
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First steps towards a generic sample preparation scheme for inorganic engineered nanoparticles in a complex matrix for detection, characterization, and quantification by asymmetric flow-field flow fractionation coupled to multi-angle light scattering and ICP-MS†
S.
Wagner
a,
S.
Legros
d,
K.
Loeschner
b,
J.
Liu
a,
J.
Navratilova
b,
R.
Grombe
c,
T. P. J.
Linsinger
c,
E. H.
Larsen
b,
F.
von der Kammer
*a and
T.
Hofmann
a
aUniversity of Vienna, Department of Environmental Geosciences, Althanstrasse 14, UZA II, 1090 Vienna, Austria. E-mail: frank.kammer@univie.ac.at; Tel: +43-1-4277-53380
bNational Food Institute, Technical University of Denmark, Mørkhøj Bygade 19, 2860 Søborg, Denmark
cEuropean Commission, JRC, Institute for Reference Materials and Measurements (IRMM), Reference Material Unit, Retieseweg 111, 2440 Geel, Belgium
dCIRAD, UPR Recyclage et risque, Avenue Agropolis, F-34398 Montpellier, France
First published on 10th February 2015
Abstract
The applicability of a multi-step generic procedure to systematically develop sample preparation methods for the detection, characterization, and quantification of inorganic engineered nanoparticles (ENPs) in a complex matrix was successfully demonstrated. The research focused on the optimization of the sample preparation, aiming to achieve a complete separation of ENPs from a complex matrix without altering the ENP size distribution and with minimal loss of ENPs. The separated ENPs were detected and further characterized in terms of particle size distribution and quantified in terms of elemental mass content by asymmetric flow-field flow fractionation coupled to a multi-angle light scattering detector and an inductively coupled plasma mass spectrometer. Following the proposed generic procedure SiO2-ENPs were separated from a tomato soup. Two potential sample preparation methods were tested these being acid digestion and colloidal extraction. With the developed method a complete SiO2-ENPs and matrix separation with a Si mass recovery >90% was achieved by acid digestion. The alteration of the particle size distribution was minimized by particle stabilization. The generic procedure which also provides quality criteria for method development is urgently needed for standardized and systematic development of procedures for separation of ENPs from a complex matrix. The chosen analytical technique was shown to be suitable for detecting SiO2-ENPs in a complex food matrix like tomato soup and may therefore be extended to monitor the existence of ENPs during production and safety control of foodstuffs, food labelling, and compliance with legislative limits.
Introduction
Labelling of consumer products containing engineered nanoparticles (ENPs) will be a future legislative requirement in the EU (“EU recommendation on the definition of nanomaterials”, 2011/696/EU) but also in many other countries which develop regulatory approaches for nanomaterials. Analytical methods to detect, characterize, and quantify these ENPs will therefore be required for the implementation and enforcement of such regulations.1 Besides, such methods are also required for the detection and quantification of target ENPs in order to provide empirical data for risk assessments of ENPs released into the environment.2 Generic procedures are not available yet. Therefore, they have to be developed in order to harmonize systematic method development procedures and apply uniform quality criteria for method optimization.The ENPs in consumer products such as personal care products or foodstuffs are usually suspended or embedded in complex matrices containing particles of sizes and/or compositions similar to the ENPs which shall be quantified. Interactions between the matrix components and the ENPs and/or the lack of specificity in measurement techniques prohibit the direct use of available sizing techniques such as nanoparticle tracking analysis (NTA). In order to overcome this problem, von der Kammer et al.3 suggested using a stepwise procedure (including several preparative and analytical steps) to obtain the desired information on particle sizes and concentrations. Following the stepwise procedure the complexity of the sample is decreased during sample preparation by separation of the ENPs from the matrix, without changing the properties of the ENPs. The separation can be based on differences between the chemical and physical properties of the ENPs and those of the matrix constituents. Quantitative information is subsequently required on particle sizes and concentrations (i.e. elemental mass concentration).
This paper extends this stepwise sample preparation by the introduction of quantitative quality criteria and it demonstrates its applicability by means of a case study. In principle this stepwise procedure can be considered as a generic methodology for development of sample preparation methods. The generic sample preparation for separation of inorganic ENPs from a complex matrix was demonstrated for a systematic method development for separation of engineered SiO2 nanoparticles (SiO2-ENPs) from a tomato soup matrix. For subsequent characterization and quantification of the separated SiO2-ENPs a combination of field flow fractionation (FFF) coupled online to multi-angle light scattering (MALS) and inductively coupled plasma mass spectrometry (ICP-MS) detectors was selected. FFF is an analytical separation technique, which is both rapid and non-destructive. For complex samples containing natural nanoparticles FFF has been proven to be a powerful technique4–6 and its application for ENP analysis in food or cosmetics has been shown to be promising7 (TiO2,8,9 Ag,10,11 SiO212). The most widely used FFF technique is currently asymmetric-flow FFF (AF4) that only separates the particles according to their diffusion coefficient or hydrodynamic diameter. Therefore, AF4 is typically coupled with online detectors such as UV-vis spectroscopy, MALS, and/or ICP-MS, in order to obtain information on the concentrations (or other characteristics) of particles eluting from the separation channel.13–16 The presence of large particles (>1 μm) interferes with the desired normal mode of AF4 separation and ENPs attached to large flocks or large particles must be removed from the sample. AF4 therefore requires the ENPs to be separated from the matrix and the extracted ENPs to be stabilized in aqueous suspension. Several proof-of-concept demonstrations have been published for the separation of different inorganic nanoparticles from organic matrices (e.g. from sunscreen or rat lung tissue).8–10,12,17–19 Methods for characterizing TiO2 nanoparticles as an ingredient of sunscreens have been reported.8,9,19 Recovery of spherical SiO2 nanoparticles from rat lung tissue by enzyme digestion was demonstrated by Deering et al.,17 but SiO2 mass recovery was less than 30%. Tadjiki et al.18 reported SiO2 mass recoveries of between 25 and 79% from biological media through acid digestion. SiO2-ENPs as a food additive were separated from coffee creamer by aqueous extraction and subsequent analysis by AF4-ICP-MS revealed possible artifacts due to sample preparation.12 The detection and characterization of Ag-ENPs in complex matrices (e.g. in wastewater) has been addressed by Poda et al.20 and Hoque et al.16 Loeschner et al.10 demonstrated the extraction of Ag-ENPs from chicken meat and their subsequent size separation by AF4. Their work revealed that the retention behaviour of the ENPs could be affected by the sample preparation; in this particular case changes in the surface properties of ENPs resulted in problems during the subsequent analysis by AF4. Most of the reported data does not include any criteria for evaluating the quality of the method presented, or provide independent size information derived from online static or dynamic light scattering measurements following FFF that could validate the size distributions determined by AF4. Only Contado & Pagnoni,8 Loeschner et al.10 and Heroult et al.12 used EM (SEM or TEM) imaging of the eluting particles to verify their separation methods. None of them provided a generic procedure, which would allow translating sample preparation methods to other complex matrices. Therefore, the objectives of this study were (1) to test and verify the applicability of a generic sample preparation procedure to isolate ENPs from a complex food matrices using the case of SiO2-ENPs contained in tomato soup, and (2) to identify and reduce artefacts of the sample preparation on the particle size distribution and particle mass recovery. These objectives were addressed by developing a method for food material, which was produced and carefully characterized in Grombe et al. (2014)21 as a proof-of-concept food reference material containing engineered nanoparticles. This material was tomato soup spiked with SiO2-ENPs. The choice of SiO2-ENPs was based on their practical relevance as an approved food additive (anti-caking agent, E551, EU no. 1129/2011), while the choice of tomato soup was also made on their practical relevance and to provide a complex matrix.
Materials and methods
Chemicals
The Milli-Q water (MQ-water) used throughout the study was prepared using a Millipore Advantage A10 system (Millipore, Billerica, USA) equipped with a Bio-Pak™ ultrafilter (5000 g mol−1 molecular mass cut-off) for final purification. Ammonium carbonate (AC, analytical grade) and sodium chloride (analytical grade) were purchased from Sigma Aldrich. The commercial surfactant mixture used was Fisherbrand™ FL-70™ Concentrate, a biodegradable detergent from Thermo Fisher Scientific (USA, New Jersey). All solutions were pre-filtered using Anodisc 0.02 μm membrane filters (Whatman, Maidstone, UK). The pH values were measured with a Metrohm 6.0234.100 electrode (Metrohm, Switzerland). Different concentrations of NaOH solution (0.01, 0.1, and 1 mol L−1 NaOH) were prepared from NaOH pellets (Merck, analytical grade, USA) and Milli-Q water which were used for pH adjustment. For acid digestion we used 65% HNO3 (Merck, Suprapure®, USA) and 30% H2O2 (Merck, Suprapure®, USA) solutions. For total digestion tests 40% HF (Merck, Suprapure®, USA), 30% HCl (Merck, Suprapure®, USA), and H3BO3 (Merck, ACS reagent, USA) were purchased from Merck.Samples
The method was developed for tomato soup containing SiO2-ENPs. The material was designed and produced by Grombe et al.21 as a proof-of-concept reference material for food products containing ENPs. The material was produced to enable the control of the accuracy of analytical methods for characterization of inorganic ENPs in complex matrices such as food. For the sake of a homogeneous material with a natural composition of the matrix and a stable reference dispersion of the originally added ENPs a number of compromises had to be made. E.g. a liquid sample was produced instead of a powdered food material and a SiO2-ENP suspension (not approved as food additive) instead of a SiO2 powder (approved food additive) was selected as additive to the tomato soup. Detailed information on the sample production and sample characterization are given by Grombe et al.21For development of the sample preparation in this study four types of samples were applied (Table 1). (1) Pure SiO2-ENP suspension (Aerodisp® W7520 N, Evonik (Hanau, DE)) which was used to spike to tomato soup. The initial pure SiO2-ENP suspension was characterized in terms of size and concentration (see ESI part 3†). This sample was used to identify the effect of sample preparation on the particle size distribution. Tomato soup without (2) and with SiO2-ENPs (3) was used to demonstrate the potential of particle matrix separation and the selectivity of the detection method. Tomato soup samples (TS + SiO2-ENPaged) were spiked with the SiO2-ENP suspension approximately one year prior to conducting the experiments, as described by Grombe et al. (2014) (where it is named NanoLyse10), in order to reflect realistic conditions since it is usually “aged” samples that are of interest in food control. (4) Blank tomato soup was spiked with a known amount of SiO2-ENPs prior (ca. 30 minutes) to the experiment (TS + SiO2-ENP), using SiO2-ENPs from the same batch as used in (3) in order to identify effects of the ageing on the sample preparation procedure. Additionally, blank tomato soup samples were run in parallel in order to determine the background level of SiO2-ENPs. The organic carbon concentration in all samples (except the pure particle suspension) was similar to that in the TS + SiO2-ENPaged sample. All samples were stored at 4 °C until analysis.
| Sample type | Abbreviation | c initial(SiO2) [g L−1] | Description |
|---|---|---|---|
| 1. SiO2-ENP suspension in pure water (pH = 8) | SiO2-ENPs | 40.4 ± 0.6 | No tomato soup matrix |
| 2. Pure tomato soup | TS | 0.23 ± 0.02 | Blank sample of tomato soup |
| 3. Tomato Soup spiked with SiO2-ENPs (aged) | TS + SiO2-ENPaged | 17.5 ± 2.3 | Spiked with SiO2-ENPs about 12 months prior to experiment |
| 4. Tomato Soup spiked with SiO2-ENPs (fresh) | TS + SiO2-ENP | 20.2 ± 0.6 | Spiked with SiO2-ENPs immediately prior to the experiment |
Generic sample preparation procedure
