acknowledgements
The authors would like to thank Dr. Cecilia Magnusson for the generous gift of the DU145 cells. The work was supported by the Swedish governmental agency for innovation systems, VINNOVA, CellCARE (grant no. 2009-00236), Knut and Alice Wallenberg Foundation (grant no. KAW 2012.0023), the Swedish Research Council (grants no. 621-2010-4389, 2012-6708, and 637-2013-444), Swedish Cancer Society (grants no. 11-0624 and 14-0722) the Sten K Johnson Foundation, the Royal Physiographic Society, the Crafoord Foundation, and the Carl Trygger Foundation, the National Cancer Institute at the National Institutes of Health (P50 CA092629); the Sidney Kimmel Center for Prostate and Urologic Cancers; David H. Koch through the Prostate Cancer Foundation; the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Program in UK; and Fundacion Federico SA The funding sources did not have any role in the design or conduct of the study; the collection, management, analysis, or interpretation of the data; or the preparation, review, or approval of the manuscript.References
- K. Christensen, G. Doblhammer, R. Rau and J. W. Vaupel, Lancet, 2009, 374, 1196–1208 CrossRef.
- P. Mehlen and A. Puisieux, Nat. Rev. Cancer, 2006, 6, 449–458 CrossRef CAS PubMed.
- K. Pantel, R. H. Brakenhoff and B. Brandt, Nat. Rev. Cancer, 2008, 8, 329–340 CrossRef CAS PubMed.
- D. C. Danila, A. Anand, N. Schultz, G. Heller, M. Wan, C. C. Sung, C. Dai, R. Khanin, M. Fleisher, H. Lilja and H. I. Scher, Eur. Urol., 2014, 65, 1191–1197 CrossRef CAS PubMed.
- M. Cristofanilli, G. T. Budd, M. J. Ellis, A. Stopeck, J. Matera, M. C. Miller, J. M. Reuben, G. V. Doyle, W. J. Allard, L. W. M. M. Terstappen and D. F. Hayes, N. Engl. J. Med., 2004, 351, 781–791 CrossRef CAS PubMed.
- A. D. Rhim, E. T. Mirek, N. M. Aiello, A. Maitra, J. M. Bailey, F. McAllister, M. Reichert, G. L. Beatty, A. K. Rustgi, R. H. Vonderheide, S. D. Leach and B. Z. Stanger, Cell, 2012, 148, 349–361 CrossRef CAS PubMed.
- T. M. Gorges, I. Tinhofer, M. Drosch, L. Röse, T. M. Zollner, T. Krahn and O. von Ahsen, BMC Cancer, 2012, 12, 178 CrossRef CAS PubMed.
- W. Zhang, K. Kai, D. S. Choi, T. Iwamoto, Y. H. Nguyen, H. Wong, M. D. Landis, N. T. Ueno, J. Chang and L. Qin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18707–18712 CrossRef CAS PubMed.
- L. Mazutis, J. Gilbert, W. L. Ung, D. A. Weitz, A. D. Griffiths and J. A. Heyman, Nat. Protoc., 2013, 8, 870–891 CrossRef CAS PubMed.
- M. G. Lee, J. H. Shin, C. Y. Bae, S. Choi and J.-K. Park, Anal. Chem., 2013, 85, 6213–6218 CrossRef CAS PubMed.
- J. P. Beech, S. H. Holm, K. Adolfsson and J. O. Tegenfeldt, Lab Chip, 2012, 12, 1048–1051 RSC.
- A. A. S. Bhagat, H. Bow, H. W. Hou, S. J. Tan, J. Han and C. T. Lim, Med. Biol. Eng. Comput., 2010, 48, 999–1014 Search PubMed.
- H. Tsutsui and C.-M. Ho, Mech. Res. Commun., 2009, 36, 92–103 CrossRef PubMed.
- J. C. Giddings, Sep. Sci. Technol., 1985, 20, 749–768 CrossRef CAS.
- P. Augustsson, J. Persson, S. Ekström, M. Ohlin and T. Laurell, Lab Chip, 2009, 9, 810–818 RSC.
