
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTechnical bias of microcultivation environments on single-cell physiology†
Christian
Dusny‡
a,
Alexander
Grünberger‡
b,
Christopher
Probst
b,
Wolfgang
Wiechert
b,
Dietrich
Kohlheyer
b and
Andreas
Schmid§
*a
aLaboratory of Chemical Biotechnology, Department of Biochemical & Chemical Engineering, TU Dortmund University, Emil-Figge-Str. 66, 44227 Dortmund, Germany. E-mail: andreas.schmid@ufz.de
bInstitute of Bio- and Geosciences, IBG-1, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
First published on 9th February 2015
Abstract
Microscale cultivation systems are important tools to elucidate cellular dynamics beyond the population average and understand the functional architecture of single cells. However, there is scant knowledge about the bias of different microcultivation technologies on cellular functions. We therefore performed a systematic cross-platform comparison of three different microscale cultivation systems commonly harnessed in single-cell analysis: microfluidic non-contact cell traps driven by negative dielectrophoresis, microfluidic monolayer growth chambers, and semi-solid agarose pads. We assessed the specific single-cell growth rates, division rates and morphological characteristics of single Corynebacterium glutamicum cells and microcolonies as a bacterial model organism with medical and biotechnological relevance under standardized growth conditions. Strikingly, the specific single-cell and microcolony growth rates, μmax, were robust and conserved for several cell generations with all three microcultivation technologies, whereas the division rates of cells grown on agarose pads deviated by up to 50% from those of cells cultivated in negative dielectrophoresis traps and monolayer growth chambers. Furthermore, morphological characteristics like cell lengths and division symmetries of individual cells were affected when the cells were grown on agarose pads. This indicated a significant impact of solid cultivation supports on cellular traits. The results demonstrate the impact of microcultivation technology on microbial physiology for the first time and show the need for a careful selection and design of the microcultivation technology in order to allow unbiased analysis of cellular behavior.
Introduction
The accurate description of cellular individuality and dynamics enables functional understanding of biological phenomena such as stress response,1,2 adaptation3–6 and robustness7 of microbial populations, traditionally investigated by measuring population-wide and averaged responses to certain environmental perturbations, hiding the fate and dynamics of single cells.8 To access physiological changes in individual cells beyond the bulk, development efforts have been carried out which resulted in different technologies that enable the cultivation and analysis of single cells.9–15 Microcultivation technologies range from simple agarose pads as growth supports to complex microfluidic single-cell trapping and cultivation systems, allowing analyses with high spatial and temporal resolution. Microfluidic microcultivation systems enable rapid removal of secreted metabolites by continuous perfusion, resulting in virtually gradient-free extracellular environments that can be precisely manipulated.16–18 However, different microscale cultivation technologies offer individual advantages. Contactless microfluidic methods like dielectrophoresis traps provide selectivity and active control during cell capture, selection and cultivation. Contact-based microfluidic methods, relying on hydrodynamic barrier structures or chambers, enable high throughput single-cell cultivation. Contact-based semi-solid growth supports like agarose pads are effective and simple to use.11,19 The application of such microscale cultivation technologies gave insight into cell-to-cell heterogeneity, cell–environment interaction and phenotypic plasticity.20,21 However, the comparison and interpretation of the results obtained for such fundamentally different cultivation principles are difficult since knowledge about intrinsic influences of the respective cultivation methods on cellular physiology is still very limited.We therefore performed a systematic evaluation of microfluidic non-contact cell traps driven by negative dielectrophoresis (nDEP), microfluidic monolayer growth chambers (MGC), and semi-solid agarose pads as growth supports (PAD). This comparative study includes detailed analyses of characteristic features of the cultivation technologies on the basis of technology-specific parameters and systematic growth and morphology analysis, using wild-type Corynebacterium glutamicum ATCC 13032. This bacterial strain is broadly used in biotechnology and exhibits a close phylogenetic relationship to pathogens like Corynebacterium diphtheria and Mycobacterium tuberculosis.22 We developed a standardized methodology to assess specific growth rates on the basis of the volume increase in single cells from microscopy images, enabling a quantitative comparison of growth in all three systems. Growth represents one of the most important and fast responding quantitative descriptors for global microbial physiology.23 Although analyses of single-cell growth have already been performed in the 1960's, a consistent methodology for quantitative measurement of specific growth rates of single microbes in microcultivation devices has still not been developed so far.24,25 Several different approaches for the description of single-cell growth have been used, like the rate of elongation, cell number and cell area.7,26–28 However, it is often desirable to compare specific growth rates of single cells with averaged population-based growth.29 Such studies require a universal methodology for growth quantification that is valid for both the single-cell and the population level. Cell volume measurements of single cells meet these requirements, since the volume of a cell can be correlated with its total mass, which enables calculation of the single-cell specific growth rate, μ.
In addition to growth analysis, we exploited the snapping fission movement of C. glutamicum which is studied as a biological 3D sensor for the characterization of the spatial degree of freedom within the respective microcultivation systems. The combination of division rate and morphological analysis with these analytical procedures resulted in an objective cross-platform comparison of the three single-cell cultivation technologies on the basis of cellular traits.
