
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mesofluidic platform integrating restricted access-like sorptive microextraction as a front end to ICP-AES for the determination of trace level concentrations of lead and cadmium as contaminants in honey†
Alexandra
Sixto
a,
Marta
Fiedoruk-Pogrebniak
b,
María
Rosende
c,
David
Cocovi-Solberg
c,
Moisés
Knochen
a and
Manuel
Miró
*c
aCátedra de Química Analítica, Departamento Estrella Campos, Facultad de Química. Universidad de la República, Av. Gral. Flores 2124, 11800 Montevideo, Uruguay
bDepartment of Chemistry, University of Warsaw, Pasteura 1, 02-093, Warsaw, Poland
cFI-TRACE Group, Department of Chemistry, University of the Balearic Islands, Carretera de Valldemossa, km 7.5, E-07122 Palma de Mallorca, Spain. E-mail: manuel.miro@uib.es; Tel: +34-971172746
First published on 4th November 2015
Abstract
An automatic programmable-flow system capitalizing upon mesofluidic Lab-On-Valve (LOV) sample processing coupled on-line to inductively coupled plasma atomic emission spectrometry (ICP-AES) is proposed in this work as a quality control tool for expedient assessment of potential contamination episodes of food safety elements (namely, Cd and Pb) in a variety of undigested ripened honeys, taken as a model of an edible matrix. On-chip micro-solid phase extraction (μSPE) is effected in a bead-injection disposable sorbent mode using a cationic exchange restricted access-like material (viz., Bond Elut Plexa PCX). We have proven that the lifetime of the miniaturized solid reactor on-chip (11 ± 0.6 mg) is limited to processing less than 6 mL of unfiltered sample suspension (5% (w/w) honey buffered at pH 4.5) after which the analytical performance is severely deteriorated because of strong adherence of the sample matrix to the bead surface. Several physicochemical parameters including the nature of the sorbent material and its ionic form were investigated in detail so as to maximize absolute recoveries and enrichment factors of Pb and Cd. Variables for the elution process were explored by means of a full factorial design. Using 4.0 mL of sample and 200 μL of 3.0 mol L−1 HNO3 as an eluent, enrichment factors of ca. 15 with extraction/elution efficiencies close to 80% and relative recoveries ranging from 90–111% in honey were obtained for both Pb and Cd. The limits of detection (LODs), based on the 3sintercept criterion, were 26 ng g−1 and 68 ng g−1 for Cd and Pb, respectively, which are far below those endorsed by current regulatory agencies (namely, 100 and 300–500 ng g−1 for Cd and Pb, respectively). Demonstrated by the analysis of a suite of off-the-shelf honey brands, the proposed LOV-μSPE platform hyphenated to ICP-AES is deemed suitable for the reliable quantitation of trace level concentrations of Pb and Cd in honey and the detection of heavy metal contamination episodes.
1. Introduction
Honey is a natural sweet substance produced by Apis mellifera bees from the nectar and secretions of plants or from excretions of plant-sucking insects on the living parts of plants.1 Honey possesses important nutritional value because of its elevated sugar content, with about 80% of carbohydrates and electrolytes in percentages from 0.02–0.4%, including Ca, K, Na, Mg, Cu, Mn, Fe and Zn as macro- and micro-nutrients.2,3 The content of metals in honey is greatly variable and relates to the botanical and the geographical origin of floral resources available for bees. Bees are deemed fit-for-purpose environmental biomonitors as a result of exposure to contamination events.4 The occurrence of harmful trace elements in honey at trace concentration levels, viz., lead, cadmium, nickel and mercury to name a few, might be indicative of air, water and soil pollution in the neighborhood of nectar collection, and of potential contamination episodes of hive materials and supplies for feeding bees or in the course of honey processing, packing and storage as well.1 It should be noted that maximum allowed concentrations (MACs) of trace metal contaminants in honey have not been set at the international level, but individual countries or regions have their own regulations. For example, MACs by Mercosur are set to 0.10 and 0.30 mg kg−1 for cadmium and lead, respectively.5,6 The Brazilian Ministry of Agriculture, Livestock, and Supply (MAPA) approved a technical protocol to define the identity of bee products and minimal parameters for their quality control with MACs of 0.1 and 0.5 mg kg−1 for cadmium and lead, respectively.7A great deal of effort has been made over the past few years to evaluate trace metal contamination in honey. As a consequence of matrix complexity, practitioners are commonly calling for appropriate sample preparation procedures involving ashing methods,8 acid digestion (usually using concentrated nitric acid or mixtures of the former with sulfuric or perchloric acids and hydrogen peroxide as the oxidizing agent)9–11 with or without prior heating and sonication for enhanced homogenization, besides alternate procedures encompassing cation-exchange sorptive separations,12,13 UV photolysis14 or microwave-assisted digestion.10,15,16 For example, the French Committee of Accreditation endorses ICP-MS for assays of Pb, Hg, Cd, and As in foods and substances of animal origin-including honey – after microwave digestion.17 Macronutrients in honey might be easily analyzed by flame atomic absorption spectrometry as demonstrated by Pohl and coworkers in a series of publications exploiting fractionation procedures with a variety of sorptive materials18–20 or by appropriate honey dilution and direct injection in inductively coupled plasma (ICP) detectors using dedicated micronebulizers or glow-discharge detectors.21 Caroli and co-workers22 demonstrated that simple dilution with distilled water was proven equally effective than acid-assisted microwave digestion for honey processing and interference-free ICP analysis with negligible cross-contamination effects. Unfortunately, sample dilution fails in assays of trace level contaminants in honey because of the lack of preconcentration and clean-up capabilities. In fact, direct injection methods of ICP are reported to provide biased results for honey for some trace elements, e.g., Pb,23 unless undertaking isotope dilution mass spectrometric assays24 though undigested honey might jeopardise the performance and reliability of the measurements by carry-over effects and/or potential clogging of the capillary in direct injection nebulization.24
In this work, a novel automatic programmable-flow platform is proposed for on-line processing of honey as a front end to inductively coupled plasma atomic emission spectrometry (ICP-AES) as a quality control tool to detect low abundance inorganic contaminants, such as Pb and Cd. Using a restricted-access material (RAM)-like cation exchanger accommodated in a mesofluidic Lab-On-Valve (LOV) manifold configuration,25–28 manual manipulations are kept to minimum while facilitating expeditious on-line sample clean-up and concomitant analyte enrichment with no need for prior sample digestion. On-line fully renewable sorptive RAM-based protocols are also evaluated in a bead-injection configuration26,28,29 using a fresh amount of sorbent in every individual assay so as to avoid potential sample cross-contamination effects, bead surface deactivation and irreversible uptake of honey matrix components. It should be noted that RAM-type mixed-mode sorbents are increasingly attracting interest as advanced copolymeric/hybrid materials for clean-up of honey samples.30–33 To the best of our knowledge, automatic mesofluidic platforms integrating micro-solid phase extraction in a renewable mode for on-chip processing of foodstuff with elevated carbohydrate content aimed at trace metal content assays have not been reported as of yet. A literature survey has in fact revealed that there is merely one paper by Wang and Hansen34 on the coupling of LOV to ICP for the on-line determination of non-radioactive trace elements but environmental samples were in all instances digested prior to analysis.
