
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A lung-on-a-chip array with an integrated bio-inspired respiration mechanism†
Andreas O.
Stucki‡
ab,
Janick D.
Stucki‡
ab,
Sean R. R.
Hall
de,
Marcel
Felder
ab,
Yves
Mermoud
a,
Ralph A.
Schmid
de,
Thomas
Geiser
ce and
Olivier T.
Guenat
*acd
aARTORG Center for Biomedical Engineering Research, Lung Regeneration Technologies, University of Berne, Switzerland. E-mail: olivier.guenat@artorg.unibe.ch
bGraduate School for Cellular and Biomedical Sciences, University of Berne, Switzerland
cDivision of Pulmonary Medicine, University Hospital of Berne, Switzerland
dDivision of Thoracic Surgery, University Hospital of Berne, Switzerland
eDepartment of Clinical Research, University of Berne, Switzerland
First published on 3rd December 2014
Abstract
We report a lung-on-a-chip array that mimics the pulmonary parenchymal environment, including the thin alveolar barrier and the three-dimensional cyclic strain induced by breathing movements. The micro-diaphragm used to stretch the alveolar barrier is inspired by the in vivo diaphragm, the main muscle responsible for inspiration. The design of this device aims not only at best reproducing the in vivo conditions found in the lung parenchyma but also at making the device robust and its handling easy. An innovative concept, based on the reversible bonding of the device, is presented that enables accurate control of the concentration of cells cultured on the membrane by easily accessing both sides of the membranes. The functionality of the alveolar barrier could be restored by co-culturing epithelial and endothelial cells that form tight monolayers on each side of a thin, porous and stretchable membrane. We showed that cyclic stretch significantly affects the permeability properties of epithelial cell layers. Furthermore, we also demonstrated that the strain influences the metabolic activity and the cytokine secretion of primary human pulmonary alveolar epithelial cells obtained from patients. These results demonstrate the potential of this device and confirm the importance of the mechanical strain induced by breathing in pulmonary research.
Introduction
The pharmaceutical sector is currently experiencing a serious efficiency crisis that forces all of the actors in this field to rethink the way research and development can be performed more efficiently.1 One of the key issues that urgently needs to be addressed is the lack of efficient and reproducible drug discovery models able to predict the toxicity and the efficiency of compounds in humans prior to launching expensive clinical trials. Animal models used in the preclinical phase often poorly predict the toxicological responses in humans2 and standard in vitro models fail to reproduce the complexity of the biophysical and cellular microenvironment found in vivo. Recent progress in microtechnologies has enabled the emergence of novel in vitro models that better reproduce the in vivo conditions.3 These models called “organs-on-chips” are widely seen as being able to better predict human responses and simultaneously to importantly reduce the ethically controversial animal testing.Lungs-on-chips aiming at mimicking the complex microenvironment of the lung alveoli have only recently been reported. In sharp contrast to standard in vitro models, such systems allow the reproduction of cyclic mechanical stress induced by respiratory movements. Takayama and colleagues investigated the mechanical stress induced by liquid plug propagation in small flexible airway models and suggested that the possibility of induced injuries on lining cells along the airways in emphysema is higher due to larger wall stresses.4 His group further studied the combined effect of mechanical and surface tension stresses that typically occur in ventilator induced lung injury.5 Using this device, they demonstrated cellular-level lung injury under flow conditions that caused symptoms characteristic of a wide range of pulmonary diseases.6 More recently, Ingber and colleagues reported a lung alveoli model that further reproduces the in vivo situation by mimicking the thin alveolar barrier being cyclically stretched.7 The barrier, made of a thin, porous and stretchable polydimethylsiloxane (PDMS) membrane on which epithelial and endothelial cells are cultured, is sandwiched between two microfluidic structures creating two superposed microchannels. Actuation resulting in the cyclic strain of the PDMS membrane is performed by varying the negative pressure in the two channels located on each side of the superposed microchannels and separated from it by thin walls. This actuation principle presents the drawback that the strain applied to the thin, porous membrane strongly depends on the viscoelastic properties of the stretched material (here PDMS) and on the dimensions, in particular the thickness, of the thin PDMS walls. Consequently, the negative pressure applied to the adjacent channels needs to be precisely controlled. In addition, the typical confined microfluidic setting used does not allow the precise control of the concentration of cells seeded on the membrane.
Furthermore, the in vivo relevance of almost all in vitro human lung alveoli models is limited by the use of lung epithelial cell lines or primary lung cells from rats. Unfortunately, the most used lung epithelial cells, A549, is a lung adenocarcinoma cell line that poorly mimics the phenotype of original pulmonary alveolar epithelial cells. On the other hand, primary lung cells from rats suffer from interspecies differences and are thus inadequate to recreate the human air–blood barrier.
We report here a novel lung-on-a-chip that does not suffer from these limitations. It mimics the lung alveolar barrier in an unprecedented way, using primary human pulmonary alveolar epithelial cells obtained from patients who underwent partial lung resection. The lung alveolar barrier can be exposed to a 3D cyclic mechanical strain using a novel bioinspired actuation mechanism. Experiments performed with those cells and lung bronchial epithelial cells revealed the significance of the mechanical strain on those cells. This robust and easy to use lung-on-a-chip is intended to be a tool for drug discovery development as well as for toxicology fields in research and industry.
Materials and methods
Fabrication of the lung-on-a-chip
The lung-on-a-chip consists of a fluidic part and a pneumatic part (Fig. 1C). The fluidic part comprises two PDMS plates between which a thin, porous and flexible PDMS membrane is sandwiched and bonded. The top plate contains a 3 mm diameter access hole on the apical side of the membrane and a 1 mm diameter hole to access the overflow chamber. The bottom plate is structured with a cell culture medium reservoir. The pneumatic part is made of a 40 μm thin PDMS layer bonded with the actuation plate in which pneumatic channels are structured. The fluidic and pneumatic parts were made by soft lithography.8 Briefly, the PDMS base and curing agents (Sylgard 184, Dow Corning) were mixed well (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w ratio), degassed in a vacuum desiccator and casted in hard plastic molds made directly from aluminum molds structured by standard machining (ki-Mech GmbH). The PDMS pre-polymer was cured at 60 °C for at least 24 hours. The porous and flexible membrane was fabricated by a microstructuring–lamination process. The PDMS pre-polymer was sandwiched between a silicon mold containing an array of micropillars structured by DRIE and a 75 μm thin PE sheet (DuPont Teijin Films, Melinex® 411). The micropillars have different heights ranging from 3.5 μm up to 10 μm and different diameters (3 μm or 8 μm) that define the final thickness and the pore size of the membrane. The silicon mold and the plastic sheet were then clamped together with the PDMS pre-polymer sandwiched in between and cured at 60 °C for at least 24 hours. The thickness of the produced membrane corresponds to the height of the micropillars, which have pores of 3 or 8 μm in diameter (Fig. 2A). After curing, the membrane was released from the mold and irreversibly bonded by O2 plasma (Harrick Plasma) onto the bottom plate. The top plate was then reversibly bonded to the bottom plate. The thin PDMS layer in which the micro-diaphragm is included was fabricated by spinning the PDMS pre-polymer onto a PE sheet attached to a silicon wafer at 1700 rpm for 60 seconds. After spinning, the membrane was allowed to cure for 24 hours at 60 °C and was then irreversibly bonded by O2 plasma to the actuation plate.