Conceptual differences in nDEP, MGC and agarose pad microcultivation systems
The microcultivation technology itself determines, but also limits, its applicability to particular biological questions. The properties of the cell trapping mechanism, mass transfer as well as device design and dimensions are illustrated in Fig. 1a–b. Details of system dimensions, materials and applications are given in Fig. 1c and S1 in the ESI.†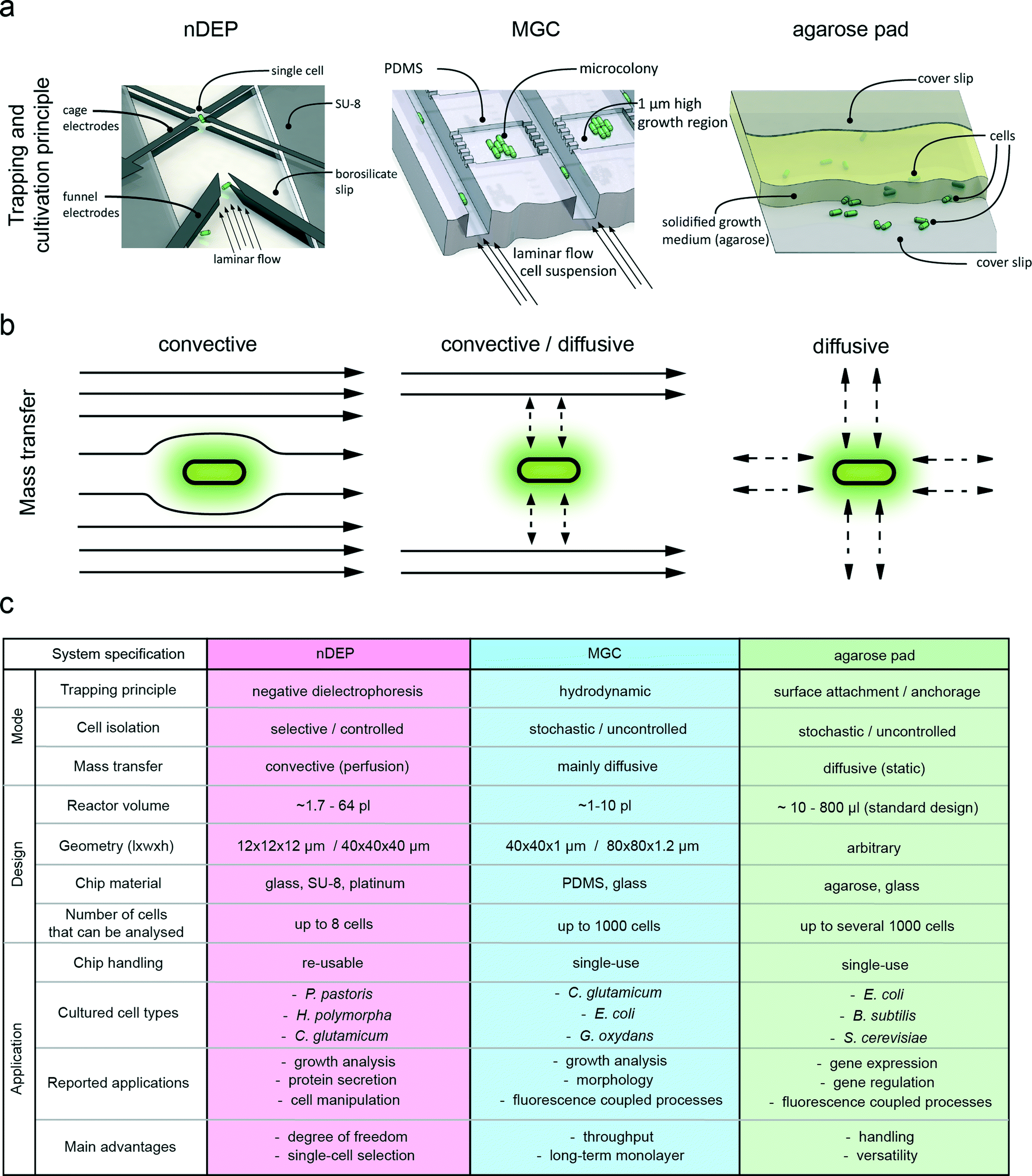 | ||
| Fig. 1 Differences in design and functional principles of the three single-cell microcultivation systems from a macroscopic and microscopic point of view. (a) Operating principle of negative dielectrophoresis traps (nDEP) for single-cell isolation and trapping, with cells guided into the electrode cage by funnel electrodes under continuous perfusion. Illustration of a PDMS-based monolayer growth chamber chip (MGC) with single-cell seeding and cultivation. Casted sandwich agarose pad with a layer of solidified growth medium between two glass cover slides, where cells are located between the agarose pad (PAD) and the bottom glass cover slide. Figures are not true to scale for the purpose of proper illustration. (b) Convective mass transfer is dominant in the nDEP system. Mass transfer inside the MGC chamber is mainly driven by diffusion with convection by a continuous medium flow that is adjacent to the growth chamber. Convection is absent in agarose pads and mass transfer is exclusively facilitated by diffusion from the surrounding agarose gel. (c) Key numbers and characteristics of nDEP, MGC and agarose pad microcultivation systems. The figure of the trapping and cultivation principle of the MGC was adapted from Binder et al.30 | ||
Cell trapping principle and application
Despite their common application purpose, single-cell analysis, the most significant difference between the evaluated microcultivation systems is the mode of cell trapping. The nDEP principle enables contactless retention of the cell within a dielectrophoretic force field. This force field is generated by an arrangement of eight equidistant electrodes (Fig. 1a). Periodical switching of the electrode drive pattern induces a dipole at the cell surface, creating a repelling force on the cell. As a result of this force, the cell is trapped in the electrode cage and levitates at the point with the lowest field intensity. This method allows highly selective isolation of cells from the whole population. The technology was shown to be applicable for cultivating a broad range of different cell types, including mammalian cells, bacteria and several different yeast species.15,16,29,31 The second technology under investigation, the MGC system, employs mechanical cell trapping in growth chambers (Fig. 1a). The chambers have a restricted height and match the average width of the investigated cell type. This spatial confinement forces the trapped cells to grow in a microcolony monolayer. The actual cell isolation process relies on the stochastic distribution of the introduced cells across the microchambers.32 Such microfluidic microchamber-based cultivation approaches have already been applied to answer numerous biological questions at a single-cell level.20,21,33–38 The third evaluated microcultivation concept is realized by using agarose pads, where single cells are entrapped between a layer of semi-solid growth medium and a glass cover slide (Fig. 1a). Cells are randomly spread on the agarose pad by manually dispensing an appropriately diluted cell suspension. As with MGC, cells are also forced to grow in one focal plane. Several studies employed agarose pads to study the dynamics of gene expression.1,2,39Mass transfer and nutrient supply
Mass transfer in nDEP systems is typically dominated by convective flow and eddy diffusion as a result of direct cell perfusion. This provides a steady supply of nutrients and also guarantees fast removal of secreted metabolites from the extracellular microenvironment. Fluctuations in concentration in the direct surroundings of the cell are therefore minimized.16 Mass transfer in MGC relies on a combination of convective and diffusive mass transfer. Each microchamber is interconnected to 10-fold deep supply channels which are continuously flushed with fresh medium, facilitating solely diffusion inside the cultivation area.30 In contrast to nDEP and MGC, agarose pads exhibit a static environment, where mass transfer is exclusively facilitated by diffusion without additional replenishment. Here, nutrient depletion and metabolite accumulation may occur when consumption or production rates exceed diffusion velocities in the agarose pad. The modes of medium supply, as well as nutrient and metabolite transport are illustrated in Fig. 1a–b.Platform design, setup and periphery
There are substantial differences in the design, fabrication and periphery of the respective cultivation technologies (ESI† Table S1). To operate nDEP devices, a rather complex periphery is required, consisting of a radio frequency generator that drives the electrodes and a temperature control system using Peltier elements for cooling (ESI† Fig. S2). The chip fabrication process is comparably complicated and involves several lithography and etching steps for creating channel structures and electrode geometries. However, robust microfluidic chips are produced that can be thoroughly cleaned after cultivation experiments, allowing repeated use.16 In contrast, the MGC fabrication process and its periphery are rather simple (ESI† Fig. S3). The microfluidic chip consists of a glass plate that adheres to a PDMS slab. Because of the cheap materials and the simple molding process, MGC systems are typically disposed after usage, avoiding laborious device cleaning and sterilization. The MGC structures are created by soft lithography, which enables production of multiple chips from one mold. The simplest technology in terms of design, fabrication and periphery is the agarose pad, which is prepared within a short time period of only minutes without any specialized equipment (ESI† Fig. S4). It is made of standard materials that are typically in stock in every standard bio(techno)logical laboratory. Advantageous features that all three cultivation technologies have in common, as well as their respective unique characteristics, are illustrated in Fig. 2. | ||
| Fig. 2 Functional description of negative dielectrophoresis traps (nDEP), monolayer growth chambers (MGC) and agarose pad microcultivation systems (PAD). The Venn diagram illustrates the common properties as intersections and unique properties of each method (nDEP = red, MGC = blue, agarose pad = green). | ||
Materials and methods
Strains and media for nDEP and agarose pad cultivations
Corynebacterium glutamicum ATCC 13032 was stored in BHI (brain heart infusion) medium supplemented with glycerol to a concentration of 20% (v/v) at −80 °C. All preculture, main culture and single-cell experiments of C. glutamicum ATCC 13032 were performed in brain heart infusion (BHI) medium, containing 37.5 g L−1 BHI extract in dH2O. The pH of the medium was adjusted to 7.0 with sodium hydroxide. The conductivity of BHI cultivation medium was adjusted to 1 S m−1 with sterile dH2O (approximate dilution of 5%) to enable optimal trapping performance. Cells from cryocultures were incubated on BHI agar plates at 30 °C and stored for no longer than 48 h at 4 °C to prevent nutrient depletion. From the agar plates, an individual colony was taken and transferred into 100 mL baffled shake flasks filled with 20 mL of sterile BHI medium. The shake-flask cultures were incubated in an Edmund Buehler shaker KS-15 at 300 rpm and 30 °C (Edmund Bühler GmbH, Germany). Cells were grown from the early to mid-exponential growth phase with an OD600 between 0.2 and 0.7, corresponding to cell titers of 4.48 × 106 cells per mL to 1.57 × 107 cells per mL, prior to cultivation in the nDEP system and on agarose pads. The culture was diluted with fresh BHI medium to a final OD600 of 0.01 (N = 2.24 × 105 cells mL−1) and introduced to the nDEP chip or seeded onto the agarose pads.Strains and media for MGC cultivations
