
Research highlights: digital assays on chip
Donghyuk
Kim
,
Qingshan
Wei
,
Janay Elise
Kong
,
Aydogan
Ozcan
and
Dino
Di Carlo
*
Department of Bioengineering, California NanoSystems Institute, Jonsson Comprehensive Cancer Center, University of California Los Angeles, 420 Westwood Plaza, 5121 Engineering V, Box 951600, Los Angeles, California 90095, USA. E-mail: dicarlo@ucla.edu
First published on 20th November 2014
Abstract
The ability to break up a volume of fluid into smaller pieces that are confined or separated to prevent molecular communication/transport is a key capability intrinsic to microfluidic systems. This capability has been used to develop or implement digital versions of traditional molecular analysis assays, including digital PCR and digital immunoassays/ELISA. In these digital versions, the concentration of the target analyte is in a range such that, when sampled into smaller fluid volumes, either a single molecule or no molecule may be present. Subsequent amplification is sensitive enough to obtain a digital readout of the presence of these target molecules. Advantages of such approaches that are claimed include quantification without calibration and robustness to variations in reaction conditions or times because the digital readout is less sensitive to absolute signal intensity levels. Weaknesses of digital approaches include a lower dynamic range of concentrations over which the assay is sensitive, which depends on the total volume that can be analyzed. We highlight recent efforts to expand the dynamic range of digital assays based on exploiting reaction/diffusion phenomena. A side-by-side study that evaluates the strengths of digital assays reveals that the majority of these claims are supported, with specific caveats. Finally, we highlight approaches to apply digital assays to analyze new types of reactions, including the active transport of protons across membranes by ATPases at the single protein level – perhaps opening up new biophysical understanding and screening opportunities, similar to widely deployed single-molecule ion channel analysis.
Digital measurement with a high dynamic range
On-chip digital assays interrogate events at the single molecule level through compartmentalizing bulk assays into an array of microscale confined volumes (or “digital units”), in which each compartment monitors a single-molecule binding event and reports binary “on” or “off” signals. Because of the limited total number of digital units integrated on a chip, conventional digital assays typically display a moderate dynamic range on the order of 103 or 104-fold. Therefore, there is a need to develop next-generation quantitative digital assays with much wider dynamic range to be able to screen biomarkers over larger concentration ranges.Instead of simply introducing more digital units, Ge et al. recently have demonstrated a strategy to extend the dynamic range of on-chip digital immunoassays by up to 1000-fold by combining the concept of Brownian trapping with drift into a microwell assay.1 Their approach is based on the observation that under flow over an adhesive surface, target molecules drift and become trapped with an exponentially decaying distribution along the direction of flow (Fig. 1a). The drift motion dominates diffusion along the channel and competes with reaction/binding to the surface in controlling the transport of molecules inside the channel when Pe ≫ 1, where Pe is the Péclet number, defined by the ratio of the advection rate to the diffusion rate. Therefore, given an unsaturated surface and a known capture rate that competes with the transport rate, the signal distribution over distance could be predicted to follow an exponential distribution with distance. At high analyte concentrations, while the digital signals are saturated in the conventional stationary digital assay (as can be seen in the beginning region of the channel with >104 compartment units), the new spatial readout of the number of positive wells induced by the drift over a certain distance could be readily fitted with an exponential decay curve to estimate much higher molecular concentrations.