:1 w/w ratio), degassed in a vacuum desiccator and casted in hard plastic molds made directly from aluminum molds structured by standard machining (ki-Mech GmbH). The PDMS pre-polymer was cured at 60 °C for at least 24 hours. The porous and flexible membrane was fabricated by a microstructuring–lamination process. The PDMS pre-polymer was sandwiched between a silicon mold containing an array of micropillars structured by DRIE and a 75 μm thin PE sheet (DuPont Teijin Films, Melinex® 411). The micropillars have different heights ranging from 3.5 μm up to 10 μm and different diameters (3 μm or 8 μm) that define the final thickness and the pore size of the membrane. The silicon mold and the plastic sheet were then clamped together with the PDMS pre-polymer sandwiched in between and cured at 60 °C for at least 24 hours. The thickness of the produced membrane corresponds to the height of the micropillars, which have pores of 3 or 8 μm in diameter (Fig. 2A). After curing, the membrane was released from the mold and irreversibly bonded by O2 plasma (Harrick Plasma) onto the bottom plate. The top plate was then reversibly bonded to the bottom plate. The thin PDMS layer in which the micro-diaphragm is included was fabricated by spinning the PDMS pre-polymer onto a PE sheet attached to a silicon wafer at 1700 rpm for 60 seconds. After spinning, the membrane was allowed to cure for 24 hours at 60 °C and was then irreversibly bonded by O2 plasma to the actuation plate.
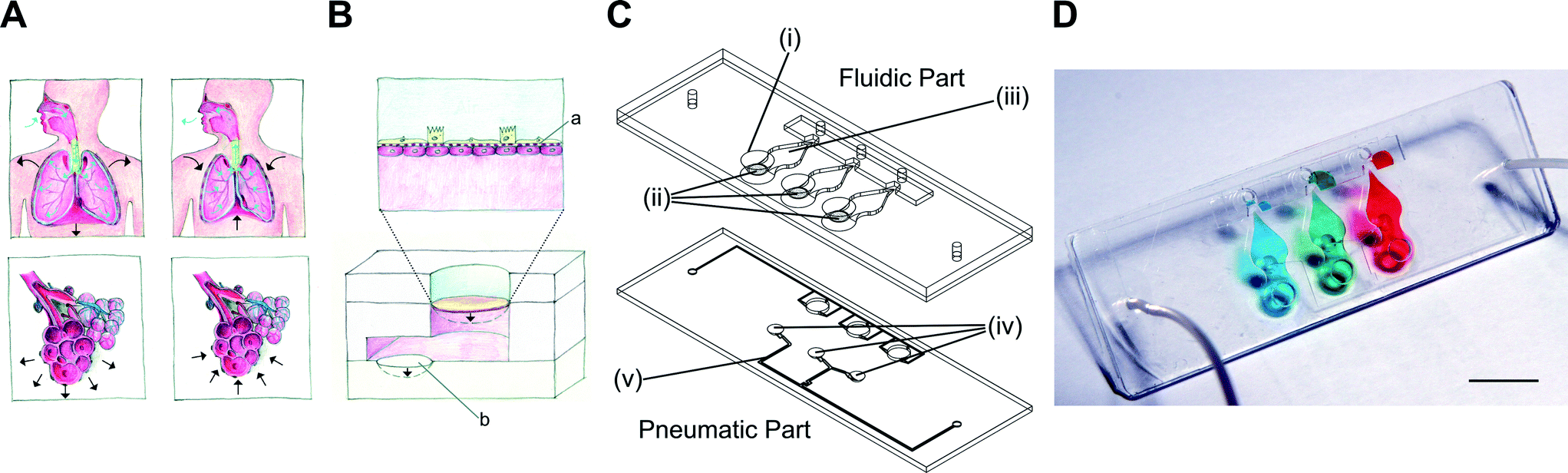 | ||
| Fig. 1 Working principle and design of the lung-on-a-chip. (A) In vivo, inspiration is controlled by the diaphragm, whose contraction leads to the three-dimensional expansion of the alveolar sacs. (B) In vitro, the 3D cyclic mechanical strain of the bioartificial alveolar membrane (a) induced by a micro-diaphragm (b) that is actuated by an electro-pneumatic set-up. The bioartificial alveolar membrane consists of a thin, porous and stretchable membrane on which epithelial and endothelial cells are cultured. (C) The lung-on-a-chip is made of 25 × 75 mm fluidic and pneumatic parts. The fluidic part consists of three alveolar cell culture wells (i) and thin, porous and flexible membranes (ii), beneath which the basolateral chambers are located (iii). The micro-diaphragms (iv) are integrated into the pneumatic part and connected to pneumatic microchannels (v). (D) Photograph of the lung-on-a-chip filled with food-dye-colored solutions inside the basolateral chambers. Scale bar: 10 mm. | ||
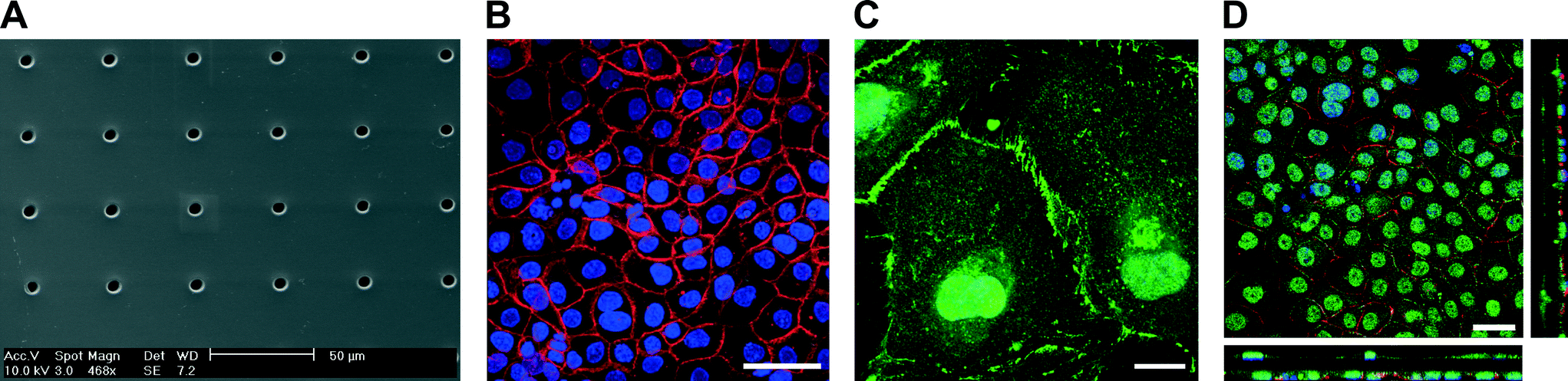 | ||
| Fig. 2 Porous and flexible PDMS membrane used as a cell culture substrate. (A) SEM image of a PDMS membrane with 8 μm pores. (B) Z-projection of a confluent layer of lung epithelial cells (16HBE14o−) stained for adherens junction E-cadherin (red) and for cell nuclei (blue). The pores of the porous membrane can be seen in the background. (C) Immunofluorescence image of endothelial cells (HUVEC) stained for adherens junction VE-cadherin (green) and for cell nuclei (blue). (D) Confocal picture of the 16HBE14o− and HUVEC co-culture, stained for E-cadherin (red), VE-cadherin (green) and cell nuclei (blue). Cell–cell contacts between the epithelial and endothelial cells can be seen through the pores. Scale bars: 50, 50, 20 and 30 μm. | ||
Cell culture protocols
Bronchial epithelial 16HBE14o− cells (from Dr. Gruenert of the University of California, San Francisco) were cultured in MEM medium (Gibco) supplemented with 10% FBS (Gibco), 1% L-glutamine (2 mM, Gibco), 1% penicillin (100 U ml−1, Gibco) and 1% streptomycin (100 U ml−1, Gibco). Primary human pulmonary alveolar epithelial cells (pHPAEC) were obtained from a lung resection from a patient undergoing pneumonectomy for lung cancer. All participants provided written informed consent. Briefly, healthy lung tissue was digested into a single cell suspension using a solution of 0.1% collagenase I/0.25% collagenase II (Worthington Biochemicals). Healthy epithelial cells were isolated using fluorescence activated cell sorting (BD FACSAria III) with an antibody that recognizes CD326, also known as the epithelial cell adhesion molecule (EpCAM, clone 1B7, eBioscience), while excluding hematopoietic (CD45, clone 2D1 and CD14, clone 61D3, eBioscience) and endothelial cells (CD31, clone WM59, eBioscience). Following sorting, EpCAM+ primary human pulmonary alveolar epithelial cells (pHPAECs) were cultured for expansion in CnT-Prime Airway epithelial culture medium (CELLnTEC, Berne, CH) supplemented with 1% penicillin (100 U ml−1, Gibco) and 1% streptomycin (100 U ml−1, Gibco), 1% Pen Strep (Gibco). Immunophenotyping of culture-expanded EpCAM+ cells was carried out using flow cytometry (BD FACSCanto LSRII) for the expression of the type I and type II epithelial markers podoplanin (clone NZ-1.3, eBioscience) and CD63 (clone H5C6, eBioscience), respectively. Primary human umbilical vein endothelial cells (pHUVEC, Lonza) were cultured in EBM-2 medium (Lonza) supplemented with 2% FBS and growth factors according to the manufacturer's protocol. All cells were maintained at 37 °C, 5% CO2 in air. Prior to cell seeding, the microfluidic devices were sterilized by ozone (CoolCLAVE, Genlantis) and the porous membranes were covalently coated with human fibronectin (2.5 μg cm−2, Merck Millipore) or 0.1% gelatin and 2 μg ml−1 collagen I as previously described.9 Briefly, the membranes were activated by O2 plasma and immediately covered with 5% (3-aminopropyl)triethoxysilane (Sigma-Aldrich) in H2O. After 20 min, the membranes were thoroughly rinsed with deionized water and covered with 0.1% glutaraldehyde (Sigma-Aldrich). After an additional 20 min incubation, the membranes were washed again with deionized water, and then coated with fibronectin and incubated overnight. Prior to cell seeding, the membranes were washed with cell culture medium.Stretching protocol
Once a confluent cell monolayer is formed on the thin membrane, a drop of 50 μl of cell culture medium is added to the basal side of the fluidic part. The fluidic part is then flipped with the drop of cell culture medium hanging and mounted onto the pneumatic part. The micro-diaphragm is able to apply a reproducible three-dimensional cyclic strain to the cells (corresponding to 10% linear strain). To cyclically stretch the membrane at a frequency of 0.2 Hz, the lung-on-a-chip is connected to an external electro-pneumatic setup. This setup controls the magnitude of the applied negative pressure as well as the frequency. The pressure-curve is modeled as a sinusoidal wave. A 10% linear stretch magnitude is within the physiological range of strain, experienced by the alveolar epithelium in the human lung.10Permeability assay
Upon confluence (after 72 hours in culture), the basal compartment was filled with cell culture medium and mounted on the pneumatic part. The cells were either preconditioned by stretching for 19 hours or kept under static conditions for the same amount of time prior to performing the assay. To assess the apical to basal permeability of the epithelial barrier, 1 μg ml−1 FITC-sodium (Sigma Aldrich) in MEM medium and 1 mg ml−1 RITC-dextran (70 kDa, Sigma-Aldrich) in MEM medium were added from the apical side of the epithelial barrier. The system was allowed to incubate for two hours under either dynamic or static conditions. After two hours of incubation, the fluid gained from the basal side of the barrier was collected and analyzed with a multiwell plate reader (M1000 Infinite, Tecan) at 460 nm and 553 nm excitation and 515 nm and 627 nm emission for FITC-sodium and RITC-dextran, respectively. The permeability was assessed in terms of relative transport across the epithelial barrier by normalizing the fluorescence intensity signal obtained from the solution sampled in the basal chamber with the fluorescence signal obtained from the standard solution initially added to the apical side of the barrier.Cell viability and proliferation
To measure cell viability and proliferation, the non-toxic alamar blue (Invitrogen) assay was used according to the manufacturer's protocol. Briefly, the alamar blue reagent was mixed with cell culture medium in a 1:10 ratio. 60 μl of the mixture was added to the cell culture well and incubated for one hour at 37 °C under static conditions. After incubation, the fluorescence intensity of the cell supernatant was measured using a multiwell plate reader at 570 nm excitation and 585 nm emission. The fluorescence intensity corresponds directly to the metabolic activity of the cells. The assay was performed at 0 h (before applying stretch) and after 24 h and 48 h of stretching. The same time points were used for the static controls.
IL-8 secretion
IL-8 secretion in the supernatant was measured using an ELISA kit (R&D Diagnostics) according to the manufacturer's protocol. The supernatant analyzed was collected after 2, 24 and 48 hours of stretching. After collection of the supernatant, new media were added. Cells kept under static conditions served as control.Immunofluorescence imaging
For immunofluorescence imaging, cells were rinsed with PBS and fixed with 4% paraformaldehyde (Sigma-Aldrich) in PBS for 12 min at room temperature. After several washing steps with PBS, the cells were permeabilized with 0.2% Triton X-100 (Sigma-Aldrich) in PBS for another 10 min. To prevent any unspecific antibody binding, a blocking solution of PBS with 5% FBS and 1% bovine serum albumin (Sigma-Aldrich) was added for 30 min. Primary antibodies (E-cadherin (67A4), Santa Cruz, and VE-cadherin (V1514), Sigma-Aldrich) were diluted 1:100 in blocking solution and incubated for 2 h at RT. The corresponding secondary antibodies (donkey anti-mouse-AlexaFluor568, Invitrogen, and donkey anti-rabbit-AlexaFluor488, Invitrogen), diluted 1:200, and Hoechst 33342 (1:10000, Invitrogen) to counterstain cell nuclei were incubated for 1 h at 20 °C in the dark. After rinsing three times with blocking solution, the specimens were embedded in Vectashield anti-fade medium (Sigma-Aldrich). Images were obtained using a confocal laser scanning microscope (CLSM, Zeiss LSM 710).
Statistics
Data are presented as mean ± standard deviation (SD). Differences between two means were determined by the two-tailed unpaired Student's t-test and p < 0.05 was taken as the level of significance.Results and discussion
Design of the lung-on-a-chip with a bioinspired respiration mechanism
The air–blood barrier of the lung, with a thickness of about 1 to 2 μm,11 is constantly exposed to cyclic mechanical stress induced at the organ level by the diaphragm, the most important muscle for inspiration. It consists of a thin dome shaped sheet of muscle that contracts, thereby increasing the volume of the thoracic cavity.12 The negative pressure created by the diaphragm is further transmitted to the complex architecture of the lung, up to its most delicate structures, the alveolar sacs (Fig. 1A). Stress concentration is particularly important in the thin alveolar septa that separate adjacent alveoli. This site comprises two monolayers of alveolar epithelial cells separated by the basal membrane and pulmonary microcapillaries.10 During normal breathing, the respiratory cycle consists of 10 to 12 breathings per minute, with mechanical strain comprising between 5 and 12% linear elongation.10 The effects of mechanical strain have been reported in a number of biological processes, for example in lung development13,14 or in the evolution of various respiratory diseases, such as acute lung injury,15 lung fibrosis16 and other interstitial lung diseases.17 Although our knowledge of the mechanobiology of the lung, in particular the mechano-responses of lung epithelial cells, has advanced significantly during the last two decades, much remains to be discovered and understood in this research field.10 This fact is due in large part to the lack of systems able to reproduce the dynamic and structurally complex environment of the alveolar barrier. Indeed, with the exception of the system recently reported by Huh and colleagues,7 standard systems used to investigate mechanical effects mimic only the respiratory movements, but not the characteristics of the thin alveolar barrier.10,18 In addition, the experimental conditions of all these systems vary considerably making cross-comparisons between studies difficult. This is particularly true for the applied strain, which is applied in one direction (cell elongation, e.g. Huh et al.7), in two dimensions (stress of the cell surface area, e.g. Flexcell) or in three dimensions, as in the case in vivo and in the present lung-on-a-chip.The design of the lung-on-a-chip presented in this study mimics the alveolar sac environment of the human lung including the mechanical stress induced by respiration movements (Fig. 1A–B). The bioartificial alveolar barrier consists of a thin, porous and flexible PDMS membrane on which cells are cultured, typically epithelial cells on the apical side and endothelial cells on the basal side of the membrane. This barrier is indirectly stretched downwards by the movements of a 40 μm thick actuation PDMS membrane that acts as a micro-diaphragm. It is cyclically deflected by a negative pressure applied in a small cavity located beneath the micro-diaphragm. The cavity volume limits the deflection of the micro-diaphragm enabling a clearly defined maximum strain when the actuation membrane reaches the bottom of the cavity. As the alveolar barrier and the micro-diaphragm are located in a closed compartment filled with an incompressible cell culture solution, the pressure applied on the micro-diaphragm is transmitted to the alveolar membrane according to Pascal's law. The maximum three-dimensional mechanical strain applied – set at 10% linear strain – to the alveolar membrane is thus accurately controlled by the volume of the micro-diaphragm cavity.
The reproduction of in vivo features is a priority to create biologically relevant organs-on-chips. However the ease of use and the robustness of such systems are parameters that are as important in view of their broader use. The design of the present lung-on-a-chip also addresses those constraints, in particular the precise control of the number of cells seeded in the culturing well, which is a typical and recurrent issue of cell-based microfluidic systems. In such systems, the cells loaded on the chip are not controlled once they enter the microfluidic network, which often results in an inhomogeneous cellular spatial distribution and the “loss” of cells in the fluidic network outside of the culturing zone. To address this issue, a semi-open design was used which allowed accurate control of the cells seeded on the apical side of the membrane (Fig. 1C–D). Cells are pipetted directly on the thin membrane like in a standard multiwell plate. The problem of cell seeding on the basal side of the membrane was solved by using an extension of the hanging drop technique (ESI† Fig. S1). Once the cell layer is confluent on the apical side of the membrane, the chip is flipped and a drop of cell culture medium with cells in suspension is added to the basal side of the membrane. After cell adhesion on the basal side of the membrane, the fluidic part of the lung-on-a-chip is flipped with the cell culture medium drop hanging. The fluidic part is then brought into contact with the lower part of the lung-on-a-chip and closed. During this step, the drop of cell culture medium is forced into the basal compartment defined between both plates. Excess solution is pushed outside of the compartment via a microvalve.
The following results demonstrate in the first phase the mechanical functionality and robustness of the lung-on-a-chip. In the second phase, the effects of physiological mechanical strain on lung epithelial cells were demonstrated. The experiments were performed under normal breathing conditions, meaning a breathing cycle of 12 cycles min−1, at a physiological level of strain corresponding to 10% linear elongation.
Characterization of the lung-on-a-chip
Fig. 1C illustrates a bioinspired lung-on-a-chip with three alveolar cell culture wells (i) each having direct access to a thin, porous and stretchable alveolar membrane (ii). The basal chambers (iii) located under each membrane are filled with dyed solutions confined between the fluidic and the pneumatic parts. The slight over deflection (about 1% linear strain) of the alveolar membrane that results from bringing the two parts together is levelled by a normally closed pneumatic microvalve located between the basal compartment and the over-flow chamber. A slight pressure exerted on the two rubber parts leads to reversible bonding (ESI† Fig. S1) that is strong enough to ensure the operation of the chip as well as to prevent any leakages. The cyclic mechanical stress of the micro-diaphragm (iv) located in the pneumatic part of the lung-on-a-chip enables the alveolar barrier to be mechanically stressed at a well-defined level. The homemade electro-pneumatic set-up, connected to the microchannels of the pneumatic part (v), generates a negative pressure with a sinusoidal function that reproduces the respiration parameters during normal breathing. The maximum strain in the alveolar membrane is evaluated by comparing two pictures taken in the center of the alveolar membrane at rest and when the micro-diaphragm is completely deflected (ESI† Fig. S2). At maximum deflection, the strain in the alveolar membrane accounts for a maximal linear elongation of 10%.The microstructuring–lamination process developed to fabricate thin, porous and flexible membranes produces reliable and reproducible features. The membranes can be produced with thicknesses of either 3.5 or 10 μm and with pore sizes and densities of either 3 μm with 800000 pores cm−2 or 8 μm with 60000 pores cm−2 with little variations in pore densities (Fig. 2A). The pore sizes and densities of the produced PDMS membrane correspond to those of commercially available cell culture inserts.
Reconstitution of the lung alveolar barrier
The integrity of the lung alveolar barrier is one of the most critical parameters of a healthy lung. Damaging it leads to fluid infiltration into the alveolar sacs that may cause lung edema and other types of pulmonary diseases. The integrity of the barrier is guaranteed by a number of proteins forming either tight junctions or adherens junctions. Tight junction proteins are responsible for the formation of functional epithelial and endothelial barriers and primarily function as a diffusion barrier.19 Adherens junctions link actin filaments between neighboring cells, maintain tissue integrity and translate mechanical forces throughout a tissue via the cytoskeleton.20To recapitulate a functional epithelial barrier, a co-culture of bronchial epithelial cells (16HBE14o−) and primary endothelial cells (HUVEC) was cultured on a 10 μm thin, porous membrane coated with fibronectin (Fig. 2D). HUVECs and 16HBE14o− bronchial epithelial cells were seeded on the basal side and on the apical side of the membrane, respectively. The epithelial and endothelial layers grew to confluence in two to four days building a homogeneous and tight barrier. Tight junction proteins (e.g. zona occludens-1 (ZO-1)) and adherens junction proteins (e.g. E-cadherin) accumulated at the cellular interface of the epithelial layer forming strong cell–cell contacts (Fig. 2B). On the endothelial side, vascular endothelial cadherin (VE-cadherin) based adherens junction expression was also observed confirming the formation of a tight endothelial barrier. The brush borders of the endothelial cells are typical of endothelial layers (Fig. 2C). Cell–cell contacts between the endothelial and epithelial layers could also be confirmed through the 8 μm pores (Fig. 2D).
Influence of mechanical stress on barrier permeability
The alveolar epithelial barrier with its huge surface in contact with air makes it one of the most important ports of entry in the human body. The alveolar barrier is constantly exposed to a variety of xenobiotics that are either cleared by the epithelium or taken up by the air–blood barrier. A portion of these molecules enter the bloodstream and are transported to other organs, which they may then affect. Although it was shown in different in vivo studies that the mechanical strain highly affects the uptake of such molecules,21 only little is known about the exact transport mechanisms taking place.22 The role of respiratory movements, in particular the dynamics taking place in the tight junctions, is unknown and requires the advent of novel devices enabling such investigations.19The lung-on-a-chip with a monolayer of bronchial epithelial cells was used to investigate the effects of physiological strain (10% linear) on the transport of specific molecules across the epithelial barrier. A monolayer of bronchial epithelial cells (16HBE14o−) was cultured on a fibronectin coated porous PDMS membrane (8 μm pores). 16HBE14o− cells have similar permeability properties to primary human alveolar epithelial cells,23 which makes them a good model for permeability studies. Furthermore, a monoculture of epithelial cells was used to model transport within the lung because endothelial cells have a much higher permeability24 and were therefore neglected in this model. The permeability assays were performed, either in static or in dynamic mode, with two different molecules dispensed simultaneously to the epithelial layer. The effect of physiological strain was assessed based on the transport of hydrophilic molecules (using FITC-sodium) and the epithelial barrier integrity (RITC-dextran). The experiments revealed that the permeability of small hydrophilic molecules is significantly (p < 0.005) increased if the cells are kept in a dynamic environment (n = 3) compared to a static environment (n = 6) (Fig. 3). The relative increase in transport is about 46% (5.68 ± 0.52% vs. 3.88 ± 0.47%). This increase cannot be explained by the increase in diffusive transport due to the stretching of the pores. In fact, the diffusive transport scales linearly with the pore surface area, which is 21% in the present case. In addition, the pore array is covered by a confluent layer of cells. On the other hand, the physiological strain did not affect the cell layer integrity, since no significant increase of the permeability was observed for RITC-dextran. This finding is supported by immunofluorescence images of the 16HBE14o− cells, which did not show any significant differences in morphology or in the tight junctions when comparing the static and dynamic conditions (ESI† Fig. S3).