2. Experimental
2.1. Reagents, sorbent materials and samples
All chemicals were of analytical reagent grade and used without further purification. Ultra-pure water (specific resistivity of 18.2 MΩ cm) obtained from a Milli-Q system (Millipore, Bedford, USA) was employed to prepare all solutions. All glassware and polyethylene containers were previously soaked in 10% (v/v) HNO3 and rinsed with deionized water prior to use.Individual elemental stock solutions of 1000 mg L−1 of Cd and Pb were obtained from Scharlab (Barcelona, Spain). Diluted working standard solutions were prepared in the range of 5–40 and 10–80 ng mL−1 for Cd and Pb, respectively, using matrix matched calibration (5% (w/w) honey at pH 4.5) by using a Cd and Pb-free honey sample.
A stock buffer solution of 0.05 mol L−1 acetic acid/acetate (also used as a carrier) was prepared by adding 0.3 mL of ultra-trace glacial acetic acid (Sigma Aldrich) to 90 mL Milli-Q water to which concentrated ammonia was added dropwise until pH 4.5, followed by making up to 100 mL with Milli-Q water.
Different types of commercially available strong cation-exchanger and chelating materials were tested in this work as sorptive surfaces for sample clean-up and uptake of Cd and Pb from honey. The physicochemical characteristics of the sorptive materials are listed in Table S1.† Dilute solutions of Trace Select grade HNO3 (69%, Sigma Aldrich, St. Louis, MO, USA) were assayed as eluents for on-line stripping of Cd and Pb from the LOV microcolumn.
Five different honey samples were purchased from local markets in Uruguay. Prior to analysis, 5.0 g of honey were dissolved in water with manual agitation, adjusted to pH 4.5, by adding 7.5 mL of 0.05 mol L−1 acetic acid/acetate buffer, and made up to 100 g with water. The honey solution was analysed directly by the LOV-μSPE assembly without prior filtration.
2.2. Analytical instrumentation
A diagrammatic description of the mesofluidic LOV system for the on-line processing and detection of trace level inorganic contaminants in honey is illustrated in Fig. 1.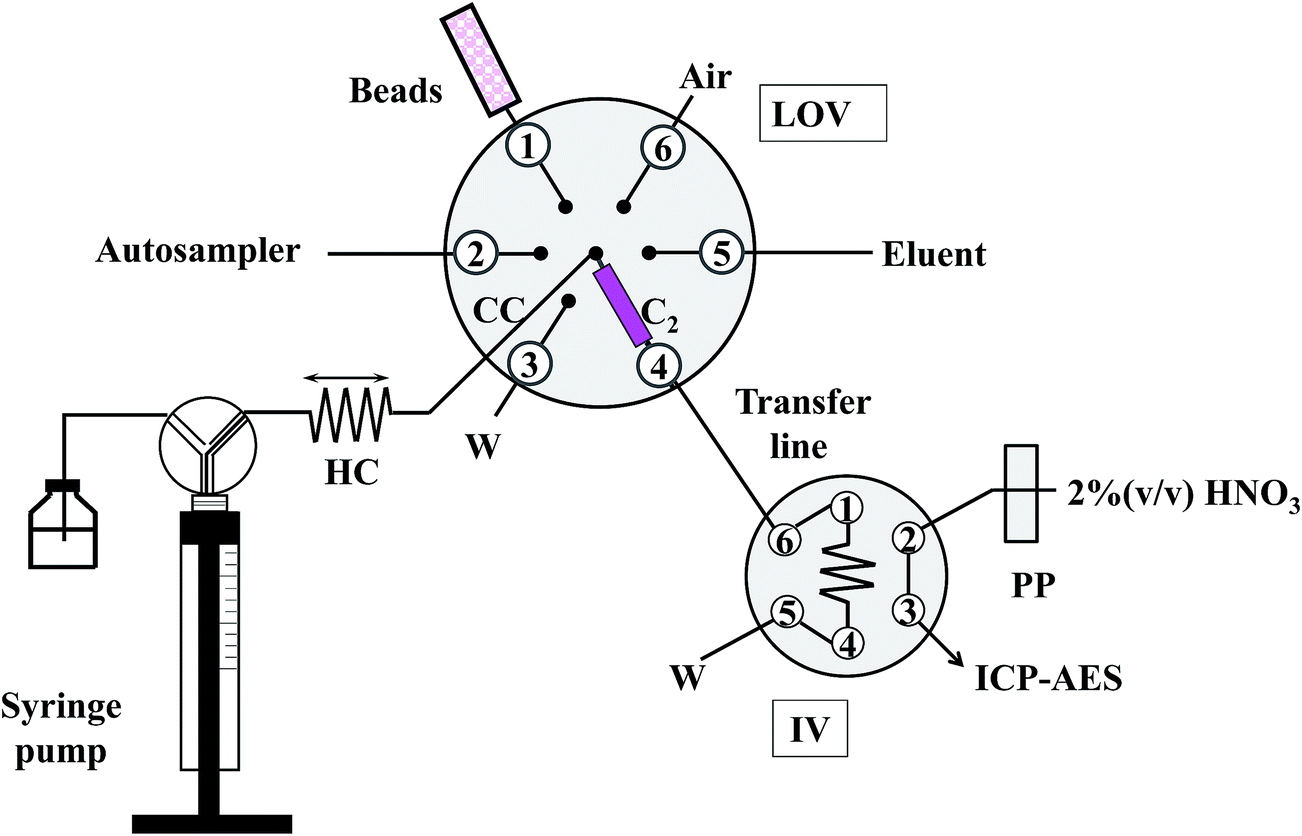 | ||
| Fig. 1 Schematic illustration of the LOV mesofluidic platform integrating RAM-type bead injection μSPE for clean-up and preconcentration of Pd and Cd as contaminants in honey as a front end to ICP-AES (HC – holding coil, PP – peristaltic pump, IV – injection valve, LOV – Lab-On-Valve, W – waste, CC – communication channel, C2 – μSPE column). The IV is activated in the figure to the load position. | ||
The flow manifold consisted of a micro-sequential injection system (μSI, FIAlab, Bellevue, Washington, USA) comprising a 3000-step syringe pump (Cavro, Sunnyvale, USA) furnished with a 5.0 mL gas-tight syringe (Cavro) for automatic fluid handling, a three-way distribution valve at its head, which allowed connection with either the manifold or the carrier (buffer at pH 4.5) solution, and a six-port selection valve on top of which is mounted a mesofluidic conduit platform, the so-called Lab-On-Valve (LOV) unit. The LOV microbore assembly (diameter, 5 cm; thickness, 1 cm) made from hard polyvinylchloride contains a central port which can communicate with the other six conduits (1.66 mm ID/12.0 mm length). The central port in LOV was connected to a holding coil (HC) made of 300 cm × 1.5 mm ID polytetrafluoroethylene (PTFE) tubing. The microchannel connecting the central port with port 4 serves as a conduit for on-chip sorptive microcolumn extraction in which a 1 mm thick polypropylene frit with a pore diameter of 10 μm (MoBiTec, Goettingen, Germany, ref. #2210) is used for retaining the beads while allowing the solution to flow freely. Bead suspensions were prepared by suspending ca. 100 mg of sorbent material in 1 mL of methanol as contained in a 1 mL glass barrelled, Teflon-lined Cavro syringe mounted onto port 1 in the LOV. The outlet of the LOV microcolumn is via a 22 cm × 0.76 mm ID PTFE tubing (transfer line in Fig. 1) allied to a secondary microprocessor-controlled 6-port injection valve (IV, IDEX V 1451 DC, Scivex, Denver, Colorado, USA) furnished with a 55 cm × 0.76 mm ID PTFE loop, which is in turn connected to the inlet tubing of the inductively coupled plasma atomic emission spectrometer (ICP-AES) (Perkin Elmer Optima 5300DV, Waltham, Massachusetts, USA) for on-line eluate analysis. The detector is equipped with a GemCone high dissolved solid nebulizer and a cyclonic spray chamber. The readouts were recorded on-line at 1 Hz with a 250 ms integration time. The area of the transient peak was used as an analytical signal, except for enrichment factor calculations and evaluation of method repeatability. Further operational conditions of the ICP-AES are given in Table S2.†
An XYZ autosampler (AIM3000, Aim Lab Automation Technologies, Brisbane, Australia) equipped with a 60 position sample rack for 12 mL vials is nested to the LOV platform (port 2) for probing of samples and standard solutions unattended.
Cocosoft 4.3. software package written in Phyton35 and the extensions for μSI and IDEX V 1541 DC are utilized in this work for user-friendly control of the syringe pump displacement and flow rate, position of the distribution and injection valves, selection of distinct ports of the LOV platform, and relay activation of the detection instrument (ICP-AES) via the contact closure output of the μSI setup.
2.3. Automated analytical procedure
The bead injection workflow for single-use RAM-type (Bond Elut Plexa PCX) μSPE microcolumn is composed of a first step in which a well-defined air plug (200 μL) is drawn in the holding coil in order to separate the beads from the carrier solution. A measured portion of methanolic bead suspension (50 μL) was whereupon introduced through port 1 and then directed to port 4 in order to pack the column on-chip with a flow rate of 8 μL s−1. In this work, a surplus of beads was brought into the LOV so as to fully load the conduit of port 4 with the sorptive material, the excess of which being discarded to waste. Under the above experimental conditions the LOV microcolumn contained a sorbent mass of 11 ± 0.6 mg (n = 10). Prior to sample loading the LOV microcolumn was rinsed subsequently with 350 μL of 3.0 mol L−1 nitric acid, and 350 μL of 0.05 mol L−1 acetic acid/ammonium acetate buffer (carrier). The syringe pump was then programmed for sample line priming by drawing 500 μL of the next sample. The μSPE protocol in a programmable-flow automatic mode involved the aspiration of 4.0 mL of honey (or alternatively standard solution) in the holding coil between two small segments of air to define a discrete sample segment, and pushed to the microcolumn at 20 μL s−1 followed by column rinsing with 300 μL buffer.The elution procedure was carried out using 200 μL of 3.0 mol L−1 nitric acid at 14 μL s−1 in air-segmentation mode so as to preclude eluate dispersion. The syringe pump was then activated to dispense 900 μL toward the IV so as to fill the injection loop with the acidic eluate, whereupon the ICP-AES was triggered via the relay and the IV activated for eluate analysis. To finalize the bead injection protocol, the spent column was rinsed with buffer solution and the entire volume of beads was aspirated back to the holding coil at 200 μL s−1 and discarded to waste, whereby the LOV system was ready to initiate a new sample analysis. To assure quantitative removal of beads the above cleansing protocols were usually repeated twice. Sample cross-contamination was proven to be negligible by washing the holding coil at 200 μL s−1 with 5 mL of 0.05% (v/v) nitric acid, 20 mL of Milli-Q water and 20 mL of buffer solution (0.05 mol L−1 acetic acid/acetate, pH 4.5), all located in autosampler vials, following each individual series of measurements (calibration and sample cohort analysis). Detailed information of the operational analytical sequence is given in Table S3 in the ESI.†
2.4. Full factorial design
A multivariate procedure36,37 was undertaken for evaluating the effects of distinct variables upon the in-line bead injection-based μSPE-LOV protocol for the uptake preconcentration of Cd and Pb and clean-up of honey samples. The criterion was to maximize the absolute recoveries of both targeted metal species. A two-level full factorial screening design was employed to detect the main factors that significantly influence the dynamic sorptive preconcentration process. The screening design was performed in a dimensionless coordinate system using factor coding. In this factor space, the highest and lowest levels are given as +1 and −1, respectively. The involved coded and uncoded levels for each factor are presented in Table S4.† Three replicates of the center of the design (center point) were also included to ensure that the variability found is on account of the factor effect rather than the random error. Along with the main effects associated with individual factors, the 3 two-term interactions were calculated to explore the potential degree of twisting of the first-order planar model.The statistical computer package StatGraphics Centurion XV (Stat Point Inc., Herndon, VA, USA, 2005) was used to build the two-level factorial design with 11 runs including center points (23 + 3).
3. Results and discussion
3.1. Selection of the sorbent material
Preliminary experiments were undertaken in a batchwise mode so as to assess the sorptive capacity of four different cationic exchange bead materials, namely, Oasis MCX, Dowex 50X8, Plexa Bond Elut PCX, and Chelex 100 for the uptake of low abundance Pb and Cd in honeys. To this end, 5 mL of 5% (w/w) honey sample spiked with known amounts of analytes at the endorsed regulation levels,6 that is, 5 ng mL−1 and 15 ng mL−1 for Cd and Pb, respectively, were loaded through 200 mg of sorbent beds in triplicate assays and eluted quantitatively in 5 mL of 2 mol L−1 HNO3. Both the effluent and eluate were collected and Pb and Cd were measured by ICP-AES.The experimental results compiled in Fig. S1† demonstrate that the retention and elution efficiencies of Cd and Pb under the abovementioned conditions, onto Chelex 100, Oasis MCX and Plexa Bond Elut PCX, were virtually the same at the 0.05 significance level with quantitative uptake and stripping of both targeted elements. On the contrary, functionalized co-polymeric styrene–divinylbenzene ion-exchange sorbents rendered recoveries down to 50%. Chelex 100 is also a cross-linked styrene–divinylbenzene resin but containing anchored iminodiacetate chelating moieties. As a result of the hydrophobic nature of the sorbent bead, slow mass transfer might be expected in dynamic flow-through SPE protocols in aqueous samples. Further, the wide particle size distribution of the chelating resin hinders repeatable handling of the reactive surfaces via programmable flow in a renewable bead-injection mode out and into the LOV system, as a result of which assay repeatabilities (RSD) were above 20%. To circumvent Chelex-100 shortcomings, Bond Elut Plexa PCX and Oasis MCX of hydrophobic/hydrophilic mixed mode nature are deemed suitable alternates for bead injection-LOV procedures because of the narrower particle size distribution, the bead spherical shape and the hydrophilic water rich component of the polymers allowing excellent phase transfer rates. On the other hand, a unique feature of Plexa PCX is the RAM-type nature of the sorbent for retaining small cationic species whilst excluding potential matrix interfering ingredients (e.g., protein traces, disaccharides and other sugars) from accessing the binding sites inside the pore structure. Hereto, the latter sorbent, previously used in the preconcentration of trace metal assays from water samples in a flow-based configuration,38 was selected for the remainder of the studies.
The feasibility of the cationic exchange sorbent for on-chip μSPE was also tested in the NH4+ form by conditioning – prior to use – the sorbent with 350 μL of acetic acid/ammonium acetate buffer (0.05 mol L−1, pH 4.5) inasmuch as previous researchers indicated the suitability of assessing distinct counter ions on sorbent performance in LOV platforms.34,39 The experimental results indicate that better enrichment factors under optimized experimental conditions were encountered for ammonium against oxonium forms. The enrichment factors calculated as the sample to eluate volume ratios for the quantitative retrieval of the target elements were estimated to be 14 and 6.7 for NH4+ and H+ forms, respectively.
Sorbent reusability in LOV was investigated by repeated loading of fortified honeys onto a single sorbent microcolumn followed by metal quantification in every assay. The shelf-life of the miniaturized solid reactor (11.0 ± 0.6 mg) was proven to be down to 10 mL of sample (regardless of the concentration of the targeted species) after which the analytical performance deteriorated significantly (absolute recoveries for Pb and Cd dropped down to 30 and 40%, respectively) because of strong adherence of sugar components from the matrix to the bead surfaces, which resulted in a progressive tighter packing of the sorbent bed and undue flow backpressure effects.
Analyte breakthrough was studied by loading the LOV microcolumn, which contained ca. 11 mg resin with fortified sample (5% (w/w) honey) volumes spanning from 2–10 mL at the 15 and 45 ng mL−1 levels for Cd and Pb, respectively. Analyte breakthrough was observed for both target metals from sample volumes of about 6 mL and onwards, whereby bead-injection protocols with a renewable sorbent material using 4 mL sample (to prevent metal pre-elution in low to moderately contaminated honeys or breakthrough in the case of highly contaminated samples) were used throughout.
3.2. Investigation of experimental variables for dynamic μSPE-LOV assays of Pd and Cd in honey
A two-level full factorial design with three replicates of the central point was used to ascertain those experimental factors with the greatest influence on the μSPE-LOV integrated system for quality control of trace element contamination in honey. Three critical factors related to the elution process, namely, the concentration and volume of nitric acid (eluent), and the elution flow rate were evaluated as experimental variables within the screening design.The eluent concentration domain ranged from 1 to 3 mol L−1, in view of the vulnerability of the LOV platform to elevated concentration of acids. The eluent flow rate varied within the range of 8–20 μL min−1 according to preliminary univariate assays in which the uptake of Cd and Pb was proven in all instances to be quantitative. Higher flow rates are not deemed applicable because of the buildup of unendurable backpressures. The eluent volume domain of 100–600 μL was selected so as to ensure quantitative elution of Cd and Pb without significant deterioration of the enrichment factors. Table S4† lists the experimental design matrix in which the highest and lowest values of each factor and the analytical response, viz., the absolute recoveries of Cd and Pb, are compiled.
Evaluation of the significance of the influence of factors and their second-order interactions on the analytical response was explored using ANOVA.36,40 The standardized factor effects for Cd and Pb can be readily visualized using Pareto charts (see Fig. S2†).
Pareto charts are histograms where the length of every individual bar is proportional to the absolute value of the estimated effect, viz., eluent concentration, eluent flow rate and eluent volume. The cross-vertical line indicates the t-critical value at the 0.05 significance level, corresponding in our case to a t of 2.78 for four degrees of freedom. An effect exceeding this vertical line should be regarded as statistically significant in the mesofluidic μSPE-LOV system for the detection of Cd and Pb in honey. The positive (light grey) or negative (navy blue) bars denote those scenarios where the absolute recovery is improved or reduced, respectively, when increasing the given factor from the lowest to the highest level in the experimental domain.
The Pareto chart for Pb revealed that the eluent concentration was statistically significant at the 0.05 significance level. The higher the eluent concentration the better was the absolute recovery of Pb, indicating a strong binding to the exchange sites within the RAM-type pores. In contrast, the eluent volume and the eluent flow rate and interactions thereof have no influence on the absolute recovery of Pb within the investigated range of experimental conditions. As to Cd, the Pareto graph demonstrated that neither the main factors nor their interactions do affect significantly the elution of Cd from Plexa PCX in an LOV-bead injection mode. These observations led us to conclude that the sorptive behavior of the RAM-type hydrophilic/hydrophobic sorbent resembles that of organic co-polymeric cation exchange resins, for which the selectivity coefficient of Pb is usually superior to that of Cd by 2.5-fold.41
Lack of fit tests for both target elements were undertaken to determine whether the first order model is adequate to describe the observed data or whether a second-order model should be used instead. The tests are performed by comparing the variability of the predicted errors by the current model against the variability between observations at replicate settings of factors, in our case, at the center point level. The p values of 0.51 and 13.18 for Pb and Cd, respectively, which are greater than 0.05, revealed that the first-order model appears to be appropriate to describe the influence of elution parameters in the LOV-bead injection system at the 0.05 significance level with no need to build further second-order multivariate designs.
Taking into account that a further increase of the eluent concentration is not feasible inasmuch as the chemical stability of the flow system components might be jeopardized along with the strong dependence of the eluent volume on the system enrichment factor, a more exhaustive univariate investigation of the effect of the eluent volume on the preconcentration capability of the sorptive procedure was carried out. The experimental range selected in this test was 100–1000 μL, with an eluent concentration and a flow rate of 3 mol L−1 HNO3 and 14 μL s−1, respectively. We have observed that faster flow rates should not be used in combination with eluent concentrations above 2.0 mol L−1 HNO3 because of increased pressure drop in the LOV assembly.
The experimental results revealed that absolute recoveries were in all instances above 60% at the 5 and 15 ng mL−1 levels for Cd and Pb, respectively, (see Fig. 2), which is in good agreement with previous results by experimental design explorations. However, repeatabilities of absolute recoveries, expressed as % RSDs, for a 100 μL-eluent volume were 13.6% and 9.3% for Cd and Pb, respectively, against <6% for both elements for a 200 μL-eluent volume. The latter was thus selected for the remainder of the studies so as to assure Pb and Cd enrichment factors of about 15 (no significant differences in absolute recoveries were found at increasing eluent volumes, see Fig. 2) with a good repeatability and absolute recoveries >75% while precluding the injection of excessive volumes of moderately concentrated nitric acid (ca. 19% (w/v)) into the ICP instrument.
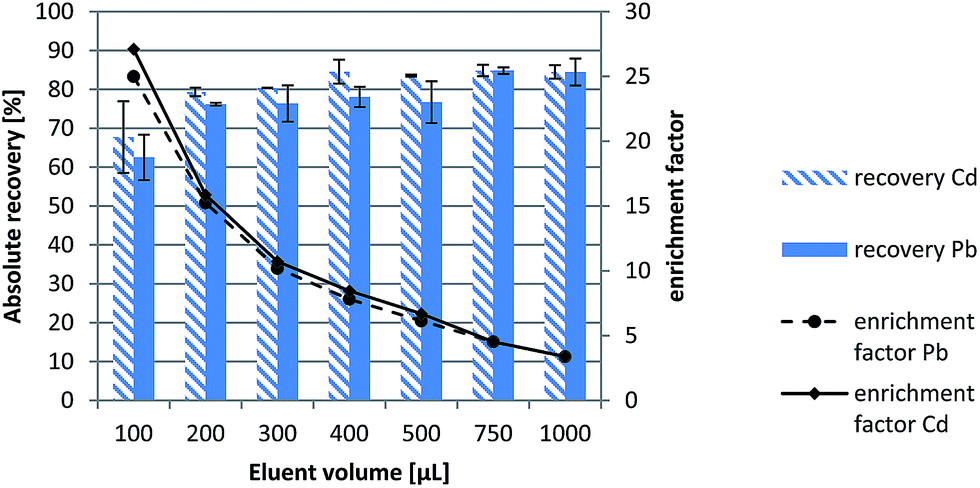 | ||
| Fig. 2 Absolute recoveries (extraction/elution efficiencies) and enrichment factors of Pb and Cd as obtained by μSPE-LOV on the basis of the volume of 3.0 mol L−1 HNO3 used as the eluent. | ||
The feasibility of the miniaturized LOV Plexa PCX-sorptive microcolumn for on-line honey clean-up was systematically assessed by evaluating analyte recoveries in fortified honey by external calibration (in acetic acid/acetate buffer, pH 4.5). To avoid potential differences in the elution profile, the eluent volume was here fixed at 1000 μL. Relative recoveries in honey ranged from 100.3–107.5% and 101.3–116.5% for Cd and Pb, respectively. Therefore, there is no appreciable matrix effect on the sorptive retention by the RAM-type Plexa PCX sorbent in an LOV configuration with quantitative uptake of both metals. However, the binding strength (distribution constant) in a dynamic flow-through mode is found to be matrix-dependent on the basis of differences in the elution profiles for external standards against fortified analyte-free honeys with narrower on-line metal stripping profiles for matrix matched standards. A matrix-matched calibration approach, previously adopted in RAM-SPE procedures for honey samples,32,33 in combination with the optimized elution protocol (200 μL of 3.0 mol L−1 HNO3 at 14 μL s−1) is thus chosen in this work for real-sample analysis.
3.3. Analytical figures of merit and real sample analysis
The analytical performance of the mesofluidic μSPE-LOV system for the sorptive preconcentration and determination of Cd and Pb as potential contaminants in off-the-shelf honey samples is shown in Table 1. The matrix-matched calibration standards were processed in LOV and analysed by ICP-AES using the very same analytical method as for honey samples. A schematic illustration of ICP-AES readouts for on-line analysis of standards and honey samples is presented in Fig. S3.† The detection (LOD) and quantification (LOQ) limits were calculated on the basis of the standard deviation of the regression line intercept, taken as an estimate of the standard deviation of the blank.42–44 The LOQs (see Table 1) are below the regulation limits set for quality control of metal contamination in honeys (100 and 300–500 ng g−1 for Cd and Pb, respectively).5–7 The absolute recovery is calculated as the ratio of the amount of analyte in the eluate under the optimized experimental conditions to the total amount loaded in the LOV microcolumn using a matrix matched standard. The enrichment factor is defined as the product of absolute recoveries and the sample to eluent volume ratio. The repeatability and intermediate precision are calculated as intra-day measurements (n = 8) at the 5 and 15 μg L−1 level for Cd and Pb, respectively, and inter-day sensitivity from the slopes of the calibration curves (n = 6), respectively, using the renewable μSPE-LOV mode.| Parameter | Lead | Cadmium |
|---|---|---|
| Calibration graph: Y as relative intensity (peak area) and X as concentration (ng mL−1) | Y = 251.51X + 436.88 | Y = 1733.70X + 845.06 |
| Correlation coefficient | 0.9968 | 0.9981 |
| Linear range (ng mL−1) | 10–80 | 5–40 |
| LOD (ng g−1) | 68 | 26 |
| LOQ (ng g−1) | 230 | 87 |
| Repeatability (based on peak height) | 5.9% | 6.4% |
| Repeatability (based on peak area) | 9.9% | 5.9% |
| Intermediate precision (based on peak area) | 10.9% | 8.1% |
| Absolute recoveries (%) | 76.0 | 79.5 |
| Enrichment factor | 15.2 | 15.9 |
Notwithstanding the increasing interest and effort in preparing reference materials for trace elements in honey via certification tests,22 to the best of our knowledge, no certified reference material is commercially available from the major manufacturers for food safety elements in honey. A variety of certified reference materials were used incorrectly in the literature to assess the trueness (lack of bias) of new methods for the determination of metal species in honey, e.g. rice flour NIST 1568a,45 tomato leaves NIST 1573a and peach leaves NIST 1547,46 just to name a few. In view of the lack of a certified reference material for trace metal contaminants in honey matrices, the analytical trueness of the dynamic RAM-like μSPE-LOV method was assessed by spike recoveries of five real honey samples at ca. the MAC by current regulatory authorities (viz., 100 and 300–500 ng g−1 of Cd and Pb, respectively).5–7 A second spike at a concentration just below the LOQ of the proposed method (namely, ca. 60 and 200 ng g−1 of Cd and Pb, respectively) was undertaken to further assess the reliability of the proposed method at the low ng mL−1 level of target elements. Experimental data compiled in Tables 2 and 3 indicated that relative recoveries spanned from 90 to 110% and 98 to 111% for Pb and Cd, respectively, with t-calculated values for the overall assays far below the t-critical value at the 0.05 significance level, thereby signaling the reliability and trueness (lack of bias) of the proposed flow method for trace level assays of Cd and Pb in honey samples.
| Honey sample* | Concentration added [μg g−1 Pb] | Concentration found [μg g−1 Pb] | SD (n = 3) | t-calculated** | Relative recovery [%] |
|---|---|---|---|---|---|
| a *Samples were purchased at local Uruguayan markets. For the sake of confidentiality we do not disclose the actual sample trade names. Note that honey no. 2 was contaminated as much as twice the MAC of lead in commercial honeys by Mercosur.6 **t-Critical: 4.30. SD: standard deviation. | |||||
| 1 | 0.000 | <LOD | |||
| 0.233 | 0.231 | 0.004 | 1.03 | 99 | |
| 0.332 | 0.34 | 0.03 | 0.65 | 102 | |
| 2 | 0.000 | 0.76 | 0.05 | ||
| 0.220 | 0.99 | 0.03 | 0.65 | 105 | |
| 0.316 | 1.060 | 0.008 | 2.46 | 96 | |
| 3 | 0.000 | <LOD | |||
| 0.213 | 0.235 | 0.006 | 0.83 | 110 | |
| 0.321 | 0.34 | 0.02 | 1.32 | 106 | |
| 4 | 0.000 | <LOD | |||
| 0.223 | 0.20 | 0.04 | 0.85 | 90 | |
| 0.314 | 0.31 | 0.01 | 0.83 | 99 | |
| 5 | 0.000 | <LOD | |||
| 0.231 | 0.24 | 0.08 | 0.23 | 104 | |
| 0.319 | 0.33 | 0.03 | 0.90 | 103 | |
| Honey sample* | Concentration added [μg g−1 Cd] | Concentration found [μg g−1 Cd] | SD (n = 3) | t-Calculated** | Relative recovery [%] |
|---|---|---|---|---|---|
| a *Samples were purchased at local Uruguayan markets. For the sake of confidentiality we do not disclose the actual sample trade names. **t-Critical: 4.30. SD: standard deviation. | |||||
| 1 | 0.000 | <LOD | |||
| 0.067 | 0.068 | 0.006 | 0.23 | 101 | |
| 0.113 | 0.118 | 0.002 | 3.92 | 104 | |
| 2 | 0.000 | <LOD | |||
| 0.060 | 0.065 | 0.009 | 0.99 | 108 | |
| 0.108 | 0.106 | 0.003 | 1.26 | 98 | |
| 3 | 0.000 | <LOD | |||
| 0.061 | 0.06 | 0.002 | 0.50 | 98 | |
| 0.117 | 0.118 | 0.001 | 0.89 | 101 | |
| 4 | 0.000 | <LOD | |||
| 0.060 | 0.063 | 0.005 | 1.06 | 105 | |
| 0.105 | 0.11 | 0.01 | 0.68 | 105 | |
| 5 | 0.000 | <LOD | |||
| 0.060 | 0.061 | 0.002 | 0.45 | 102 | |
| 0.118 | 0.131 | 0.007 | 3.2 | 111 | |
4. Conclusions
A novel approach based on mesofluidic LOV and on-chip renewable μSPE concepts is herein proposed for automatic handling of honey samples without clogging of the sorptive microcolumn while fostering the identification of potential cases of trace metal contamination. The enrichment factors (ca. 15) and LODs (68 and 26 ng g−1 for lead and cadmium, respectively) of the optimized LOV sorptive preconcentration procedure using RAM-type hydrophilic/hydrophobic cation exchangers do suffice for the sensitive determination of Pb and Cd at concentration levels below those regulated in honey. By user-friendly programmable-flow control of the automatic setup and the autosampler, and activation of ICP-AES using the very same software package, manual sample handling is limited to a mere preparation of a honey suspension (5% (w/w)) in buffer with no need for ashing methods,8 acid digestion,9–11 microwave digestion15,16 or filtration as is the case with previous methods reported in the literature. It should be noted that direct analysis methods involving honey dilution and injection into ICP-based instruments do not usually cope with the sensitivity and reliability required for trace metal analysis23 and assays are usually restricted to inorganic macronutrient detection.21,22 As compared to a recent paper dealing with dispersive liquid phase microextraction (DLPME) for trace metal assays in honey47 our work showcases improved repeatability and intermediate precision values for Pb and Cd (<11% against up to 20% by DLPME47) and superior relative recoveries in honeys ranging from 90–111% against 61–115% by DLPME.47Current work is underway in our lab to extend the bead injection-RAM type LOV protocol to environmental assays for sample clean-up and preconcentration of inorganic and organic pollutants in troublesome matrices as a front end to column separation systems.
Acknowledgements
Alexandra Sixto acknowledges financial support from PEDECIBA and CSIC (Uruguay). The authors are grateful to the Spanish Ministry of Economy and Competitiveness (MINECO) for financial support via projects CTM2014-56628-C3-3-R and CTM2014-61553-EXP. David Cocovi-Solberg extends his appreciation to MINECO for an FPI scholarship. Marta Fiedoruk-Pogrebniak thanks the Faculty of Chemistry, University of Warsaw and the Erasmus + Programme for allocation of a PhD stipendium and mobility grant to visit UIB, respectively. Special thanks are given to Mr Damian Colas from Agilent SA for the gift of Plexa Bond Elut type sorbent materials. Technical assistance provided by Dr José González in setting on-line ICP-AES detection is greatly appreciated.References
- Council Directive 2001/110/EC of 20 December 2001 relating to honey, Offic. J. Eur. Communities, 2002, L10, 47–52.
- P. Pohl, I. Sergiel and H. Stecka, Crit. Rev. Anal. Chem., 2009, 39, 276 CrossRef CAS.
- P. Pohl, TrAC, Trends Anal. Chem., 2009, 28, 117 CrossRef CAS.
- S. Ruschioni, P. Riolo, R. Minuz, M. Stefano, M. Cannella, C. Porrini and N. Isidoro, Biol. Trace Elem. Res., 2013, 154, 226 CrossRef CAS PubMed.
- Codex Stan 12-1981, Codex Standard for Honey, Codex Alimentarius, World Health Organization, Food and Agriculture Organization of the United Unions, 2nd Revision, 2001.
- MERCOSUR/GMC/RES No. 12/11-Technical specifications for maximum allowed inorganic contaminants in foodstuff, Asuncion, Paraguay, 2011, http://www.aladi.org/nsfaladi/normasTecnicas.nsf/09267198f1324b64032574960062343c/13da2f42f7b7219b032579e6005d0fb6/$FILE/Resoluci%C3%B3n%20N%C2%B0%2012-2011.pdf, last accessed date on August 31, 2015.
- MAPA, Brazilian Ministry of Agriculture, Livestock, and Supply, “Instruction No. 17 of 29 May 2013, Control program of residues and contaminants in honey—PNCRC/2013,” IOP Publishing Physics, http://www.agricultura.gov.br.
- O. M. Hernández, J. M. G. Fraga, A. I. Jiménez, F. Jiménez and J. J. Arias, Food Chem., 2005, 93, 449 CrossRef.
- M. Madejczyk and D. Baralkiewicz, Anal. Chim. Acta, 2008, 617, 11 CrossRef CAS PubMed.
- S. Caroli, G. Forte, A. L. Iamiceli and B. Galoppi, Talanta, 1999, 50, 327 CrossRef CAS PubMed.
- N. Czipa, D. Andrási and B. Kovács, Food Chem., 2015, 175, 536 CrossRef CAS PubMed.
- Handbook of Mineral Elements in Food, ed. M. de la Guardia and S. Garrigues, John Wiley & Sons Ltd, Chichester, UK, 2015 Search PubMed.
- P. Pohl, H. Stecka and P. Jamroz, Anal. Methods, 2012, 4, 125 RSC.
- P. L. Buldini, S. Cavalli, A. Mevoli and J. L. Sharma, Food Chem., 2001, 73, 487 CrossRef CAS.
- Z. Ajtony, L. Bencs, R. Haraszi, J. Szigeti and N. Szoboszlai, Talanta, 2007, 71, 683 CrossRef CAS PubMed.
- L. P. Vanhanen, A. Emmertz and G. P. Savage, Food Chem., 2011, 128, 236 CrossRef CAS PubMed.
- S. Millour, L. Noël, A. Kadar, R. Chekri, C. Vastel, V. Sirot, J.-C. Leblanc and T. Guérin, Food Chem., 2011, 126, 1787 CrossRef CAS PubMed.
- P. Pohl, I. Sergiel and B. Prusisz, Food Chem., 2011, 125, 1504 CrossRef CAS.
- P. Pohl and I. Sergiel, Microchim. Acta, 2010, 168, 9 CrossRef CAS.
- P. Pohl and I. Sergiel, Anal. Lett., 2011, 44, 2265 CrossRef CAS.
- K. Greda, P. Jamroz, A. Dzimitrowicz and P. Pohl, J. Anal. At. Spectrom., 2015, 30, 154 RSC.
- S. Caroli, G. Forte, M. Alessandrelli, R. Cresti, M. Spagnoli, S. D'Ilio, J. Pauwels and G. N. Kramer, Microchem. J., 2000, 67, 227 CrossRef CAS.
- M. D. Ioannidou, G. A. Zachariadis, A. N. Anthemidis and J. A. Stratis, Talanta, 2005, 65, 92 CAS.
- A. P. Packer and M. F. Giné, Spectrochim. Acta, Part B, 2001, 56, 69 CrossRef.
- M. Miró and E. H. Hansen, Anal. Chim. Acta, 2012, 750, 3 CrossRef PubMed.
- M. Miró and E. H. Hansen, Anal. Chim. Acta, 2007, 600, 46 CrossRef PubMed.
- Y.-L. Yu, Y. Jiang, M.-L. Chen and J.-H. Wang, TrAC, Trends Anal. Chem., 2011, 30, 1649 CrossRef CAS.
- M. Miró, TrAC, Trends Anal. Chem., 2014, 62, 154 CrossRef.
- M. Miró, S. Kradtap-Hartwell, J. Jakmunee, K. Grudpan and E. H. Hansen, TrAC, Trends Anal. Chem., 2008, 27, 749 CrossRef.
- J. He, L.-X. Song, S. Chen, Y.-Y. Li, H.-L. Wei, D.-X. Zhao, K.-R. Gu and S.-S. Zhang, Food Chem., 2015, 187, 331 CrossRef CAS PubMed.
- Y.-K. Lv, Z.-Y. Guo, J.-Z. Wang, M.-M. Guo, L.-K. Yu and H. Fang, Anal. Methods, 2015, 7, 1563 RSC.
- E. Rodríguez-Gonzalo, D. García-Gómez and R. Carabias-Martínez, Anal. Bioanal. Chem., 2010, 398, 1239 CrossRef PubMed.
- E. Rodríguez-Gonzalo, J. Domínguez-Álvarez, D. García-Gómez, M. G. García-Jiménez and R. Carabias-Martínez, Electrophoresis, 2010, 31, 2279 CrossRef PubMed.
- J.-H. Wang and E. H. Hansen, J. Anal. At. Spectrom., 2001, 16, 1349 RSC.
- D. Cocovi-Solberg and M. Miró, Anal. Bioanal. Chem., 2015, 407, 6227 CrossRef CAS PubMed.
- D. C. Montgomery, Design and Analysis of Experiments, John Wiley and Sons, New York, 7th edn, 2009 Search PubMed.
- L. A. Sarabia and M. C. Ortiz, Response surface methodology, in Comprehensive chemometrics, ed. S. Brown, R. Tauler and R. Walczak, Elsevier, Oxford, UK, 2009, pp. 345–390 Search PubMed.
- A. N. Anthemidis, S. Xidia and G. Giakisikli, Talanta, 2012, 97, 181 CrossRef CAS PubMed.
- J.-H. Wang and E. H. Hansen, Anal. Chim. Acta, 2000, 424, 223 CrossRef CAS.
- B. Dejaegher and Y. Vander Heyden, LC·GC Eur., 2008, 21, 96 Search PubMed.
- Ion Exchange Technology; Advances in Pollution Control, ed. A. K. Sengupta, Technomic Publishing Company, Inc., Lancaster, PA, USA, 1995 Search PubMed.
- D. L. Massart, B. G. Vandeginste, L. M. C. Buydens, P. J. Lewi, J. Smeyers-Verbeke and S. de Jong, Handbook of Chemometrics and Qualimetrics: Part A, Elsevier Science Inc., Amsterdam, The Netherlands, 1997 Search PubMed.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Pearson/Prentice Hall, Harlow, England, 2005 Search PubMed.
- International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use, Validation of analytical procedures: Text and methodology, ICH-Q2B, Geneva, 1996 Search PubMed.
- B. L. Batista, L. R. S. da Silva, B. A. Rocha, J. L. Rodrigues, A. A. Berretta-Silva, T. O. Bonates, V. S. D. Gomes, R. M. Barbosa and F. Barbosa, Food Res. Int., 2012, 49, 209 CrossRef CAS.
- A. B. P. Leme, S. R. Bianchi, R. L. Carneiro and A. R. A. Nogueira, Food Anal. Methods, 2014, 7, 1009 CrossRef.
- F. C. Rosa, F. A. Duarte, J. N. G. Paniz, G. M. Heidrich, M. A. G. Nunes, E. M. M. Flores and V. L. Dressler, Microchem. J., 2015, 123, 211 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ja00387c |
| This journal is © The Royal Society of Chemistry 2016 |