The tested generic procedure was based on von der Kammer et al.3 and claims that ENP matrix separation can be achieved by stepwise sample preparation. The generic procedure was used in this study for the optimization and development of a sample preparation method for separation of SiO2-ENPs from a food matrix (tomato soup). For this purpose additional quality criteria such as recovery and particle size distribution were included in the generic procedure in order to evaluate the development and optimization of the sample preparation. Besides the optimization of the sample preparation for separation of ENPs from the complex matrix the generic procedure includes tests with pure ENPs in order to identify possible alteration of the ENP size distribution due to the preparation procedure. The selected example of SiO2 in tomato soup is regarded as a first proof-of-concept for this generic sample preparation procedure (Fig. 1). The procedure involved four steps prior to AF4 analysis. These steps and the quality criteria can be considered as generic. However, in each step various treatments were tested and optimized based on test criteria which are described in detail in the ESI part 1†. These treatments are sample specific and have to be selected for depending on the properties (e.g. liquid or solid) of a sample. Fig. 1 summarizes the treatments which were tested for the separation of SiO2-ENPs from tomato soup. To improve readability of the work, detailed descriptions of these treatments and their optimization were presented in the ESI (part 2†).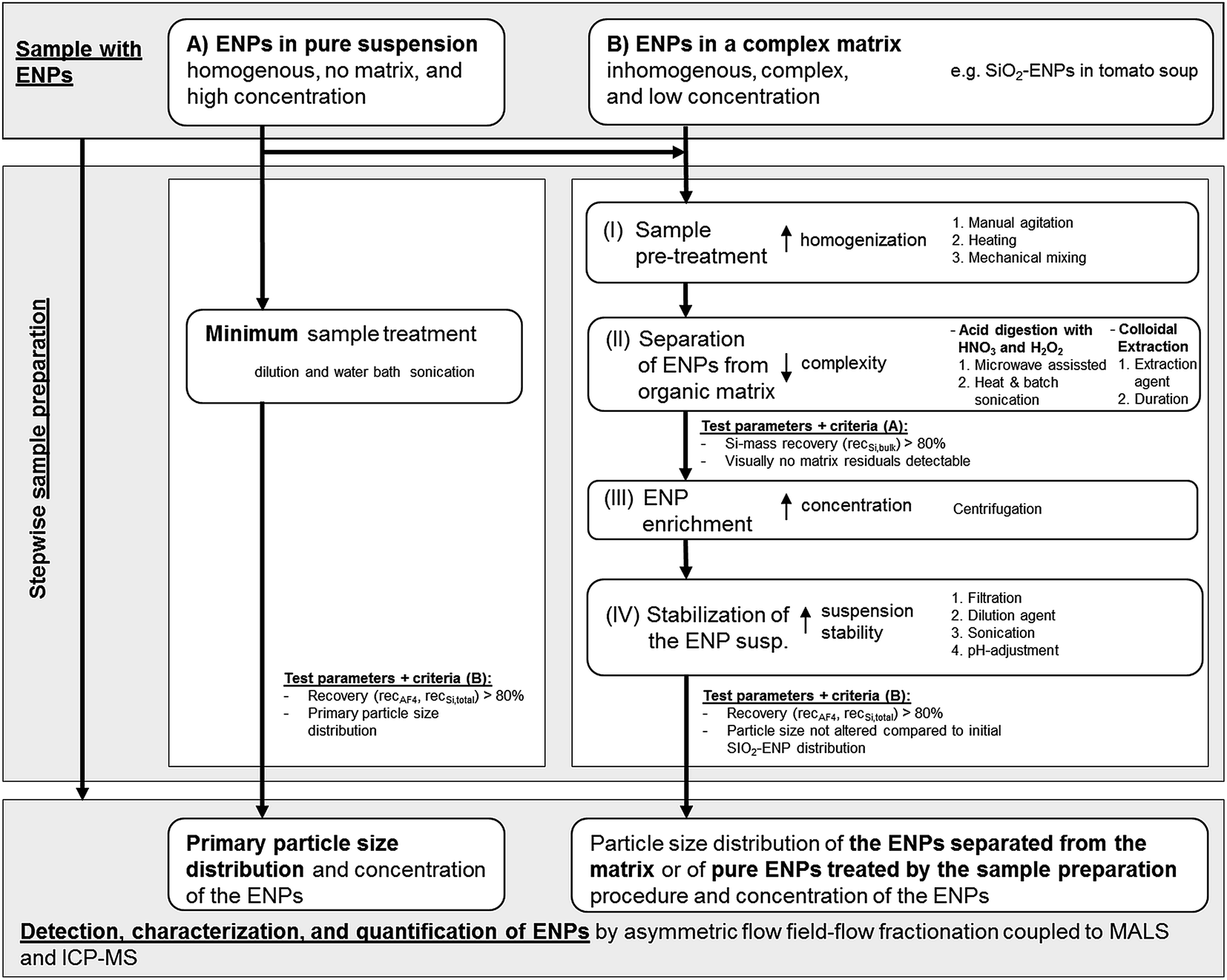 | ||
| Fig. 1 Generic multi-step procedure for development of a sample preparation method to extract ENPs from a complex matrix. Specific details for the example separation of SiO2-ENPs from tomato soup are given on the right side of the scheme (numbered sub-steps can be performed as stand-alone or in combination with other listed sub-steps). | ||
Step I: homogenization of the sample. The effects of manual agitation, heating to 50 °C for 30 minutes, and mechanical mixing were tested.
Step II: ENP separation from the matrix. Both acid digestion and colloidal extraction were investigated for the removal of the organic matrix. Based on physicochemical properties of SiO2-ENPs and the tomato soup matrix both methods are potentially suitable to fully separate SiO2-ENPs and tomato soup matrix. In case of ENPs (e.g. Ag ENPs) which are not stable at acidic conditions acid digestion would not be a suitable separation method. The efficiency of the sample preparation was evaluated after step II (test criteria A in Fig. 1). This evaluation was based on the calculation of bulk Si mass recovery (recSi,bulk see ESI part 1† for detailed calculation) and the particle separation efficiency from the matrix. Sample preparation only continued if both criteria matched (see Fig. 1).
Step III: ENP enrichment. This step was required to increase the ENP concentration in order to obtain particle mass concentrations, which were suitable for the subsequent analysis by AF4 coupled to MALS and ICP-MS detectors.
Step IV: ENP stabilization. Particles had to be stabilized in order to avoid aggregation, which would affect the particle size distribution. Subsequently, the stabilized particle suspension was characterized using AF4 coupled to MALS and ICP-MS detectors. Since details of the analytical method development has been described in von der Kammer et al.,22 herein only the conditions are described. The efficiency of the total sample preparation was evaluated after step IV (test criteria B in Fig. 1). This evaluation was based on the particle size distribution, and the calculation of Si mass recovery of the entire sample preparation (recSi,total). For the example of SiO2-ENPs separated from tomato soup, it was decided to additionally determine the recovery of the AF4 separation method (recAF4) based on the unspecific light-scattering signal in order to provide a measure for the quality of the separation which can be obtained easily (without ICP-MS instrument calibration which saves significant analysis time and resources). This approach, however, was only valid because the light scattering signal from a blank tomato soup (no SiO2-ENPs were spiked) after extraction by acid digestion did not indicate the presence of any particles. In case particle impurities can be expected in the sample, it is recommended to calculate the AF4 recovery not based on the MALS signal but on the element specific ICP-MS signal. Detailed calculation of recSi,total and recAF4 are provided in ESI part 1.†
The application of the generic sample preparation procedure and its quality criteria requires knowledge about the target ENP (i.e. compound, size, and possibly concentration). In case these parameters are not know, which would be true for unknown ENPs, the effect of the sample preparation on the ENP size distribution cannot be identified based on the generic sample preparation. To identify and quantify “unknown” ENPs in a complex matrix an adapted generic sample preparation procedure has to be applied, which e.g. considers unique features of the target particles (e.g. elemental ratios, or homogeneity in elemental composition compared to matrix components).
Measurements and instrumentation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:2:1 (sample:HCl:HNO3:HF) followed by complexation of the remaining HF with H3BO3 (350 mg boric acid/15 mL of MQ-water).
:4:2:1 (sample:HCl:HNO3:HF) followed by complexation of the remaining HF with H3BO3 (350 mg boric acid/15 mL of MQ-water).
| Unit | Value | |
|---|---|---|
| a Size calibrations of the AF4 channel were performed under similar run conditions, with the only exception being for a carrier composition of 0.025% (v/v) FL-70™ and 3 mmol L−1 NaCl. As already pointed out by Neubauer et al.6 in case that no particle size reference material of similar composition as the sample is available it might be necessary to run the AF4 calibration with a different carrier composition as the sample. The mass of injected polystyrene latex beads (PS size standards 50, 100, and 150 nm) was 0.5, 0.25, and 0.1 μg, respectively. | ||
| AF 4 | ||
| Tip to tip channel length | [cm] | 27.5 |
| Spacer | [μm] | 250 |
| Focus flow rate | [mL min−1] | 0.75 |
| Injection flow | [mL min−1] | 0.1 |
| Injection time | [min] | 10 |
| Focus time | [min] | 2 |
| Elution time | [min] | 35 |
| Detector flow rate | [mL min−1] | 1 |
| Cross flow rate | [mL min−1] | 0.75 |
| Membrane | Regenerated cellulose, 10 kDa, Nadir | |
| Carriera | Mixture of 0.025% (v/v) FL-70™ and 0.25 mM NaCl | |
| Injection massa | [μg] | 5 |
|
||
| ICP-MS parameters | ||
| RF power | [W] | 1600 |
| Sample depth | [mm] | 10 |
| Gas flow rates | ||
| Carrier | [L min−1] | 1.06 |
| Dilution | [L min−1] | 0.35 |
| Collision gas He | [mL min−1] | 4.0 |
| Sample uptake rate | [mL min−1] | 0.3 |
| Nebulizer | MICROMIST (glass expansion) | |
| Spray chamber | Scott double-pass | |
| Isotopes monitored | 28Si | |
| Dwell time | [ms] | 100 |
The size distributions were evaluated using the modes and the medians (d50) of the distributions. A mode/median ratio (peak shape factor) <1 indicates a tailing of the size distribution, while a ratio >1 indicates a fronting of the distribution. Where the ratio is equal to 1 the distribution is symmetric. The mode/median ratios were calculated for each sample and compared with each other. The independent determination of particle radii using MALS and hydrodynamic radii by AF4 size calibration allowed us to calculate the ratio of the rrms to rh. This ratio is a direct expression of particle shape.23 A solid, homogeneous, spherical shaped particle has an rrms/rh ratio of 0.775. Any deviation from such a spherical particle shape would cause the rrms/rh ratio to increase up to a maximum of 1 for oblate spheroids, and to a maximum of 2 for prolate spheroids (at an 1/100 aspect ratios).
The ICP-MS measurements were calibrated using dissolved Si standards. According to Prestel et al.,25 SiO2-ENPs smaller than 500 nm are completely ionized within the plasma. By comparing the ICP-MS 28Si signal intensities for 100, 500, and 1000 nm SiO2-ENPs (Postnova, Landsberg am Lech, Germany) at identical mass concentrations (see ESI part 6†), even 1000 nm SiO2-ENPs were shown to be quantitatively detected by the ICP-MS system used in this study. A background mixture of 0.025% (v/v) FL-70™ and 0.25 mmol L−1 NaCl were used during Si calibration of the ICP-MS in order to take into account possible interferences and matrix effects arising from the organic carbon content of the AF4 carrier mixture when it contained FL-70™ surfactant. The Si calibration range was between 5 and 200 μg L−1. The ICP-MS calibration was recorded using the full quantitative mode (R2 = 0.999). Instead of using an internal standard the calibration was repeated at regular intervals following the sample analysis in order to check for any loss of sensitivity in the detection system. The detection limit (3× standard deviation of blank run) for Si analysis by ICP-MS was 2.60 μg L−1 (or 1.3 × 10−4 μg 50 μL−1) in the measured solutions. The limit of quantification was 26 μg L−1 (10× standard deviation of blank run).
Results and discussion
From the regulatory point of view the analytical methodology has to provide size and concentration data of the primary ENPs added to the matrix of interest (e.g. foodstuff, information provision EU 1169/2011 and cosmetics, product regulation EU 1223/2009). Therefore, the developed method must be able to extract the particles without introducing artefacts by the sample preparation procedure, and be independent of any ageing of the ENPs in the complex matrix. The method development procedure must allow the identification of alterations of the ENPs concentration. Since current regulations demand number-based size distributions and the analytical methods applied in this study provide a mass-based particle size distribution a conversion algorithm has to be used to calculate number-based size distribution from mass-based input data. This conversion would result in false size distributions if the mass based signal is affected by artefacts from the sample preparation. Future work needs to focus on possible conversion algorithms and the error-prone of such conversions.In the framework of the generic sample preparation many alternative sample preparation procedures were tested (Fig. 1). However, in the following section only the optimized sample preparation procedure is presented in detail i.e. both test criteria (A) and (B) were achieved and it is demonstrated which parameters had the most significant impact on Si bulk mass recovery or particle size distribution. Details on preparation procedures which did not pass the test criteria are summarized in the ESI part 4 and 5.† Main results and conclusions are shortly summarized at the end of this section.
Si mass recovery after step II (test criterion A)
For example in step II, several types of colloidal extraction, acid digestion assisted by heat and sonication (as described in Tadjiki et al.18) and acid digestion achieved by applying microwave-assisted pressurised digestion were evaluated. Prior to the extraction the tomato soup sample was pre-treated by heating and manual agitation. It was found that microwave-assisted pressurised acid digestion results in higher recovery rates (recSi,bulk > 90%) and a more complete separation of SiO2-ENPs from the tomato soup matrix compared to colloidal extraction (recSi,bulk < 15%) (see ESI part 5†). However, acid digestion assisted by heat and sonication was not able to completely remove the tomato soup matrix, this was only achieved by applying microwave-assisted pressurised digestion (see ESI part 4†). Therefore, only microwave assisted acid digestion in combination with the various sample pre-treatment procedures (Fig. 1, step (I)) was tested in order to identify the optimum combination of pre-treatment procedure which yield maximum recovery and minimum alteration of the particle size distribution. For these tests the pristine particle suspension in MQ-water (SiO2-ENPs) and freshly spiked and aged SiO2-ENPs in tomato soup were deployed. The pristine SiO2-ENPs sample was included in the tests as a control, in order to understand the effect of sample preparation on the particles. The Si bulk recovery for SiO2-ENPs was usually greater than 85% (Table 3) for all of the pre-treatment procedures tested. Similar results were obtained for tomato soup freshly spiked with SiO2-ENP (TS + SiO2-ENP), which yielded recSi,bulk greater than 80% for each of the pre-treatment procedures. However, for the aged soup (TS + SiO2-ENPaged) the recSi,bulk dropped to less than 10% when the sample was only agitated manually prior to acid digestion (procedure I.1 in Table 3). It only exceeded 50% when the sample pre-treatment also included heating of the sample at 50 °C for 30 minutes and mechanical homogenization (procedures I.2 and I.3 in Table 3) prior to acid digestion. The differences in recovery between the samples TS + SiO2-ENP and TS + SiO2-ENPaged was likely to be due to the longer contact time between the SiO2-ENPs and the tomato soup matrix in the aged samples (more than a year, compared to a few hours) causing changes in the ENP interaction with the matrix (organic fibers) or a change in the ENP surface properties. These changes in surface properties may have resulted in the formation of ENP aggregates or agglomerates greater than 1 μm, which were not available for ICP-OES analysis due to settling. The presence of large particles was suggested by qualitative DLS analysis, which indicated the presence of particles >3 μm. This effect was however not further investigated because it was beyond the scope of this study. A further increase in recSi,bulk from 52% (I.2 + I.3) to 93% was achieved when additional tip sonication (I.2 + soni) of the particle suspension was applied after the acid digestion. The procedure I.2 + I.3 + soni was selected to provide sufficiently homogenized samples for sample preparation steps (III–IV).| Sample | Pre-treatment | c(Si) [mg L−1] | recSi,bulk [%] |
|---|---|---|---|
| a I.1: manual agitation; I.2: heating for 30 min; I.3: mechanical homogenisation; +soni: additional tip sonication of the sample prior to ICP-OES analysis. | |||
| SiO2-ENP | I.1 | 16.6 ± 4.1 | 86 ± 22 |
| I.2 | 17.4 ± 1.3 | 96 ± 9 | |
| I.2 + I.3 | 20.4 ± 1.8 | 104 ± 9 | |
| I.2 + soni | 21.2 ± 0.3 | 114 ± 2 | |
| I.2 + I.3 + soni | 15.7 ± 0.8 | 84 ± 4 | |
| TS + SiO2-ENP | I.1 | 14.5 ± 2.6 | 78 ± 14 |
| I.2 | n/a | n/a | |
| I.2 + I.3 | 17.7 ± 2.8 | 95 ± 15 | |
| I.2 + soni | 21.8 ± 0.2 | 117 ± 3 | |
| I.2 + I.3 + soni | 16.8 ± 1.5 | 90 ± 8 | |
| TS + SiO2-ENPaged | I.1 | 1.3 ± 0.4 | 8 ± 2 |
| I.2 | 7.1 ± 0.3 | 44 ± 2 | |
| I.2 + I.3 | 8.0 ± 1.0 | 52 ± 6 | |
| I.2 + soni | 15.2 ± 0.9 | 93 ± 5 | |
| I.2 + I.3 + soni | 13.2 ± 1.2 | 81 ± 7 | |
Colloidal extraction aims at separating ENPs and matrix components by physical separation e.g. by centrifugation or filtration. Separation of SiO2-ENPs from tomato soup resulted in lower recoveries and incomplete separation of ENPs and matrix compared to microwave assisted digestion. Silica recovery after colloidal extraction without any sample pre-treatment (I.1), recSi,bulk values were greater than 85% from both SiO2-ENPs and TS + SiO2-ENPs samples for all of the extraction agents tested (see ESI, section 5.1†). There was virtually no recovery (1 ± 1%) from TS + SiO2-ENPaged samples with extraction for 30 min by MQ-water. In order to improve the Si mass recovery from TS + SiO2-ENPaged the extraction period was extended to 72 hours, but the maximum recSi,bulk (20%) was already reached after 16 hours of agitation in 0.25 mM AC solution. Sample pre-treatment prior to liquid extraction was optimized through the use of mechanical homogenization (I.2) and heat treatment (I.3). Si mass recoveries from TS + SiO2-ENPaged increased to 40 ± 9% after applying the I.2 pre-treatment procedure. Where fatty constituents were dissolved or dispersed in the aqueous solution by the application of heat (I.3), the Si mass recovery was 10 to 40% lower than for the unheated sample. The surface area of the boundary layer between water and non-aqueous solution increased during heating, and particles tended to accumulate at this boundary or even to migrate into the fatty phase due to their hydrophobic properties. A well separated fatty phase reformed during the extraction, which was carried out at 20 °C. A considerable quantity of SiO2-ENPs may remain at this boundary or within the fatty phase (which was not subsequently sampled), resulting in significantly lower recoveries. Generally, colloidal extraction yielded significantly lower Si mass recoveries and incomplete separation of SiO2-ENPs and matrix (criteria A, for details see ESI, part 5†).
Particle concentration enrichment (step III)
Since AF4 separation has a broad operating range in terms of particle concentration, particle enrichment is only necessary for low concentrated samples. Particle enrichment can be achieved e.g. by centrifugation or cloud point extraction. Despite the high enrichment factors (up to 100) which can be achieved by cloud point extraction this methodology is strongly influence by matrix components and particle surface properties.26 Therefore, it was not applied to enrich SiO2-ENPs concentration, but it might be considered for other particle types and matrices. In the case of SiO2-ENPs, enrichment of the particle concentration (III) was done immediately after microwave digestion by centrifugation (4500 rpm, 15 min) in order to reach concentrations which were suitable for further AF4-MALS-ICP-MS analysis. The analysis of Si concentration in the supernatant and in the residual indicated that SiO2-ENP concentration could be increased by the factor of 2.4 in the remaining solution, without significant loss of particles in the supernatant (<5% of the total SiO2-ENP mass). However, particle enrichment by centrifugation introduces the risks of particle loss, due to incomplete sedimentation, or particle alteration. Considering that the enrichment step only increased the concentration by the factor of 2.4 alternatively the amount of sample injected in the AF4 system could be increased. The AF4 system equipped with the large volume injection loop allows injection volumes that range between 0.1 and 900 μL. An increase of the injection volume of the sample by the factor of 2.4, which means an injection of 120 μL instead of 50 μL, would substitute the particle enrichment by centrifugation. Increasing the injection volume results in both, a higher load of ENPs of interest as well as a higher load of possible remaining particles originated from the matrix. Generally, it is of course intended to remove most of the matrix components from the sample during sample preparation in order to avoid the injection of matrix components into the AF4 channel. In case of SiO2-ENPs in tomato soup it was demonstrated that blank tomato soup (no SiO2-ENPs) did not exhibit any significant MALS signal after microwave assisted acid digestion (data not shown). The required pre-concentration can also be estimated by simple calculation which is done in the following for the sample TS + SiO2-ENPaged. For the suggested analytical procedure a SiO2-ENPs concentration of >50 mg L−1 was required in suspension. The initial SiO2-ENPs concentration in the presented example was 17.5 g L−1 (Table 1). Without particle enrichment (step III) this concentration was reduced by a factor of 500 during the sample preparation and stabilization (dilution factors: microwave assisted acid digestion 1:50; stabilization 1:10, see ESI part 2.2 and 2.5†) resulting in a concentration of 35 mg L−1. For quantification of SiO2-ENPs slightly higher SiO2 concentration were required. Therefore, an increase in concentration or injection volume by the factor of 2 would result in sufficiently high SiO2-ENP concentration (70 mg L−1) for detection by MALS and ICP-MS.
Particle size distributions after step IV (test criterion B)
Several authors8–10,19 previously stated that the final measured particle size distribution is strongly dependent on the sample preparation procedure and results presented herein support this statement. It is, however possible to minimize the effect by careful development of the sample preparation procedure, especially with respect to particle stabilization. A sequence of treatment steps is required in order to obtain an unaltered stable particle suspension for AF4 separation and analysis. These steps (IV.2, IV.3 and IV.4) were essential in order to break down aggregates that were formed during digestion and to produce a particle suspension that would be stable for several days. After acid digestion the matrix was completely removed and filtration as suggested in Fig. 1 could be omitted. The acid digested sample was stabilized by pH adjustment in the range between 8 and 9 which equals the pH range of the original SiO2-ENP suspension.21 Furthermore, dilution in a suitable dilution agent was necessary (e.g. 0.025% FL-70™ as detergent or 0.25 mM ammonium carbonate as a buffer medium) in order to adjust the ionic strength. The authors refer to the ESI part 2† which depicts each single optimization step according to Fig. 1.The described sample preparation procedure and subsequent analysis were applied to SiO2-ENP, TS + SiO2-ENP and TS + SiO2-ENPaged samples. Resulting size distributions were compared to the size distribution of the undigested SiO2-ENPs (details on the characterization of undigested SiO2-ENPs are summarize in ESI part 3†) in order to find out if the sample preparation procedure affects the size distribution and to quantify its bias (Table 4). In order to distinguish a possible effect of the tomato soup matrix from effects of sample preparation on the SiO2-ENP size distribution particle size distribution obtained for TS + SiO2-ENP and TS + SiO2-ENPaged were compared. Since SiO2-ENPs were spiked shortly (ca. 30 minutes) prior to the sample preparation to TS + SiO2-ENP sample it can be assumed that SiO2-ENPs in the freshly spiked soup will not be altered by the matrix components.
| Sample | r h (mode) [nm] | r h (median) [nm] | Peak shape factor, [—] | Sample peak area [mV min] | Void peak area [mV min] | Release peak area [mV min] |
|---|---|---|---|---|---|---|
| SiO2-ENP (no acid digestion) | 63 ± 2 | 70 ± 5 | 0.90 | 0.33 | 4 × 10−3 | 3.7 × 10−2 |
| SiO2-ENP | 76 ± 3 | 81 ± 6 | 0.94 | 0.34 | 5 × 10−3 | 4.5 × 10−2 |
| TS + SiO2-ENP | 71 ± 3 | 76 ± 2 | 0.95 | 0.37 | 4 × 10−3 | 3.9 × 10−2 |
| TS + SiO2-ENPaged | 74 ± 11 | 81 ± 9 | 0.92 | 0.37 | 4 × 10−3 | 4.0 × 10−2 |
The mode of rh distribution derived from AF4 calibration was slightly increased (maximum increase 21%) for all samples than for the undigested SiO2-ENP sample. There was a less pronounced increase in median values (maximum increase 16%) resulting in less tailing and higher peak shape factors.
As for the intensity-based size distributions, the mass-based particle size distributions determined by AF4 with the ICP-MS 28Si signal intensity as a concentration signal, were shifted towards larger particle sizes for all digested samples relative to the size distribution of not digested SiO2-ENPs (the mode of the size distribution of SiO2-ENPs is indicated by a vertical line in Fig. 2 together with the SiO2-ENP size distribution for pure particle suspension).
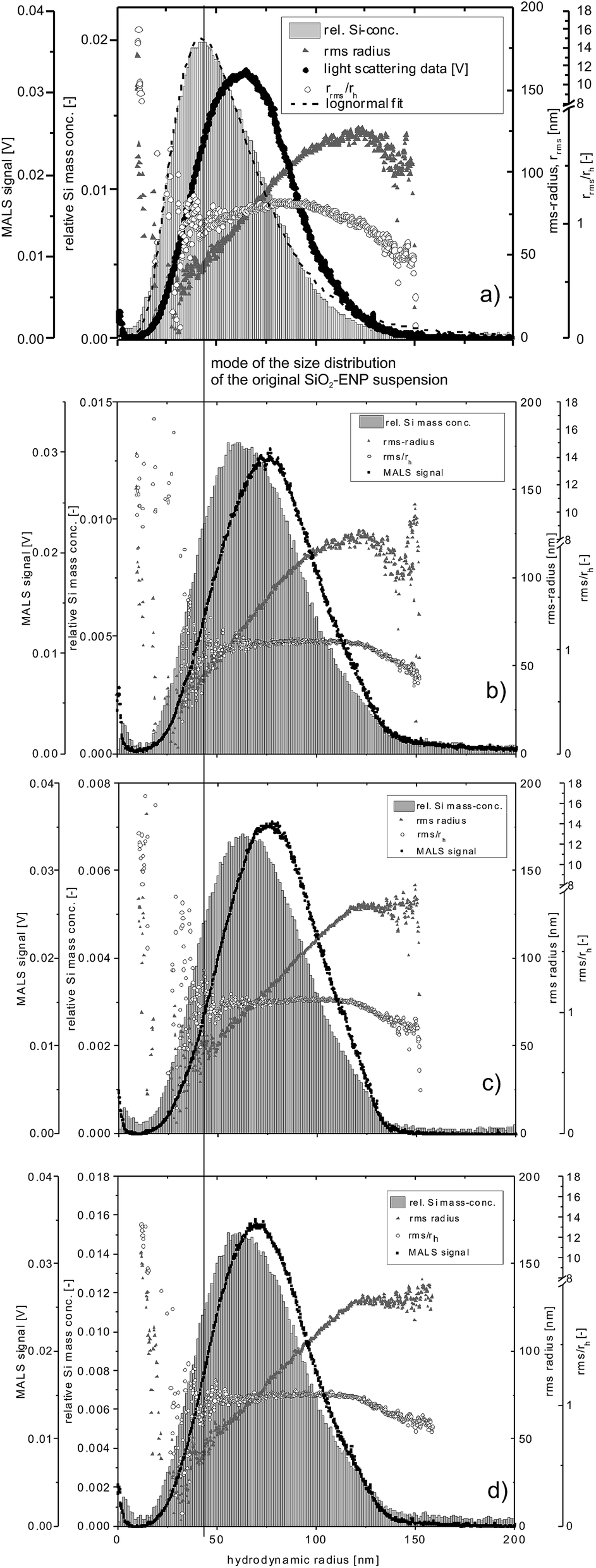 | ||
| Fig. 2 Particle size distribution of (a) original SiO2-ENP suspension and after digestion of samples (b) SiO2-ENP, (c) TS + SiO2-ENP, and (d) TS + SiO2-ENPaged, MALS data for a detector angle of 90°. | ||
The rrms/rh ratios (i.e. the peak shape factor) remained stable at values close to 1 over the elution time irrespective of the sample type, indicating a small deviation from an ideal spherical particle,4 which was expected since the particles in question are aggregates of smaller primary particles.21 Data for the particles with rh < 30 nm (based on MALS data) shows larger rms radii, indicating incomplete void peak separation. Due to limitations of the mathematical model, it is likely that the rrms derived from MALS does not reflect the real particle size in this region of the fractogram, and the rrms/rh ratio can therefore, only be interpreted for radii between 40 and 120 nm.
Despite the careful adjustment of the stabilization conditions a slight shift in the size distribution of SiO2-ENPs was inevitable. In order to explain this shift, stabilization parameters such as energy input, ionic strength conditions and AF4 separation have been considered. As a first indicator for the impact of acid digestion and the subsequent particle stabilization TEM images of the pure SiO2-ENPs and the SiO2-ENPs, extracted from the tomato soup with subsequent tip sonication, were recorded. The images indicated no alteration of the particle size distribution and particle shape (see ESI part 4†). However, TEM observation performed in this study were not appropriate to provide a quantitative particle size distribution. As an attempt to explain the slight shift in particle size distribution, the effects of energy input by sonication, ionic strength, and AF4 separation conditions on the particle size distribution were investigated.
27) during acid digestion. The increase of the pH value to the alkaline range (pH between 8 and 9), where SiO2-ENP are stable, did not lead to a break-down of the formed aggregates. Mechanical energy input in form of tip-sonication may support such a break-down. It was ensured that the primary SiO2-ENP size distribution remained unaffected by tip sonication treatment by the similarity between size distribution patterns obtained from SiO2-ENP sample following sonication for 135 seconds (calculated energy transfer 0.33 kJ mL−1), and those obtained from the untreated sample (data not shown). Tip sonication of the SiO2-ENPs extracted from the tomato soup resulted in a shift of the mode of the size distribution towards smaller sizes with increasing sonication time and the peak shape factor decreased from 1.09 to 0.95 (Fig. 3, ESI part 4.3†). Ninety seconds of sonication (calculated energy transfer 0.22 kJ mL−1) provided sufficient energy input to re-establish a particle size distribution with similar patterns to the initial size distribution of SiO2-ENPs (ESI part 4.3†). However, it was not possible to re-establish a completely similar size distribution applying mechanical energy input.
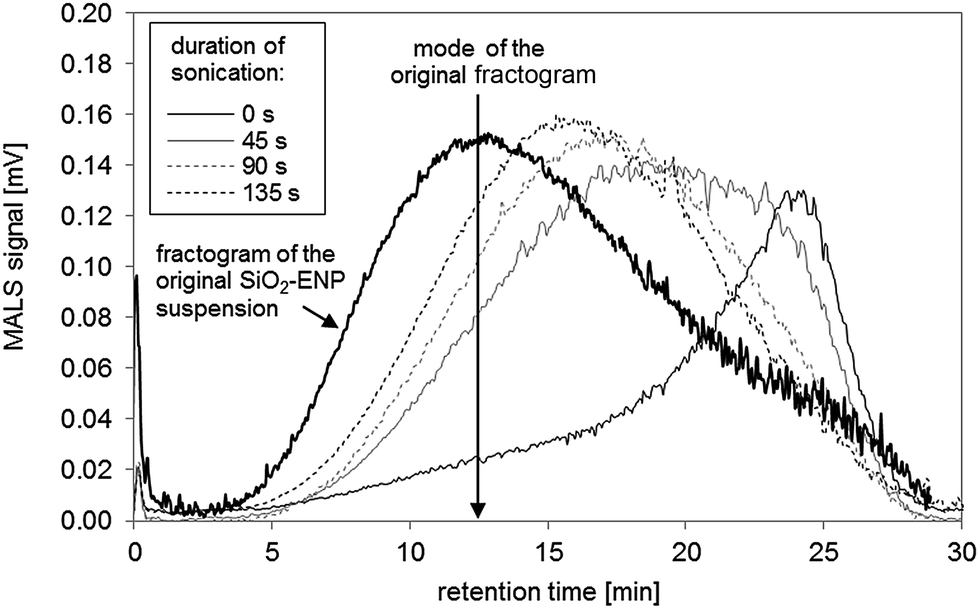 | ||
| Fig. 3 Effect of increasing time of sonication after particle stabilisation in 0.025% FL-70™ and pH adjustment on AF4 fractogram. | ||
| Sample | recAF4 [%] | recSi,tot [%] |
|---|---|---|
| SiO2-ENP (no digestion) | 90 | 97 |
| SiO2-ENP | 87 | 82 |
| TS + SiO2-ENP | 114 | 89 |
| TS + SiO2-ENPaged | 101 | 93 |
The AF4 recoveries were greater than the total recoveries, which is reasonable because the total recoveries covered the complete sample preparation and analysis procedure (see eqn (3) in ESI, part 1.1†) whereas the AF4 recoveries only covered mass loss during AF4 procedure.
Conclusions
The generic concept of systematic method development was successfully tested for the analysis of SiO2-ENPs in a complex matrix. The introduced and applied quality criteria proved to be applicable for the method development and optimization. As a next step in the direction of more routine method development the presented generic sample preparation procedure has to be transferred and tested for other ENP-matrix combination in order to prove its validity. As required by the generic procedure the method development for SiO2-ENPs in a food matrix has been thoroughly tested in terms of nanoparticle size and concentration. For quality control, Si mass recovery data and an independently acquired SiO2-ENP size distribution (e.g. using MALS) need to be determined for each run. Sample homogenization (step I) was identified as one of the most critical parameters for the recovery, while the stabilization procedure (step IV) is critical for the particle size distribution. As a result of the optimization procedure the following sample preparation is suggested: sample pretreatment (step I) by heating (60 °C) and mechanical mixing was required to sufficiently homogenize the soup. Successful SiO2-ENP separation from the matrix (step II) was achieved by microwave-assisted acid digestion with HNO3 and H2O2. After particle enrichment (step III) by centrifugation, particle stabilization is suggested (step IV) using an appropriate stabilizing agent (in this case 0.025% (v/v) FL-70™), pH adjustment to values between 8 and 9 and tip sonication for 90 seconds (0.22 kJ mL−1). The slight shift of the size distribution after acid digested of SiO2-ENPs was independent of the type of matrix (SiO2-ENP, TS + SiO2-ENP, TS + SiO2-ENPaged) and could not be explained by particle aggregation or a change in elution behaviour of SiO2-ENPs. It remained unclear to what parameter this slight shift could be attributed.The major difficulty for the direct application of this method on products, available on the market, is the lower ENP concentrations typically present in products. E.g. Dekkers et al.29 estimated concentrations of nano-sized SiO2-ENPs between <0.1 and 6.9 mg g−1. Based on the generic sample preparation procedure, a sample preparation method for lower concentrations ranges can be designed and tested e.g. by increasing the enrichment factor after particle-matrix separation or simply increasing the injection volume in the AF4.
Acknowledgements
The work leading to these results was carried out in the course of the NanoLyse project, which received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 245162.References
- H. Stamm, N. Gibson and E. Anklam, Food Addit. Contam., Part A, 2012, 29, 1175–1182 CrossRef CAS PubMed.
- F. Gottschalk and B. Nowack, J. Environ. Monit., 2011, 13, 1145–1155 RSC.
- F. von der Kammer, P. L. Ferguson, P. A. Holden, A. Masion, K. R. Rogers, S. J. Klaine, A. A. Koelmans, N. Horne and J. M. Unrine, Environ. Toxicol. Chem., 2012, 31, 32–49 CrossRef CAS PubMed.
- F. v. d. Kammer, M. Baborowski and K. Friese, Anal. Chim. Acta, 2005, 552, 166–174 CrossRef PubMed.
- S. Dubascoux, F. Von Der Kammer, I. Le Hecho, M. P. Gautier and G. Lespes, J. Chromatogr. A, 2008, 1206, 160–165 CrossRef CAS PubMed.
- E. Neubauer, F. D. von der Kammer and T. Hofmann, Water Res., 2013, 47, 2757–2769 CrossRef CAS PubMed.
- F. von der Kammer, S. Legros, E. H. Larsen, K. Loeschner and T. Hofmann, TrAC, Trends Anal. Chem., 2011, 30, 425–436 CrossRef CAS PubMed.
- C. Contado and A. Pagnoni, Anal. Chem., 2008, 80, 7594–7608 CrossRef CAS PubMed.
- V. Nischwitz and H. Goenaga-Infante, J. Anal. At. Spectrom., 2012, 27, 1084–1092 RSC.
- K. Loeschner, J. Navratilova, C. Kobler, K. Molhave, S. Wagner, F. von der Kammer and E. H. Larsen, Anal. Bioanal. Chem., 2013, 405, 8185–8195 CrossRef CAS PubMed.
- K. Loeschner, J. Navratilova, S. Legros, S. Wagner, R. Grombe, J. Snell, F. von der Kammer and E. H. Larsen, J. Chromatogr. A, 2013, 1272, 116–125 CrossRef CAS PubMed.
- J. Heroult, V. Nischwitz, D. Bartczak and H. Goenaga-Infante, Anal. Bioanal. Chem., 2014, 406, 3919–3927 CrossRef CAS PubMed.
- B. Stolpe, M. Hassellov, K. Andersson and D. R. Turner, Anal. Chim. Acta, 2005, 535, 109–121 CrossRef CAS PubMed.
- E. Bolea, J. Jimenez-Lamana, F. Laborda and J. R. Castillo, Anal. Bioanal. Chem., 2011, 401, 2723–2732 CrossRef CAS PubMed.
- H. Hagendorfer, R. Kaegi, M. Parlinska, B. Sinnet, C. Ludwig and A. Ulrich, Anal. Chem., 2012, 84, 2678–2685 CrossRef CAS PubMed.
- M. E. Hoque, K. Khosravi, K. Newman and C. D. Metcalfe, J. Chromatogr. A, 2012, 1233, 109–115 CrossRef CAS PubMed.
- C. E. Deering, S. Tadjiki, S. Assemi, J. D. Miller, G. S. Yost and J. M. Veranth, Part. Fibre Toxicol., 2008, 5, 18 CrossRef PubMed.
- S. Tadjiki, S. Assemi, C. E. Deering, J. M. Veranth and J. D. Miller, J. Nanopart. Res., 2009, 11, 981–988 CrossRef CAS.
- A. Samontha, J. Shiowatana and A. Siripinyanond, Anal. Bioanal. Chem., 2011, 399, 973–978 CrossRef CAS PubMed.
- A. R. Poda, A. J. Bednar, A. J. Kennedy, A. Harmon, M. Hull, D. M. Mitrano, J. F. Ranville and J. Steevens, J. Chromatogr. A, 2011, 1218, 4219–4225 CrossRef CAS PubMed.
- R. Grombe, J. Charoud-Got, H. Emteborg, T. P. Linsinger, J. Seghers, S. Wagner, F. von der Kammer, T. Hofmann, A. Dudkiewicz, M. Llinas, C. Solans, A. Lehner and G. Allmaier, Anal. Bioanal. Chem., 2014, 406, 3895–3907 CAS.
- F. Wagner von der Kammer, S., K. Loeschner, J. Navratilova, S. Legros, E. H. Larsen and T. Hofmann, Final report of the FP7 project "NanoLyse Nanoparticles in Food: Analytical methods for detection and characterisation", Collaborative project 245162, 2013.
- F. V. D. Kammer, M. Baborowski and K. Friese, Anal. Chim. Acta, 2005, 552, 166–174 CrossRef PubMed.
- H. Hagendorfer, R. Kaegi, J. Traber, S. F. L. Mertens, R. Scherrers, C. Ludwig and A. Ulrich, Anal. Chim. Acta, 2011, 706, 367–378 CrossRef CAS PubMed.
- H. Prestel, L. Schott, R. Niessner and U. Panne, Water Res., 2005, 39, 3541–3552 CrossRef CAS PubMed.
- G. Hartmann, T. Baumgartner and M. Schuster, Anal. Chem., 2014, 86, 790–796 CrossRef CAS PubMed.
- M. Kosmulski, J. Colloid Interface Sci., 2006, 298, 730–741 CrossRef CAS PubMed.
- S. Simovic and C. A. Prestidge, Langmuir, 2003, 19, 8364–8370 CrossRef CAS.
- S. Dekkers, P. Krystek, R. J. B. Peters, D. P. K. Lankveld, B. G. H. Bokkers, P. H. van Hoeven-Arentzen, H. Bouwmeester and A. G. Oomen, Nanotoxicology, 2011, 5, 393–405 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ja00471j |
| This journal is © The Royal Society of Chemistry 2015 |