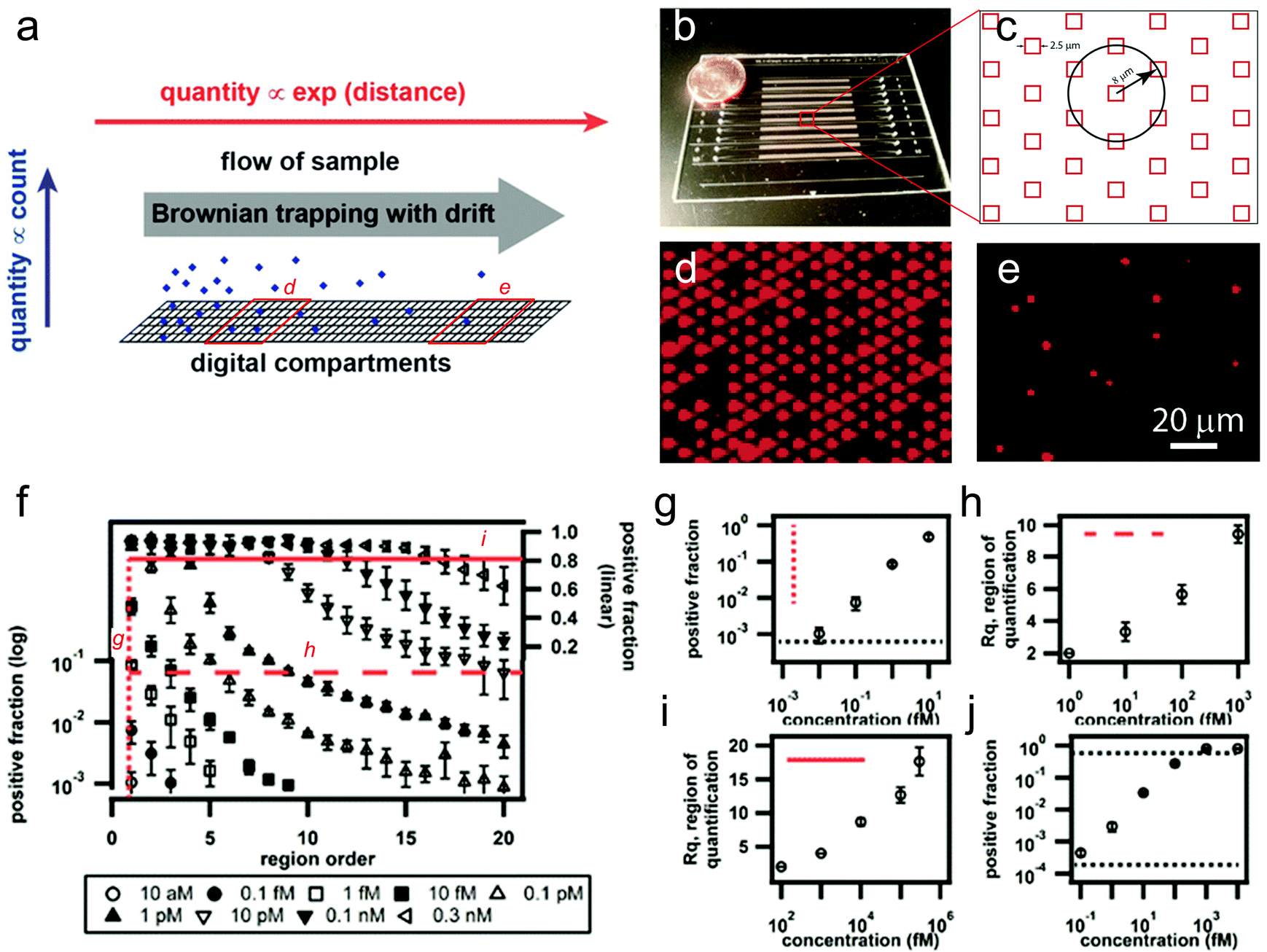 | ||
| Fig. 1 A large dynamic range digital assay. (a) Conceptual schematic of a digital measurement using Brownian trapping with drift. (b) A photograph of the microfluidic device. (c) Schematic illustration of the microwell size and pattern shape. (d, e) Two representative fluorescence images captured from different regions along the channel, using fluorogenic enzymes as the model analyte. (f) Experimental measurements of β-galactosidase with concentrations ranging from 10 aM to 0.3 nM using the digital assay based on Brownian trapping with drift. (g) Positive fraction plot at the first region, showing a detection sensitivity of ~9 aM. (h, i) The demonstration of a large combined dynamic range of ~4 × 107-fold in the drifted digital assay. (j) A comparable dynamic range of ~2 × 104-fold obtained from the conventional stationary assay performed in the same device. Figures adapted from the work by Ge et al.1 with permission. | ||
In a proof-of-concept experiment, the authors created 10 parallel channels in a single chip (Fig. 1b). The channels were divided into 22 quantification regions, each of which contained 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 604 individual microwells for counting on/off signals (Fig. 1c, in total 541250 wells for each channel). Each well was loaded with a single magnetic bead coated with streptavidin as the capture agent. Biotinylated β-galactosidase, an enzyme often used as the readout for digital immunoassays, was used as the model analyte, and was drifted through the microfluidic channel using a gentle controlled flow, followed by the injection of a fluorogenic substrate (resorufin β-D-galactopyranoside) and fluorinated oil to segment the volumes (Fig. 1d, e). Capture curves of the biotinylated β-galactosidase with concentrations ranging from 10 aM to 0.3 nM were obtained by measuring the positive fraction of wells at each quantification region (Fig. 1d–f). While the Brownian trapping based device showed a similar detection sensitivity of 9 aM or ~20 molecules (Fig. 1g), the dynamic range of the new method was found to be ~4 × 107-fold (Fig. 1h, i), which is 1000-fold larger than the stationary digital assay in the same device (~2 × 104-fold, Fig. 1j).
604 individual microwells for counting on/off signals (Fig. 1c, in total 541250 wells for each channel). Each well was loaded with a single magnetic bead coated with streptavidin as the capture agent. Biotinylated β-galactosidase, an enzyme often used as the readout for digital immunoassays, was used as the model analyte, and was drifted through the microfluidic channel using a gentle controlled flow, followed by the injection of a fluorogenic substrate (resorufin β-D-galactopyranoside) and fluorinated oil to segment the volumes (Fig. 1d, e). Capture curves of the biotinylated β-galactosidase with concentrations ranging from 10 aM to 0.3 nM were obtained by measuring the positive fraction of wells at each quantification region (Fig. 1d–f). While the Brownian trapping based device showed a similar detection sensitivity of 9 aM or ~20 molecules (Fig. 1g), the dynamic range of the new method was found to be ~4 × 107-fold (Fig. 1h, i), which is 1000-fold larger than the stationary digital assay in the same device (~2 × 104-fold, Fig. 1j).
Additionally, the authors also demonstrated the detection of a more clinically relevant target, a human protein, TNF-α, in 25% serum. A similar degree of dynamic range improvement was achieved (1000-fold), although the absolute measurement range is slightly smaller than that with the model enzymes.
In summary, by introducing Brownian trapping with drift to conventional digital assays, the authors have successfully converted a large change in the input (analyte concentration) into logarithmically smaller changes in the spatially resolved output (lateral distance or number of grouped digital regions). Such a strategy seems more efficient than simply increasing the number of digital units (e.g. 1000-fold increase in dynamic range with only 22 times increase in total well number). While further parameter optimization is needed as the authors have indicated, this approach appears very promising for broad dynamic range digital measurements for digital PCR or immunoassays, while maintaining high sensitivity at the low end of the concentration range.
Are digital nucleic acid assays more robust?
Digital nucleic acid amplification techniques (NAATs) can provide potential advantages over the conventional quantitative polymerase chain reaction (qPCR).2 Specifically, the digital format is able to provide precise, reproducible, and sensitive quantification without the need for a standard curve. Among these digital formats are isothermal amplification techniques such as loop mediated isothermal amplification (LAMP).3 Nixon et al. conducted a side-by-side performance analysis on the performance and robustness of qPCR, digital PCR (dPCR), qLAMP, and dLAMP in detecting and quantifying human cytomegalovirus (hCMV).4The performance of these NAATs were assessed with both clinical samples and laboratory derived hCMV strains. For the clinical samples, 20 prescreened, blinded DNA samples, with 5 high (>104 copies mL−1), 5 moderate (>2 × 103 and <104 copies mL−1), 5 low (<2 × 103 copies mL−1), and 5 negative samples, were provided by University College London Hospital (UCLH). Quantitative PCR was then performed at UCLH (qPCR-UCLH) and at LGC (qPCR-LGC). Both qPCR runs were correctly able to identify the 15 positive samples and the 5 negative samples. Although the two assays were highly correlated (r2 = 0.94, p < 0.001), the quantification of hCMV levels assigned by UCLH was approximately twice that assigned by LGC (Fig. 2a). This demonstrates the need for universal calibration standards across all qPCR assays when quantifying viral loads. Digital LAMP and digital PCR were also run on the same clinical samples, and strong correlation was found between qPCR-LGC and dPCR (r2 = 0.96, p < 0.001, Fig. 2b), despite the digital array chips only being able to analyze ~18% of the sample volume. The dPCR assay was correctly able to detect hCMV in four of the five low viral load samples, but the dLAMP assay was only able to detect nine out of ten of the highest viral load samples (Fig. 2c). The dPCR quantitative results of copy number were approximately half of the values given by qPCR-LGC (without calibration), demonstrating that dPCR is able to provide hCMV values within 4-fold that of standard curve calibrated qPCR assays. Comparatively, dLAMP gave results that were >10-fold lower than that of qPCR-LGC, however, undercounting could be accounted for if consistent.
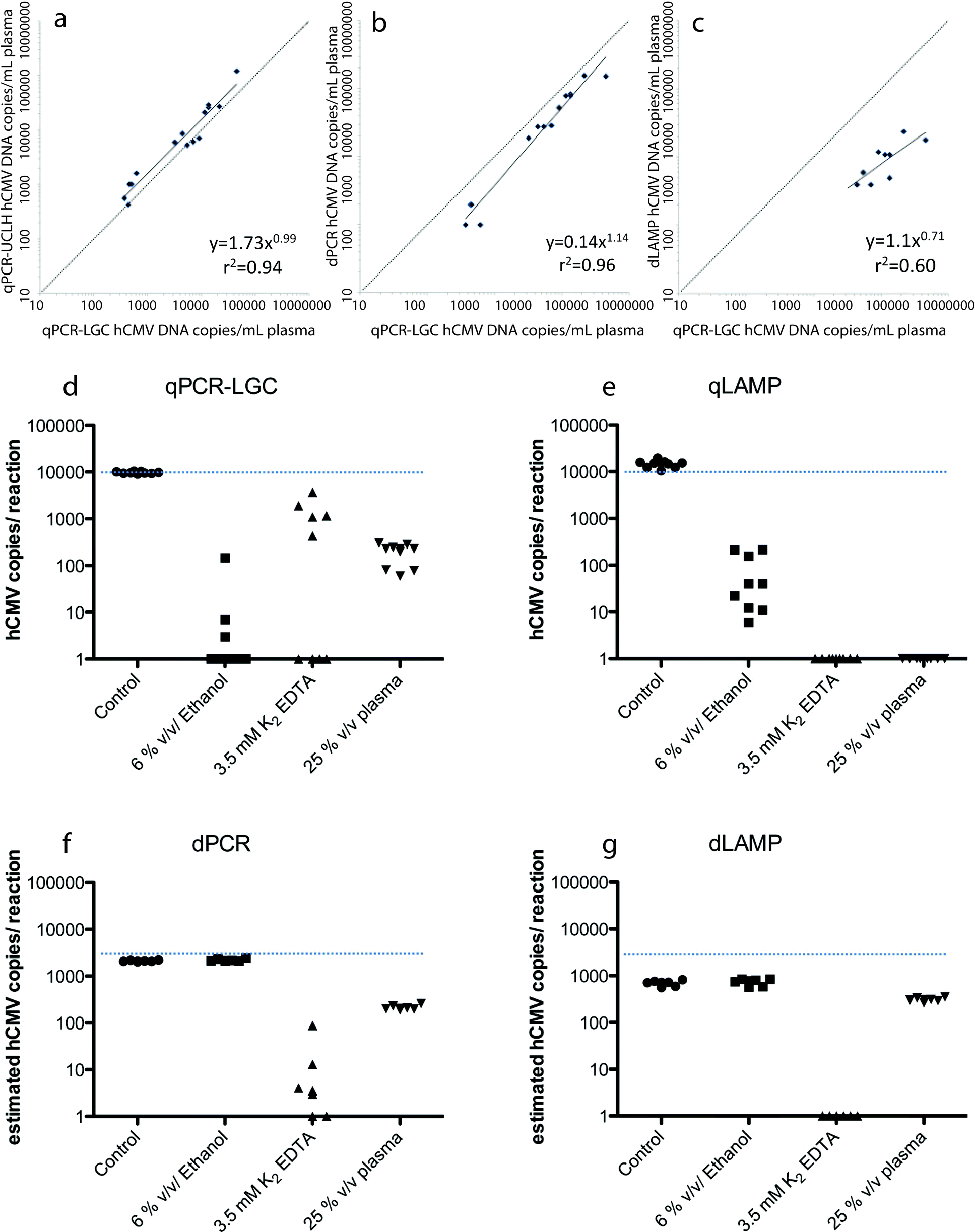 | ||
| Fig. 2 (a–c) Comparison of hCMV assays performed at LGC and (a) qPCR performed at UCLH, (b) dPCR and (c) dLAMP. (d–g) Effect of inhibitors on (d) qPCR, (e) qLAMP, (f) dPCR and (g) dLAMP. Figures adapted from the work by Nixon et al.4 with permission. | ||
To characterize the dynamic range of the techniques, qLAMP and qPCR were compared using laboratory derived hCMV. The quantitative dynamic range for qLAMP was found to be much smaller (103–106 copies per rxn) than that of qPCR (1–106 copies per rxn). dPCR and dLAMP were then tested focusing on the area outside of the dynamic range of qLAMP (50, 100, and 200 copies per rxn). This study found that dLAMP was able to extend the dynamic range of qLAMP to 50 copies, but that it also had a reduced estimated hCMV copy number value compared to that of dPCR.
The addition of inhibitors to the reaction mixes was also examined to determine the robustness of the four NAATs. qLAMP was found to be more robust than qPCR-LGC with 6% v/v ethanol contamination, but not with 25% v/v plasma or 3.5 mM K2 EDTA (Fig. 2d and e). In most cases, the digital formats proved to be even more robust (6% v/v ethanol and 25% v/v plasma), but not in every case, as shown with the use of 3.5 mM K2 EDTA in dPCR (Fig. 2f and g).
Overall, digital LAMP improved quantification, and in most cases both digital formats were more robust. Additionally, dPCR correlated well with calibrated qPCR without the need for external calibration. These digital formats show promise in quantifying viral load, but higher throughput (larger volume) devices (≥50 μL) may be required to improve dPCR sensitivity.
Digital measurement of membrane transporter proteins
Watanabe et al. have recently reported a system where the transport activity of membrane proteins can be monitored at a single molecule level. Membrane proteins are the key to the transport of biochemical mediators in cellular organisms, and thus, play pivotal roles in cellular communication.5,6 There has been a significant research effort, especially measuring ionic flux/current, to delineate the mechanisms of transport through membrane protein-containing liposomes. However, high-throughput measurements of transport or detailed investigation of the transport of different sized molecules and actively transporting membrane proteins remains difficult. Watanabe et al. created an arrayed lipid bilayer chamber (ALBiC) system that contains hundreds of thousands of microchambers, each sealed with a stable 4 μm diameter lipid bilayer membrane.6To test the versatility of their platform, they tested lipid-sealed microchamber formation using different lipids and organic solvent, where they achieved over 99% sealing efficiency for all four of the lipids (Fig. 3a–c). To examine the stable retention of solutes inside the chambers, they photo-bleached fluorescent dyes encapsulated in the microchambers and monitored the recovery of fluorescence intensity – photobleached spots did not recover over a 2 hour time period, confirming that the lipids hermetically sealed the microchambers and maintained the stable retention of solutes inside the chambers. Lastly, they tested the potential ion leakage of their system using fluo-3 dye and confirmed that the lipid membrane successfully retained Ca2+ ions within the microchambers for 2 hours.
 | ||
| Fig. 3 (a) Schematic of the lipid membrane formation process. (b) Fabricated through-hole structure illustration (through-holes: 4 μm wide, 500 nm deep). (c) Fluorescence images obtained from microchambers sealed by different lipid–solvent compositions (Dye: Alexa 488). (d) Schematics of active proton pumping by F0F1. (e) Fluorescence images at different time points (0 second, 6000 second, and the difference between the two time points) validating proton pumping by F0F1. (f) Fluorescence intensity recording over time. 12 representative chambers containing 0, 1, and 2 active F0F1 molecules in grey, blue, and red, respectively. The black solid line is the linear fit and the error bars are standard deviations. (g) Histogram representation of the same data in (f) but only at the time from 1500 s to 4000 s. The peaks were fitted to a sum of Gaussians, and the inset is the average slope of fluorescence increase against the number of active F0F1 molecules in a single microchamber. Figures adapted from the work by Watanabe et al.6 with permission. | ||
They then used their system to study membrane transport at the single molecule level. A common method in this field is to employ α-hemolysin, a membrane protein that forms heptameric rings and creates passive transmembrane nanopores. When α-hemolysin is injected into their ALBiC system assembled with fluorescent dye-containing microchambers, decay of the fluorescence intensity was observed with a time course of minutes. Such a decay was not observed with Qdot 605, a fluorescent dye that is bigger than α-hemolysin-formed nanopores, confirming that such fluorescence decay is due to dye transport through the nanopores. More importantly, the rate constant for the fluorescence decay was [α-hemolysin] dependent. Especially when [α-hemolysin] was low enough (lower than 1 μg mL−1), the response from individual chambers was no longer spontaneous but quantized. When distributed, the rate constants exhibited distinctive peaks and the intervals between peaks were constant, suggesting a correlation of each peak to a certain number of α-hemolysin pores incorporated into the membrane. The rate constant obtained from their investigation fell into an acceptable range from the literature, indicating that their ALBiC system achieves highly sensitive detection of passive transport through α-hemolysin.
They then expanded their system to a study of active membrane transport of protons through the F0F1-ATP synthase (F0F1). The fundamental concept of study stayed the same as the α-hemolysin study except that they used F0F1 as the membrane protein. This enzyme governs energy conversion between ATP and proton motive force (PMF) – the activity of F0F1 is PMF-dependent and the outcome is proton pumping from inside of the confined volumes. Thus, they assembled an ALBiC system with F0F1 incorporated into the lipid membrane, and tried to initiate proton pumping by injecting ATP (Fig. 3d, e). They fluorescently monitored the proton pumping process using RhP-M proton indicator. Once ATP was injected, the fluorescence intensity from the microchambers increased, indicating that the membrane-bound F0F1 converted ATP to ADP and actively pumped protons into the chamber. The rate constant of the fluorescence signal increase showed [F0F1] dependency, and was quantized (Fig. 3f and g). Injection of nigericin, an ionophore of protons, resulted in recovery of fluorescence intensity to its original level (i.e. active transporting of protons), while they observed complete block of proton-pumping by F0F1 once the system was exposed to N,N′-dicyclohexylcarbodiimide, an F0F1 inhibitor. The above results supported successful monitoring of active proton pumping through F0F1 at the single molecule level.
There is still room to improve the assay – the reaction occurs over a long time period, which may disturb the sensitivity of the system or un-intentionally include potential artifacts like photobleaching in the reading. However, considering the stability and capability of the ALBiC system, and the importance of transmembrane proteins in human biology, their system definitely broadens the versatility of microchamber-based digital assays. It has proven its potential in its throughput and reproducibility, and exploring signal transduction schemes for further development may facilitate better understanding of transporter proteins and membrane systems, just as single molecule measurement of ion channels has led to significant understanding of these proteins.
References
- S. Ge, W. Liu, T. Schlappi and R. F. Ismagilov, Digital, Ultrasensitive, End-Point Protein Measurements with Large Dynamic Range via Brownian Trapping with Drift, J. Am. Chem. Soc., 2014, 136, 14662–14665 CrossRef CAS PubMed.
- R. Sanders, J. F. Huggett, C. A. Bushell, S. Cowen, D. J. Scott and C. A. Foy, Evaluation of digital PCR for absolute DNA quantification, Anal. Chem., 2011, 83, 6474–6484 CrossRef CAS PubMed.
- F. Shen, E. K. Davydova, W. Du, J. E. Kreutz, O. Piepenburg and R. F. Ismagilov, Digital isothermal quantification of nucleic acids via simultaneous chemical initiation of recombinase polymerase amplification reactions on SlipChip, Anal. Chem., 2011, 83, 3533–3540 CrossRef CAS PubMed.
- G. Nixon, J. A. Garson, P. Grant, E. Nastouli, C. A. Foy and J. F. Huggett, Comparative Study of Sensitivity, Linearity, and Resistance to Inhibition of Digital and Nondigital Polymerase Chain Reaction and Loop Mediated Isothermal Amplification Assays for Quantification of Human Cytomegalovirus, Anal. Chem., 2014, 86, 4387–4394 CrossRef CAS PubMed.
- International Transporter Consortium, et al., Membrane transporters in drug development, Nat. Rev. Drug Discovery, 2010, 9, 215 CrossRef PubMed.
- R. Watanabe, N. Soga, D. Fujita, K. V. Tabata, L. Yamauchi, S. H. Kim, D. Asanuma, M. Kamiya, Y. Urano, H. Suga and H. Noji, Arrayed lipid bilayer chambers allow single-molecule analysis of membrane transporter activity, Nat. Commun., 2014, 5, 4519–4526 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |