
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-Amplified Density Assays, (MADAs), including a Density-Linked Immunosorbent Assay (DeLISA)†
Anand Bala
Subramaniam
a,
Mathieu
Gonidec
a,
Nathan D.
Shapiro
a,
Kayleigh M.
Kresse
a and
George M.
Whitesides
*abc
aDepartment of Chemistry & Chemical Biology, Harvard University, 230 Mallinckrodt Bldg., 12 Oxford St., Cambridge, MA 02138, USA. E-mail: gwhitesides@gmwgroup.harvard.edu; Tel: +1 617 495 9430
bWyss Institute for Biologically Inspired Engineering, Harvard University, 230 Mallinckrodt Bldg., 12 Oxford St., Cambridge, MA 02138, USA
cThe Kavli Institute for Bionano Science, Harvard University, 230 Mallinckrodt Bldg., 12 Oxford St., Cambridge, MA 02138, USA
First published on 4th December 2014
Abstract
This paper reports the development of Metal-amplified Density Assays, or MADAs – a method of conducting quantitative or multiplexed assays, including immunoassays, by using Magnetic Levitation (MagLev) to measure metal-amplified changes in the density of beads labeled with biomolecules. The binding of target analytes (i.e. proteins, antibodies, antigens) to complementary ligands immobilized on the surface of the beads, followed by a chemical amplification of the binding in a form that results in a change in the density of the beads (achieved by using gold nanoparticle-labeled biomolecules, and electroless deposition of gold or silver), translates analyte binding events into changes in density measureable using MagLev. A minimal model based on diffusion-limited growth of hemispherical nuclei on a surface reproduces the dynamics of the assay. A MADA – when performed with antigens and antibodies – is called a Density-Linked Immunosorbent Assay, or DeLISA. Two immunoassays provided a proof of principle: a competitive quantification of the concentration of neomycin in whole milk, and a multiplexed detection of antibodies against Hepatitis C virus NS3 protein and syphilis T. pallidum p47 protein in serum. MADAs, including DeLISAs, require, besides the requisite biomolecules and amplification reagents, minimal specialized equipment (two permanent magnets, a ruler or a capillary with calibrated length markings) and no electrical power to obtain a quantitative readout of analyte concentration. With further development, the method may be useful in resource-limited or point-of-care settings.
Introduction
Immunoassays that do not require electricity or specialized equipment, that are capable of providing quantitative readouts, and that can detect multiple analytes in a single sample (i.e. multiplexed assays), are among the objectives of the field of point-of-care (POC) diagnostics.1 Low costs and simple procedures are especially important for assays intended for resource-limited settings.2,3 Polymeric particles or beads, used initially to improve the sensitivity of flocculation assays,4 are useful alternatives to macroscopic planar surfaces (e.g. microtiter plates) for immobilizing biomolecules for a number of types of immunoassays.5,6 The high ratio of surface-to-volume,7 the improved kinetics of mass transport,8 the ability to multiplex,6,9–11 the ability to separate paramagnetic beads with bound biomolecules magnetically,12–14 and the development of mechanisms to automate steps by using microfluidics14,15 or centrifugation16 has led to successful commercial adoption17–19 of bead-based assays, and to their deployment in the field.20Bead-based assays are typically read out by fluorimetry,6 chemiluminescence,21 or colorimetry.22,23 These methods rely on measuring the intensity of light, and require electronics and signal processing to obtain a quantitative readout. Qualitative colorimetric readouts that use the naked eye are subjective, and thus subject to error. They may also be biased due to variations in the intensity or the quality of the ambient light.21 Alternatively, an electrochemical method, which requires electrical power, can be used to read out a bead-based assay quantitatively.20 A fundamentally new technique of reading bead-based assays – one that provides an alternative to electrochemical methods, and that does not measure the intensity of light – has the potential to be useful, especially if it offers a means of quantitation that does not require electronics or electricity.
This paper reports a bead-based assay – a Metal-amplified Density Assay (MADA) – that is based on detecting a change in the density of a bead; it is read by monitoring the levitation height of polystyrene beads presenting biomolecules (e.g. proteins, antigens, antibodies, or nucleic acids) on their surfaces in a Magnetic Levitation (MagLev) device, following a step in which analyte binding-events cause electroless deposition of metal on the bead.
The MagLev device consists of two permanent magnets arranged coaxially with like-poles facing.24 Due to opposing buoyant and magnetic forces, diamagnetic beads – when suspended in a paramagnetic solution in the MagLev device – levitate at a static ‘levitation height’ against gravity.24–34 The binding of target analytes (i.e. proteins, antibodies, antigens) to their cognate ligands immobilized on the surface of the beads causes a change in density that is too small to be detected conveniently. Chemical amplification of the binding in a form that changes the density of the bead (achieved by using gold nanoparticle-labeled proteins and electroless deposition of gold or silver on these nanoparticles), translates analyte binding events into measurable changes in the levitation height of the bead. This change in levitation height (the signal of the assay) depends on the concentration of analyte that was present in the sample.
To understand the processes that lead to the changes in density, we examined a model system comprising of gold-labeled streptavidin (as analyte) and polystyrene beads with biotin on their surfaces. A minimal mathematical model, based on the diffusion-limited growth of independent hemispherical nuclei (i.e. the gold nanoparticles) on a surface, adequately described the kinetics of the change in density. Measurements of the number of adsorbed gold-labeled streptavidin molecules on the surfaces of the beads using inductively-coupled plasma mass spectrometry (ICP-MS) confirmed matched the predictions of the minimal model. The addition of free gold nanoparticles in solution modified the kinetics of deposition of gold on the beads, and suggests a design for these assays in which the results would be independent of the time of development.
We use the description Density-Linked Immunosorbent Assay, or DeLISA, when those assays are performed with antigen and antibodies. As proof-of-concept experiments, we chose two different immunoassay formats – a competitive immunoassay to quantify the concentration of neomycin (an aminoglycoside antibiotic) in whole milk, and a multiplexed indirect immunoassay (including a positive and negative control) to detect antibodies against Hepatitis C virus NS3 protein and syphilis T. pallidum p47 protein in serum.
Background
We have described MagLev in other papers.24,28,32 Finite-element simulations based on the parameters (dimensions, strength of the magnetic field, magnetic susceptibility of the solution) of this device show that, to a good approximation, the gradient of the magnetic field between the magnets is linear and the magnetic field is zero at the center of the device.18 In this configuration, eqn (1) describes the relation between the density and the levitation height of the center of volume of homogenous spherical objects (i.e. beads) in the MagLev device.24,28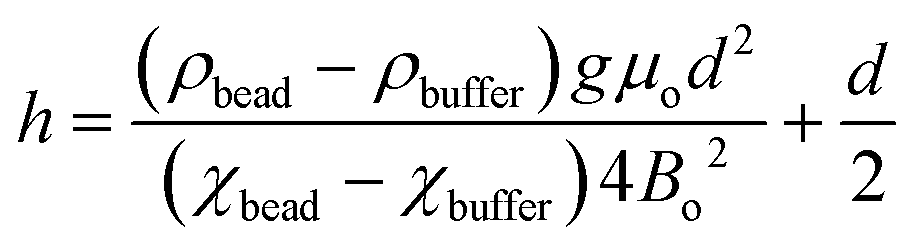 | (1) |
In this equation, ρbead (kg m−3) and ρbuffer (kg m−3) are the densities of the bead and the buffer, χbead (χbead ~ −1 × 10−5 for diamagnetic objects) and χbuffer (χbuffer ~ 1.8 × 10−4 for 1.00 M MnCl2 (ref. 24)) are the magnetic susceptibilities (dimensionless) of the bead and the buffer, d (m) is the separation between the magnets, Bo (T) is the magnitude of the magnetic field at the surface of the magnets, μ0 = 4π × 10−2 (N A−2) is the magnetic permeability of free space, and g (m s−2) is the acceleration due to gravity.
We have used MagLev to measure the binding and release of bovine carbonic anhydrase (BCA) to (aryl)sulfonamide ligands on porous beads.30,35 In one version of these assays, BCA diffuses into the beads and binds to the immobilized ligands. The accumulation of protein in the beads due to this process results in a change in their density and levitation height. Although the use of gel-immobilized ligands in MagLev provides a label-free method to detect the concentration of protein in solution, the method worked best with small (<60 kDa) proteins having high diffusion constants in the pores of the beads, and required relatively long reaction times (hours to days). These limitations made the method, without modification, unsuitable for most applications of immunoassay. In this paper, we use a chemical reaction on the surface of non-porous beads to measure the concentration of proteins in solution. As we describe in the next section, the general principles of DeLISA use some procedures developed for existing immunoassays,21 and thus allow the adoption of established protocols and reagents.