C. glutamicum ATCC 13032 for MGC experiments was obtained from the same cryoculture used for nDEP and PAD experiments and cultivated as described before. For the preculture, 20 mL of sterile cultivation medium in a 100 mL baffled shake flask were inoculated with a cryogenic culture bead (Roti-Store, Carl Roth GmbH) and incubated in a rotary shaker at 30 °C and 120 rpm overnight. The main culture was prepared by inoculation with 500 to 1000 μL of the preculture. The main culture was used for microfluidic seeding with an OD600 between 0.1 (N = 1.01 × 107 cells per mL) and 0.61 (N = 2.44 × 107 cells per mL) in the early exponential growth phase.Preparation of agarose pads
5 mL of freshly prepared BHI medium was supplemented with 75 mg of low-melt agarose (1.5% (w/v)) and repeatedly heated until agarose was dissolved. 800 μL of warm agarose solution was pipetted onto a clean standard microscope glass cover slip (18 mm2, 175 μm thickness or round slips with a diameter of d = 10 mm) and immediately covered with another glass slide to create an even and bubble-free agarose layer of 4–5 mm thickness between the two cover slides. After cooling for 45 min at room temperature, the top cover slide was carefully removed. 2 μL of the cell suspension with a cell density of OD600 = 0.01 were pipetted onto the agarose layer to seed single cells. The cell suspension on the pad was allowed to dry for 15 min at room temperature. The agarose pad was subsequently flipped and placed on a μ-Dish 35 mm microscope dish equipped with a glass bottom (thickness 175 μm) (ibidi GmbH, Germany). The petri dish was covered and mounted onto the microscope stage to monitor bacterial growth via time-lapse microscopy. All experiments were performed at 30 °C in a laboratory chamber.Single-cell trapping and cultivation with nDEP
The microfluidic nDEP chip was used for contactless cultivation of isolated single cells and isogenic micropopulations in precisely controlled environments.15 Single cells were trapped in a continuous medium flow of 150 pL s−1 to ensure immediate removal of metabolites and unlimited availability of growth substrates. The medium was pumped using a Cetoni Nemesys syringe pump system (Cetoni, Germany). The cultivation temperature was kept at 30 °C with a customized temperature control system, a TED 200C digital temperature controller (Thorlabs, Germany). Joule heating effects were compensated by appropriately adjusting the cultivation temperature. The electrode cage was operated in ROTX (rotating) mode at 3.8 V and 6.25 MHz. For device design, details of chip manufacturing, system setup, cleaning and cell seeding procedures, see Dusny et al.29Single-cell trapping and cultivation with the MGC
Isogenic microcolonies were cultivated in a single-use polydimethylsiloxane (PDMS) microfluidic chip. The fabrication procedure, setup and operation were reported previously by Gruenberger et al.40 Details about the design and characterization of the used single-cell bioreactors were recently described by Probst et al. and Binder et al.30,32System-independent determination of specific volume growth rates and division rates
Cells were observed with 100× oil immersion objectives. Time-lapse images were used to derive specific volume growth rates and division rates. For the analysis of volume growth, a universally applicable model for the description of the specific cell volume at the single-cell level was developed. It is based on the assumption that the density ρcell [kg m−3] of a single-cell is constant, which is expressed by the ratio of cell mass Mcell [kg] to cell volume Vcell [m3].41,42 | (1) |
With this, the specific volume growth rate μ can thus be described by:
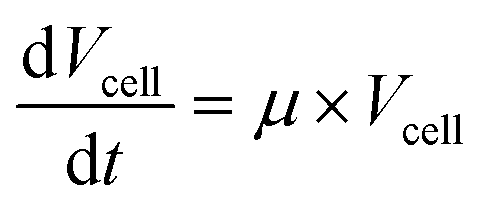 | (2) |
 | (3) |
 | (4) |
The division rate ν [h−1] was also derived using time-lapse microscopy by counting the number of cells N [−] at the respective time-points. The rate of division can be denoted by:
 | (5) |
 | (6) |
Results
Quantitative comparison of specific growth rates and division behavior
We cultivated C. glutamicum starting from 1 cell (or 1 cell pair) under standard growth conditions (BHI medium, T = 30 °C). Growth immediately commenced without any detectable lag-phase upon introduction of the cells into the respective cultivation systems. Measured specific volume growth rates were remarkably conserved for individual single cells within the respective micropopulation (Fig. 3). The increase in individual cell volume was continuous, independent of the employed microcultivation technology and biological characteristics like cell size and cell morphology. Fitting of the temporal volume increase data of single cells in the surface-based cultivation systems MGC and PAD by an exponential function resulted in coefficients of determination R2 above 0.99, with consistently higher coefficients of determination compared to linear or multilinear fit functions (ESI† Fig. S6). For nDEP cultivations, the volume increase could be fitted with coefficients of determination R2 above 0.97, while linear fitting resulted in values below 0.97. This observation reveals an exponential growth mechanism for the cell volume and mass of single C. glutamicum cells. The reduced accuracy of growth rate determination in the nDEP system was probably attributed to the constant rotation of cells during trapping with negative dielectrophoresis, hampering exact measurements of cellular dimensions. Daughter cells displayed identical growth rates to the mother cell for at least 3 generations. Consequently, the specific growth rates of the individual single cells matched the growth rates of the respective micropopulations. Identical growth characteristics were observed to be independent of the applied microcultivation technology (Fig. 4). Measured maximal specific growth rates of micropopulations were consistent in nDEP and MGC with a mean value of![[small mu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0cd.gif) max,nDEP = 0.6 h−1 ± 0.03 for nDEP cultivations and max,MGC = 0.61 h−1 ± 0.06 for cells cultivated in the MGC (CV,nDEP = 5.9%, CV,MGC = 4.4%). Specific volume growth rates of micropopulations cultivated on agarose pads were insignificantly lower with max,PAD = 0.58 h−1 ± 0.05 (CV,PAD = 4.8%). Throughout all performed growth experiments, measured specific micropopulation growth rates were comparable or exceeded those of populations cultivated in shake flasks, indicating that the growth of the cultivated cells was rather limited by the biological growth capacity than by technological limitations.29 The frequency plots of the measured specific growth rates revealed normal distributions of measured growth rates for all three systems, indicating a mere natural biological variation of growth rates without an external systematic influence. Despite substantial differences in the trapping principle and hence microscopic measurement of cell dimensions, specific growth rates of micropopulations could be determined with high accuracy (R2nDEP > 0.97; R2MGC > 0.99; R2agarose pad > 0.99) (ESI† Fig. S7).
max,nDEP = 0.6 h−1 ± 0.03 for nDEP cultivations and max,MGC = 0.61 h−1 ± 0.06 for cells cultivated in the MGC (CV,nDEP = 5.9%, CV,MGC = 4.4%). Specific volume growth rates of micropopulations cultivated on agarose pads were insignificantly lower with max,PAD = 0.58 h−1 ± 0.05 (CV,PAD = 4.8%). Throughout all performed growth experiments, measured specific micropopulation growth rates were comparable or exceeded those of populations cultivated in shake flasks, indicating that the growth of the cultivated cells was rather limited by the biological growth capacity than by technological limitations.29 The frequency plots of the measured specific growth rates revealed normal distributions of measured growth rates for all three systems, indicating a mere natural biological variation of growth rates without an external systematic influence. Despite substantial differences in the trapping principle and hence microscopic measurement of cell dimensions, specific growth rates of micropopulations could be determined with high accuracy (R2nDEP > 0.97; R2MGC > 0.99; R2agarose pad > 0.99) (ESI† Fig. S7).
 | ||
| Fig. 3 Exemplary determination of specific volume growth rates of single cells derived from time-lapse microscopy data for negative dielectrophoresis trap (nDEP), monolayer growth chambers (MGC) and agarose pad (PAD) microcultivation systems. The growth experiment started with a single mother cell. Individual cell volume (mother cell and descendants) and total biomass volume of the population were continuously monitored at maximum intervals of 2 min. The total colony volume resembles the total volume of connected cells in a micropopulation. Mother and daughter cells exhibited consistent specific volume growth rates that were identical to the specific volume growth rate of the population. | ||
 | ||
| Fig. 4 Cultivation of C. glutamicum with negative dielectrophoresis trap (nDEP), monolayer growth chambers (MGC) and agarose pad (PAD) microcultivation systems. (a) Trapped and growing cells in the center of the octupole cage by nDEP, in the MGC and on the agarose pad. Image sequence of a typical experiment (t = 0 until t = tend, tend,nDEP = 3 h, tend,MGC = 6 h, tend,PAD = 3 h). (b) Frequency distribution and scatter plot of specific single-cell growth kinetics (μmax) derived from image analysis. (c) Frequency distribution and scatter plot of division rates (νmax) of single cells and micropopulations cultivated with nDEP, in MGC and on agarose pads. | ||
All experiments were conducted under the same growth conditions (T = 30 °C, BHI growth medium). The total number of measured microcolonies originating from a single-cell was n = 14 (nDEP), n = 26 (MGC) and n = 14 (PAD). For device-specific parameters see the online methods section in the ESI† file.
In addition to specific growth rates, we also assessed specific cell division rates of individual C. glutamicum cells and averaged division rates of micropopulations among the different microcultivation systems (Fig. 4c). Interestingly, cells displayed significantly scattered division rates between νPAD = 0.41 h−1 and νPAD = 0.79 h−1 (CV,PAD = 19.8%) on agarose pads, while mean values of specific volume growth rates and specific division rates were matched (![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) PAD = 0.59 h−1 ± 0.11 and max,PAD = 0.58 h−1 ± 0.05). Consistent mean specific division rates and mean specific volume growth rates were also observed for nDEP (nDEP = 0.63 h−1 ± 0.03, max,nDEP = 0.6 h−1 ± 0.03) and MGC cultivations (MGC = 0.59 h−1 ± 0.05, max,MGC = 0.61 h−1 ± 0.06), but with a significantly less variation of specific division rates (CV,nDEP = 4.5%, CV,MGC = 7.7%) (Fig. 4b–c).
PAD = 0.59 h−1 ± 0.11 and max,PAD = 0.58 h−1 ± 0.05). Consistent mean specific division rates and mean specific volume growth rates were also observed for nDEP (nDEP = 0.63 h−1 ± 0.03, max,nDEP = 0.6 h−1 ± 0.03) and MGC cultivations (MGC = 0.59 h−1 ± 0.05, max,MGC = 0.61 h−1 ± 0.06), but with a significantly less variation of specific division rates (CV,nDEP = 4.5%, CV,MGC = 7.7%) (Fig. 4b–c).
These irregularities in terms of growth and division rate consistency could be repeatedly observed in independently performed cultivation experiments and indicate an inherent influence of the agarose pad technology on the divisome of C. glutamicum, which does not, however, affect the specific growth rate of single cells.
Cell lengths, division angles and division symmetries
During cultivation experiments on agarose pads, occasional cell elongation was observed, posing a possible explanation for the irregular division rates of C. glutamicum. Therefore, the morphology of single C. glutamicum cells was characterized in terms of cell dimensions (cell length measured as the line connecting the midpoint of the pole tips and cell width measured as the diameter of the hemispherical pole caps at both cell pole ends) during the cultivations (ESI† Fig. S8). For nDEP and MGC cultivations, the average pre- and post-divisional cell lengths match with![[l with combining macron]](https://www.rsc.org/images/entities/char_006c_0304.gif) nDEP,before = 4.7 μm ± 0.4 (CV,nDEP,before = 8.5%) and MGC,before = 4.4 μm ± 0.4 (CV,MGC,before = 12.8%) just before the division event and nDEP,after = 2.5 μm ± 0.2 (CV,nDEP,after = 8.7%) and MGC,after = 2.7 ± 0.4 μm (CV,MGC,after = 13.4%) immediately after the division event, respectively (Fig. 5). With nDEP, cells consistently divided before reaching 6 μm in length, while approximately 2% of the cells cultivated in the MGC exhibited cell lengths above 6 μm before cell division. Cell lengths were normally distributed in nDEP and MGC microcultivation technologies, indicating an undisturbed natural biological variation in cell division length. For cells grown on agarose pads, a significantly higher frequency of cells with an elongated shape in comparison with that for nDEP and MGC cultivations was observed. This inclination towards cell elongation on agarose pads was pronounced, with 23% of the cells reaching lengths of more than 6 μm and up to 7 μm right before cell division. The average cell length before division was PAD,before = 5.5 μm ± 1.2 (CV,PAD,before = 21.6%) and PAD,after = 3 μm ± 0.8 (CV,PAD,after = 24.7%) after division, with cell length distributions exhibiting significantly more variance than the respective distributions for the nDEP and MGC microcultivation technologies. Cell length distributions of cells grown on agarose pads were heavy-sided towards increased cell lengths rather than following a normal distribution. The widths of cultivated single cells were normally distributed and did not show significant differences between the three microcultivation systems (ESI† Fig. S9).