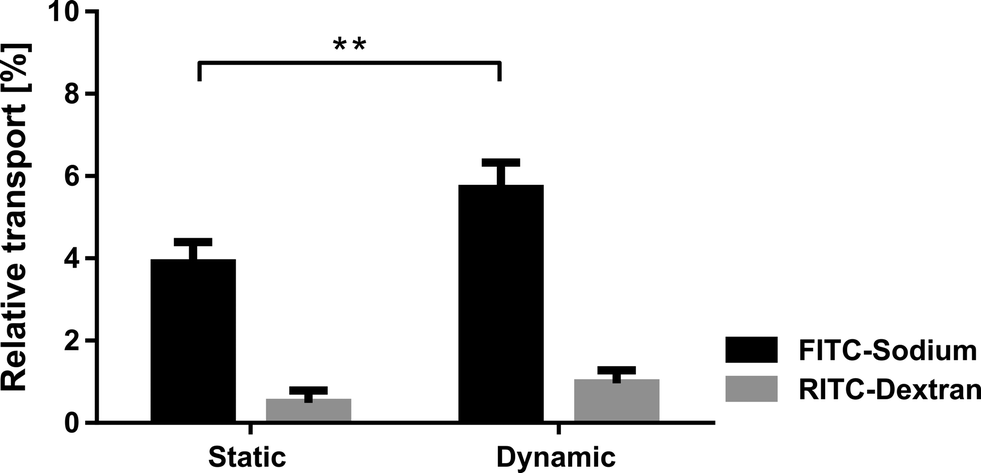 | ||
| Fig. 3 The effect of cyclic strain on the permeability of lung epithelial 16HBE14o− cells. The relative transport of a small molecule (FITC-sodium) across the monolayer significantly increased upon a physiological cyclic strain whereas the barrier integrity was not affected by the strain (no significant transport increase for RITC-dextran). Cyclic stretch was applied for 21 hours (dynamic, n = 3) and a control was kept under static conditions (static, n = 6) (mean values ± SD, p < 0.005). | ||
This experiment showed that the epithelial barrier permeability is significantly affected by the physiological strain produced in the lung-on-a-chip. These results are in good agreement with the increased permeability reported for hydrophilic solutes in an in vivo study upon distention of human lungs.25 The transport mechanism taking place is not fully understood. The most accepted theory is that due to the stretching of the cells, the intercellular junction pores are also stretched, which then leads to an enhanced permeability of hydrophilic molecules.26 These results illustrate the importance of investigating the effects of breathing motions on the epithelial barrier permeability. Such issues are of prime relevance to toxicology questions, as well as to inhalable formulations that are expected to be developed in the near future.27,28
Influence of mechanical stress on the activity of primary human pulmonary alveolar epithelial cells
Primary human pulmonary alveolar epithelial cells were selected using the common epithelial marker EpCAM (CD326). Following sorting, culture-expanded EpCAM+ cells demonstrated a cuboidal morphology and expressed markers that are typically found on type I and type II alveolar epithelial cells in the lower airway (Fig. 4A, C). Expanded EpCAM+ pHPAECs were then cultured on thin, porous and flexible membranes. The cells reached confluence after 24 h (Fig. 4B). Further, the cells could be cultured for at least 21 days on the membranes.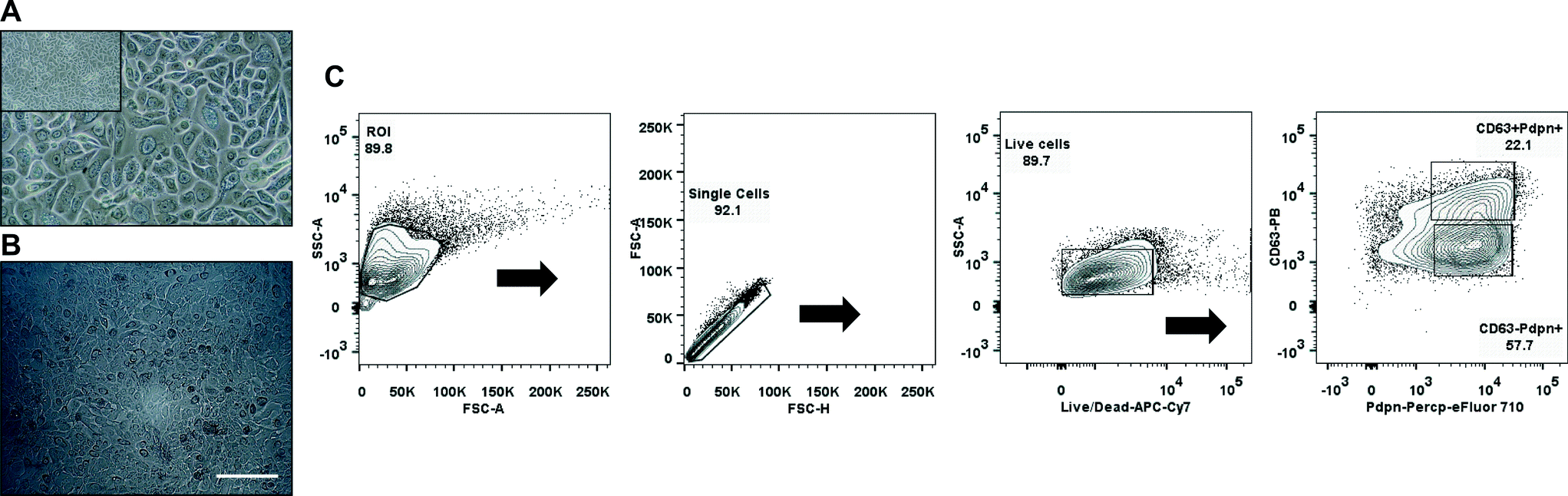 | ||
| Fig. 4 Primary human pulmonary alveolar epithelial cells. (A) Representative phase contrast image of culture expanded EpCAM+ pHPAECs. (B) Phase-contrast picture of a confluent monolayer of EpCAM+ pHPAECs cultured on the alveolar membrane (scale bar 200 μm). (C) Flow cytometric density plots showing the expression of both type I (Pdpn-Percp-eFluor 710) and type II (CD63-PB) surface markers on culture-expanded EpCAM+ cells. Pdpn, podoplanin; PB, Pacific Blue; ROI, region of interest. | ||
To determine the influence of strain on the metabolic activity of the pHPAECs, an alamar blue assay was performed with static cells and cells before and after stretching (Fig. 5A). Alamar blue measures the reductive potential of the cells and is a measure of both cell proliferation and cell viability. The fluorescence intensity of the cells under static conditions almost doubled in the first 24 h, suggesting that the cells are still proliferating (5239.5 ± 685.6 vs. 9391.7 ± 1513.3 a.u., n = 6). Similarly, the cells that were grown under static conditions followed by 24 h of stretching almost doubled their fluorescence intensity (5569.7 ± 655.6 vs. 10728.7 ± 1147.3 a.u., n = 6). Therefore, 10% linear cyclic stretch does not interfere with the proliferation of pHPAECs. Furthermore, cyclic stretch of this magnitude does not increase cell injury or cell death in primary human pulmonary alveolar epithelial cells. However, if the cells are stretched for 48 h the metabolic activity is significantly higher compared to the static control (9860.2 ± 471 vs. 8164.8 ± 831.4 a.u., n = 6).
