
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A droplet microfluidic system for sequential generation of lipid bilayers and transmembrane electrical recordings†
Magdalena A.
Czekalska‡
a,
Tomasz S.
Kaminski‡
a,
Slawomir
Jakiela
a,
K.
Tanuj Sapra§
b,
Hagan
Bayley
*b and
Piotr
Garstecki
*a
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: garst@ichf.edu.pl
bChemical Biology Sub-Department, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford OX1 3TA, UK. E-mail: hagan.bayley@chem.ox.ac.uk
First published on 6th November 2014
Abstract
This paper demonstrates a microfluidic system that automates i) formation of a lipid bilayer at the interface between a pair of nanoliter-sized aqueous droplets in oil, ii) exchange of one droplet of the pair to form a new bilayer, and iii) current measurements on single proteins. A new microfluidic architecture is introduced – a set of traps designed to localize the droplets with respect to each other and with respect to the recording electrodes. The system allows for automated execution of experimental protocols by active control of the flow on chip with the use of simple external valves. Formation of stable artificial lipid bilayers, incorporation of α-hemolysin into the bilayers and electrical measurements of ionic transport through the protein pore are demonstrated.
Introduction
Efficient screening of the function of membrane proteins against physical and chemical factors demands a reproducible and cost-effective method for generating lipid bilayers and for measuring electrical currents through channels and pores inserted in them. The widely used patch clamp analysis enables recordings of electric currents through protein channels in their natural cellular environment or in artificial lipid vesicles. However, a classical patch clamp demands skilled manual operation, thereby limiting its throughput in routine drug screening assays. The high demand for screens of protein function has prompted several attempts to increase throughput and to automate the measurements.1 These methods require control of protein expression in cells, which can be challenging,2 and provide limited information on activity, usually limited to IC50 values. Methods for in vitro formation of bilayer lipid membranes (BLMs) – the variations of the Montal–Mueller3 or painting technique – are, in turn, limited by the low stability of the bilayers.4 Formation of bilayers on a solid support significantly increases the stability; however, the format is prone to the formation of incomplete membranes.5,6 Moreover, proteins may interact unfavorably with the solid substrate and their mobility and function can be modified or hampered, making electrical measurements difficult, especially at the single-molecule level.6A convenient method for forming a stable lipid bilayer is by bringing the lipid monolayer-coated interfaces of two aqueous droplets into contact in oil.7–9 Compared to the classical planar bilayer method, droplet interface bilayers (DIBs) show critical advantages of exceptional stability that supports long-term measurements and minute, microliter consumption of samples.4 The stability of the interface and the ability to easily re-form the bilayer make the DIB method an attractive strategy for implementation in robust inhibitor screening schemes.9,10 The main challenge is in automation of the technique as implementing automation may evolve the technique to a high-throughput screening platform. Here we show, for the first time, repetitive formation of DIBs, separation of the droplets and exchange of at least one droplet of a pair to form a new DIB, all in an automated sequence.
Droplet microfluidics has successfully been used in high-throughput screening to address various problems in biochemistry11,12 with primary applications in protein crystallization,13 single cell screening,14 directed evolution of enzymes15 and the clonal selection of antibodies.16 Droplet microfluidics is an ideal tool for reproducible formation of droplets with controlled composition of their interfaces,17 including lipid monolayers.
Still, research on droplet microfluidic techniques for in vitro reconstitution and electrophysiological studies with lipid bilayers has so far been limited. In a pioneering report, Funakoshi et al.8 built microfluidic chips in which a bilayer is formed between two aqueous streams surrounded by an oily fluid. The formation of a true DIB was confirmed by capacitance measurements of the bilayer and voltage-clamp recording indicating incorporation of α-hemolysin pores. The DIB system was proved to be useful in investigation of other proteins, for example, bacterial potassium channels,10,18,19 eukaryotic mammalian menthol receptors20,21 or equinatoxin.22
So far, many microfluidic platforms and other chip technologies have been developed, usually lacking the possibility of automated serial formation of lipid bilayers.18,20,21,23–40 Highly parallelized systems capable of the creation of arrays of lipid bilayers usually rely solely on measurements of fluorescence32,35,41–43 that does not provide for complete characterization of the function of the pores.
Here, we present an automated microfluidic system that combines the active manipulation of droplets44,45 with passive trapping46–48 and the capability to insert electrodes into the droplets for in situ electrical measurements on membrane proteins incorporated into a DIB.
For screening applications it is essential that the microfluidic system is capable of automated generation and manipulation of multiple droplets. Droplet on demand systems49 provide a high degree of control over the composition and location of each droplet. Although the generation of DIBs and networks of droplets has been demonstrated on a chip,32,35,50,51 a remaining challenge was to interface electrodes for repetitive measurements of bilayer capacitance and of conductance through single channels and pores.
Results
Microfluidic device
The microfluidic device (Fig. 1) comprises 2 polycarbonate plates, milled with an Ergwind msc4025 CNC milling station and subsequently thermally bonded in a hydraulic press. The width of the channels is 400 μm (excluding channels no. 14 and 15 [Fig. S1†] which have a width of 200 μm). The depth of all channels is 400 μm. The device consists of i) a microfluidic trap (N, Fig. 1a) in which droplets are brought into contact to form a DIB, ii) two microfluidic T-junctions (D and G) with additional sample ports, and iii) an inlet port for a sequence of droplets containing various inhibitors (F). Two pieces of silver wire coated with AgCl and a thin layer of agarose gel serve as an electrode pair. One electrode is inserted into each of the two chambers of the trap prior to running an assay. | ||
| Fig. 1 Geometry of the microfluidic device. a) Schematic diagram of the microfluidic chip: A, E, J, K, P – inlets for oil, B, I – inlets for aqueous samples, C – aspiration module with pure buffer for washing electrodes, D, G – T-junctions, F – tubing with a sequence of droplets containing inhibitors, H – aspiration module with a solution of proteins, M – ground electrode, N – hydrodynamic trap, O – working electrode, S – inlet for oil in a control channel, L, R – outlets. b) Schematic picture of the microfluidic trap. The DIB is formed at the interface of two droplets locked in the trap filled with oil. The light-grey area represents so-called ‘bypasses’ – shallow channels on the sides of the trap (dark grey). The continuous liquid (oil) passes through the bypasses while the droplets are locked in the position. (For details of the trap architecture see Fig. S2.†) c) Fragment of current trace (20 s) reflecting the incorporation of α-hemolysin into a lipid bilayer clamped at +50 mV. Stepwise increase in current from 0 to 50 pA reflects the incorporation of a single pore. The presence of inhibitor (γ-cyclodextrin, 5 μM) within the pore is seen as a decrease in current by approximately 60%. The presence of the next αHL heptamer in a lipid bilayer is seen as a further stepwise increase in current. BL – baseline, O – open pore level, 2× O – 2 open pores, B – blocked pore, 2× b – 2 blocked pores. | ||
Operation of a chip
We first filled the whole system with a continuous organic phase containing lipids (1 mg mL−1 1,2-diphytanoyl-sn-glycero-3-phosphocholine [DPhPC] in 75% hexadecane and 25% silicone oil AR20). We then opened the port (B) and transferred a small volume (tens of microliters) of buffer (1 M KCl, 10 mM Tris–HCl, pH 7.0) from a glass microsyringe connected to the chip via a capillary. We used the port (I) to introduce a small volume of the protein solution (α-hemolysin (αHL) wild type, 5 μg mL−1, diluted in the same buffer). After closing the deposition ports we opened the flow of oil from inlets (A and J) into the channel containing the sample to push the front of the aqueous aliquot to the T-junction. A sequence of droplets containing inhibitors was deposited on the chip in a similar manner (F and E).We used valves to control the flow of oil from pressurized containers (Fig. S3†). Droplets were generated in T-junctions – a defined volume of the aqueous sample was pushed into the orthogonal channel, then broken off with the flow of oil52 (Fig. 1a, K and P) and transported into the trap (N). The system (see Fig. 1b) comprises two cavities (dark grey in Fig. 1b) that lock the droplets by capillary force in place. The shallow bypasses (marked with light grey in Fig. 1b) allow for a small flow of oil around the droplets without distorting them. In the experiments, we first generated and placed in one trap a droplet containing α-hemolysin, and then sequentially pushed droplets containing different concentrations of an inhibitor into the second trap.
The pair of chambers in the hydrodynamic traps allows us to lock in place two droplets and observe the kinetics of formation of the DIB (Fig. 2). In addition to tracking the process visually with a video camera we measured the capacitance of the DIB in the absence of any membrane proteins. To this end we applied a triangular potential (10 Hz, 50 mV peak to peak) and recorded the resulting square wave current at a 1 kHz sampling rate. After the droplets first touch each other, it takes about 60 s for the bilayer to form.
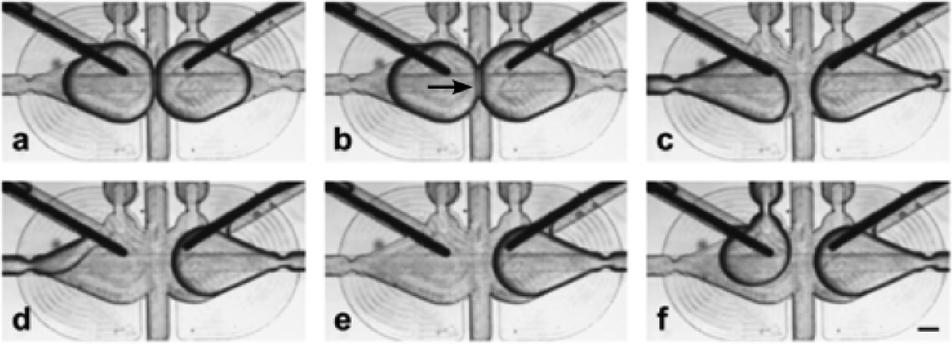 | ||
| Fig. 2 Micrographs illustrating the orientation of the electrodes in the microfluidic trap and the exchange of one of the droplets. a) Two droplets are trapped. b) Bilayer (indicated with an arrow) is formed at the interface. c–e) Removal of one of the droplets. f) Introduction of the new droplet. The droplet wets the electrode immediately after being placed in a trap (see details in the ESI†). The scale bar is 200 μm. | ||
Characterization of the bilayer
Formation of a bilayer causes a rapid increase in the capacitive current IC that reaches typically 394 ± 13 pA for two droplets of volume 300 nL each.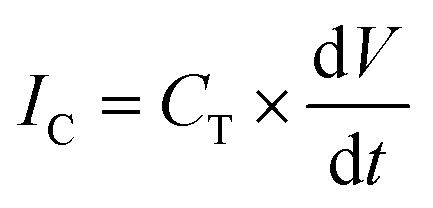 | (1) |
Capacitive current IC (eqn (1)) is directly proportional to the capacitance CT and the rate of change of potential, which was set to 1 V s−1. Therefore, we find the value of the capacitance to be 394 ± 13 pF for 300 nL droplets. The area of the bilayer can be determined through the measurement of the capacitive current.53 Using the specific capacitance for a bilayer formed in pure hexadecane53 (0.64 μF cm−2), we estimated the contact area between the 300 nL droplets to be 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 ± 2500 μm2. Our system allows us to trap droplets with volumes ranging between 250 nL and 350 nL and form lipid bilayers between them. This range of volumes allows for a nearly 4-fold range in the surface area of the DIB (Fig. 3). Internal flows of oil (e.g. convection) cause fluctuations of the capacitive current which is proportional to bilayer size, yet these changes are small and do not impact the process of pore insertion. Relative standard deviation (RSD) of a single bilayer area is in the range of 1–6%. Additionally, subsequent pairs of droplets can have slightly different positions during formation of the bilayer; thus the overall RSD for a given droplet volume is slightly higher (in the range of 4–10%, see detailed analysis in the ESI† Table S1).
500 ± 2500 μm2. Our system allows us to trap droplets with volumes ranging between 250 nL and 350 nL and form lipid bilayers between them. This range of volumes allows for a nearly 4-fold range in the surface area of the DIB (Fig. 3). Internal flows of oil (e.g. convection) cause fluctuations of the capacitive current which is proportional to bilayer size, yet these changes are small and do not impact the process of pore insertion. Relative standard deviation (RSD) of a single bilayer area is in the range of 1–6%. Additionally, subsequent pairs of droplets can have slightly different positions during formation of the bilayer; thus the overall RSD for a given droplet volume is slightly higher (in the range of 4–10%, see detailed analysis in the ESI† Table S1).
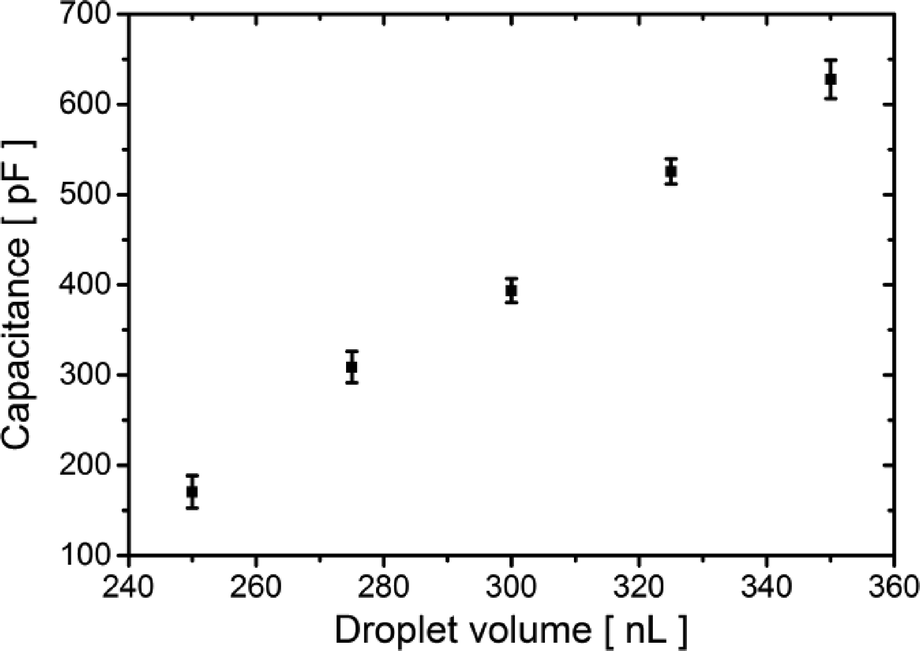 | ||
| Fig. 3 Dependence of the measured bilayer capacitance on the volume of droplets. Each data point represents the average capacitance values of five droplet pairs; error bars represent the standard deviation (SD). The trend in the capacitance reflects the systematic dependence of the surface area of the bilayer on the volume of the droplets. The standard deviation of the recorded capacitance roots in small differences in the positioning of the droplets in the traps. | ||
α-Hemolysin measurements
In order to test the functionality of the DIB we trapped a pair of ~300 nL droplets, one containing a solution of WT α-hemolysin and the second with only the buffer. After formation of a DIB, we conducted a voltage clamp measurement at +50 mV. We observed the incorporation of single heptameric protein pores as stepwise (50 pA) increases in the current. Subsequently, we replaced the buffer droplet with a droplet containing 50 μM γ-cyclodextrin (γCD), which is a reversible non-covalent blocker of the α-hemolysin pore. Pore blockades were observed as transient decreases in the current to ~40% of its open-pore value. Next, we checked whether the residual inhibitor remained on the electrode after the droplet containing γCD was replaced by a droplet with buffer alone. Rare blockades (on average 1.5 events per minute compared to ~80 min−1 prior to washing) were recorded. Repetition of the exchange of the droplet completely eliminated the blockades (Fig. S5†). These results illustrate efficient washing of the electrodes.Control of the surface area
We are able to change the surface area of the DIB formed between the two droplets by varying the rate of flow of oil injected from the perpendicularly oriented additional channel (Fig. 1a, S). This flow pushed the interfaces apart with the magnitude of the effect depending on the rate of flow (Fig. 4). The control over the surface area of the DIB allows us to influence the surface concentration of the inserted pores. After a high number (~45) of αHL pores incorporated into the bilayer, we decreased the contact area. Despite decreasing the area of the bilayer, the current produced by the inserted pores remained the same (Fig. 4d, S8 and S9†). Therefore, the number of functional protein molecules per unit area of the DIB increased.51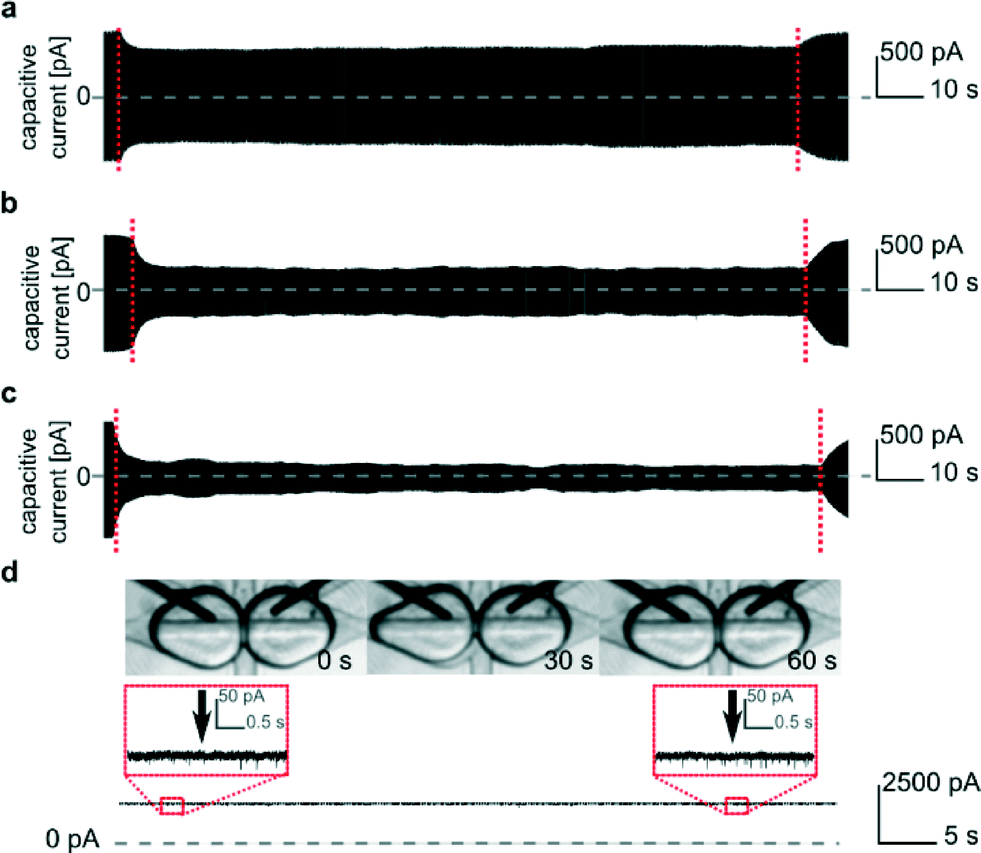 | ||
| Fig. 4 Measurements of capacitive current reflecting the size of the bilayer. The application of a constant flow of oil a) 5 nL s−1, b) 10 nL s−1 and c) 15 nL s−1 results in a decrease in the bilayer surface area. The vertical dotted red lines indicate the moment when the flow of oil was applied and when it was stopped. d) We were able to reduce the surface area of the bilayer from about 85 900 μm2 to 31 400 μm2. The areas were estimated by the analysis of images with ImageJ with the assumption that the contact area is circular. The ion current across the bilayer was constant and equal to ~2330 pA at a potential of +50 mV, which indicated that the number of pores in the membrane remained constant throughout the measurement. The vertical arrows in inset d) indicate the beginning and ending of a period of 45 s during which the droplets were partially separated by the flow of oil (15 nL s−1). | ||
Screening experiment
We demonstrated the ability of our system to perform automated screening. We first prepared a sequence of 12 droplets presenting 6 different concentrations (duplicates of 1, 10, 20, 30, 40 and 50 μM) of the inhibitor γ-cyclodextrin. The droplets were formed inside a 0.3 mm diameter PTFE tube using a syringe pump by alternating the aspiration of portions (300 nL) of aqueous solutions of different concentrations of inhibitor and oil admixed with lipids (700 nL) stored in a standard 96-well plate54 (as shown in Fig. S6†), demonstrating compatibility of the system with standard screening interfaces. Routing of the droplets containing inhibitors on the chip was controlled by optical feedback and external valves.44,45In the screening experiments, we first trapped a droplet containing α-hemolysin heptamers. Then we introduced the first droplet from the sequence of inhibitors. We voltage-clamped the bilayer at +50 mV and performed measurements of the current through the pore. Then we exchanged the droplet containing the inhibitor with a fresh one, presenting a new concentration (preceded by washing the electrode with a droplet of pure buffer). An exemplary trace of 4 subsequent exchanges is depicted in Fig. S7.† It is important to note that the exchange of the droplets disrupts the bilayer and dislodges the protein. As a result, most likely different molecules are studied in the measurements on subsequent droplet pairs. We observed, with a judiciously chosen concentration of protein (5 μg mL−1), clear intervals between incorporation of the first and subsequent pores. This allows for the analysis of kinetics of inhibition at a single-pore level. After the recording, we removed the droplet containing the inhibitor and washed the electrode with a droplet comprising buffer alone generated automatically at a T-junction. The single cycle of removing a droplet, washing the electrode, placing a fresh droplet with a new concentration of the inhibitor and performing the measurements took 210 s. Screening experiments in which we measured the current through the pores in the presence of inhibitors at different concentrations lasted a multiplicity of the interval.
We were able to perform 2 repetitions of the screening with 6 concentrations of the blocker in each screening (Fig. 5a) by using a single droplet containing α-hemolysin. In about 50% of individual DIBs we observed the successful incorporation of at least one pore, which is consistent with the results of a previous study.38 Over time we noticed a decreased rate of pore insertion. We suppose that the decrease in the insertion rate is associated with aggregation of the diluted protein. After the 2 screening repetitions (40 min) we observed almost no insertion of pores within the arbitrarily set interval of 210 s.
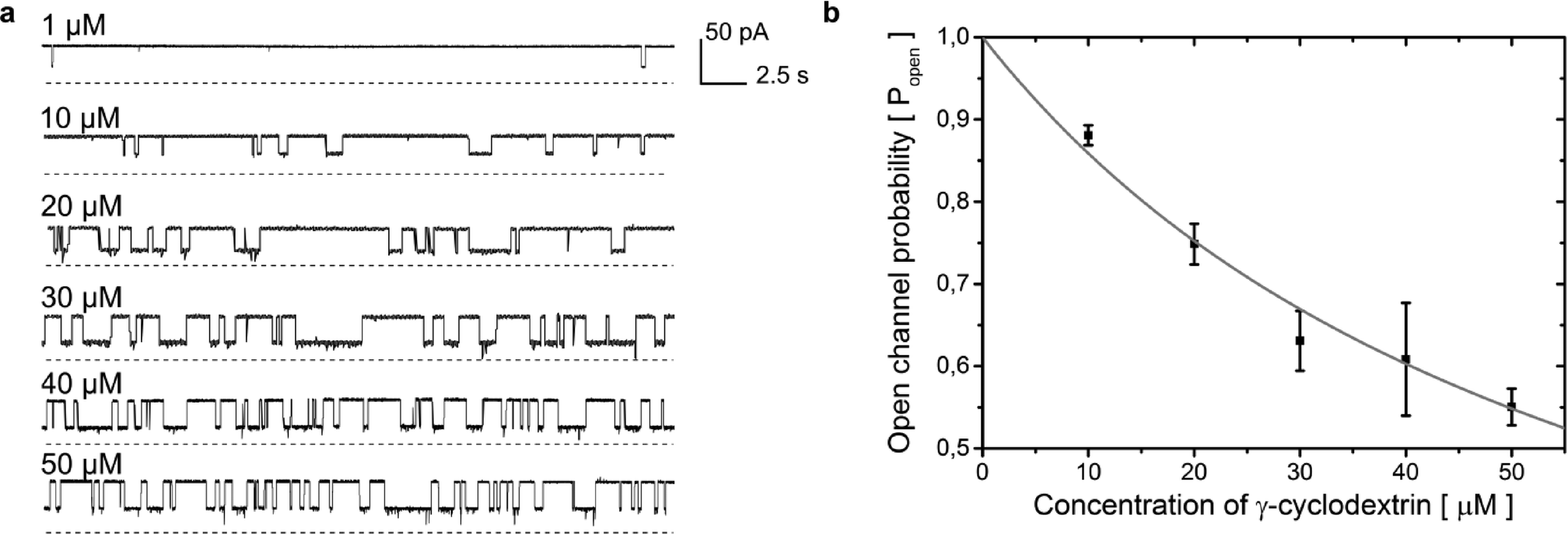 | ||
Fig. 5 a) Rapid screening of inhibitors against a single α-hemolysin channel. Short segments of current traces in the presence of γ-cyclodextrin at various concentrations (1–50 μM). b) Concentration dependence of the inhibition of a single αHL pore by γ-cyclodextrin. Popen was plotted versus the concentration of the blocker (c). The error bars represent the SD from at least three trials. The sum of all the measurement intervals (recordings of current) included in the calculation of the probability of blocking of the pore at each concentration was at least 145 s (see details in Table S2†). The curve was plotted using the saturation function (eqn (2)) and the dissociation constant (Kd) was found to be 61 ± 7 μM. 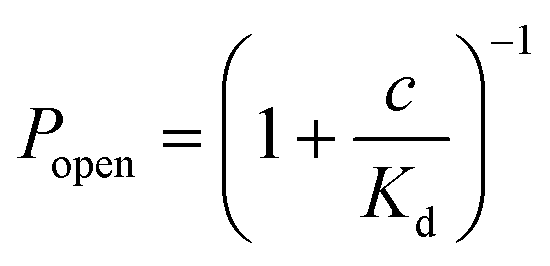 (2). (2). | ||
Our system allows the replacement of the droplet containing the protein with a fresh one. In order to demonstrate this, we repeated the whole screening procedure five times, giving in total ten repetitions (2 repetitions in each screening experiment) of the sequence of six different concentrations (a total of 60 measurements). Using the current traces obtained for 10–50 μM γCD, we analyzed the duration and frequency of blocking and calculated the dissociation constant (Kd) value for the binding of γ-cyclodextrin to α-hemolysin to be 61 ± 7 μM (ref. 55, 47 ± 9; see Fig. 5b and description in the ESI†).
Discussion
We demonstrated for the first time an automated microdroplet system that enables fast, simple and reproducible screening of inhibitors against membrane proteins.Up to now, droplet microfluidics has been used to generate droplet interface bilayers in high yield32,35 but without the ability to perform electrophysiological measurements on samples encapsulated in the droplets. In comparison to high-throughput systems relying on the assay of diffusion of fluorescent dye through the pore,41–43 our system is able to carry out reproducible single-pore electrophysiological recordings with high resolution including the on–off kinetics. The technique that we report here allows for automation of these measurements and for additional operations, such as washing and exchange of both the inhibitor solution and the protein. The exclusion of manual handling of droplets is an important feature that increases the reproducibility of the protocol.
The droplet microfluidic system also allows the use of a minimal volume of sample. In our system this is achieved by the direct aspiration of tiny aqueous droplets (300 nL) from a microwell plate and by modules that accept the microliter batches of liquids for further processing on chip. Our system is open for easy exchange and for automated processing of sequences of samples, an outstanding challenge that inspired a number of studies in the recent years.18–21,23,25,30,36,39
In addition, we demonstrated control of the area of a bilayer, both statically by tuning the volume of the droplets and dynamically by adjusting the rate of flow of an additional stream of oil. Interestingly, the decrease in bilayer area did not result in a decrease in the number of inserted pores, consistent with the results reported by Gross et al.56 In the future, our system may be improved to permit the precise, automated control of the bilayer area and its use in the control of protein density in a bilayer.56
Additionally, the electrodes in our device did not show loss of activity despite their intensive use – including repetitive wetting and de-wetting of electrodes with droplets – during a period of 3 weeks. Neither the layer of agarose gel nor the layer of AgCl on the surface of silver wire was depleted. We did not observe any changes in the performance of the chip over extended periods of operation; in particular we did not observe any wetting of the walls of the channels with the aqueous phase.
The increasing number of studies on various membrane proteins incorporated into droplet interface bilayers10,18–22 suggests that the DIB may constitute an alternative to the BLM method, with the cautionary note that each type of protein must be tested for compatibility with the droplet system.
Experimental
Reagents
1,2-Diphytanoyl-sn-glycero-3-phosphocholine (DPhPC, Avanti Polar Lipids), hexadecane (Sigma Aldrich), silicone oil AR20 (Sigma Aldrich), and γ-cyclodextrin (Cyclolab) were used as received. The buffer for washing electrodes and protein and γ-cyclodextrin dilution consisted of 1 M KCl (Sigma Aldrich) and 10 mM Tris–HCl (Roth), pH 7.0. The lipid solution was prepared by dissolving DPhPC (200 mg) in chloroform (10 mL, Chempur). The chloroform was evaporated under vacuum and the lipid film was re-solubilized in a mixture of 75% (v/v) hexadecane and 25% (v/v) silicone oil AR20.Protein expression and purification
WT αHL was expressed in Staphylococcus aureus, converted to a heptamer with deoxycholate and purified by preparative SDS-polyacrylamide gel electrophoresis as described.57 The stock solution (50 μg mL−1) was kept on ice at all times and diluted 10-fold in buffer immediately before introduction onto the microfluidic chip. The rate of protein incorporation into a bilayer decreased with time. We did not notice any difference in performance when a diluted sample was kept at room temperature (20 °C) for 2 h. We suppose that the decrease in activity is due to the aggregation of protein molecules after dilution.Electrical recordings
Ag/AgCl electrodes were used for electrical measurements. Pieces of silver wire 100 μm in diameter (Sigma Aldrich) were treated overnight (12 h) with sodium hypochlorite solution (Sigma Aldrich). The tips of the electrodes were submerged in melted agarose (1% w/v, Roth) and diluted in 1 M KCl and 10 mM Tris–HCl (pH 7.0) buffer. The AgCl and agarose coatings allowed for sufficient wetting and electrical connectivity when a chip was tested intensively over a period of 3 weeks.An Axopatch200B patch-clamp amplifier (Molecular Devices) was used for recording the electrical current, which was acquired with a 1 kHz low-pass Bessel filter at a sampling rate of 10 kHz. The electrical recordings were post-filtered with a 400 Hz Gaussian filter for the analysis of single channels and capacitive current and with 100 Hz filter for display only.
Microchip fabrication
The polycarbonate chip was fabricated from 5 mm thick plates (Makrolon, Bayer) using a CNC milling machine (MSG4025, Ergwind). The two milled plates were then thermally bonded by compressing them together for 30 min at 130 °C. 11 steel needles (~4 cm long, O.D. 0.82 mm, I.D. 0.65 mm, Fishman Corporation) served as inlets. 7 of them were connected to resistive steel capillaries (O.D. 400 μm, I.D. 205 μm, length 100 cm, Mifam) using segments of Tygon tubing (~2 cm, O.D. 0.91 mm, I.D. 0.25 mm, Ismatec). We used the capillaries to connect the device to the external electromagnetic valves. The use of steel capillaries of high fluidic resistance allows for precise control of the flow on the chip and the performance of iterative operations, including generation of droplets on demand. One of the capillaries was extended with a short piece of HPLC tubing (10 cm, PEEK Tubing 1/16 × 0,0025, Upchurch Scientific) in order to increase the resistance to flow. 2 out of 9 needles were connected through 10 cm long capillaries with 500 μl syringes, each equipped with a built-in valve (1750SL, Gastight Hamilton), which were used to store and introduce onto the chip the pure buffer and the protein solution. 2 of the ports, which served as outlets, were connected with valves via 50 cm PTFE tubing (O.D. 1.6 mm, I.D. 0.8 mm, Bola).We generated the sequence of droplets containing inhibitors off the chip (see the procedure in the ESI†). We then introduced these droplets onto the chip through PTFE tubing, rather than a needle. Therefore, a connection was made directly between the channel and the tubing: 3 cm of PTFE tubing (O.D. 1.0 mm, I.D. 0.5 mm, Bola) was sealed to the chip with Hysol glue.
Calculations of Kd
All measurements of αHL activity were performed at +50 mV. 5 concentrations (10–50 μM) of γCD were used. We discarded the results of αHL blocking by 1 μM γCD since the number of blockades was very low. The protein was contained in a droplet positioned at the working electrode. The buffer was 1 M KCl and 10 mM Tris–HCl, pH 7.0. For more detailed explanation see the ESI.†Conclusions
The system presented here provides numerous opportunities for further development. The use of in vitro transcription and translation (IVTT) for protein expression directly in the droplets may allow avoidance of the steps of purification, freezing and dilution.10 Alternatively the addition of a microfluidic module for the dilution48 of protein stock on chip will allow for more efficient screening protocols. The use of on-demand droplet screening techniques49,52 may provide for fast screening of the composition of buffers and their effects on the function of protein pores. We hope that the technique reported here is a step towards the construction of systems for high-throughput in vitro tests of the activity of membrane proteins, in which sequential screens might be combined with parallelization.19,58,59Acknowledgements
The authors declare no competing financial interest.We thank S. Mantri, L. Höfler, G. Villar, T. Mach, M. Laskowski and L. Kozon for helpful discussions. The αHL was made by E. Mikhailova. The project is co-financed by the European Research Council Starting Grant 279647 (to P.G.) and the European Regional Development Fund under the Operational Programme Innovative Economy within the Foundation for Polish Science grant VENTURES/2012-10/4 (to T.S.K.) and the START fellowship (to T.S.K.). S.J. acknowledges financial support from the Polish Ministry of Science under the grant Iuventus Plus – IP2012 015172. K.T.S. was supported by the Marie-Curie Intra-European fellowship.
References
- J. Dunlop, M. Bowlby, R. Peri, D. Vasilyev and R. Arias, Nat. Rev. Drug Discovery, 2008, 7, 358–368 CrossRef CAS PubMed.
- T. J. Dale, C. Townsend, E. C. Hollands and D. J. Trezise, Mol. BioSyst., 2007, 3, 714–722 RSC.
- M. Montal and P. Mueller, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 3561–3566 CrossRef CAS.
- H. Bayley, B. Cronin, A. Heron, M. A. Holden, W. L. Hwang, R. Syeda, J. Thompson and M. Wallace, Mol. BioSyst., 2008, 4, 1191–1208 RSC.
- L. K. Tamm and H. M. McConnell, Biophys. J., 1985, 47, 105–113 CrossRef CAS.
- E. T. Castellana and P. S. Cremer, Surf. Sci. Rep., 2006, 61, 429–444 CrossRef CAS PubMed.
- L. Tsofina, E. Liberman and A. Babakov, Nature, 1966, 212, 681–683 CrossRef CAS.
- K. Funakoshi, H. Suzuki and S. Takeuchi, Anal. Chem., 2006, 78, 8169–8174 CrossRef CAS PubMed.
- M. A. Holden, D. Needham and H. Bayley, J. Am. Chem. Soc., 2007, 129, 8650–8655 CrossRef CAS PubMed.
- R. Syeda, M. A. Holden, W. L. Hwang and H. Bayley, J. Am. Chem. Soc., 2008, 130, 15543–15548 CrossRef CAS PubMed.
- Y. Schaerli and F. Hollfelder, Mol. BioSyst., 2009, 5, 1392–1404 RSC.
- A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. S. Huck, Angew. Chem., Int. Ed., 2010, 49, 5846–5868 CrossRef CAS PubMed.
- L. Li, D. Mustafi, Q. Fu, V. Tereshko, D. L. Chen, J. D. Tice and R. F. Ismagilov, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19243–19248 CrossRef CAS PubMed.
- E. Brouzes, M. Medkova, N. Savenelli, D. Marran, M. Twardowski, J. B. Hutchison, J. M. Rothberg, D. R. Link, N. Perrimon and M. L. Samuels, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14195–14200 CrossRef CAS PubMed.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004–4009 CrossRef CAS PubMed.
- B. El Debs, R. Utharala, I. V. Balyasnikova, A. D. Griffiths and C. A. Merten, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11570–11575 CrossRef CAS PubMed.
- J. E. Kreutz, L. Li, L. S. Roach, T. Hatakeyama and R. F. Ismagilov, J. Am. Chem. Soc., 2009, 131, 6042–6043 CrossRef CAS PubMed.
- S. Aghdaei, M. E. Sandison, M. Zagnoni, N. G. Green and H. Morgan, Lab Chip, 2008, 8, 1617–1620 RSC.
- R. Kawano, Y. Tsuji, K. Sato, T. Osaki, K. Kamiya, M. Hirano, T. Ide, N. Miki and S. Takeuchi, Sci. Rep., 2013, 3, 1995 Search PubMed.
- A. M. El-Arabi, C. S. Salazar and J. J. Schmidt, Lab Chip, 2012, 12, 2409–2413 RSC.
- S. A. Acharya, A. Portman, C. S. Salazar and J. J. Schmidt, Sci. Rep., 2013, 3, 3139 Search PubMed.
- N. Rojko, B. Cronin, J. S. H. Danial, M. A. B. Baker, G. Anderluh and M. I. Wallace, Biophys. J., 2014, 106, 1630–1637 CrossRef CAS PubMed.
- N. Malmstadt, M. A. Nash, R. F. Purnell and J. J. Schmidt, Nano Lett., 2006, 6, 1961–1965 CrossRef CAS PubMed.
- M. Zagnoni, M. E. Sandison, P. Marius, A. G. Lee and H. Morgan, Lab Chip, 2007, 7, 1176–1183 RSC.
- M. Zagnoni, M. E. Sandison and H. Morgan, Biosens. Bioelectron., 2009, 24, 1235–1240 CrossRef CAS PubMed.
- J. L. Poulos, W. C. Nelson, T.-J. Jeon, C.-J. Kim and J. J. Schmidt, Appl. Phys. Lett., 2009, 95(1), 013706 CrossRef PubMed.
- M. Zagnoni, M. E. Sandison, P. Marius and H. Morgan, Anal. Bioanal. Chem., 2009, 393, 1601–1605 CrossRef CAS PubMed.
- S. A. Sarles and D. J. Leo, Lab Chip, 2010, 10, 710–717 RSC.
- J. L. Poulos, S. A. Portonovo, H. Bang and J. J. Schmidt, J. Phys.: Condens. Matter, 2010, 22, 454105 CrossRef CAS PubMed.
- R. Kawano, T. Osaki, H. Sasaki and S. Takeuchi, Small, 2010, 6, 2100–2104 CrossRef CAS PubMed.
- M. Takinoue, H. Onoe and S. Takeuchi, Small, 2010, 6, 2374–2377 CrossRef CAS PubMed.
- C. E. Stanley, K. S. Elvira, X. Z. Niu, A. D. Gee, O. Ces, J. B. Edel and A. J. deMello, Chem. Commun., 2010, 46, 1620–1622 RSC.
- T. Thapliyal, J. L. Poulos and J. J. Schmidt, Biosens. Bioelectron., 2011, 26, 2651–2654 CrossRef CAS PubMed.
- G. Baaken, N. Ankri, A.-K. Schuler, J. Rühe and J. C. Behrends, ACS Nano, 2011, 5, 8080–8088 CrossRef CAS PubMed.
- Y. Elani, A. J. deMello, X. Niu and O. Ces, Lab Chip, 2012, 12, 3514–3520 RSC.
- V. C. Stimberg, J. G. Bomer, I. van Uitert, A. van den Berg and S. Le Gac, Small, 2013, 9, 1076–1085 CrossRef CAS PubMed.
- M. Lein, J. Huang and M. A. Holden, Lab Chip, 2013, 13, 2749–2753 RSC.
- Y. Tsuji, R. Kawano, T. Osaki, K. Kamiya, N. Miki and S. Takeuchi, Anal. Chem., 2013, 85, 10913–10919 CrossRef CAS PubMed.
- Y. Tsuji, R. Kawano, T. Osaki, K. Kamiya, N. Miki and S. Takeuchi, Lab Chip, 2013, 13, 1476–1481 RSC.
- T. Tonooka, K. Sato, T. Osaki, R. Kawano and S. Takeuchi, Small, 2014, 10(16), 3197–3420 CrossRef.
- M. Urban, A. Kleefen, N. Mukherjee, P. Seelheim, B. Windschiegl, M. Vor der Brüggen, A. Koçer and R. Tampé, Nano Lett., 2014, 14, 1674–1680 CrossRef CAS PubMed.
- I. Kusters, A. M. van Oijen and A. J. M. Driessen, ACS Nano, 2014, 8, 3380–3392 CrossRef CAS PubMed.
- O. K. Castell, J. Berridge and M. I. Wallace, Angew. Chem., Int. Ed., 2012, 51, 3134–3138 CrossRef CAS PubMed.
- M. E. Dolega, S. Jakiela, M. Razew, A. Rakszewska, O. Cybulski and P. Garstecki, Lab Chip, 2012, 12, 4022–4025 RSC.
- S. Jakiela, T. S. Kaminski, O. Cybulski, D. B. Weibel and P. Garstecki, Angew. Chem., Int. Ed., 2013, 52, 8908–8911 CrossRef CAS PubMed.
- Y. Bai, X. He, D. Liu, S. N. Patil, D. Bratton, A. Huebner, F. Hollfelder, C. Abell and W. T. S. Huck, Lab Chip, 2010, 10, 1281–1285 RSC.
- M. Zagnoni and J. M. Cooper, Lab Chip, 2010, 10, 3069–3073 RSC.
- P. M. Korczyk, L. Derzsi, S. Jakiela and P. Garstecki, Lab Chip, 2013, 13, 4096–4102 RSC.
- K. Churski, P. Korczyk and P. Garstecki, Lab Chip, 2010, 10, 816–818 RSC.
- P. H. King, G. Jones, H. Morgan, M. R. R. de Planque and K.-P. Zauner, Lab Chip, 2014, 14, 722–729 RSC.
- S. Thutupalli, J.-B. Fleury, A. Steinberger, S. Herminghaus and R. Seemann, Chem. Commun., 2013, 49, 1443–1445 RSC.
- K. Churski, T. S. Kaminski, S. Jakiela, W. Kamysz, W. Baranska-Rybak, D. B. Weibel and P. Garstecki, Lab Chip, 2012, 12, 1629–1637 RSC.
- L. C. M. Gross, A. J. Heron, S. C. Baca and M. I. Wallace, Langmuir, 2011, 27, 14335–14342 CrossRef CAS PubMed.
- T. S. Kaminski, S. Jakiela, M. A. Czekalska, W. Postek and P. Garstecki, Lab Chip, 2012, 12, 3995–4002 RSC.
- S. Mantri, K. T. Sapra, S. Cheley, T. H. Sharp and H. Bayley, Nat. Commun., 2013, 4, 1725 CrossRef PubMed.
- L. C. M. Gross, O. K. Castell and M. I. Wallace, Nano Lett., 2011, 11, 3324–3328 CrossRef CAS PubMed.
- S. Cheley, L.-Q. Gu and H. Bayley, Chem. Biol., 2002, 9, 829–838 CrossRef CAS.
- B. Lu, G. Kocharyan and J. J. Schmidt, Biotechnol. J., 2014, 9, 446–451 CrossRef CAS PubMed.
- B. Le Pioufle, H. Suzuki, K. V. Tabata, H. Noji and S. Takeuchi, Anal. Chem., 2007, 80, 328–332 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Description of the microchip design and operation, screening experiments, analysis of bilayer size and videos. See DOI: 10.1039/c4lc00985a |
| ‡ M. A. Czekalska and T. S. Kaminski contributed equally to this work. |
| § Present address: Biochemisches Institut, Universität Zürich, Winterthurerstrasse 190, 8057, Zürich. |
| This journal is © The Royal Society of Chemistry 2015 |