nDEP,before = 4.7 μm ± 0.4 (CV,nDEP,before = 8.5%) and MGC,before = 4.4 μm ± 0.4 (CV,MGC,before = 12.8%) just before the division event and nDEP,after = 2.5 μm ± 0.2 (CV,nDEP,after = 8.7%) and MGC,after = 2.7 ± 0.4 μm (CV,MGC,after = 13.4%) immediately after the division event, respectively (Fig. 5). With nDEP, cells consistently divided before reaching 6 μm in length, while approximately 2% of the cells cultivated in the MGC exhibited cell lengths above 6 μm before cell division. Cell lengths were normally distributed in nDEP and MGC microcultivation technologies, indicating an undisturbed natural biological variation in cell division length. For cells grown on agarose pads, a significantly higher frequency of cells with an elongated shape in comparison with that for nDEP and MGC cultivations was observed. This inclination towards cell elongation on agarose pads was pronounced, with 23% of the cells reaching lengths of more than 6 μm and up to 7 μm right before cell division. The average cell length before division was PAD,before = 5.5 μm ± 1.2 (CV,PAD,before = 21.6%) and PAD,after = 3 μm ± 0.8 (CV,PAD,after = 24.7%) after division, with cell length distributions exhibiting significantly more variance than the respective distributions for the nDEP and MGC microcultivation technologies. Cell length distributions of cells grown on agarose pads were heavy-sided towards increased cell lengths rather than following a normal distribution. The widths of cultivated single cells were normally distributed and did not show significant differences between the three microcultivation systems (ESI† Fig. S9).
 | ||
| Fig. 5 (a) Determination of cell lengths and division angles based on cytometric image data. (b) Frequency distribution of cell lengths before and after cell division with nDEP (n = 35/70), MGC (n = 151/302) and agarose pad (n = 51/102). (c) Comparison of the division angle distribution of C. glutamicum ATCC 13032 cell poles cultivated with nDEP (n = 35), in MGC (n = 152) and on agarose pad (n = 51). The respective division angles were measured immediately after snapping division events. The dotted lines represent the Gaussian distribution of the determined cell lengths and division angles. | ||
Besides influences on the cell length and division behavior, cells grown on agarose pads exhibited a stronger tendency towards asymmetric division compared to cells grown in nDEP and MGC microcultivation systems, indicating a disturbance in the placement of the division septum and the regulatory machinery responsible for the coordination of cell division (Fig. 6). Mean division symmetries, expressed as ratios of daughter cell lengths, were measured to be ![[small delta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c5.gif) nDEP = 0.93 and MGC = 0.88 directly after the cell division event. On agarose pads, the mean division symmetry was reduced to PAD = 0.77. Coherent cell pairs in shaken liquid suspensions featured virtually identical cell lengths, resulting in a mean division symmetry of suspension = 0.95 (ESI† Fig. S10). We concluded from these results that the physiology of cells cultivated on agarose pads, and especially their divisome, is subjected to stress caused by the mode of trapping, spatial constriction, environmental conditions like local depletion of oxygen and nutrients or accumulation of metabolic products.
nDEP = 0.93 and MGC = 0.88 directly after the cell division event. On agarose pads, the mean division symmetry was reduced to PAD = 0.77. Coherent cell pairs in shaken liquid suspensions featured virtually identical cell lengths, resulting in a mean division symmetry of suspension = 0.95 (ESI† Fig. S10). We concluded from these results that the physiology of cells cultivated on agarose pads, and especially their divisome, is subjected to stress caused by the mode of trapping, spatial constriction, environmental conditions like local depletion of oxygen and nutrients or accumulation of metabolic products.
 | ||
| Fig. 6 Division symmetry of C. glutamicum is influenced by microcultivation technology. Cells tend to divide in a more asymmetric fashion on agarose pads. Division symmetry was measured as the ratio of short to long cell poles directly after the cell division event (n = 200). | ||
In addition to growth, morphology and division characteristics, as well as cell lengths, we measured the division angles of single cells directly after the division event. The daughter cells of a recently divided C. glutamicum mother cell exhibit a distinct V-formed shape after cell division as a result of the snapping post-fission movement (snapping division), which we exploited as a biological 3D-sensor for the assessment of spatial confinement in the MGC or PAD system or strength of the negative dielectrophoresis force on cells cultivated in the nDEP system.43,44 Species-specific snapping movements upon cell division are particularly well suited for evaluating available cultivation space with the microcultivation technologies. For a quantitative description, the angular arrangement of the cells immediately after the division event was used. With an average angle of ![[small alpha, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c2.gif) nDEP = 73.3° ± 6.6 (CV,nDEP = 9%), the division angle of cells cultured in the nDEP system is 22% smaller than that of cells cultured in the MGC system with a mean angle of MGC = 96.1° ± 30.5 (CV,MGC = 31.7%) (Fig. 5). A tendency towards higher division angles was observed for cells cultivated on agarose pads, with cells exhibiting a mean division angle of PAD = 107.8° ± 22.2 (CV,PAD = 20.6%). The frequency distribution of division angles was very sharp and normally distributed in the nDEP system. A broader distribution of division angles in comparison with that in the nDEP system was observed in the MGC system and agarose pads, which may have resulted from inhomogeneous adhesion of the cells to adjacent surfaces and local variations in the extent of spatial constriction. In general, the division angles of cells cultivated in contact-based microcultivation systems (MGC and PAD) were increased in comparison with cell pair angles measured from C. glutamicum cells in suspension cultures (ESI† Fig. S11). In order to investigate the dynamics of cell pair angles, we also monitored the cell pair angle development during cultivations (ESI† Fig. S12). We repeatedly observed a sudden decrease in cell pair angles during the first 30 to 45 minutes of cell cultivation on agarose pads, most likely as a result of agarose gel movement due to initial water evaporation from the pad edges. After this equilibration period and saturation of the internal volume of the μ-dish with water, sudden changes in cell pair angles were no longer observable. Pad movement may be a central aspect influencing cellular parameters like division and cell morphology, since inhomogeneous movement of the agarose gel results in mechanical stress and strain on the cell, possibly disturbing these sensitive biological processes.
nDEP = 73.3° ± 6.6 (CV,nDEP = 9%), the division angle of cells cultured in the nDEP system is 22% smaller than that of cells cultured in the MGC system with a mean angle of MGC = 96.1° ± 30.5 (CV,MGC = 31.7%) (Fig. 5). A tendency towards higher division angles was observed for cells cultivated on agarose pads, with cells exhibiting a mean division angle of PAD = 107.8° ± 22.2 (CV,PAD = 20.6%). The frequency distribution of division angles was very sharp and normally distributed in the nDEP system. A broader distribution of division angles in comparison with that in the nDEP system was observed in the MGC system and agarose pads, which may have resulted from inhomogeneous adhesion of the cells to adjacent surfaces and local variations in the extent of spatial constriction. In general, the division angles of cells cultivated in contact-based microcultivation systems (MGC and PAD) were increased in comparison with cell pair angles measured from C. glutamicum cells in suspension cultures (ESI† Fig. S11). In order to investigate the dynamics of cell pair angles, we also monitored the cell pair angle development during cultivations (ESI† Fig. S12). We repeatedly observed a sudden decrease in cell pair angles during the first 30 to 45 minutes of cell cultivation on agarose pads, most likely as a result of agarose gel movement due to initial water evaporation from the pad edges. After this equilibration period and saturation of the internal volume of the μ-dish with water, sudden changes in cell pair angles were no longer observable. Pad movement may be a central aspect influencing cellular parameters like division and cell morphology, since inhomogeneous movement of the agarose gel results in mechanical stress and strain on the cell, possibly disturbing these sensitive biological processes.
In summary, these results demonstrate the significant effects of microcultivation technologies on key cellular parameters. Division rates and cell morphology, including division symmetry and spatial cellular arrangement, were heavily affected for cells cultivated on agarose pads, while, in contrast, no irregularities could be observed during nDEP and MGC cultivations. Interestingly, specific growth rates were not affected by the microcultivation technology and were highly consistent in all three systems.
Discussion
Microcultivation technologies for single-cell analysis are an integral part of the next generation toolbox for systems, biology, bio(techno)logy and synthetic biology, enabling studies of individual cellular microbial physiology beyond the bulky average of populations with precise control of the extracellular microenvironment. However, accompanied by the increasing importance of these technologies, knowledge about the technical bias on cellular functions and hence on the obtained results of single-cell experiments is required. Here, we address this issue and present a comparative study that reveals the distinct effects of different microcultivation technologies on fundamental microbial parameters like the rate of reproduction, cell division and cell morphology.The analysis initially focused on the quantitative measurement of the rate of bacterial reproduction, one of the most important and commonly employed biological readouts for the determination of cellular fitness and physiological response to physicochemical extracellular conditions.45,46 During comparison of specific growth rates of single C. glutamicum cells, no apparent effects of the different microcultivation technologies were measurable. Despite distinct differences in the trapping principle and the type of environment (nDEP: levitation – liquid medium – continuous perfusion; MGC: surface contact – liquid medium – diffusion; agarose pad: surface adhesion/embedding – semi-solid medium – diffusion), specific growth rates of single cells were consistent in all three investigated systems. The rate of biogenesis, hence the increase in biomass of single cells, was found to be best described by fitting with an exponential function. According to this observation, the biomass increase in single cells is a tightly regulated process and not only a simple biological parameter that linearly scales with environmental parameters.47 Furthermore, the robustness in growth indicated that an optimal supply of nutrients was provided by all three systems, allowing the cells to exploit their maximal biological capacity in terms of the growth rate. It can be deduced from these results that stress, originating from an adaptation to changing environmental conditions, inevitably occurs during bulk cultivations as a result of metabolic population activity. It was previously shown to impair growth and was minimized in all three systems.20,29 Moreover, these comparative results also imply that the electrical field during nDEP trapping did not influence the cellular physiology of C. glutamicum with the chosen trapping parameters. The origin of previously reported adverse effects of nDEP trapping on cellular viability in E. coli is thus to be sought elsewhere, for example, in the insufficient compensation of temperature effects induced by resistive joule heating.48 In addition to elevated and robust specific volume growth rates, none of the cultivated single cells showed a detectable lag-phase after introduction into the cultivation devices. This observation was especially surprising for agarose pad cultivations, since recent population-based studies on bacteria reported that a change from liquid medium to semi-solid medium involves stress response entailing temporary growth arrest.49 Despite the differences of agarose pads and agar plates in terms of cell exposure to ambient air and cell density during the seeding process, our results suggest studying this phenomenon in greater detail to understand the underlying mechanisms. This is highly important for single-cell cultivations based on mechanical cell trapping and surface contact of the cells. We could prove that all three investigated microcultivation systems are capable of providing a steady state between the cell and its extracellular environment, ultimately enabling reproducible analysis of balanced growth. This substantiates the superiority of single-cell cultivation systems in comparison with bulk cultivation approaches when it comes to an accurate description of fundamental biological parameters and their response to external physicochemical stimuli. It should be also noted that, although single-cell growth analysis is becoming increasingly widespread, a standardized methodology for measuring single-cell growth rates has not been developed so far. Instead of determining specific single-cell growth rates, cell length and area increase, as well as division rates based on cell counting, are commonly employed to assess growth rates, precluding comparison of single-cell measurement with population measurement. Newly developed methods for the accurate determination of biomass in live cells, like spatial light interference microscopy or pedestal resonator sensors, are difficult to apply on suspended cells like those in the nDEP system and therefore cannot satisfy these needs.50 Measurements of single-cell DNA content could be also used as a growth standard, but usually require the employment of fluorophores for a quantitative readout. However, the methodology presented here for single-cell growth quantification is simple and represents a universal method for the description of biomass increase from time-lapse images based on cell volume increase. This is highly relevant for virtually all microfluidic single-cell investigations, as confirmed by recent studies, providing evidence that the biomass density of single cells is constant during balanced growth. The measurement of specific growth rates based on cell volume can hence be regarded as valid and applicable to compare specific growth rates between single cells and populations.42 Besides specific growth rates, we also investigated cell division of individual cells cultivated with the three microcultivation technologies. Although specific growth rates were highly consistent for the investigated systems, single-cell division rates significantly deviated on agarose pads. This observation illustrates a significant influence of the microcultivation technology and the respective local environments on cellular functions. The results also reflect the fundamental difference between the specific growth rate and division rate of single cells, which can only match under balanced growth conditions and regular cell morphology. This allows the conclusion that the rate of biomass formation is, in contrast to the division rate, a tightly controlled biological process that is maintained even under stressful cultivation conditions. Quantitative analyses of single-cell proliferation (DNA synthesis) and nuclear division should be used in the future to shed light on the relationship between biogenesis, cell division and DNA number and content. However, the onset of the observed cellular responses may possibly remain hidden in the bulk average behind the average of billions of cells, but becomes evident with microfluidic single-cell analysis. Differences in single-cell division rates on agarose pads could be clearly assigned to premature or delayed cell division, leading to small or elongated daughter cells. Cell division is accurately and reproducibly initiated at distinct cell lengths of Corynebacterium, implying that this process is disturbed by external influences.51 A possible explanation for these processes can be found in the response to stress of C. glutamicum. Cell elongation in bacteria was shown to be induced by various stresses, for example DNA damage, sub-optimal pH or nutrient starvation during the stationary phase.52,53 Cells on agarose pads possibly experience nutrient limitation due to rapid consumption or changes in the chemical composition of the extracellular microenvironment by secreted metabolites. However, cell elongation occasionally occurs shortly after cell seeding on agarose pads. It is unlikely that such short time periods are sufficient for nutrient exhaustion or metabolite accumulation in the vicinity of the cells. Elongation and the resulting delayed cell division in bacteria have been shown to constitute a cellular emergency response to extracellular stress like osmotic shock or antibiotic stress, as well as a disturbance in gene expression.54–56 Considering the severity of these stress factors that trigger cell elongation, it can be concluded that the cellular physiology of C. glutamicum is significantly impaired when it is cultivated on agarose pads. The degree of spatial constriction might be a possible extracellular stress that affected the cell division process. Bacterial cells grown in constricted environments exhibited deformation and irregular shapes, but were able to maintain their rate of biomass increase.57,58
In addition to cell elongation, C. glutamicum displayed non-symmetric cell division on agarose pads, resulting in daughter cells of different lengths. Such non-symmetric cell division is abnormal in C. glutamicum, which usually exhibits a symmetric type of cell division.59 We also verified the symmetric type of cell division in C. glutamicum by microscopic analysis of cells grown in suspension. The loss of accurate cell partitioning on agarose pads indicates a severe disturbance of regulatory processes, most likely in the chromosome segregation machinery, responsible for tightly regulated midcell divisome positioning.60 Considering these facts, it can be concluded that mechanical influences and spatial constriction on agarose pads may be key factors disturbing divisome regulation in C. glutamicum. Interestingly, the sum of the daughter cell lengths immediately after the division event consistently exceeded the length of the mother cell before cell division, which might be a result of the tension that facilitates the snapping division movement.44 This aspect might be of vital interest for further biophysical studies on post-fission movement in mycobacteria.
We exploited the snapping movement of C. glutamicum upon cell division as a three dimensional biological sensor to quantitatively evaluate the degree of spatial constriction in the respective microcultivation systems. Cell division angles were increased on agarose pads in comparison with those in the nDEP system and suspended cells. Cells cultivated in the MGC system also featured increased division angles, probably due to friction on the growth chamber bottom, but were less pronounced than those of cells grown on agarose pads.
Even though the origin of elongation and deviating division symmetry of C. glutamicum on agarose pads cannot be unambiguously explained by our data, they point to a central aspect distinguishing agarose pads from the other two systems: the static environment with spatial constriction of cells in between the agarose layer and the glass cover slip. We speculate that the spatial constriction of the cells is the origin of stress that triggers elongation in Corynebacterium on agarose pads.
Conclusions
This study extends our knowledge about the technical bias of single-cell microcultivation technologies on cellular traits. Such knowledge is highly important for modern biology, encompassing all disciplines from systems to synthetic biology, ultimately permitting the approach for questions requiring undisturbed analyses of single-cell features. The microcultivation technology itself influences the functionality of single Corynebacterium cells. Division rates and cellular morphology were disturbed by spatial constriction when cells were grown on agarose pads, while biomass increases of single cells were balanced and tightly controlled, regardless of the technology employed. Our findings indicate that cellular traits can be much more affected by technological peculiarities of different microcultivation systems than previously assumed. This might have far-reaching consequences, questioning specific findings about single cells that were obtained with solid growth supports like agarose pads. Considering that the microcultivation technology, as demonstrated for agarose pads, influences such crucial biological parameters like cell division control and cell morphology, it is most probable that several other processes of the complex cellular machinery are affected as well. This might not only be true for coryneform bacteria, but also for many other microorganisms. There is thus a need to evaluate the chosen technology on the possible technical bias by the method of single-cell cultivation. Only if such influences are known, it is possible to understand the complex functions of the individual cell as a basic biological unit without methodological bias and exploit the full potential of single-cell analysis using specifically designed microscale cultivation systems.Acknowledgements
We gratefully thank Frederik Fritzsch for co-initiating this study and Katrin Rosenthal for fruitful scientific discussions about this manuscript. The German Helmholtz Association is gratefully acknowledged for providing funding within the framework of the Helmholtz Young Investigators program and the German Research Foundation (DFG) is gratefully acknowledged for providing funding within the framework of the priority program SPP 1617. The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this article and declare that there is no conflict of interest.References
- J. C. W. Locke, J. W. Young, M. Fontes, M. J. H. Jimenez and M. B. Elowitz, Science, 2011, 334, 366–369 CrossRef CAS PubMed.
- J. W. Young, J. C. Locke and M. B. Elowitz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4140–4145 CrossRef CAS PubMed.
- N. Q. Balaban, J. Merrin, R. Chait, L. Kowalik and S. Leibler, Science, 2004, 305, 1622–1625 CrossRef CAS PubMed.
- N. Dhar and J. D. McKinney, Curr. Opin. Microbiol., 2007, 10, 30–38 CrossRef CAS PubMed.
- K. Lewis, Nat. Rev. Microbiol., 2007, 5, 48–56 CrossRef CAS PubMed.
- T. K. Wood, S. J. Knabel and B. W. Kwan, Appl. Environ. Microbiol., 2013, 79, 7116–7121 CrossRef CAS PubMed.
- P. Wang, L. Robert, J. Pelletier, W. L. Dang, F. Taddei, A. Wright and S. Jun, Curr. Biol., 2010, 20, 1099–1103 CrossRef CAS PubMed.
- M. E. Lidstrom and M. C. Konopka, Nat. Chem. Biol., 2010, 6, 705–712 CrossRef CAS PubMed.
- F. S. Fritzsch, C. Dusny, O. Frick and A. Schmid, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 129–155 CrossRef CAS PubMed.
- A. Schmid, H. Kortmann, P. S. Dittrich and L. M. Blank, Curr. Opin. Biotechnol., 2010, 21, 12–20 CrossRef CAS PubMed.
- A. Grunberger, W. Wiechert and D. Kohlheyer, Curr. Opin. Biotechnol., 2014, 29C, 15–23 CrossRef PubMed.
- J. C. Love, AIChE J., 2010, 56, 2496–2502 CrossRef CAS.
- V. Lecault, A. K. White, A. Singhal and C. L. Hansen, Curr. Opin. Chem. Biol., 2012, 16, 381–390 CrossRef CAS PubMed.
- B. Okumus, S. Yildiz and E. Toprak, Curr. Opin. Biotechnol., 2014, 25, 30–38 CrossRef CAS PubMed.
- F. S. Fritzsch, K. Rosenthal, A. Kampert, S. Howitz, C. Dusny, L. M. Blank and A. Schmid, Lab Chip, 2013, 13, 397–408 RSC.
- H. Kortmann, P. Chasanis, L. M. Blank, J. Franzke, E. Y. Kenig and A. Schmid, Lab Chip, 2009, 9, 576–585 RSC.
- J. W. Young, J. C. Locke, A. Altinok, N. Rosenfeld, T. Bacarian, P. S. Swain, E. Mjolsness and M. B. Elowitz, Nat. Protoc., 2012, 7, 80–88 CrossRef CAS PubMed.
- A. Grunberger, N. Paczia, C. Probst, G. Schendzielorz, L. Eggeling, S. Noack, W. Wiechert and D. Kohlheyer, Lab Chip, 2012, 12, 2060–2068 RSC.
- T. Schnelle, R. Hagedorn, G. Fuhr, S. Fiedler and T. Muller, Biochim. Biophys. Acta, 1993, 1157, 127–140 CrossRef CAS.
- A. Grunberger, J. van Ooyen, N. Paczia, P. Rohe, G. Schiendzielorz, L. Eggeling, W. Wiechert, D. Kohlheyer and S. Noack, Biotechnol. Bioeng., 2013, 110, 220–228 CrossRef PubMed.
- S. Unthan, A. Grunberger, J. van Ooyen, J. Gatgens, J. Heinrich, N. Paczia, W. Wiechert, D. Kohlheyer and S. Noack, Biotechnol. Bioeng., 2014, 111, 359–371 CrossRef CAS PubMed.
- S. Kinoshita, S. Udaka and M. Shimono, J. Gen. Appl. Microbiol., 2004, 50, 331–343 Search PubMed.
- S. Klumpp and T. Hwa, Curr. Opin. Biotechnol., 2014, 28, 96–102 CrossRef CAS PubMed.
- M. Schaechter, J. P. Williamson, J. R. Hood Jr. and A. L. Koch, J. Gen. Microbiol., 1962, 29, 421–434 CrossRef CAS PubMed.
- C. Dusny and A. Schmid, Environ. Microbiol., 2014 DOI:10.1111/1462-2920.12667 , accepted and published online.
- J. R. Moffitt, J. B. Lee and P. Cluzel, Lab Chip, 2012, 12, 1487–1494 RSC.
- J. Dai, S. H. Yoon, H. Y. Sim, Y. S. Yang, T. K. Oh, J. F. Kim and J. W. Hong, Anal. Chem., 2013, 85, 5892–5899 CrossRef CAS PubMed.
- S. Boulineau, F. Tostevin, D. J. Kiviet, P. R. ten Wolde, P. Nghe and S. J. Tans, PLoS One, 2013, 8, 1–9 Search PubMed.
- C. Dusny, F. S. Fritzsch, O. Frick and A. Schmid, Appl. Environ. Microbiol., 2012, 78, 7132–7136 CrossRef CAS PubMed.
- D. Binder, A. Grunberger, A. Loeschcke, C. Probst, C. Bier, J. Pietruszka, W. Wiechert, D. Kohlheyer, K. E. Jaeger and T. Drepper, Integr. Biol., 2014, 6, 755–765 RSC.
- H. Kortmann, F. Kurth, L. M. Blank, P. Dittrich and A. Schmid, Lab Chip, 2009, 9, 3047–3049 RSC.
- C. Probst, A. Grunberger, N. Braun, S. Helfrich, K. Noh, W. Wiechert and D. Kohlheyer, Anal. Methods, 2015, 7, 91–98 RSC.
- W. Mather, O. Mondragon-Palomino, T. Danino, J. Hasty and L. S. Tsimring, Phys. Rev. Lett., 2010, 104, 208101 CrossRef.
- N. Mustafi, A. Grunberger, D. Kohlheyer, M. Bott and J. Frunzke, Metab. Eng., 2012, 14, 449–457 CrossRef CAS PubMed.
- N. Mustafi, A. Grunberger, R. Mahr, S. Helfrich, K. Noh, B. Blombach, D. Kohlheyer and J. Frunzke, PLoS One, 2014, 9, e85731 Search PubMed.
- C. Probst, A. Grunberger, W. Wiechert and D. Kohlheyer, J. Microbiol. Methods, 2013, 95, 470–476 CrossRef PubMed.
- G. Ullman, M. Wallden, E. G. Marklund, A. Mahmutovic, I. Razinkov and J. Elf, Philos. Trans. R. Soc., B, 2013, 368, 20120025 CrossRef CAS PubMed.
- S. S. Lee, I. Avalos Vizcarra, D. H. Huberts, L. P. Lee and M. Heinemann, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4916–4920 CrossRef CAS PubMed.
- J. W. Young and M. B. Elowitz, Mol. Cell, 2011, 42, 405–406 CrossRef CAS PubMed.
- A. Gruenberger, C. Probst, A. Heyer, W. Wiechert, J. Frunzke and D. Kohlheyer, J. Visualized Exp., 2013, 82, 50560 Search PubMed.
- C. L. Woldringh, P. G. Huls and N. O. Vischer, J. Bacteriol., 1993, 175, 3174–3181 CAS.
- Y. Zhao, H. S. Lai, G. Zhang, G. B. Lee and W. J. Li, Lab Chip, 2014, 14, 4426–4434 RSC.
- M. Letek, M. Fiuza, E. Ordonez, A. F. Villadangos, A. Ramos, L. M. Mateos and J. A. Gil, Anton. Leeuw. Int. J. G., 2008, 94, 99–109 CrossRef CAS PubMed.
- N. R. Thanky, D. B. Young and B. D. Robertson, Tuberculosis, 2007, 87, 231–236 CrossRef CAS PubMed.
- J. Volmer, C. Neumann, B. Buhler and A. Schmid, Appl. Environ. Microbiol., 2014, 80, 6539–6548 CrossRef CAS PubMed.
- B. Li, Y. Qiu, A. Glidle, J. Cooper, H. Shi and H. Yin, Analyst, 2014, 139, 3305–3313 RSC.
- K. Abner, T. Aaviksaar, K. Adamberg and R. Vilu, J. Theor. Biol., 2014, 341, 78–87 CrossRef PubMed.
- S. S. Donato, V. Chu, D. M. Prazeres and J. P. Conde, Electrophoresis, 2013, 34, 575–582 CrossRef CAS PubMed.
- C. Cuny, M. Lesbats and S. Dukan, Appl. Environ. Microbiol., 2007, 73, 885–889 CrossRef CAS PubMed.
- G. Popescu, K. Park, M. Mir and R. Bashir, Lab Chip, 2014, 14, 646–652 RSC.
- G. Joyce, K. J. Williams, M. Robb, E. Noens, B. Tizzano, V. Shahrezaei and B. D. Robertson, PLoS One, 2012, 7, e44582 CAS.
- S. S. Justice, D. A. Hunstad, L. Cegelski and S. J. Hultgren, Nat. Rev. Microbiol., 2008, 6, 162–168 CrossRef CAS PubMed.
- A. M. Nanda, A. Heyer, C. Kramer, A. Grunberger, D. Kohlheyer and J. Frunzke, J. Bacteriol., 2014, 196, 180–188 CrossRef PubMed.
- C. C. Chung, I. F. Cheng, H. M. Chen, H. C. Kan, W. H. Yang and H. C. Chang, Anal. Chem., 2012, 84, 3347–3354 CrossRef CAS PubMed.
- B. Sieger, K. Schubert, C. Donovan and M. Bramkamp, Mol. Microbiol., 2013, 90, 966–982 CrossRef CAS PubMed.
- J. Bos, Q. Zhang, S. Vyawahare, E. Rogers, S. M. Rosenberg and R. H. Austin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 178–183 CrossRef CAS PubMed.
- J. Mannik, F. Wu, F. J. Hol, P. Bisicchia, D. J. Sherratt, J. E. Keymer and C. Dekker, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6957–6962 CrossRef PubMed.
- J. Mannik, R. Driessen, P. Galajda, J. E. Keymer and C. Dekker, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14861–14866 CrossRef PubMed.
- A. Neumeyer, T. Hubschmann, S. Muller and J. Frunzke, Microb. Biotechnol., 2013, 6, 157–167 CrossRef PubMed.
- C. Donovan, A. Schauss, R. Kramer and M. Bramkamp, PLoS One, 2013, 8, e55078 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc01270d |
| ‡ These authors contributed equally to this work. |
| § Present address: Department of Solar Materials, Helmholtz Centre for Environmental Research - UFZ, Permoserstr. 15, 04318 Leipzig, Germany. |
| This journal is © The Royal Society of Chemistry 2015 |