Results and discussion
Principles of Metal-amplified Density Assays (MADAs)
Fig. 1 shows schematically the principles of a Metal-amplified Density Assay (MADA). Like other bead-based assays, MADAs require that ligands be covalently immobilized on the surfaces of the beads.21 The beads with bound ligands are then exposed to a sample of interest (primary incubation). The cognate analyte, if present in the sample, binds to the ligands on the beads. To obtain analyte-dependent electroless deposition of metal (typically gold, AuMADA, shown in main text, and silver, AgMADA, shown in ESI†), a catalyst (metal nanoparticles, e.g. gold nanoparticles) must be attached to the analyte bound to the surface of the bead. For a DeLISA, this objective is achieved after the primary incubation step by exposing the beads to secondary antibodies conjugated to gold nanoparticles. We demonstrate this procedure using immunoassays, but the same principles should apply to small molecules, nucleic acids36,37 or sugars.38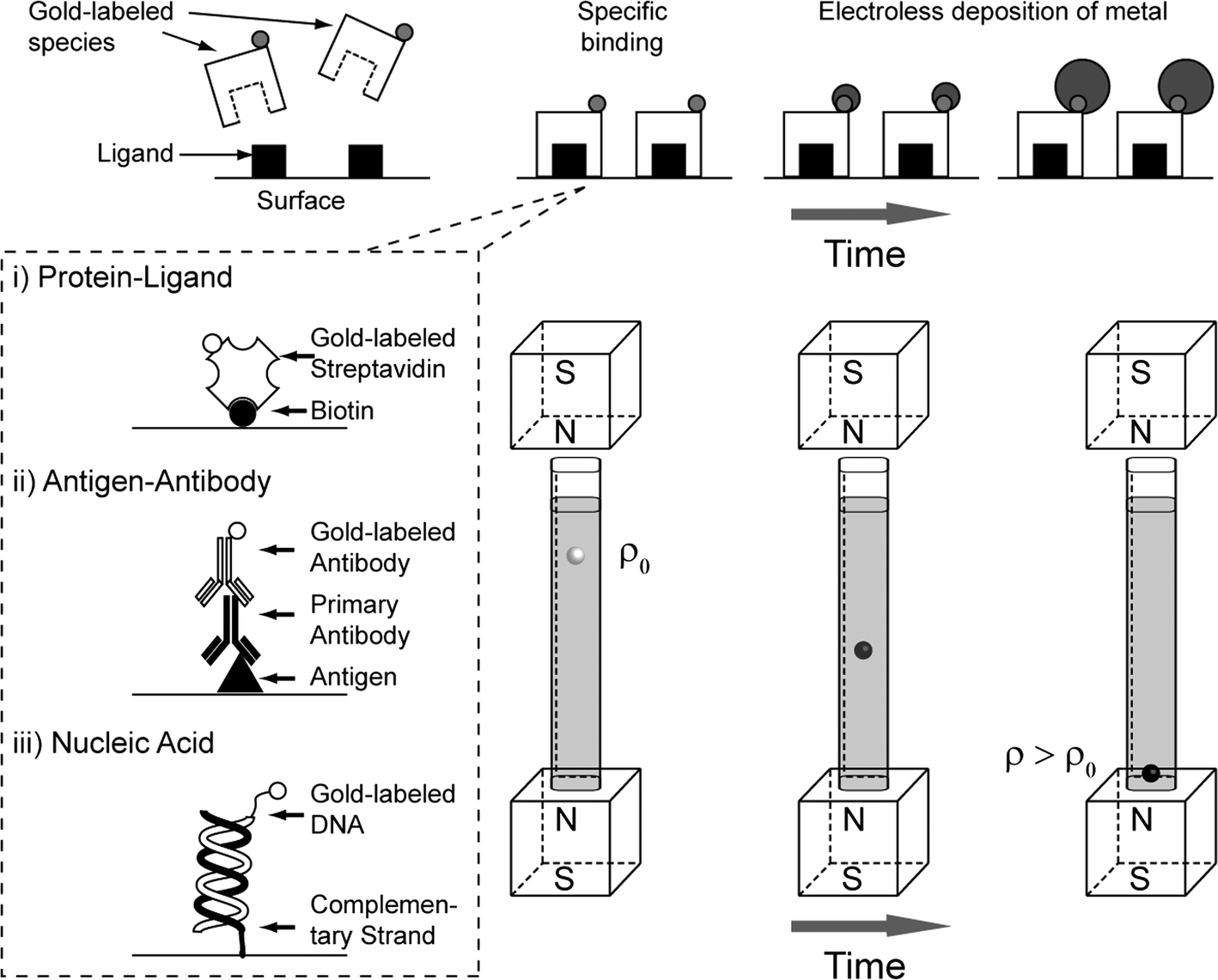 | ||
| Fig. 1 Schematic illustrating the principles of a Metal-amplified Density Assay (MADA). Gold-labeled analyte in the sample bind to ligands immobilized on the surface of the bead. The bead is placed in a developing buffer and then placed in a MagLev device. The gold nanoparticles catalyze the deposition of metallic gold from solution, and causes the growth of the nanoparticles. The increase in the density of the bead causes the bead to decrease in levitation height in the device. The rate of change of the levitation height of the beads should depend on the number of gold-linked analytes that are bound to its surface. The schematics in the dotted rectangular box shows examples of MADA with i) gold-labeled proteins binding to their ligand, ii) gold-labeled secondary antibodies bound to primary antibodies and antigens (a Density-Linked Immunosorbent Assay, DeLISA), and iii) gold-labeled DNA bound to a DNA strand with a complementary sequence. We implemented, i) and ii) in this paper. | ||
After the primary and secondary incubation steps, the beads are then loaded into a capillary containing an aqueous solution of a paramagnetic salt (MnSO4) dissolved in a commercial gold amplification solution (the developing buffer). The gold amplification solution, which contains gold ions, a reducing agent, and various proprietary stabilizers, causes gold ions in solution to be reduced to elemental gold on the surface of the gold nanoparticles.39 Gold and silver amplification solutions are used, conventionally, to increase the size, and hence the scattering cross-section, of gold nanoparticle labels in tissue slices for optical and electron microscopy,39–41 and to conduct assays with optical readouts.42–44 We reasoned that the deposition of elemental gold onto the surface of a polymeric bead would also increase the density of the bead.
The rate at which gold is deposited on the bead (and hence the change in density and levitation height of the bead) depends on the number of bound gold nanoparticles on the surface of the bead, and the kinetics of the electroless deposition. This dependence allows a density-based assay for the quantitation of a target analyte in the sample.
A Gold Metal-amplified Density Assay, AuMADA, for streptavidin as a validation of principle
To test the concept of a MADA, we designed a model assay that quantified the concentration of gold-labeled streptavidin in a buffer solution using electroless deposition of gold. We used polystyrene beads with biotin covalently immobilized on their surface. We chose this model system because: i) the reagents were commercially available and well-characterized, ii) the binding of gold-labeled streptavidin to antibodies conjugated with biotin is a standard mechanism of detection in many immunoassays,21 iii) the binding of biotin to streptavidin in solution is well-characterized,45–47 and the binding of biotin to streptavidin-modified surfaces has been studied.48–50 The dissociation constant for biotin and streptavidin in solution is Kd ~ 10−14 M−1; this reaction is generally considered a good model for irreversible binding. On a surface, binding is claimed to be less favorable – conjugation of streptavidin with fluorophores, DNA, or solid surfaces increases the Kd by about 4–5 orders of magnitude.48,49 In contrast, antibodies have Kd that range from 10−6 M−1 to 10−12 M−1.21 Thus, the dissociation constant of the gold-labeled streptavidin is close to that expected for a tight-binding antigen–antibody pair. Antibodies with appropriate Kd are selected for heterogeneous immunoassays, as part of the design of the assay, to ensure that the antibodies do not desorb significantly during wash steps. Generally, antibodies with Kd above 10−8 M−1 are unsuitable for immunoassay,21 due to the rapid unbinding of the antibodies during wash steps.Fig. 2A shows time-lapse photographs of a single bead in the developing buffer in the MagLev device. The bead had previously been exposed to a buffer solution containing 3000 pM gold-labeled streptavidin (see Materials and methods for the detailed procedure). The bead, which was initially colorless, turned black, and then golden, and decreased in levitation height before reaching the bottom of the capillary after 21 minutes. The change in levitation height of the bead corresponded to an increase in the density of the bead from 1.050 g cm−3 to over 1.055 g cm−3. A bead previously exposed to a 30 pM solution of gold-labeled streptavidin also changed color and decreased in levitation height. The decrease in levitation height, however, was slower (Fig. 2B): at 21 minutes its levitation height was 27 mm and after one hour its levitation height was 7 mm, i.e. 3 mm above the bottom of the capillary.
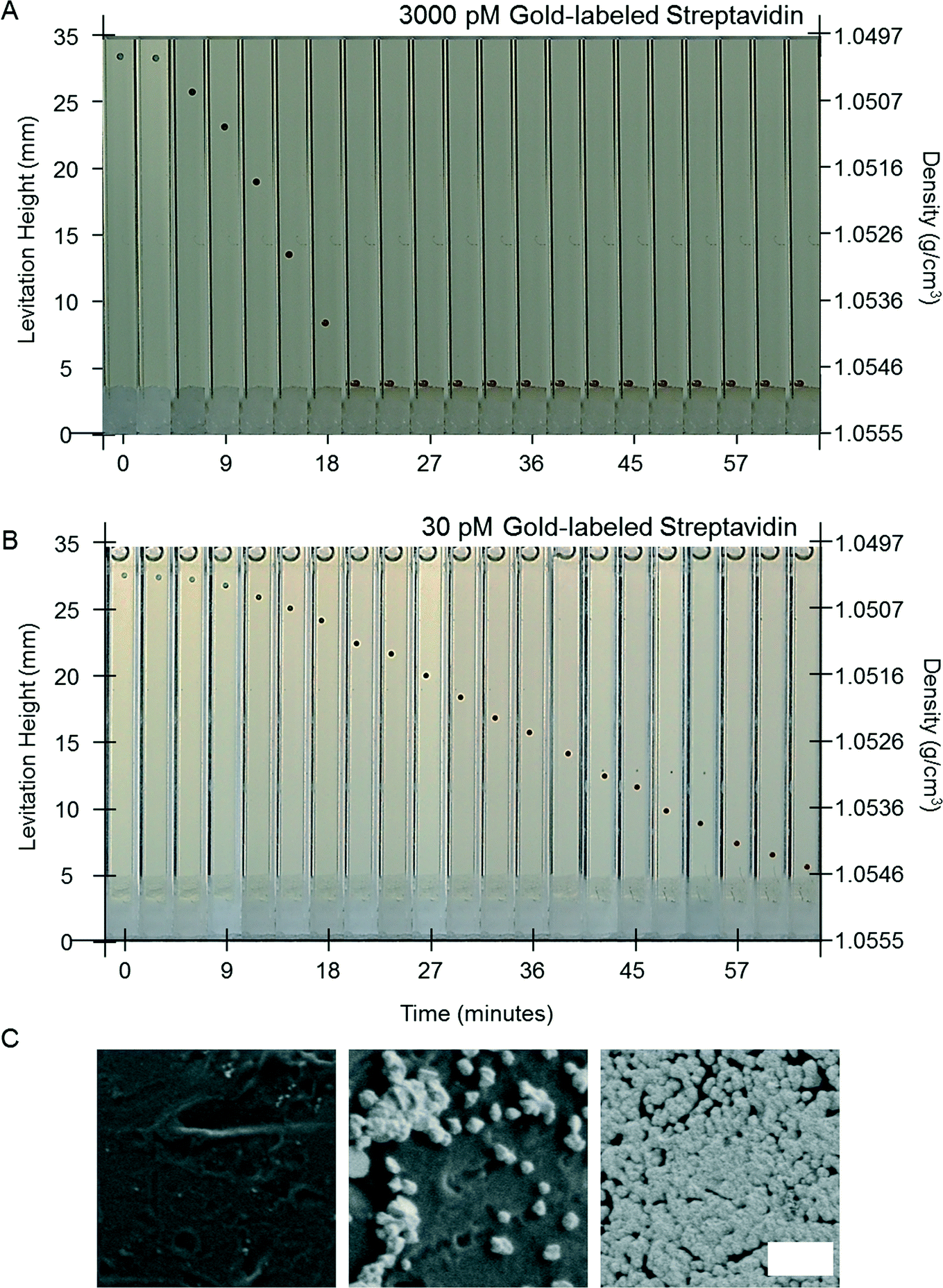 | ||
| Fig. 2 A MADA to detect the concentration of gold-labeled streptavidin. A) Time lapse images of a biotin-labeled polystyrene bead in the developing buffer in the MagLev device. The bead was previously exposed to a sample containing 3000 pM of gold-labeled streptavidin. The bead, which was initially colorless, changed color and decreased in levitation height, reaching the bottom of the capillary at 21 minutes. B) A bead that was previously exposed to 30 pM of gold-labeled streptavidin was still 3 mm above the bottom of the capillary after 1 hour in the developing buffer. The white substance at the bottom of the capillaries was Tacky Wax that was used to seal the capillaries. C) SEM images of the surface of the bead (exposed to 3000 pM gold-labeled streptavidin) at t = 0, 30, and 60 minutes. The gold nanoparticles, which were initially too small to be observed, grew and formed an almost continuous film of gold at 1 hour. Scale bar 500 nm. | ||
The change in the color of the beads is characteristic of the growth of the nanoparticles on the surfaces of the beads. The appearance of the beads in reflection depends of the ratio of their scattering cross-section and their optical density in the visible spectrum. Despite their strong optical density in the visible spectrum, the initial 10 nm gold nanoparticles immobilized on the polymeric beads have a small scattering cross-section, and therefore have a small albedo, which makes them nearly undetectable with the naked eye (hence the beads appear colorless). As they grow in size, they maintain a high optical density in the visible and their cross-section increases which makes the beads appear darker and darker. After some time, a continuous film is formed that does not significantly absorb in the visible and the scattering of the gold-covered beads is reminiscent of that of bulk gold (i.e. the beads appear golden).