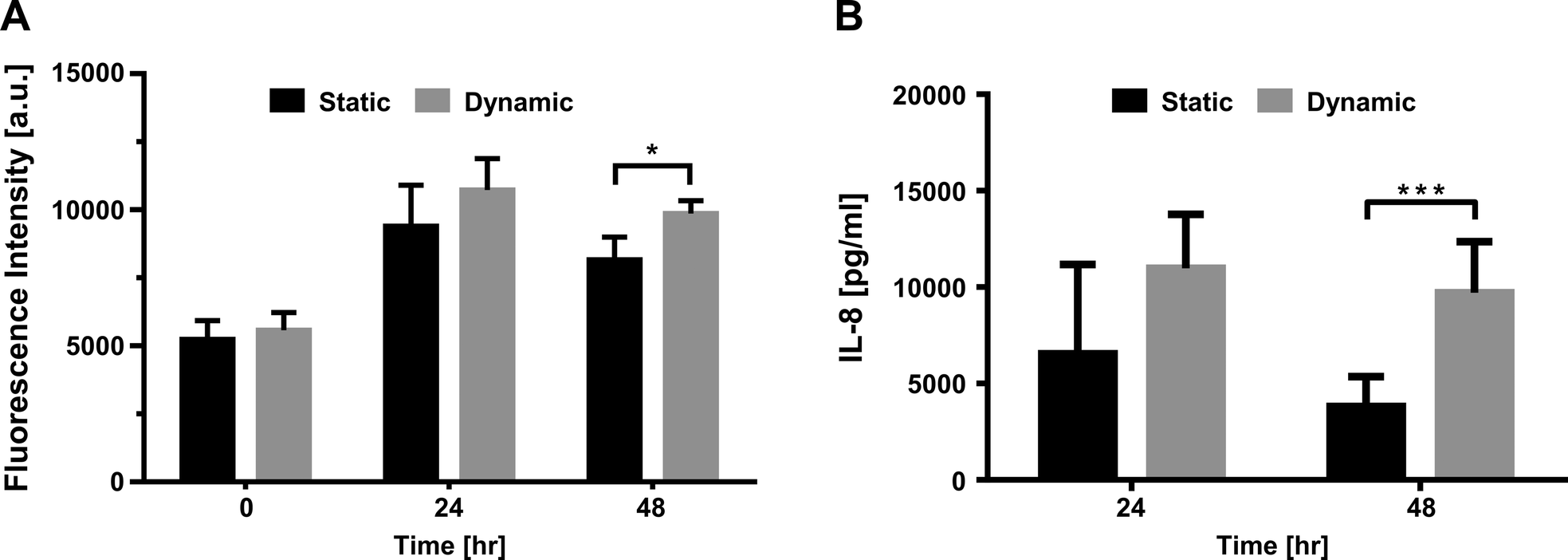 | ||
| Fig. 5 The effect of cyclic strain on the activity of primary human pulmonary alveolar epithelial cells. (A) The fluorescence intensity of alamar blue increases similarly in the first 24 h under both conditions, static and stretched (dynamic), showing that the proliferation of primary alveolar epithelial cells is not affected by cyclic stretch. However, after 48 h of stretching the metabolic activity is significantly higher in the stretched cells than in the static control (n = 6, mean values ± SD, *p < 0.05). (B) The IL-8 secretion of primary alveolar cells being stretched (dynamic) compared to that of a static control (static). After 24 hours of cyclic strain there is a tendency to an increased secretion of IL-8. After 48 hours the secretion of IL-8 is significantly higher compared to that of the static control (n = 3), mean values ± SD, p < 0.001. | ||
These findings are supported by the study of McAdams et al., in which they exposed the human alveolar-like adenocarcinomic cell line A549 to 16% linear strain.29 Similar to our findings, they did not observe any significant difference between cells stretched for 24 h and cells cultured under static conditions. However, after 48 h of stretching, the proliferation of the stretched cells was significantly enhanced.29 They also showed that cyclic strain with a magnitude of 16% linear elongation did not change the percentage of dead cells compared to a non-stretched control over 48 h. In contrast, several studies with primary rat ATII cells showed a significant increase in apoptosis and cell death even at linear stretch as low as 6%.30,31 However, with our primary human pulmonary alveolar cells we did not observe such a behavior. It is not known whether these differences in the effect of stretching on cell proliferation and viability are due to interspecies differences or not.
The supernatant from pHPAEC cells under static and dynamic conditions was further sampled at different time points and analyzed for their cytokine release patterns. Interleukin-8 (IL-8), a pro-inflammatory cytokine known to be upregulated in cell lines upon mechanical stretching,32,33 was measured by ELISA (Fig. 5B). After 2 h of stretching, no difference between dynamic and static conditions was observed (0.71 ± 0.08 ng ml−1vs. 0.58 ± 0.15 ng ml−1, n = 3). After 24 h of stretching, a higher tendency of IL-8 secretion was seen compared to the static control (10.97 ± 2.8 ng ml−1vs. 6.54 ± 4.64 ng ml−1, n = 3). However, after 48 h of stretching, the IL-8 concentration found in the supernatant of the stretched cells was 2.5× higher than the IL-8 concentration in the static control (9.7 ± 2.65 ng ml−1vs. 3.81 ± 1.55 ng ml−1). The knowledge of the effect of stretch on IL-8 production in the lung is controversial and restricted to A549 cells only. Two studies showed that IL-8 secretion is increased in A549 already after 5 min to 4 h with low linear stretch of 2% and 5%, respectively.34,35 In contrast, several studies did not see this increase in IL-8 production in A549 cells in the first few hours even with a stretch magnitude of 10%.36,37 In our study, we did not observe an increase after 2 h in primary human pulmonary alveolar cells either. Jafari et al. only found higher IL-8 production when stretching the cells with a linear elongation of 15%.36 Ning & Wang further showed stretch magnitude dependent IL-8 secretion, which does not depend on the stretch frequency.35 The only study looking at longer periods of stretching (up to 48 h) observed an increase of IL-8 production in A549 when stretched at 30% linear stretch, but not when stretched at 20% linear stretch.33 To our knowledge, our study shows for the first time that over longer periods of stretching, IL-8 secretion is enhanced in primary human pulmonary alveolar epithelial cells.
Conclusion
To better model the in vivo conditions of the biophysical and cellular microenvironment, new and more accurate in vitro models are needed. Unlike standard cell cultures, organs-on-chips are widely seen as promising candidates capable of predicting human responses to drugs.This bioinspired lung-on-a-chip mimics the microenvironment of the lung parenchyma by reproducing the thin alveolar barrier constantly exposed to respiratory movements. A flexible, thin and porous membrane, on which a co-culture of epithelial and endothelial cells is cultured, is cyclically deflected by a micro-diaphragm, whose function is similar to that of the in vivo diaphragm. The effects of breathing movements were investigated using a bronchial epithelial cell line as well as primary human lung epithelial cells from patients. With this device we could demonstrate that the mechanical stress profoundly and significantly affects the epithelial barrier permeability. In addition, the metabolic activity of primary human pulmonary alveolar epithelial cells cultured in dynamic mode was found to be significantly higher than that of cells cultured in static mode. Similarly, a significantly higher production of the inflammation marker IL-8 was found in these cells. To the best of our knowledge, this is the first time that the effects of mechanical strain on healthy human primary cells derived from patients have been investigated.
Although the main challenge of organs-on-chip systems is how to best reproduce the in vivo conditions, a second challenge is how to make such a device as robust and reproducible as possible. This aspect is central in view of a wider acceptance of those systems by cell biologists, toxicologists and pharmacologists. The strategy followed during the development of the present device was therefore aimed at designing a system that would combine ease of handling (e.g. compatible with a multi-pipette), reproducible control of the cultured conditions (the number of cells cultured on the membrane and the defined level of mechanical strain) and recapitulation of the main in vivo features. Such systems are widely expected to better predict human responses to drugs and present a great possibility of improving the selection of drug candidates early in the drug discovery process.