- D. R. Gossett, H. T. K. Tse, J. S. Dudani, K. Goda, T. A. Woods, S. W. Graves and D. Di Carlo, Small, 2012, 8, 2757–2764 CrossRef CAS PubMed.
- F. Petersson, A. Nilsson, H. Jönsson and T. Laurell, Anal. Chem., 2005, 77, 1216–1221 CrossRef CAS PubMed.
- J. Persson, P. Augustsson, T. Laurell and M. Ohlin, FEBS J., 2008, 275, 5657–5666 CrossRef CAS PubMed.
- B. D. Plouffe, M. Mahalanabis, L. H. Lewis, C. M. Klapperich and S. K. Murthy, Anal. Chem., 2012, 84, 1336–1344 CrossRef CAS PubMed.
- S. H. Ling, Y. C. Lam and K. S. Chian, Anal. Chem., 2012, 84, 6463–6470 CrossRef CAS PubMed.
- J. Takagi, M. Yamada, M. Yasuda and M. Seki, Lab Chip, 2005, 5, 778–784 RSC.
- Z. Wu, B. Willing, J. Bjerketorp, J. K. Jansson and K. Hjort, Lab Chip, 2009, 9, 1193–1199 RSC.
- A. Lenshof, C. Magnusson and T. Laurell, Lab Chip, 2012, 12, 1210–1223 RSC.
- M. Burguillos, C. Magnusson, M. Nordin, A. Lenshof, P. Augustsson, M. Hansson, E. Elmér, H. Lilja, P. Brundin, T. Laurell and T. Deierborg, PLoS One, 2013, 8, e64233 CAS.
- M. Wiklund, Lab Chip, 2012, 12, 2018–2028 RSC.
- J. Dykes, A. Lenshof, I.-B. Åstrand-Grundström, T. Laurell and S. Scheding, PLoS One, 2011, 6, e23074 CAS.
- P. Augustsson, C. Magnusson, M. Nordin, H. Lilja and T. Laurell, Anal. Chem., 2012, 84, 7954–7962 CrossRef CAS PubMed.
- F. Petersson, L. B. Åberg, A.-M. K. Swärd-Nilsson and T. Laurell, Anal. Chem., 2007, 79, 5117–5123 CrossRef CAS PubMed.
- Y. Ai, C. K. Sanders and B. L. Marrone, Anal. Chem., 2013, 85, 9126–9134 CrossRef CAS PubMed.
- J. Nam, H. Lim, D. Kim and S. Shin, Lab Chip, 2011, 11, 3361–3364 RSC.
- X. Ding, Z. Peng, S.-C. S. Lin, M. Geri, S. Li, P. Li, Y. Chen, M. Dao, S. Suresh and T. J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12992–12997 CrossRef CAS PubMed.
- S. Deshmukh, Z. Brzozka, T. Laurell and P. Augustsson, Lab Chip, 2014, 14, 3394–3400 RSC.
- G. R. Goddard and G. Kaduchak, J. Acoust. Soc. Am., 2005, 117, 3440 CrossRef CAS PubMed.
- O. Manneberg, J. Svennebring, H. M. Hertz and M. Wiklund, J. Micromech. Microeng., 2008, 18, 095025 CrossRef.
- O. J. E. Jakobsson, C. Grenvall, M. Nordin, M. Evander and T. Laurell, Lab Chip, 2014, 14, 1943–1950 RSC.
- M. Nordin and T. Laurell, Lab Chip, 2012, 12, 4610–4616 RSC.
- Z. I. Mandralis and D. L. Feke, Chem. Eng. Sci., 1993, 48, 3897–3905 CrossRef CAS.
- R. Barnkob, P. Augustsson, T. Laurell and H. Bruus, Lab Chip, 2010, 10, 563–570 RSC.
- R. Barnkob, P. Augustsson, T. Laurell and H. Bruus, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 056307 CrossRef.
- M. Antfolk, P. B. Muller, P. Augustsson, H. Bruus and T. Laurell, Lab Chip, 2014, 14, 2791–2799 RSC.
- R. Barnkob, P. Augustsson, T. Laurell and H. Bruus, Lab Chip, 2010, 10, 563–570 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5lc00078e |
| This journal is © The Royal Society of Chemistry 2015 |