Scanning electron micrographs of the surface of a bead exposed to 3000 pM of gold-labeled streptavidin at 0, 30 and 60 minutes confirmed that the nanoparticles on the surfaces of the beads, which were not visible prior to amplification, increased in size and eventually coalesced to form a film (Fig. 2C). Both these observations confirm that the amount of gold that was deposited on the surface of the bead increased with time in the developing buffer.
Fig. 3A shows the evolution with time of the levitation height of the beads exposed to 3000 pM, 300 pM, 30 pM, and 3 pM of gold-labeled streptavidin. The rate of change of the levitation height of the beads depended on the concentration of the gold-labeled streptavidin in solution. This dependence is seen clearly in the dose-response plot in Fig. 3B which shows the levitation height of beads (averaged over seven replicates) plotted against the concentration of gold-labeled streptavidin in the samples, at 30 and 60 minutes.
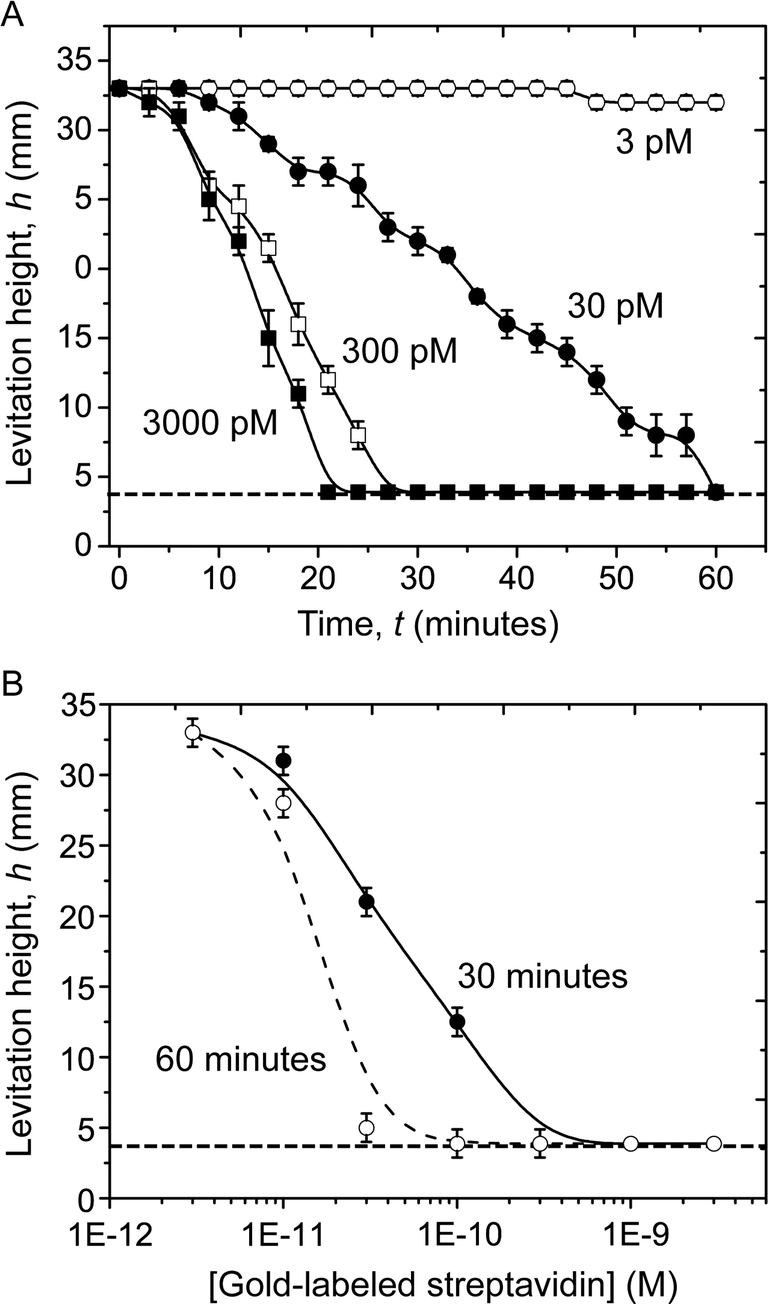 | ||
| Fig. 3 Change in levitation height of the beads as a function of gold-labeled streptavidin. A) Plots of the levitation height of the beads exposed to 3000 pM (■, filled squares), 300 pM (□, open squares), 30 pM (●, filled circles) and 3 pM (○, open circles) of gold-labeled streptavidin versus time. The black continuous lines are guides to the eyes. B) Dose-response plot of the levitation height of beads (averaged over seven replicates) against the concentration of gold-labeled streptavidin in the samples, at 30 (●, filled circles) and 60 minutes (○, open circles). The dashed and solid lines are guides to the eye. The dashed horizontal line indicates the height of the wax plug at the bottom of the capillary on which the beads come to rest. | ||
A model for MADA
The decrease in levitation height of the beads is due to the increase in their density. In the developing buffer, the 10 nm diameter gold nanoparticles that are adsorbed on the surface of the bead – due to analyte binding events – grow in size with time as gold deposits (Fig. 4A). As they increase in size, they eventually touch and form a continuous film on the surface of the bead (Fig. 4A). The electrochemical deposition of metal from solution onto an electrode has been studied extensively.51–54 The initial stages of the process of deposition is typically modeled as the nucleation and growth of hemispherical nanoparticles (nuclei) on the electrode surface. The kinetics of deposition and growth in an electroless deposition system, such as in a MADA, is expected to be more complicated since the surfaces of the nanoparticles serve as both anode and cathode and as a catalyst for the deposition.55 Chemicals such as stabilizers and growth modulators likely further complicate the system.39,55,56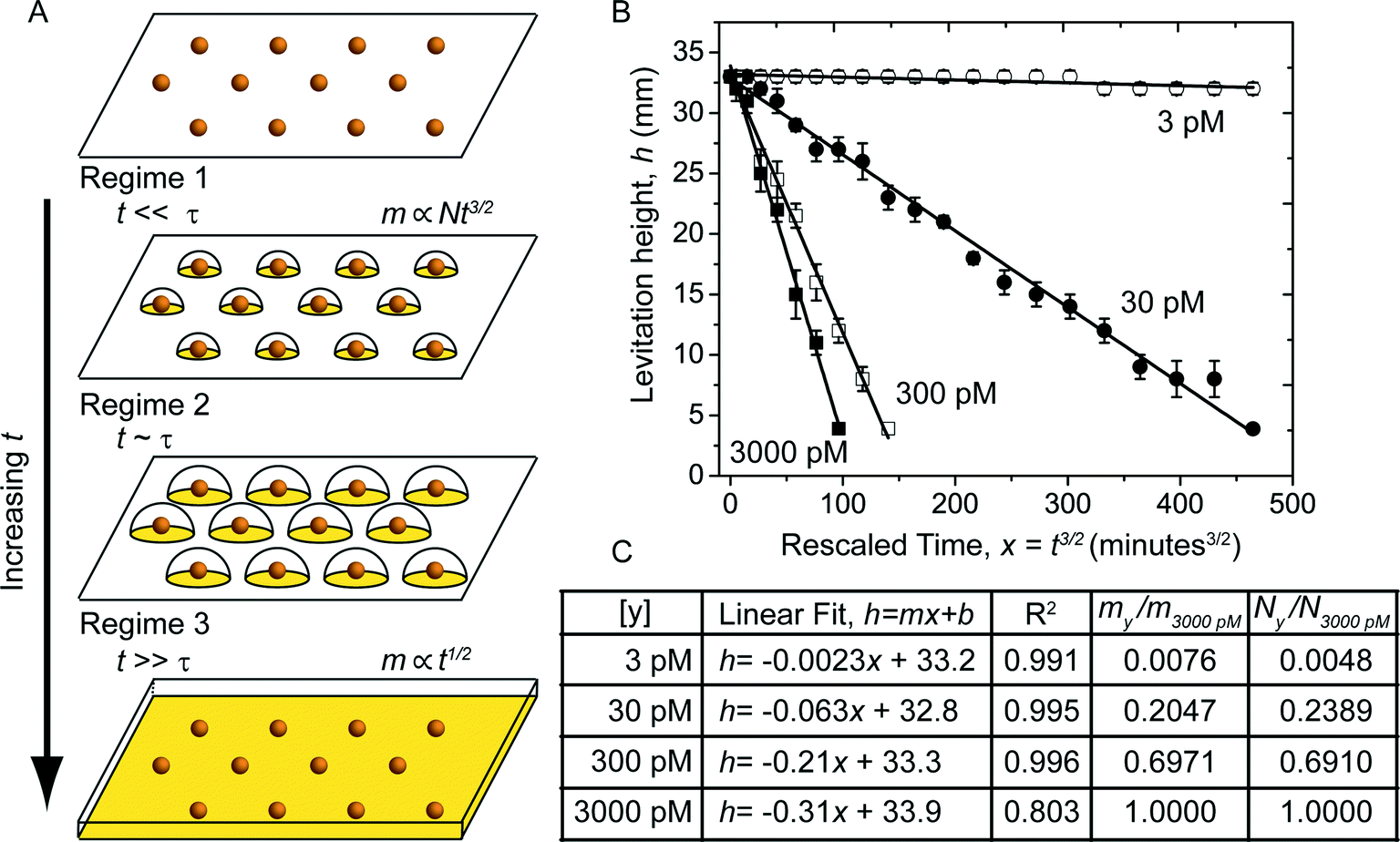 | ||
| Fig. 4 Model of a MADA. A) Schematic of the three growth regimes of the gold nanoparticles on the surface of the bead in the developing buffer. When t ≪ τ (regime 1), the nanoparticles are far apart and the mass of gold on the bead, m ∝ Nt3/2. When t ~ τ (regime 2) m can be obtained by integrating eqn (2) numerically. When t ≫ τ (regime 3) the gold has fused to form a continuous layer; m ∝ t1/2. B) Plot of the levitation height, h (Fig. 3A) versus rescaled time, x = t3/2. The black lines are linear fits to the data. C) Table showing the concentration of free gold-labeled streptavidin that the beads were exposed to ([y]), the coefficients of the linear fit, and the values of the coefficient of determination, R2. The fits are good, indicating that the MADA can be modeled in the regime where t ≪ τ. The two leftmost columns report the ratio of the slope relative to the slope for the 3000 pM line, and the ratio of the number of nanoparticles on the surface of the beads relative to the number of nanoparticles for beads exposed to 3000 pM of gold-labeled streptavidin. The values are in good agreement. | ||
Eqn (2) describes the rate of deposition of metal, dm/dt (kg s−1), on a flat (electrode) surface of area S (m2) due to the diffusion-limited growth of N surface-adsorbed hemispherical nuclei (nanoparticles) per unit area (number m−2) with an initial radius, r = 0 (m) at t = 0 (s).51–53
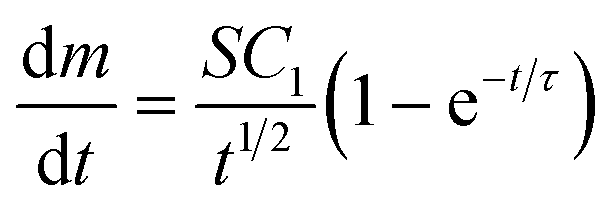 | (2) |
In this equation, C1 is a constant that depends on experimental conditions and 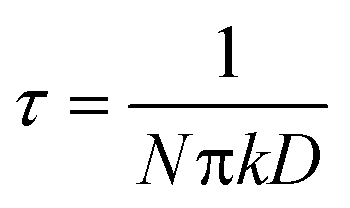 is the ‘characteristic time’ of the process, D is the diffusion coefficient (m2 s−1), and k is a unitless numerical constant (see ESI† for full discussion and derivation). Integrating eqn (2) with respect to time gives the mass of gold deposited on the bead, i.e.
is the ‘characteristic time’ of the process, D is the diffusion coefficient (m2 s−1), and k is a unitless numerical constant (see ESI† for full discussion and derivation). Integrating eqn (2) with respect to time gives the mass of gold deposited on the bead, i.e.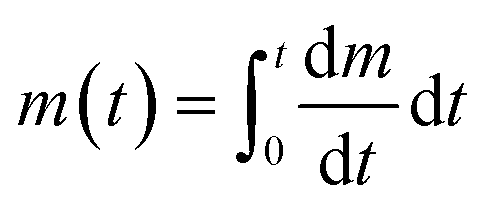 . This integral is readily solved numerically for all t.
. This integral is readily solved numerically for all t.
Simplified analytical expressions of eqn (2), obtained by considering limiting cases, provide physical insight. We divided the process of the growth of the nanoparticles on the surface of the bead into three regimes. When t ≪ τ (regime 1), the nanoparticles are sufficiently far apart that they grow independently of each other (i.e. the nanoparticles do not know that they are surrounded by neighbors on the surface). When t ~ τ (regime 2) the nanoparticles have grown to a size that their growth rate is influenced by their neighbors, which are also consuming reactants. When t ≫ τ (regime 3) the nanoparticles have coalesced to form a continuous layer. In regime 1, eqn (2) simplifies to 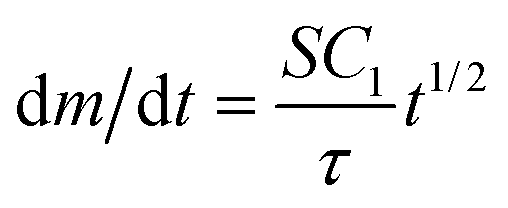 and m(t) ∝ t3/2. In regime 3, eqn (2) simplifies to
and m(t) ∝ t3/2. In regime 3, eqn (2) simplifies to 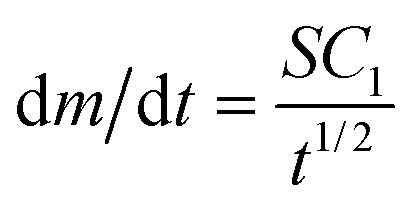 and m(t) ∝ t1/2. There is no closed-form expression for m(t) in regime 2. Note that the transition from regime 1 to the other regimes is controlled by the characteristic time, τ. Since τ ∝ 1/N, a higher density of adsorbed nanoparticles on the surface results in an earlier transition from regime 1 to the other regimes.
and m(t) ∝ t1/2. There is no closed-form expression for m(t) in regime 2. Note that the transition from regime 1 to the other regimes is controlled by the characteristic time, τ. Since τ ∝ 1/N, a higher density of adsorbed nanoparticles on the surface results in an earlier transition from regime 1 to the other regimes.
Substituting the expression for m(t) in regime 1 into eqn (1) and collecting the constants leads to a (relatively) simple expression for the change in levitation height of the bead as function of time
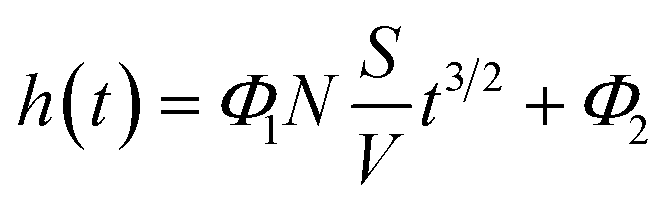 | (3) |
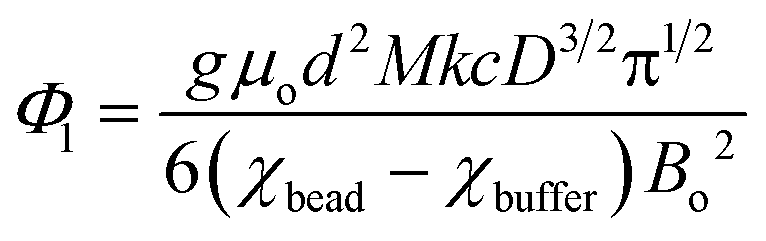 | (4) |
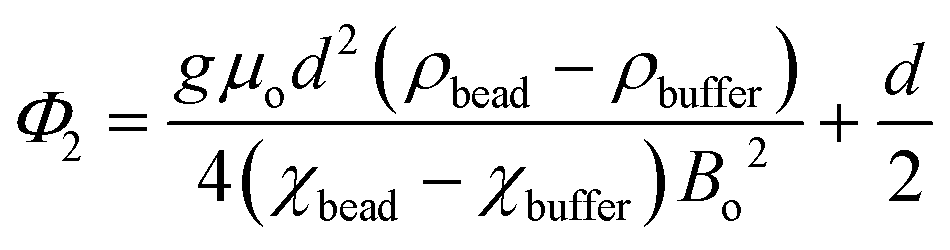 | (5) |
In eqn (3), 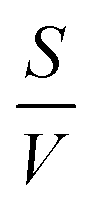 is the surface-to-volume ratio of the bead, M is the molar mass (kg mol−1) of the metal and c (mol m−3) is the concentration of reactants. Inspecting eqn (3) the levitation height, h is linearly related to the surface-to-volume ratio, everything else being equal, the use of beads with higher surface-to-volume ratio should lead to a change in levitation height in the developing buffer that is greater than when beads with a lower surface to volume ratio are employed.
is the surface-to-volume ratio of the bead, M is the molar mass (kg mol−1) of the metal and c (mol m−3) is the concentration of reactants. Inspecting eqn (3) the levitation height, h is linearly related to the surface-to-volume ratio, everything else being equal, the use of beads with higher surface-to-volume ratio should lead to a change in levitation height in the developing buffer that is greater than when beads with a lower surface to volume ratio are employed.
Fig. 4B shows the levitation height of the beads plotted versus the rescaled time, t3/2. The continuous black lines are linear regressions to the rescaled data. Fig. 4C shows a table with the equations for the linear regression and the coefficient of determination, R2. The linear fits are generally good, with the exception of the beads that were exposed to 3000 pM solution of gold-labeled streptavidin. We presume that N was sufficiently high in this case that the system transitioned to regime 2 or 3 during the course of the experiment.
Since the experiments were carried out under similar conditions, the ratio of the slopes of the lines should correspond to the ratio of the number of nanoparticles, N, that were adsorbed on the surface of the beads (eqn (3)). We used ICP-MS to measure quantitatively the amount of metallic gold on the beads (Fig. S1†). ICP-MS is sufficiently sensitive to detect the presence of gold on the beads due to the binding of gold-labeled proteins57,58 prior to amplification. This exquisite sensitivity allowed us to obtain the binding isotherm of gold-labeled streptavidin to the biotin-labeled beads (i.e. N vs. [free gold-labeled streptavidin]). Fig. S2† shows the equilibrium binding of gold-labeled streptavidin to the surface of the biotin-labeled beads versus the concentration of free gold-labeled streptavidin. Each datum is an average of seven beads. We assumed that due to sterics, there is one available streptavidin molecule per gold nanoparticle, we obtain a dissociation constant, Kd = 1.3 × 10−10 M−1 (we can also use the average number of streptavidin molecules on the gold nanoparticles specified by the manufacturer ~6.5. In that case, we find Kd = 8.0 × 10−10 M−1). This value compares favorably with measurements of the surface binding of biotin to streptavidin modified surfaces48,49 in other systems. The ratio of the number of gold nanoparticles adsorbed on the surfaces of the beads obtained through ICP-MS, N/N3000 pM agrees with the ratio of the slopes from the linear regression, m/m3000 pM (Fig. 4C).
Changing the dynamics of an AuMADA by adding free gold nanoparticles in solution
One goal of immunoassays is to obtain a readout that is independent of time after an initial (ideally short) period of development. This feature is particularly valuable in the field, or in busy settings where careful control over developing times might be difficult. Current assays often have strict time limits within which the assay must be read,21 and the MADA, due to the time-dependent deposition of metal, also has similar limitations.We hypothesized that AuMADA might be made less dependent on time by adding free gold nanoparticles in the developing buffer. These free nanoparticles would compete for gold ions in the solution, and serve as sinks for unreduced metal ions. Once all the gold ions are depleted from the developing buffer, the levitation height of the beads should stop changing. Fig. 5A and B shows the evolution of the levitation height of beads that had been previously exposed to 3000 pM of gold-labeled streptavidin (similar to the bead in Fig. 2A). The developing buffer in Fig. 5A was doped to a final concentration of 1 × 1010 gold nanoparticles mL−1 and the buffer in Fig. 5B was doped to 2 × 1010 20 nm diameter gold nanoparticles mL−1. As expected, with larger concentrations of free nanoparticles in solution, the change in levitation height slowed after a shorter time (Fig. 5B). The levitation height of the bead, however, does not stop changing completely, at least within the timescale of a 2 hour experiment. We attribute these observations to residual low concentrations of reactants that continue the deposition process on the surfaces of the beads. For example, free gold nanoparticles that increase in size sufficiently that they sediment to the bottom of the capillary (black arrow in Fig. 5A) effectively stop acting as sinks. Further optimization of the procedure demonstrated here, which would require a study of the interaction between free and surface-bound gold nanoparticle catalysts and the concentrations of various reactants on the kinetics of electroless deposition, could eventually lead to the development of time-independent assays. Such a study is beyond the scope of this work.
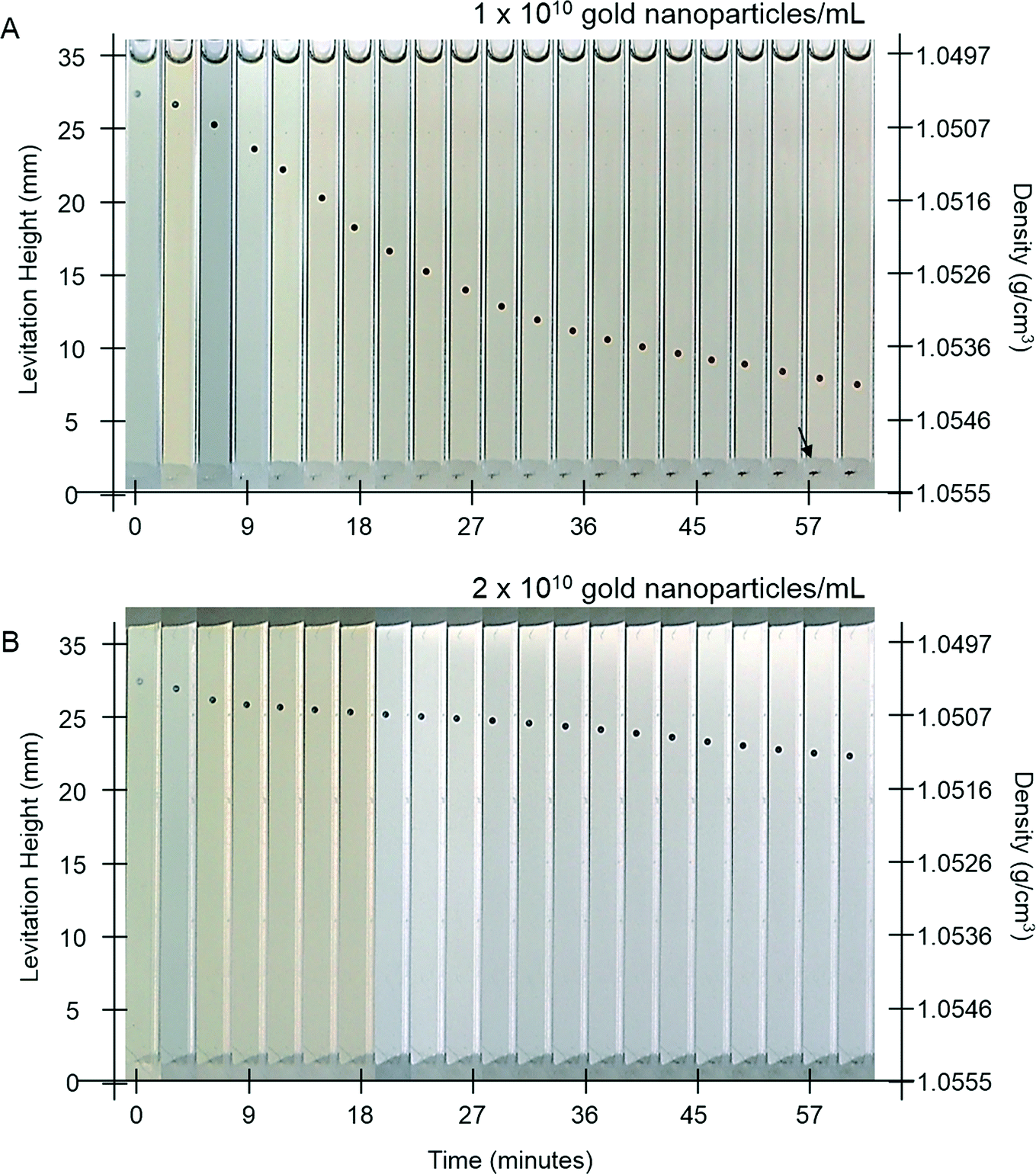 | ||
| Fig. 5 Addition of free gold nanoparticles to the developing buffer to modify the dynamics of MADA. Time lapse images of polystyrene beads with biotin on their surfaces in the developing buffer in the MagLev device. The beads were previously exposed to a sample containing 3000 pM of gold-labeled streptavidin. A) The developing buffer contained 1 × 1010 20 nm gold nanoparticles mL−1. B) The developing buffer contained 2 × 1010 20 nm gold nanoparticles mL−1. The black arrow indicates the brownish gold sediment of free gold particles. | ||
Density-linked Immunosorbent Assays, DeLISAs
In the next two sections, we demonstrate AuMADAs applied to the binding of antigens and antibodies (immunoassays), i.e. DeLISAs. Fig. 6A and B shows schematically the steps of the two immunoassays that we implemented. Both these assays are heterogeneous immunoassays, and thus have brief wash steps to remove unbound and non-specifically bound antibodies after the incubation steps.21 Modifications of assay procedures, for example by using a gold-labeled analyte (a homogenous immunoassay format),21 might eliminate the necessity for washing. Like other heterogeneous immunoassays, improper or excessive washing can decrease the final signal21 of these assays.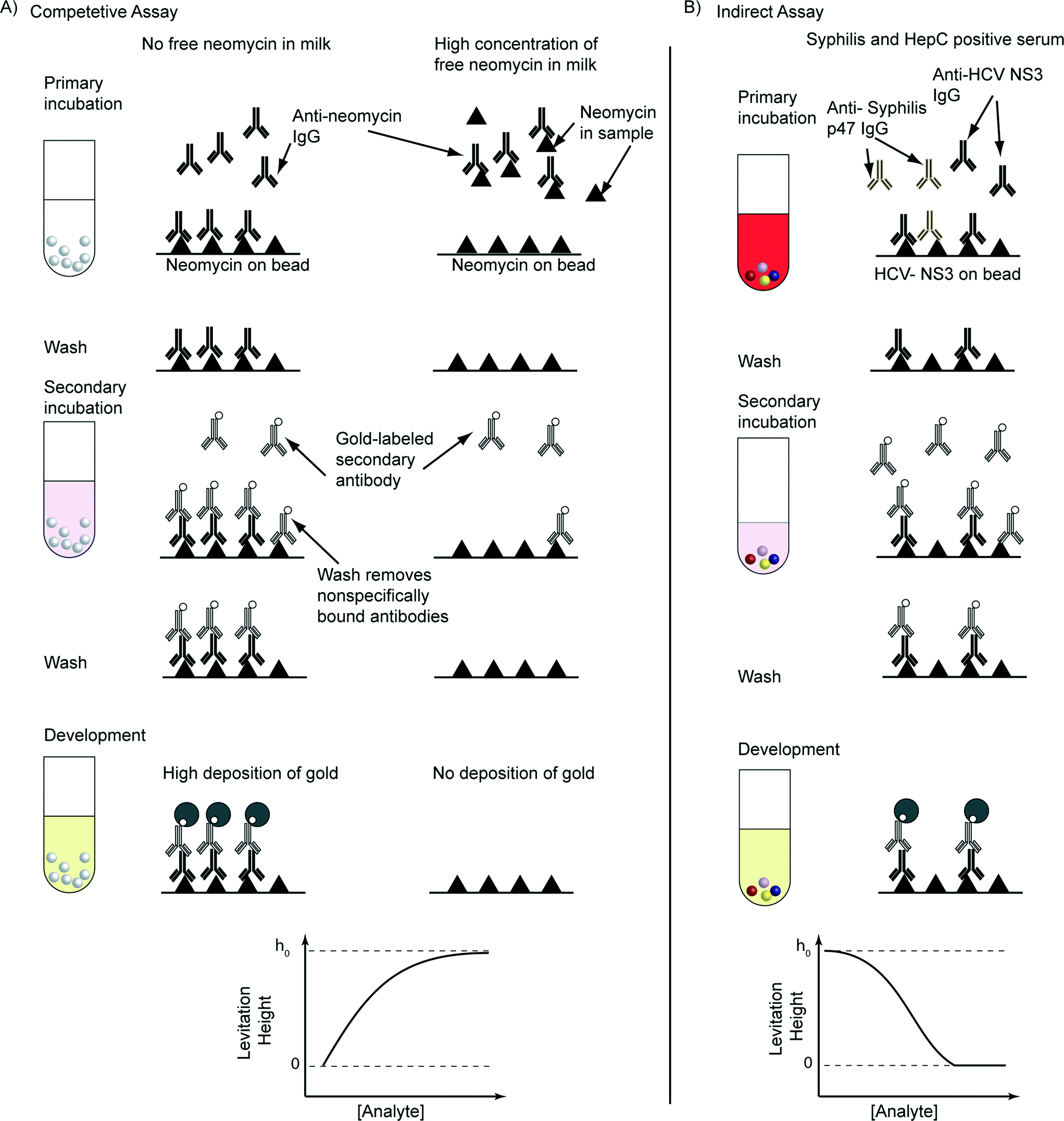 | ||
| Fig. 6 Schematic of the two immunoassay formats that we implemented with DeLISA. A) A competitive immunoassay for the antibiotic neomycin. B) A multiplexed indirect immunoassay. We drew the schematic to illustrate the case of serum that was positive for syphilis and for Hepatitis C. The surface of the bead was functionalized with HCV-NS3 protein and thus only binds anti-HCV NS3 proteins specifically. | ||
The first assay was a quantitative competitive assay for the concentration of the small molecule antibiotic neomycin (an aminoglycoside) in milk (Fig. 6A). In this assay, the antigen was neomycin, the primary antibody was a monoclonal mouse anti-neomycin immunoglobulin G (IgG), and the secondary antibody was a polyclonal goat anti-mouse-IgG conjugated to 10 nm diameter gold nanoparticles. In a competitive immunoassay for neomycin, a fixed concentration of mouse anti-neomycin IgG was added to the samples of milk before the addition of polystyrene beads with neomycin on their surfaces. Free neomycin, if present in the milk, binds to the anti-neomycin IgG, and thus reduces the amount of anti-neomycin antibody available for binding to the neomycin on the surfaces of the beads. The higher the concentration of free neomycin in the sample of milk, the less anti-neomycin IgG is available to bind to the neomycin on the beads. Hence, the strength of the signal that is generated from the bound molecules on the beads correlates inversely with the concentration of free neomycin in the sample of milk, that is a sample with no neomycin will generate the strongest signal, and samples with higher concentrations of neomycin will generate weaker signals.
The second assay was a multiplexed indirect immunoassay for antibodies against syphilis and Hepatitis C (HepC) in serum (Fig. 6B). The antigens were Treponema pallidum p47 protein on polystyrene beads (which were colored blue), and Hepatitis C virus NS3 protein on polystyrene beads (which were colored pink). These experiments include two controls: bovine serum albumin (BSA) on polystyrene beads (which were colored red) as a negative control, and human IgG on polystyrene beads (which were colored yellow) as a positive control. The primary antibodies were the targeted analytes assayed in the human serum. In a sample of serum, they consist of polyclonal antibodies with a range of avidities and affinities.21 The secondary antibody was polyclonal goat anti-human IgG (γ-chain specific) conjugated to 10 nm gold nanoparticles. The presence of the primary antibody is – in the absence of non-specific adsorption – required for binding of the gold-labeled secondary antibody to the beads. Thus, in this assay, the signal (i.e. the electroless deposition of gold and the change in levitation height) correlates positively with the concentration of primary antibody in a sample of serum.
A quantitative competitive DeLISA for neomycin residues in whole milk
We chose this particular assay since, i) quantitation of antibiotic residues is important in the milk industry, and tables of Maximum Residue Limits (MRL) in milk are published,59,60 ii) the assay must work on whole milk, which is a complex suspension of proteins, lipids and sugars, and iii) the antigen–antibody pair had been optimized by the manufacturer for ELISA. The MRL for neomycin in milk is 500 ppb.59,60Fig. 7A shows a photograph of seven beads that were exposed to a sample of milk containing no neomycin (left), and a sample of milk spiked with 500 ppb of neomycin (right), after 30 minutes in the developing buffer. Fig. 6A shows the steps of the assay; the details are in the materials and methods section. To reduce the amount of reagents consumed, and to allow parallelization, we loaded the beads into cylindrical capillaries with an inner diameter of 1 mm. Since we performed a competitive assay, we expected an inverse relationship between the levitation height and concentration of free antibiotic in the milk. Indeed, beads that were exposed to the antibiotic-free milk had a levitation height of 27 mm, while the beads exposed to the sample with 500 ppb of neomycin levitated at 35 mm (that is, they did not change levitation height). Fig. 7B shows a plot of the levitation height of beads exposed to a range of neomycin concentrations. At 30 minutes, milk samples with residues above the MRL can be distinguished from samples with residues below the MRL. The sample of milk that was at the MRL could not be distinguished from those that were above the MRL. After a further 30 minutes in the levitation buffer (i.e. after a total time of 60 minutes), the beads decreased in levitation height further (Fig. 7C) and the sample of milk that was at the MRL could be distinguished from the samples with antibiotic residues above the limit (Fig. 7D).
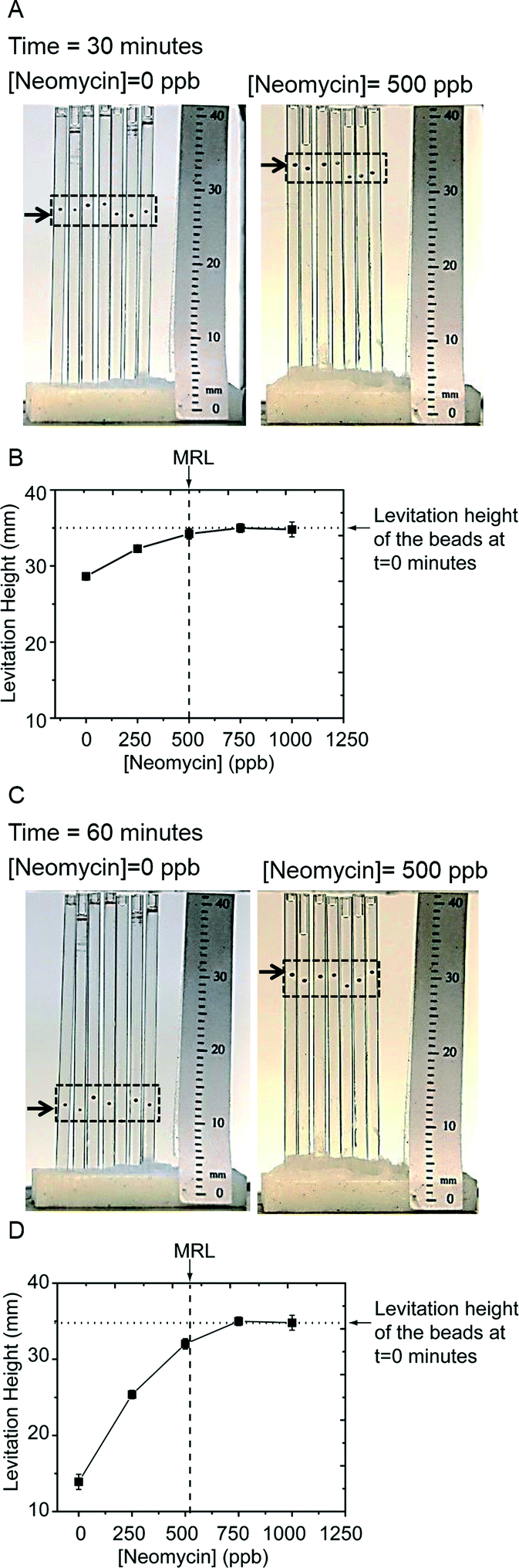 | ||
| Fig. 7 A DeLISA for neomycin in whole milk. Seven beads were exposed to samples of milk that were spiked with various concentrations of neomycin. A) Photographs of the levitation height of the beads after 30 minutes in the developing buffer. The left image is of beads exposed to a sample containing 0 ppb and the right image is of beads exposed to a sample containing 500 ppb of neomycin. The black arrows and the dashed rectangular boxes are guides to the eye showing the location of the beads. B) Plot of the levitation height versus the concentration of neomycin in the samples of milk at 30 minutes. The beads exposed to samples with no antibiotic residues and residues below the MRL (concentration marked with a dashed vertical line in the plots) have sunk below the beads exposed to samples with antibiotic residues above the MRL. The dotted horizontal line is a guide that shows the average levitation height of all these beads at the beginning of the development period. C, D) At 60 minutes, the milk sample at the MRL can be distinguished from the samples that exceed the MRL, while beads exposed to the milk samples with a high concentration of neomycin did not change levitation height. | ||
A multiplexed indirect DeLISA for syphilis and Hepatitis C (HepC) in blood serum
Bead-based assays are particularly suited for multiplexing. Immobilizing different antigens onto distinguishable beads allows the simultaneous detection of many different disease targets from a single sample.6,11Fig. 8 shows photographs of a set of the red, pink, blue, and yellow beads exposed to two simulated diseased serum samples and one simulated normal serum sample. The steps for carrying out the assays are shown schematically in Fig. 6B and provided in detail in the materials and methods section. Sample 1, the normal sample, was negative for HepC and syphilis. Sample 2 was positive for HepC and sample 3 was positive for both HepC and syphilis. Fig. 8A shows the configuration of the beads at the start of the developing period. Each of the colored beads levitated at approximately the same height in the capillaries. Fig. 8B–D shows beads after 30 minutes in the developing buffer. We observed that for the set of beads that were exposed to sample 1, only the yellow bead (human IgG, positive control) decreased in levitation height (Fig. 8B). For the beads exposed to sample 2, the pink (HCV NS3) and yellow bead (human IgG) decreased in levitation height (Fig. 8C). For beads exposed to sample 3, the pink (HCV NS3), blue (T. pallidum p47) and yellow (human IgG) beads changed levitation height (Fig. 8D). As expected, the red beads (BSA) which served as negative controls did not change levitation height for all the assays. Thus, the beads only changed levitation height when their cognate antibody was present in the serum sample. The positive and negative controls assured that the assay conditions were appropriate and served as an additional check that the deposition of gold and the change in levitation height were specific to the presence of the disease antibodies in the samples.
 | ||
| Fig. 8 Multiplexed immunoassay for syphilis and Hepatitis C (HepC). Beads exposed to samples containing antibodies against the immobilized antigens on the respective beads caused the beads to decrease in levitation height. A) Configuration of the beads in the MagLev device immediately after placement in the developing buffer. After 30 minutes of development, B) only the yellow negative control bead decreased in levitation height for the beads exposed to sample 1 which was negative for both syphilis and HepC, C) the pink and yellow beads decreased in levitation height for the beads exposed to sample 2 which was positive for HepC, D) the pink, blue and yellow beads decreased in levitation height for the beads exposed to sample 3 which was positive for both syphilis and HepC. The red beads were negative controls which should not (and did not) change in levitation height for all samples. The dashed black circles are guides to the eye showing the location of the beads. Note that the coloring of the bead disrupts the optical appearance (e.g. the apparent color, by eye) of the gold that is deposited on the surface. | ||
Conclusions
Current bead-based assays are read out using colorimetry, fluorimetry,21 chemiluminescence, and electrochemistry.20 MagLev, along with a combination of gold-labeled proteins and electroless deposition of metal, allows a density-based readout of bead-based assays. In MagLev, the change in density of the beads translates quantitatively to a change in levitation height. The method thus does not require instruments with moving parts, or a source of power to obtain a quantitative readout. The metal-amplified density assay (MADA) is compatible with conventional immunoassay formats such as competitive assays and indirect assays, can be multiplexed, can be performed on complex fluids, and can potentially be made time-insensitive. Although we have not attempted to optimize the performance of the assay, DeLISA, at its current level of development, is sufficiently sensitive and specific to detect clinically relevant concentrations of antibodies in standardized serum samples. Clinical samples from patients would likely present greater variability.DeLISA, like other bead-based assays, has three advantages over typical plate-based assays. i) It requires a small volume of reagents and sample. ii) The high ratio of surface area to volume allows short (<1 h) incubation times. iii) Multiplexing can be carried out by using distinguishable (for example, differently colored) beads. DeLISA at its present level of development also has several limitations. i) The assay still requires multiple steps, including washing steps, to obtain a result. ii) The gold amplification reagents and processes might be sensitive to large variations in temperature or to other factors (such as decomposition at tropical temperatures, T ≥ 35 °C). We have not characterized these sensitivities. Use of antibodies with higher avidity and specificity, and incorporation of centrifugation16 or other automation14,15 may circumvent some of these limitations.
Materials and methods
Materials
Manganese(II) sulfate monohydrate (United States Pharmacopeia (USP) grade), biotin-labeled bovine serum albumin (BSA), sucrose (molecular biology grade), neomycin sulfate (USP reference standard), goat anti-mouse immunoglobulin G (IgG) conjugated to 10 nm gold nanoparticles, and goat anti-human IgG (γ-chain specific) conjugated to 10 nm gold nanoparticles were purchased from Sigma-Aldrich. Neomycin-conjugated BSA and monoclonal murine anti-neomycin antibodies were purchased from Silver Lake Research Corp (Monrovia, CA). Virotrol Syphilis Total (single level control), Virotrol 1 (single level control), and Viroclear (single level control) were purchased from Bio-Rad Clinical Diagnostics (Hercules, CA). Monodisperse polystyrene beads (PS) cross-linked with divinylbenzene (600 μm mean diameter, C.V. 5%) were purchased from Polysciences Inc. Streptavidin conjugated to 10 nm gold nanoparticles was purchased from Cytodiagnostics. GoldEnhance/LM gold amplification solution was purchased from Nanoprobes. Treponema pallidum p47 full-length protein (ab51323, Lot GR141137-1), Hepatitis C Virus NS3 protein (ab49022, Lot GR20412-3) were purchased from AbCam (Cambridge, MA). Human IgG was purchased from Arista Biologicals (Allentown, PA). Trace-analysis grade concentrated hydrochloric acid (37%, OmniTrace) and nitric acid (70%) were purchased from EMD Millipore. We purchased 20 nm gold nanoparticles from British Biocell International (BBI). Square glass capillaries with an inner diameter of 2 mm were purchased from VitroCom, 1 mm inner diameter untreated micro-hematocrit polycarbonate capillaries (SafeCrit) were purchased from VWR. Certified antibiotic-free organic whole milk (Shaw Farm Dairy, Dracut, MA) was purchased from a local store.Procedure for dyeing the polystyrene beads
For the multiplex assay, we dyed the colorless polystyrene beads to allow visual discrimination of the various disease targets. Four milligrams of dye (Sudan Red, Reactive Blue, Alazarin Yellow, or Fat Brown) was dissolved in 1.5 mL of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 toluene:ethanol. After ten minutes, the dye solutions were passed through cotton filters to remove any remaining particulates. Approximately 300 mg of polystyrene beads were then added to the dye solution and gently rocked for one hour. The beads were then thoroughly rinsed with ethanol and dried in vacuo for at least four hours (typically overnight).
:1 toluene:ethanol. After ten minutes, the dye solutions were passed through cotton filters to remove any remaining particulates. Approximately 300 mg of polystyrene beads were then added to the dye solution and gently rocked for one hour. The beads were then thoroughly rinsed with ethanol and dried in vacuo for at least four hours (typically overnight).
Functionalization and covalent grafting of proteins onto polystyrene beads
To functionalize the polystyrene beads with carboxylic groups, we oxidized them with a dilute solution of potassium permanganate and sulfuric acid in water.61We stress that mixing concentrated aqueous solutions of potassium permanganate with concentrated aqueous solution of sulfuric acid is highly hazardous.62 In contrast, handling dilute solutions of those same reagents has not been reported to be a source of hazard. For those reasons, we first prepared a dilute aqueous solution of sulfuric acid (1.2 N, 1/30 dilution, 10 mL) to which we slowly added potassium permanganate (5 w/V%) with constant stirring, thus avoiding variations in the concentration of permanganate in the reaction mixture. The polystyrene beads (200 mg) were suspended in this mixture (10 mL), which was maintained at 60 °C for 30 minutes in a water bath. During the course of the reaction, the surface of the polystyrene beads was partially oxidized to yield surface carboxylic groups, while potassium permanganate was reduced to insoluble brown manganese(IV) dioxide which deposited on the surface of the carboxylated beads. We then washed the beads by vortexing in 6 N hydrochloric acid (10 mL) to dissolve this brown manganese oxide layer. We repeated the washing step until the final solution was colorless (typically 5–6 times).We then proceeded with grafting the proteins of interest (a BSA–biotin conjugate for the MADA for gold-labeled streptavidin, a BSA–neomycin conjugate for the quantitative detection of neomycin in milk, and the proteins Treponema pallidum p47, Hepatitis C NS3, BSA, and human IgG for the multiplex immunoassays) following the procedure from the technical note from Bangs Labs.63 Briefly, we washed the beads twice with 10 mL of 200 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer at pH 6.0, and resuspended them in 10 mL of the same. We added 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluenesulfonate (100 mg) while mixing and allowed the mixture to react at room temperature for 15 minutes with continuous mixing. We then washed the beads with a 50 mM borate buffer at pH 8.3 (2 × 10 mL) and resuspended them in 10 mL of the same. We added an aqueous solution of the proteins of interest (200 μL of the stock solutions containing 1 mg mL−1 of protein) to this suspension, and incubated it at room temperature for 4 hours with constant mixing. We then washed the beads with phosphate-buffered saline (PBS) and allowed the beads to react with a solution containing 0.05 w/V% of BSA (to reduce non-specific adsorption of protein onto the surface of the beads) in 10 mL of a 40 mM aqueous solution of hydroxylamine (to react with the remaining activated carboxylic acids on the beads) at room temperature with continuous mixing for 30 minutes. Finally, we washed the protein-functionalized beads with PBS (3 × 10 mL) and stored the functionalized beads in PBS containing 0.05 w/V% of BSA.
Procedure for the AuMADA for gold-labeled streptavidin
We transferred seven polystyrene beads with biotin immobilized on their surfaces to a vial containing 100 μL of gold-labeled streptavidin of different concentrations. We incubated the beads for 30 minutes in the solution. The gold-labeled streptavidin solution was decanted, and the beads washed briefly with 100 μL of deionized water. This procedure was sufficient to remove non-specifically bound gold-labeled streptavidin while still minimizing the desorption of bound protein from the surface. The beads were immediately placed in fresh developing buffer and loaded into square glass capillaries with dimensions 35 × 2 × 2 mm through capillary action (i.e. the liquid containing the bead spontaneously entered the capillary when the hydrophilic capillaries were contacted with the developing buffer). The buffer was prepared by mixing equal parts of the four components of the gold enhancing solution before adding 300 mM manganese(II) sulfate and 600 mM sucrose (to adjust the density of the solution). We sealed both end of the capillaries with Tacky Wax (Bard's Products, Inc.) and placed them into a MagLev device and visualized the change in density of the beads by taking sequential photographs with a Nikon Digital Camera equipped with a macrolens.Procedure for the quantitative competitive immunoassay for neomycin in milk
We prepared milk samples spiked with neomycin by making a stock solution of neomycin in 10 mL of whole organic certified antibiotic-free milk at a concentration of 10000 ppb. We then diluted this stock solution with milk to 1000 ppb, and serially diluted the 1000 ppb solution to obtain a range of neomycin concentrations from 250 ppb to 1000 ppb. The MRL of neomycin is 500 ppb, and samples with concentrations far from this value are not relevant practically. We performed the competitive assay by following the manufacturer's (Silver Lake Research) protocol with minor modifications. Briefly, we diluted the monoclonal murine anti-neomycin antibodies 1:1500 times in PBS from the stock obtained from the manufacturer. We added 25 μL of the milk sample and 25 μL of the diluted antibody to an Eppendorf tube. We added seven beads functionalized with a BSA–neomycin conjugate to the Eppendorf tube and incubated for 30 minutes at room temperature. We removed the milk sample from the Eppendorf, added 100 μL of PBS and agitated the beads gently for less than 1 min before removing the buffer with a pipette. We then added 50 μL of 10 nm gold-labeled goat anti-mouse-IgG that was diluted 1:100 times in PBS as recommended by the manufacturer. We incubated the mixture for 30 minutes at room temperature. We removed the gold-labeled antibody, and washed the beads briefly by adding 100 μL of deionized water, and immediately transferred the beads (with a minimal amount of liquid) into an Eppendorf with developing buffer. We loaded the beads into capillaries through capillary action and placed them in the MagLev device.
Procedure for preparing simulated diseased and normal serum samples
Antibody concentrations can often vary widely from patient to patient and also depends strongly on the progression of the disease.21 To ensure that we were assaying for clinically relevant concentrations, we obtained certified human serum diagnostic standards from the Clinical Diagnostics Division of Biorad. These standards are used to calibrate commercially available immunoassays. For sample 1, we used 100 μL of ViroClear. For sample 2 we used 100 μL of Virotrol 1. For sample 3 we combined 50 μL Virotrol 1, and 50 μL of Virotrol Syphilis Total to mimic serum of an individual co-infected with syphilis and Hepatitis C. The procedure that we used for performing the multiplexed indirect assay is shown schematically in Fig. 6B. After 15 minutes, the serum samples were removed with a pipette, and 100 μL of PBS buffer was added and discarded right away. This step removed any remaining sample from the supernatant and removed any non-specifically bound antibodies from the surface of the beads. The beads were then exposed to gold-labeled goat anti-human IgG, and incubated for 15 minutes. The beads were transferred into 100 μL of deionized water and then immediately into the developing buffer and loaded into individual capillaries.Preparation of the samples for ICP-MS analysis
A dilute solution of aqua regia was prepared by mixing trace analysis grade nitric acid, hydrochloric acid and MilliQ water in a 1:3:2 ratio. Seven beads exposed to samples of differing concentrations of gold-labeled streptavidin were placed in 0.87 mL of freshly prepared dilute aqua regia for 20 minutes. To each sample, we added 50 μL of a 1000 ppb solution of bismuth (prepared by a dilution of a 1000 ppm ICP grade standard, which was prepared by dissolving high purity bismuth metal in 5% nitric acid) in MilliQ water to serve as an internal standard. We diluted our samples to a total volume of 5 mL with MilliQ water before analyzing the samples with ICP-MS. To obtain a calibration curve we also conducted ICP-MS measurements on serially diluted gold standards (ICP grade) from (0.01–10000 ppb).
Competing financial interests
The authors declare no competing financial interests.Acknowledgements
This work was supported by the Bill and Melinda Gates Foundation award # 51308, and a subcontract from LLNL under award # DE-AC52-07NA27344. M. G. acknowledges financial support from the Marie Curie project SAM-TunEGaIn:IOF-2012-328412. The electron microscopy experiments were performed at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Infrastructure Network (NNIN), which is supported by NSF award no. ECS-0335765. CNS is part of Harvard University. We thank Dr. Zhongxing Chen for help with the ICP-MS measurements, and Dr. Arvind Gopinath for helpful conversations.Notes and references
- D. Issadore and R. M. Westervelt, Point-of-Care Diagnostics on a Chip, Springer, Heidelberg, New York, 2013 Search PubMed.
- P. K. Drain, E. P. Hyle, F. Noubary, K. A. Freedberg, D. Wilson, W. R. Bishai, W. Rodriguez and I. V. Bassett, Lancet Infect. Dis., 2014, 14, 239–249 CrossRef.
- A. H. B. Wu, Clin. Chim. Acta, 2006, 369, 119–124 CrossRef CAS PubMed.
- J. M. Singer and C. M. Plotz, Am. J. Med., 1956, 21, 888–892 CrossRef.
- J. H. W. Leuvering, P. J. H. M. Thal, M. Van Der Waart and A. H. W. M. Schuurs, J. Immunoassay, 1980, 1, 77–92 CrossRef CAS PubMed.
- D. A. A. Vignali, J. Immunol. Methods, 2000, 243, 243–255 CrossRef CAS.
- E. Verpoorte, Lab Chip, 2003, 3, 60N–68N RSC.
- W. de Jager and G. T. Rijkers, Methods, 2006, 38, 294–303 CrossRef CAS PubMed.
- X. W. Zhao, Z. B. Liu, H. Yang, K. Nagai, Y. H. Zhao and Z. Z. Gu, Chem. Mater., 2006, 18, 2443–2449 CrossRef CAS.
- Y. J. Zhao, X. W. Zhao, C. Sun, J. Li, R. Zhu and Z. Z. Gu, Anal. Chem., 2008, 80, 1598–1605 CrossRef CAS PubMed.
- A. M. E. Whitelegg, J. Birtwistle, A. Richter, J. P. Campbell, J. E. Turner, T. M. Ahmed, L. J. Giles, M. Fellows, T. Plant, A. J. Ferraro, M. Cobbold, M. T. Drayson and C. A. MacLennan, J. Immunol. Methods, 2012, 377, 37–46 CrossRef CAS PubMed.
- D. R. Baselt, G. U. Lee, M. Natesan, S. W. Metzger, P. E. Sheehan and R. J. Colton, Biosens. Bioelectron., 1998, 13, 731–739 CrossRef CAS.
- M. A. M. Gijs, Microfluid. Nanofluid., 2004, 1, 22–40 CAS.
- R. S. Sista, A. E. Eckhardt, V. Srinivasan, M. G. Pollack, S. Palanki and V. K. Pamula, Lab Chip, 2008, 8, 2188–2196 RSC.
- N. Pamme, Lab Chip, 2006, 6, 24–38 RSC.
- D. I. Walsh III, G. J. Sommer, U. Y. Schaff, P. S. Hahn, G. J. Jaffe and S. K. Murthy, Lab Chip, 2014, 14, 2673–2680 RSC.
- R. J. Fulton, R. L. McDade, P. L. Smith, L. J. Kienker and J. R. Kettman, Clin. Chem., 1997, 43, 1749–1756 CAS.
- C. T. Lim and Y. Zhang, Biosens. Bioelectron., 2007, 22, 1197–1204 CrossRef CAS PubMed.
- S. A. Dunbar, Clin. Chim. Acta, 2006, 363, 71–82 CrossRef CAS PubMed.
- L. Krejcova, L. Nejdl, D. Hynek, S. Krizkova, P. Kopel, V. Adam and R. Kizek, Molecules, 2013, 18, 15573–15586 CrossRef CAS PubMed.
- The Immunoassay Handbook, ed. D. Wild, Elsevier Ltd., Oxford, 2005 Search PubMed.
- Y. S. Xing, P. Wang, Y. C. Zang, Y. Q. Ge, Q. H. Jin, J. L. Zhao, X. Xu, G. Q. Zhao and H. J. Mao, Analyst, 2013, 138, 3457–3462 RSC.
- F. Deiss, C. N. LaFratta, M. Symer, T. M. Blicharz, N. Sojic and D. R. Walt, J. Am. Chem. Soc., 2009, 131, 6088–6089 CrossRef CAS PubMed.
- K. A. Mirica, S. S. Shevkoplyas, S. T. Phillips, M. Gupta and G. M. Whitesides, J. Am. Chem. Soc., 2009, 131, 10049–10058 CrossRef CAS PubMed.
- N. Hirota, M. Kurashige, M. Iwasaka, M. Ikehata, H. Uetake, T. Takayama, H. Nakamura, Y. Ikezoe, S. Ueno and K. Kitazawa, Phys. B, 2004, 346, 267–271 CrossRef PubMed.
- Y. Ikezoe, T. Kaihatsu, S. Sakae, H. Uetake, N. Hirota and K. Kitazawa, Energy Convers. Manage., 2002, 43, 417–425 CrossRef CAS.
- T. Kimura, S. Mamada and M. Yamato, Chem. Lett., 2000, 11, 1294–1295 CrossRef.
- K. A. Mirica, F. Ilievski, A. K. Ellerbee, S. S. Shevkoplyas and G. M. Whitesides, Adv. Mater., 2011, 23, 4134–4140 CrossRef CAS PubMed.
- K. A. Mirica, S. T. Phillips, S. S. Shevkoplyas and G. M. Whitesides, J. Am. Chem. Soc., 2008, 130, 17678–17680 CrossRef CAS PubMed.
- N. D. Shapiro, K. A. Mirica, S. Soh, S. T. Phillips, O. Taran, C. R. Mace, S. S. Shevkoplyas and G. M. Whitesides, J. Am. Chem. Soc., 2012, 134, 5637–5646 CrossRef CAS PubMed.
- S. S. Shevkoplyas, A. C. Siegel, R. M. Westervelt, M. G. Prentiss and G. M. Whitesides, Lab Chip, 2007, 7, 1294–1302 RSC.
- A. B. Subramaniam, D. Yang, H.-D. Yu, A. Nemiroski, S. Tricard, A. K. Ellerbee, S. Soh and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12980–12985 CrossRef CAS PubMed.
- A. Winkleman, R. Perez-Castillejos, K. L. Gudiksen, S. T. Phillips, M. Prentiss and G. M. Whitesides, Anal. Chem., 2007, 79, 6542–6550 CrossRef CAS PubMed.
- K. Yokoyama, N. Hirota and M. Iwasaka, IEEE Trans. Appl. Supercond., 2007, 17, 2181–2184 CrossRef CAS.
- N. D. Shapiro, S. Soh, K. A. Mirica and G. M. Whitesides, Anal. Chem., 2012, 84, 6166–6172 CrossRef CAS PubMed.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- H. Cai, Y. Q. Wang, P. G. He and Y. H. Fang, Anal. Chim. Acta, 2002, 469, 165–172 CrossRef CAS.
- C. L. Schofield, A. H. Haines, R. A. Field and D. A. Russell, Langmuir, 2006, 22, 6707–6711 CrossRef CAS PubMed.
- G. Danscher, Histochemistry, 1984, 81, 331–335 CAS.
- P. M. Lackie, Histochem. Cell Biol., 1996, 106, 9–17 CrossRef CAS.
- I. Zehbe, G. W. Hacker, H. C. Su, C. Hauser-Kronberger, J. F. Hainfeld and R. Tubbs, Am. J. Pathol., 1997, 150, 1553–1561 CAS.
- C. D. Chin, T. Laksanasopin, Y. K. Cheung, D. Steinmiller, V. Linder, H. Parsa, J. Wang, H. Moore, R. Rouse, G. Umviligihozo, E. Karita, L. Mwambarangwe, S. L. Braunstein, J. van de Wijgert, R. Sahabo, J. E. Justman, W. El-Sadr and S. K. Sia, Nat. Med., 2011, 17, 1015–U1138 CrossRef CAS PubMed.
- S. K. Sia, V. Linder, B. A. Parviz, A. Siegel and G. M. Whitesides, Angew. Chem., Int. Ed., 2004, 43, 498–502 CrossRef CAS PubMed.
- S. Gupta, S. Huda, P. K. Kilpatrick and O. D. Velev, Anal. Chem., 2007, 79, 3810–3820 CrossRef CAS PubMed.
- M. Srisa-Art, E. C. Dyson, A. J. deMello and J. B. Edel, Anal. Chem., 2008, 80, 7063–7067 CrossRef CAS PubMed.
- M. Wilchek and E. A. Bayer, in Methods in Enzymology, ed. W. Meir and A. B. Edward, Academic Press, 1990, vol. 184, pp. 5–13 Search PubMed.
- D. M. Mock and P. Horowitz, in Methods in Enzymology, ed. W. Meir and A. B. Edward, Academic Press, 1990, vol. 184, pp. 234–240 Search PubMed.
- T. Buranda, G. M. Jones, J. P. Nolan, J. Keij, G. P. Lopez and L. A. Sklar, J. Phys. Chem. B, 1999, 103, 3399–3410 CrossRef CAS.
- S. C. Huang, M. D. Stump, R. Weiss and K. D. Caldwell, Anal. Biochem., 1996, 237, 115–122 CrossRef CAS PubMed.
- X. Duan, Y. Li, N. K. Rajan, D. A. Routenberg, Y. Modis and M. A. Reed, Nat. Nanotechnol., 2012, 7, 401–407 CrossRef CAS PubMed.
- S. Fletcher, J. Chem. Soc., Faraday Trans., 1983, 79, 467–479 RSC.
- G. Gunawardena, G. Hills, I. Montenegro and B. Scharifker, J. Electroanal. Chem., 1982, 138, 225–239 CrossRef CAS.
- B. Scharifker and G. Hills, Electrochim. Acta, 1983, 28, 879–889 CrossRef CAS.
- S. Abbasi, S. Kitayaporn, M. J. Siedlik, D. T. Schwartz and K. F. Bohringer, Nanotechnology, 2012, 23, 305301 CrossRef PubMed.
- M. Paunovic and M. Schlesinger, Fundamentals of Electrochemical Deposition, John Wiley & Sons, Hoboken, 2006 Search PubMed.
- G. Festag, A. Steinbrueck, A. Csaki, R. Moeller and W. Fritzsche, Nanotechnology, 2007, 18, 015502 CrossRef.
- A. Scheffer, C. Engelhard, M. Sperling and W. Buscher, Anal. Bioanal. Chem., 2008, 390, 249–252 CrossRef CAS PubMed.
- C. Zhang, Z. Zhang, B. Yu, J. Shi and X. Zhang, Anal. Chem., 2001, 74, 96–99 CrossRef.
- J. Adrian, D. G. Pinacho, B. Granier, J. M. Diserens, F. Sanchez-Baeza and M. P. Marco, Anal. Bioanal. Chem., 2008, 391, 1703–1712 CrossRef CAS PubMed.
- B. G. Knecht, A. Strasser, R. Dietrich, E. Martlbauer, R. Niessner and M. G. Weller, Anal. Chem., 2004, 76, 646–654 CrossRef CAS PubMed.
- N. Zammatteo, C. Girardeaux, D. Delforge, J.-J. Pireaux and J. Remacle, Anal. Biochem., 1996, 236, 85–94 CrossRef CAS PubMed.
- Fire Protection Guide to Hazardous Materials, National Fire Protection Association, Quincy, MA, 12th edn, 1997 Search PubMed.
- Bang's Laboratories Technical Note 205: Covalent Coupling, http://www.bangslabs.com/sites/default/files/bangs/docs/pdf/205.pdf.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc01161a |
| This journal is © The Royal Society of Chemistry 2015 |