Acknowledgements
The authors are very grateful to the Gebert Rüf Stiftung (GRS-066/11), the Swiss Commission for the Technology and Innovation (CTI 15794.1 PFLS-LS), the Swiss National Science Foundation (315230_141127) and the Lungenliga Bern for their generous financial support. They would also like to thank Prof. Dr. Robert Rieben (University of Berne) for generously providing primary human umbilical vein endothelial cells (pHUVEC). We thank Dr. Marco Alves and Aline Schoegler for providing advice and materials for ELISA. We also acknowledge the Flow Cytometry Core of the Department of Clinical Research of the University of Berne. Images were acquired using equipment supported by the Microscopy Imaging Center of the University of Berne. A patent on the microfluidic device described is pending.References
- F. Pammolli, L. Magazzini and M. Riccaboni, Nat. Rev. Drug Discovery, 2011, 10, 428–438 CrossRef CAS PubMed.
- H. Olson, G. Betton, D. Robinson, K. Thomas, A. Monro, G. Kolaja, P. Lilly, J. Sanders, G. Sipes, W. Bracken, M. Dorato, K. Van Deun, P. Smith, B. Berger and A. Heller, Regul. Toxicol. Pharmacol., 2000, 32, 56–67 CrossRef CAS PubMed.
- J. H. Sung, M. B. Esch, J. Prot, C. J. Long, A. Smith, J. J. Hickman and M. L. Shuler, Lab Chip, 2013, 13, 1201–1212 RSC.
- Y. Zheng, H. Fujioka, S. Bian, Y. Torisawa, D. Huh, S. Takayama and J. B. Grotberg, Phys. Fluids, 1994, 2009(21), 71903 Search PubMed.
- N. J. Douville, P. Zamankhan, Y.-C. Tung, R. Li, B. L. Vaughan, C.-F. Tai, J. White, P. J. Christensen, J. B. Grotberg and S. Takayama, Lab Chip, 2011, 11, 609–619 RSC.
- D. Huh, H. Fujioka, Y.-C. Tung, N. Futai, R. Paine, J. B. Grotberg and S. Takayama, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18886–18891 CrossRef CAS PubMed.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- J. C. McDonald and G. M. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS PubMed.
- C. P. Ng and M. A. Swartz, Am. J. Physiol., 2003, 284, H1771–H1777 CAS.
- C. M. Waters, E. Roan and D. Navajas, Compr. Physiol., 2012, 2, 1–29 Search PubMed.
- P. Gehr, M. Bachofen and E. R. Weibel, Respir. Physiol., 1978, 32, 121–140 CrossRef CAS.
- J. B. West, Adv. Physiol. Educ., 2008, 32, 177–184 CrossRef PubMed.
- V. D. Varner and C. M. Nelson, Development, 2014, 141, 2750–2759 CrossRef CAS PubMed.
- T. Mammoto, A. Mammoto and D. E. Ingber, Annu. Rev. Cell Dev. Biol., 2013, 29, 27–61 CrossRef CAS PubMed.
- M. Plataki and R. D. Hubmayr, Expert Rev. Respir. Med., 2010, 4, 373–385 CrossRef PubMed.
- M. E. Blaauboer, T. H. Smit, R. Hanemaaijer, R. Stoop and V. Everts, Biochem. Biophys. Res. Commun., 2011, 404, 23–27 CrossRef CAS PubMed.
- B. Suki, D. Stamenović and R. Hubmayr, Compr. Physiol., 2011, 1, 1317–1351 Search PubMed.
- D. J. Tschumperlin and J. M. Drazen, Annu. Rev. Physiol., 2006, 68, 563–583 CrossRef CAS PubMed.
- E. Steed, M. S. Balda and K. Matter, Trends Cell Biol., 2010, 20, 142–149 CrossRef CAS PubMed.
- C. M. Niessen, D. Leckband and A. S. Yap, Physiol. Rev., 2011, 91, 691–731 CrossRef CAS PubMed.
- G. Taylor, Adv. Drug Delivery Rev., 1990, 5, 37–61 CrossRef CAS.
- H. Smyth and A. Hickey, Controlled Pulmonary Drug Delivery, 2011, p. 31 Search PubMed.
- B. Forbes and C. Ehrhardt, Eur. J. Pharm. Biopharm., 2005, 60, 193–205 CrossRef CAS PubMed.
- S. Braude, K. B. Nolop, J. M. Hughes, P. J. Barnes and D. Royston, Am. Rev. Respir. Dis., 1986, 133, 1002–1005 CAS.
- J. D. Marks, J. M. Luce, N. M. Lazar, J. N. Wu, A. Lipavsky and J. F. Murray, J. Appl. Physiol., 1985, 59, 1242–1248 CAS.
- G. R. Mason, A. M. Peters, E. Bagdades, M. J. Myers, D. Snook and J. M. Hughes, Clin. Sci., 2001, 100, 231–236 CrossRef CAS.
- M. Haghi, H. X. Ong, D. Traini and P. Young, Pharmacol. Ther., 2014, 144, 235–252 CrossRef CAS PubMed.
- Z. Liang, R. Ni, J. Zhou and S. Mao, Drug Discovery Today, 2014, 1–10 Search PubMed.
- R. M. McAdams, S. B. Mustafa, J. S. Shenberger, P. S. Dixon, B. M. Henson and R. J. DiGeronimo, Am. J. Physiol., 2006, 291, L166–L174 CAS.
- D. Tschumperlin and S. Margulies, Am. J. Physiol., 1998, 275, L1173–L1183 CAS.
- S. P. Arold, E. Bartolák-Suki and B. Suki, Am. J. Physiol., 2009, 296, L574–L581 CAS.
- D. Quinn, Chest, 1999, 116, 89S CrossRef CAS.
- N. E. Vlahakis, M. A. Schroeder, A. H. Limper and R. D. Hubmayr, Am. J. Physiol., 1999, 277, L167–L173 CAS.
- L.-F. Li, B. Ouyang, G. Choukroun, R. Matyal, M. Mascarenhas, B. Jafari, J. V. Bonventre, T. Force and D. A. Quinn, Am. J. Physiol., 2003, 285, L464–L475 CAS.
- Q. Ning and X. Wang, Respiration, 2007, 74, 579–585 CrossRef CAS PubMed.
- B. Jafari, B. Ouyang, L.-F. Li, C. A. Hales and D. A. Quinn, Respirology, 2004, 9, 43–53 CrossRef PubMed.
- A. Tsuda, B. K. Stringer, S. M. Mijailovich, R. A. Rogers, K. Hamada and M. L. Gray, Am. J. Respir. Cell Mol. Biol., 1999, 21, 455–462 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: A movie of real-time stretching of primary cells, chip handling, strain characterization and immunofluorescence images. See DOI: 10.1039/c4lc01252f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |