
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tracking the transformation and transport of arsenic sulfide pigments in paints: synchrotron-based X-ray micro-analyses†
Katrien
Keune
*a,
Jennifer
Mass
b,
Florian
Meirer
c,
Carol
Pottasch
d,
Annelies
van Loon
ad,
Alyssa
Hull
e,
Jonathan
Church
f,
Emeline
Pouyet
g,
Marine
Cotte
gh and
Apurva
Mehta
i
aFaculty of Science, University of Amsterdam, 1090 GD Amsterdam, The Netherlands. E-mail: k.keune@rijksmuseum.nl
bScientific Research and Analysis Laboratory, Conservation Department, Winterthur Museum, Winterthur, Delaware, USA
cInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
dRoyal Picture Gallery Mauritshuis, The Hague, The Netherlands
eUniversity of Delaware, Department of Chemistry, Newark, Delaware, USA
fUniversity of Delaware, Department of Materials Science and Engineering, Newark, Delaware, USA
gEuropean Synchrotron Radiation Facility, 71 av. des Martyrs, 38043 Grenoble, France
hLaboratoire d'archéologie moléculaire et structurale, LAMS, UMR 8220, CNRS, F-75005, Paris, France
iStanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, 2575 Sand Hill Rd., Menlo Park, California, USA
First published on 18th February 2015
Abstract
Realgar and orpiment, arsenic sulfide pigments used in historic paints, degrade under the influence of light, resulting in transparent, whitish, friable and/or crumbling paints. So far, para-realgar and arsenic trioxide have been identified as the main oxidation products of arsenic sulfide pigments. This paper shows that after photo-degradation, various oxidation and migration processes take place. Synchrotron radiation (SR) micro-X-ray fluorescence (μ-XRF) reveals arsenic to be distributed throughout the whole multi-layered paint system. Arsenic (As) K-edge micro-X-ray absorption near edge structure (μ-XANES) analyses indicate the presence of an intact AsxSy pigment, arsenite compounds (As3+; As2O3), and arsenate compounds (As5+); the latter are certainly present as calcium, lead, aluminium and iron arsenates. Sulfur (S) K-edge μ-XANES points to the conversion of the sulfide (S2−) group to a sulfate (SO42−) group, probably via an elemental sulfur (S0) or sulfoxide (S2+) compound. Principal Component Analysis (PCA) and subsequent k-means clustering of multi-energy SR μ-XRF maps and μ-XANES were performed to identify the various arsenic species and visualize their distribution. The arsenates (As5+) are spread throughout the entire paint system and dominate the photo-degraded paint and ground layers, while the arsenite compounds (As3+) are located close to the intact arsenic sulfide pigment. The oxidation of arsenic trioxide into arsenates likely takes place in aqueous solutions. The presence of As5+ compounds in the paint systems indicates that the arsenic trioxide is dissolved by ambient water present in the paint. Arsenite and arsenate compounds are water soluble and are transported by water throughout the paint system. This knowledge is crucial for the conservation field, as this is the first time that (indirect) evidence of water transport within paintings has been given.
Introduction
The arsenic sulfide-based yellow and red-orange pigments, orpiment (As2S3) and realgar (As4S4), have been used by artists from different cultures since antiquity. In the 17th and 18th centuries, they were less commonly used in easel paintings, with the exception of still-life paintings at the turn of the 18th century, but are observed more frequently in historic interiors and furniture. The early 19th century saw gradual replacement of the arsenic pigments by new pigments such as chrome yellow that were created by a revolution in synthetic inorganic chemistry.Arsenic sulfide pigments are well known to shift color under exposure to visible light. Realgar undergoes photo-induced polymorphism and becomes friable, assuming hues from bright yellow (AsxSy, para-realgar) to colorless (As2O3, arsenolite), whereas the photo-oxidation of orpiment results only in a colorless product (arsenolite).1–8 The degradation products of arsenic sulfide pigments have been observed and identified in painted works of art by visual observation, Raman spectroscopy, and X-ray diffraction.9–17 Spatially resolved elemental analysis (scanning electron microscopy-energy dispersive spectroscopy) on embedded and polished paint samples, so-called paint cross-sections, has shown that arsenic is not limited to its original layer but rather distributed throughout the entire multi-layered paint system and is even found in the varnish layer and panel.18 As the sulfur content is low or absent in these areas and arsenic-rich varnishes are not found in areas far from the arsenic-containing paint, it does not appear that the original pigment has migrated. This points towards migration of the photo-degradation products of arsenic sulfide pigments throughout the multi-layered paint system. Displaced arsenic is not only found in paintings and furniture with photo-degraded arsenic sulfide pigments, but also in a 19th century oil painting containing a chemically degraded emerald green (Cu(C2H3O2)2·3Cu(AsO2)2, copper acetoarsenite) pigment.19
The migration of arsenic in painted works of art described above has not been encountered before within the conservation field. To understand the migration mechanism, it is important to characterize the chemical speciation of the mobile arsenic-containing products. The low relative concentration of the dispersed arsenic-based degradation products makes it difficult to identify these compounds with conventional micro-analytical imaging techniques, such as Raman spectroscopy, Fourier transform infrared (FTIR) spectroscopy, or scanning electron microscopy studies in combination with energy dispersive X-ray analysis (SEM-EDX). This problem can be overcome with the use of synchrotron radiation (SR) micro-X-ray fluorescence (μ-XRF) analysis and micro-X-ray absorption near edge structure (μ-XANES) measurements at the arsenic K-edge and sulfur K-edge, which have much better sensitivity. When performed with a micro-probe, these measurements provide information about the spatial distribution of elemental concentrations and oxidation states of As and S at the necessary resolution.
For this study, two objects have been selected that contain photo-degraded arsenic sulfide paints from the early 18th century. Still Life with Five Apricots (1704, oil on canvas) was painted by the Dutch master Adriaen Coorte (Royal Picture Gallery Mauritshuis, The Hague, The Netherlands). The second object is one of the earliest surviving examples of colonial American polychromed furniture, a Saybrook, Connecticut chest on stand (c. 1710–1727) that is attributed to Charles Guillam, a cabinetmaker from the Channel Islands who emigrated to the colonies in the early 18th century and worked in Guilford and Saybrook, Connecticut (Winterthur Museum, Winterthur, USA) (Fig. 1). The small oil painting on canvas is painted on a thick yellow ground pigmented with yellow and red earths (i.e. particles containing iron, aluminium, and silicon), chalk (CaCO3), and umber (FeO(OH) + MnO2). The apricots are generally composed of a light orange layer that contains realgar mixed with gypsum (CaSO4·2H2O). On top of the orange layer is a bright red vermilion (HgS)-containing layer either with white highlights or with a red glaze finishing layer. The paint is very crumbly, especially along the cracks, and appears to be vulnerable to further degradation when exposed to moisture and solvents.20 The Saybrook chest on stand is painted in distemper, a proteinaceous-binding medium based on animal skin glue, and is decorated with painted foliate, crown and thistle, and fleur-de-lis motifs based on the English Royal Arms. It has an overall red-toned ground composed of iron ochre and is painted with lead white (2PbCO3·Pb(OH)2), copper green pigment (now converted to copper stearate), vermilion, blue bice (2CuCO3·Cu(OH)2), orpiment, and realgar. This painted surface, as is common with American furniture, is covered with multiple layers of natural resin varnish, which has helped to protect the fragile distemper paint. Extensive photo-degradation of the painted surface has occurred and the copper-based pigments now have altered to black, while the AsxSy pigments range in color from mottled brown to ivory white hue.
 | ||
| Fig. 1 Still Life with Five Apricots by the Dutch master Adriaen Coorte (1704) (Royal Picture Gallery Mauritshuis, The Hague, The Netherlands) (a); a colonial American polychromed chest on stand attributed to Charles Guillam painted in Saybrook, Connecticut c. 1710–1727 (Winterthur Museum, Winterthur, USA) (b). | ||
In this paper, we use SR-based μ-X-ray techniques to determine the spatial resolution of arsenic species at the micrometer level in photo-degraded paint samples of Still Life with Five Apricots and the Saybrook chest on stand. Characterizing the chemical speciation of the arsenic provides insight for the first time into the full degradation mechanism of arsenic sulfide pigments and the mechanism of migration of their products. This knowledge helps to explain the observed paint defects and current appearance of the painted works. It also brings a better understanding of the forces that drive migration processes inside oil and distemper paints. This in turn supports decision-making in conservation practice as well as providing guidelines for display and storage conditions. Future studies of the kinetics of the phenomena involved may better inform the care of these objects.
Results
Coorte sample – preliminary investigation with laboratory-based techniques
The visible light microscopy image reveals a paint layer with a translucent orange upper part (20 μm) and thicker yellow-orange opaque lower region (Fig. 2A and C). Under UV light, the upper part is highly fluorescent with few orange particles, while the lower part shows a densely packed layer of orange particles (Fig. 2D). Backscattered electron imaging reveals a mixture of large clastic or angular para-realgar particles (up to 20 μm) and smaller flake-like orpiment particles (Fig. 2B and E). Both phases were identified by Raman spectroscopy (characteristic peaks for para-realgar are found at 233 and 228 cm−1 and for orpiment at 311 and 294 cm−1, assignment of peaks is based on ref. 9). There is a lack of para-realgar/orpiment particles in the upper region, while in the lower part these particles are still intact (Fig. 2B and E). Based on this difference, it is concluded that the upper part of the paint layer is photo-degraded. This conclusion is supported by other samples taken from Still Life with Five Apricots, where the orpiment/realgar paint layer was covered by a vermilion-containing paint layer and thus shielded from the light. In this case, the orpiment and para-realgar pigment particles were still intact and found throughout the paint layer (see ref. 20, page 103). | ||
| Fig. 2 Light microscopic image of the paint cross-section Still life with Five Apricots by Coorte, under normal light conditions (A) and BSE image (B). Details of the paint cross-section, under normal (C) and UV light (D) conditions and BSE image (F). The details are indicated with the dashed box in image A. The upper part of the paint layer is photo-degraded (layer 2b), while the lower part is still intact (layer 2a). The BSE image visualizes a thin deposit layer between the ground (layer 1) and paint layer (layer 2), indicated with the arrow. The solid box in (A) indicates the area of the μ-XRF map displayed in (F–H), the BSE image (F) and atomic distribution of As (green)/S (red)/Pb (blue) (G) and As (green)/Si (red)/Al (blue) (H) obtained with μ-XRF (data collection at ESRF ID21; μ-XRF maps excited at 3 keV, beam size 0.4 v × 0.7 h μm2; step size: 0.5 v × 0.7 h. Map size: 160 μm v × 100 μm h). The points represent the location of μ-XANES spectra presented in Fig. 4. The large crack above the upper paint layer, visible in the backscatter electron image, occurred after the synchrotron experiments. The dotted box in (A) indicates the area of the μ-XRF map displayed in (I–J): atomic distribution of As (green)/S (red)/Fe (blue) (I) and As (green)/Ca (red) (J) obtained with μ-XRF (data collection at SSRL BL 2–3; 300 × 160 μm2). The points represent the location of μ-XANES spectra presented in Fig. 4. The sample is briefly dry hand-polished in between the analyses of (A–E), (F–H), and (I–J), which resulted in a slightly different particle distribution. | ||
Remarkably, SEM-EDX analyses reveal an arsenic-rich thin deposit (around 2–3 μm) at the interface between the ground and paint layer (white arrow, Fig. 2E). Imaging ATR-FTIR spectra taken from the ground layer reveal a medium broad band at 873 cm−1 indicative of an arsenate vibration, suggesting the presence of calcium arsenate [Ca3(AsO4)2] (Fig. 3). The position of the ν(As–O) band is strongly determined by the identity of the cation bound to the arsenate and the type of coordination of the arsenate-group to the cation; sorption on the surface of minerals can influence the As–O vibration and cause a shift in the wavelength.21,22
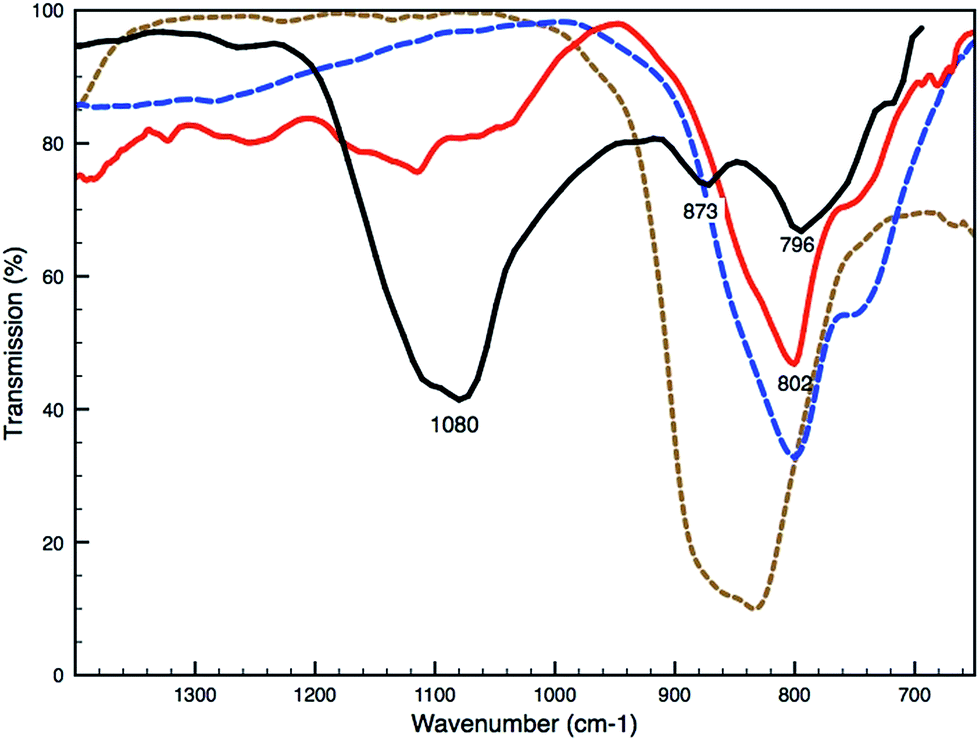 | ||
| Fig. 3 Part of the ATR-FTIR spectrum of the ground of the Coorte sample (black solid line), the peaks at 1080 and 796 cm−1 are allocated to the silicate-groups present in the earth pigment. Part of transmission FTIR spectra of the degraded yellow paint of the Saybrook sample (red solid line) and the reference spectra of calcium arsenate (brown dotted line) and lead arsenate (blue dotted line). | ||
Coorte sample – synchrotron-based techniques
Composite elemental distribution maps show highly heterogeneous paint layers for the Coorte sample (Fig. 2G–J). A sub-region of the sample was mapped at ID21 at the ESRF using an X-ray energy of 3 keV and a lateral resolution of 0.4 (v.) × 0.7 (h.) μm2 (Fig. 2G and H). An XRF map of the entire sample was gathered at BL 2–3 at SSRL using an X-ray energy of 13 keV and a spatial resolution of 2 × 2 μm2 (Fig. 2I and J).Arsenic is present throughout the paint sample. Higher intensities occur at the intact para-realgar and orpiment particles in the lower part of the paint layer and where arsenic is deposited at the interface between the paint and ground layers. Sulfur is more or less homogeneously distributed in the paint layer and is absent in the ground layer. Besides the intact arsenic sulfide pigments, the sulfur correlates with the calcium, indicating a gypsum filler (CaSO4·2H2O), which was also confirmed by SEM-EDX and FTIR. The calcium found in the ground layer is representative of chalk (CaCO3), confirmed by FTIR. In the paint layer, tiny particles of aluminium and silicon are present, suggestive of a clay, and a few tiny particles of lead are present at the top of the paint layer. The ground layer contains, in addition to chalk, red earth pigments (i.e. particles containing iron, aluminium, and silicon), umber (FeO(OH) + MnO2), and traces of lead- and chlorine-containing compounds. The components of red earths are clay minerals (the observed aluminosilicates) and iron oxides as well as iron hydroxides. The XRF maps point to a paint mixture for the apricot of arsenic sulfide pigments, gypsum, and possibly a lead drier. The ground/priming layer consists of chalk and earth pigments comprised of iron oxides, aluminosilicates, and quartz.
As K-edge XANES analyses carried out on the Coorte sample identified species containing both As3+ and As5+. Fig. 4a–d show the XANES spectra taken from an intact arsenic sulfide particle, at the interface between the ground and the paint layer, from the photo-degraded upper part of the paint, and from within the ground. The reference spectra for the relevant arsenic species are shown in Fig. 4g–k. The most reduced arsenic species in the paint sample, arsenic sulfide, is found, as expected, in the intact orange pigment particle (white-line at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 869.7 eV, spectrum a). Although some self-absorption is present due to the high As concentration in the particle, the main XANES features and the edge position match reasonably well with the XANES data for realgar and orpiment, which are the most reduced forms of the arsenic species in this paint sample. However, due to the damping of the XANES features caused by self-absorption, we cannot unambiguously identify the As species in the particle as either orpiment or realgar. The most oxidized arsenic species, arsenate, is detected in the photo-degraded paint matrix and in the ground (white-line at 11875.2 eV, spectra c and d, respectively). While the XANES spectrum recorded in the ground (Fig. 4d) matches the reference spectrum of arsenate (As2O5), the XANES spectrum of the photo-degraded paint matrix (Fig. 4c) reveals an additional shoulder at lower energies (around 11871 eV). This indicates the presence of an arsenite compound (As3+), most likely arsenic trioxide. A very small contribution of arsenite is also seen in the XANES spectrum of the ground (Fig. 4d). The XANES spectrum recorded at the interface between the paint and the ground (Fig. 4b) shows two peaks, one at ∼11870 eV, representative of arsenite (As3+), and one at ∼11875.5 eV, representative of arsenate (As5+), indicating a mixture of states and pointing to a combination of arsenite- and arsenate-containing compounds.
869.7 eV, spectrum a). Although some self-absorption is present due to the high As concentration in the particle, the main XANES features and the edge position match reasonably well with the XANES data for realgar and orpiment, which are the most reduced forms of the arsenic species in this paint sample. However, due to the damping of the XANES features caused by self-absorption, we cannot unambiguously identify the As species in the particle as either orpiment or realgar. The most oxidized arsenic species, arsenate, is detected in the photo-degraded paint matrix and in the ground (white-line at 11875.2 eV, spectra c and d, respectively). While the XANES spectrum recorded in the ground (Fig. 4d) matches the reference spectrum of arsenate (As2O5), the XANES spectrum of the photo-degraded paint matrix (Fig. 4c) reveals an additional shoulder at lower energies (around 11871 eV). This indicates the presence of an arsenite compound (As3+), most likely arsenic trioxide. A very small contribution of arsenite is also seen in the XANES spectrum of the ground (Fig. 4d). The XANES spectrum recorded at the interface between the paint and the ground (Fig. 4b) shows two peaks, one at ∼11870 eV, representative of arsenite (As3+), and one at ∼11875.5 eV, representative of arsenate (As5+), indicating a mixture of states and pointing to a combination of arsenite- and arsenate-containing compounds.
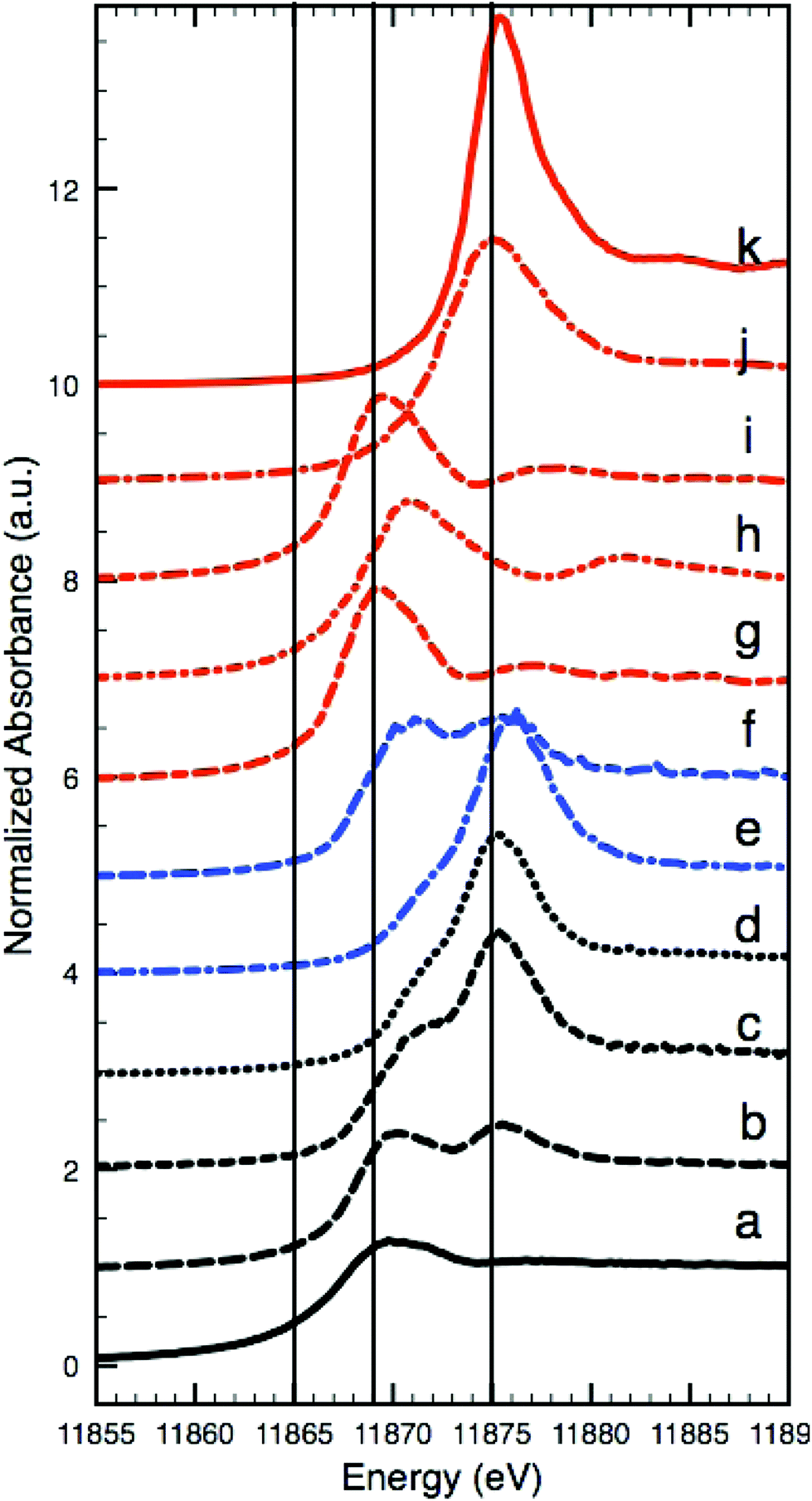 | ||
| Fig. 4 Arsenic K-edge XANES: intact arsenic sulfide particles in the Coorte sample, see point 1 in Fig. 2I (a), at the interface between the ground and the paint layer in the Coorte sample, see point 2 in Fig. 2I (b), in the photo-degraded upper part of the paint in the Coorte sample, see point 3 in Fig. 2I (c), in the ground layer of the Coorte sample, see point 4 in Fig. 2I (d), in the ground layer of Saybrook sample X8 (e), in the paint layer of Saybrook sample X8 (f) and references: realgar (g), As2O3 (h), orpiment (i), As2O5 (j) and Ca3(AsO4)2 (k). The vertical lines indicate the energies selected for multiple energy XRF mapping (Fig. 5), because they show the largest differences in XRF intensities with respect to the different As oxidation states. | ||
To gain a better understanding of the diffuse nature of the arsenic photo-alteration products, multiple μ-XRF maps were collected while tuning the X-ray incidence energy across the As K-edge. This generates XANES data for each pixel in the resulting map while using a reduced number of energy points in comparison to the data reported in Fig. 4, resulting in significant time saving during measurement. These multi-energy maps clearly indicate the presence of multiple As species. In total, 20 maps were collected, with 10 of them being separated by 1 eV from 11867 to 11877 eV. Four of these maps are presented in Fig. 5A–D. Map A displays the As XRF intensities recorded at 11865 eV, i.e. in the As K-edge pre-edge region. In this region, As XRF is exclusively generated by the As3+ (or even more reduced) species because this energy indicates the rising edge of the As3+ species (see the corresponding vertical line at 11865 eV in Fig. 4). At 11869 eV (map B), the XRF intensity originating from the As3+ species is the strongest, while the contribution from the As5+ species is still small (see the corresponding vertical line at 11869 eV in Fig. 4). In map C, which was recorded at 11875 eV, the opposite is true: the As5+ contribution is the largest while the contribution from the As3+ species has dropped to an almost constant value above the white-line. Map D, which was recorded at 11901 eV, i.e. in the post-edge region of the As K-edge, the contributions from both species are identical because there are no specific XANES features in the post-edge region. This map therefore represents the total As intensity distribution. It is interesting to note that a clear line of a high As5+ intensity appears in map C when compared to map B, which indicates an oxidation front at the interface between the ground and paint layer. In order to put this qualitative analysis on an unbiased and more quantitative basis, we performed a standard XANES image analysis with this limited XANES dataset using the XANES-Wizard software, which is part of the TXM-Wizard software package.23 After XANES normalization, single pixel XANES data with noise levels that were too high or showed extreme self-absorption (at the intact realgar particles) were removed before further processing (resulting in white pixels in maps F, G, and I of Fig. 5). In the first step, the edge energy, which is indicative of the oxidation state of As (Fig. 4), has been determined for each single pixel in the map by identifying the energy at which the normalized XANES intensity was equal to half of the edge jump. The result is displayed in Fig. 5F, where red colors indicate more oxidized species, while blue represents more reduced species. In agreement with maps B and C, it becomes clear that the central part of the photo-degraded region of the paint layer contains mainly As3+ sandwiched between oxidation fronts containing mixed states, while the ground layer is clearly dominated by As in the 5+ state.
 | ||
| Fig. 5 XRF maps taken at different X-ray incidence energies: 11865 (A), 11869 (B), 11875 (C) and 11901 (D) eV. The As(III)-species dominates in the X-ray map taken at 11869 eV and the As(V)-species dominates in the X-ray map taken at 11875 eV. A light microscopic image of the Coorte sample corresponding to the area measured for the multi-energy maps (E). The distribution of the As K-half edge position where blue pixels indicate more reduced and orange/red pixels more oxidized As (F). Image segmentation based on PCA and k-means clustering (G), average XANES profile of each cluster (H) and the phase image representing arsenic sulfide (red), arsenic trioxide (green) and calcium arsenate (blue) (I). The reference data on which the phase image is built are shown in Fig. 4. The dotted line indicates in all images the interface between the ground and paint layer. | ||
To further confirm this observation, we performed a Principal Component Analysis (PCA) and the subsequent k-means clustering of the XRF-XANES datasets (as described for example by Meirer et al. and Boesenberg et al.).24,25 The first three principal components (PC) covered 84% of the variance in the data and were therefore used for image segmentation by k-means clustering. k-means clustering was performed in this 3-dimensional PC space, using seven clusters (Fig. 5G and H), intentionally over-clustering the dataset to obtain an image segmentation into 7 regions of XANES similarity. This resulted in a finer separation of the phase transformations present in the sample by visualizing five statistically significant and distinct regions, each representing a different degree of phase mixing (Fig. 5G). This natural reduction to five clusters occurs because clusters 2 and 6 contain very few pixels, and their spatial distribution correlates with the embedding medium of the sample. These clusters were therefore attributed to noise contributions and their average XANES spectra have not been used in the following discussion. The average XANES spectra for the remaining clusters are reported in Fig. 5H. Cluster 1 (dark blue) represents the most reduced arsenic species in the paint sample (at 11870 eV). The spatial distribution of this cluster fits with the lower part of the paint layer (the remaining intact arsenic sulfide paint), as well as with the degraded paint region. The most oxidized arsenic-containing species are found in cluster 4 (light blue). This cluster is mainly found in the ground layer and with small contributions in the top part of the cross-section. The XANES spectra of clusters 3 and 7 are dominated by the white-line at 11875 eV, the arsenate group, but both spectra also show a shoulder originating from reduced arsenite (As3+). Cluster 3 (red) corresponds to the photo-degraded paint layer and the region immediately below the paint-ground interface. Cluster 7 (grey) is mainly present at this same paint–ground layer interface and at the interface of the photo-degraded and intact paint regions. The image segmentation-based clustering in the Principal Component (PC) space thus shows a layered microstructure of the As species.
High energy-resolution μ-XANES spectra covering 40 eV below and 300 eV above the As K-edge, with a 0.25 eV step size over the critical white-line region were collected at 77 locations on the sample. PC analysis of these μ-XANES spectra indicates that there must be at least 3 and perhaps as many as 5 As species present. Comparison of the XANES spectra and their PCs with published spectra and spectra collected from As reference compounds [Sigma Aldrich] was carried out (Fig. 4g, h and k). These analyses suggest that As2S3 (most reduced), Ca3(AsO4)2 (most oxidized), and As2O3 can adequately represent the majority of the As speciation in the sample. After identifying these compounds, reference XANES spectra of As2S3, Ca3(AsO4)2, and As2O3 were used in a Linear Combination Least Squares (LCLS) fit of the μ-XRF-XANES dataset. This resulted in the phase distribution map displayed in Fig. 5I. In this map, the total As intensity in every pixel measured as the edge-jump height of the XANES has been incorporated as a different transparent color, where opaque pixels represent high intensity and transparent pixels represent low intensity. This phase map shows the presence of arsenic trioxide at the interface between the paint and ground layer, and in the lower part of the paint layer. Calcium arsenate is predominantly present in the ground layer as well as in the upper photo-degraded part of the paint layer. The arsenic sulfide pigment is, as expected, found in the intact paint layer, but is difficult to locate precisely due to the relatively large number of null pixels in the map. The intact pigment is also present in the degraded paint, which is not unexpected, given that the backscattered electron image reveals remnants of AsxSy particles (Fig. 2B and E).
The S K-edge XANES spectrum (Fig. 6d) from an arsenic sulfide particle in the lower ‘intact’ part of the paint layer of the Coorte sample (point 1 in Fig. 2G) fits best with the orpiment spectrum using the Linear Combination Least Squares (LCLS) fit (ESI, Fig. 1†). The spectrum (Fig. 6e) taken from an arsenic sulfide particle positioned in the upper part of the seemingly degraded paint layer (point 2 in Fig. 2G) reveals an additional peak at 2482.6 eV, along with the feature characteristics for the S2− group of the orpiment particle at 2471.7 eV. The peak at 2482.6 eV is representative of an oxidized sulfur species, most likely the sulfate group (see for comparison the spectrum of gypsum, Fig. 6i). The XANES spectra collected outside the particle match, as expected, the S K-edge XANES spectrum for gypsum. S K-edge XANES spectra collected at the interface between the paint and ground layers in the Coorte sample show the presence of a sulfate peak (at 2482.3 eV, no clear fine structure) (Fig. 6f). Additional features observed are at 2473.7 eV with a shoulder at 2471.8 eV, and a local maximum at 2478.2 eV.
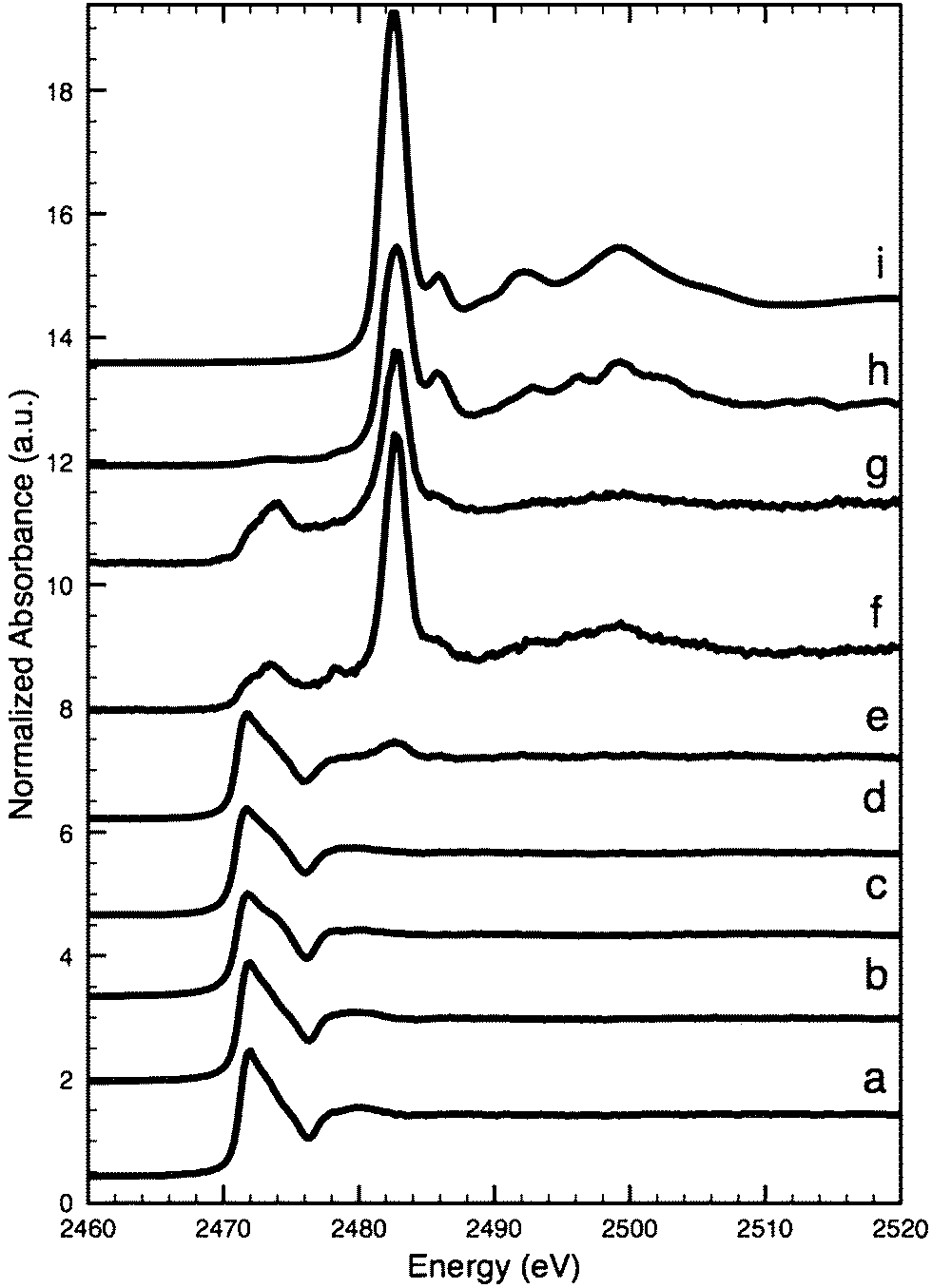 | ||
| Fig. 6 Sulfur K-edge XANES: para-realgar (a), realgar (b), orpiment (c), intact (para-)realgar particles in the lower part of the paint layer in the Coorte sample, see point 1 in Fig. 5B (d), (para-)realgar particles in the upper part of the paint layer in the Coorte sample, see point 2 in Fig. 5B (e), at the interface between the ground and the paint layer in the Coorte sample, see point 3 in Fig. 5B (f), an arsenic rich area in the paint layer of Saybrook X4, see point 4 in Fig. 8B (g), next to the arsenic rich area in the paint layer of Saybrook X4, see point 5 in Fig. 8B (h) and reference of gypsum (i). | ||
Saybrook sample – preliminary investigation using laboratory-based techniques
Cross-section samples removed from the arsenic sulfide-painted leaves on the proper left and right sides of the Saybrook chest on stand were studied using visible and UV light microscopy. The visible light microscopy image of sample X4 (Fig. 7A) reveals fragments of a compact lead white-containing ground layer and a yellow-orange paint layer. This paint layer is in a more advanced state of alteration than the Coorte sample and has few remaining intact orpiment pigment particles, all of which are located toward the bottom of the paint layer. The particles toward the top of the paint layer are beginning to turn white and the others are translucent. Under UV illumination, multiple campaigns of plant resin-containing varnish layers are also observed, in addition to a central crack in the section into which the plant resin has flowed (white arrow, Fig. 7B). The overall bright fluorescence of the paint layers is suggestive of a proteinaceous binding medium, and this is confirmed by μ-FTIR and GC-MS analyses of samples from this work. The upper half of the pigmented layer, when viewed under UV illumination, shows two large relatively homogeneous regions on either side of the crack that fluoresce brightly and appear almost particle-free. This may be a result of the larger degree of photo-alteration and migration of the end products occurring in the Saybrook samples, as opposed to what is observed in the Coorte sample.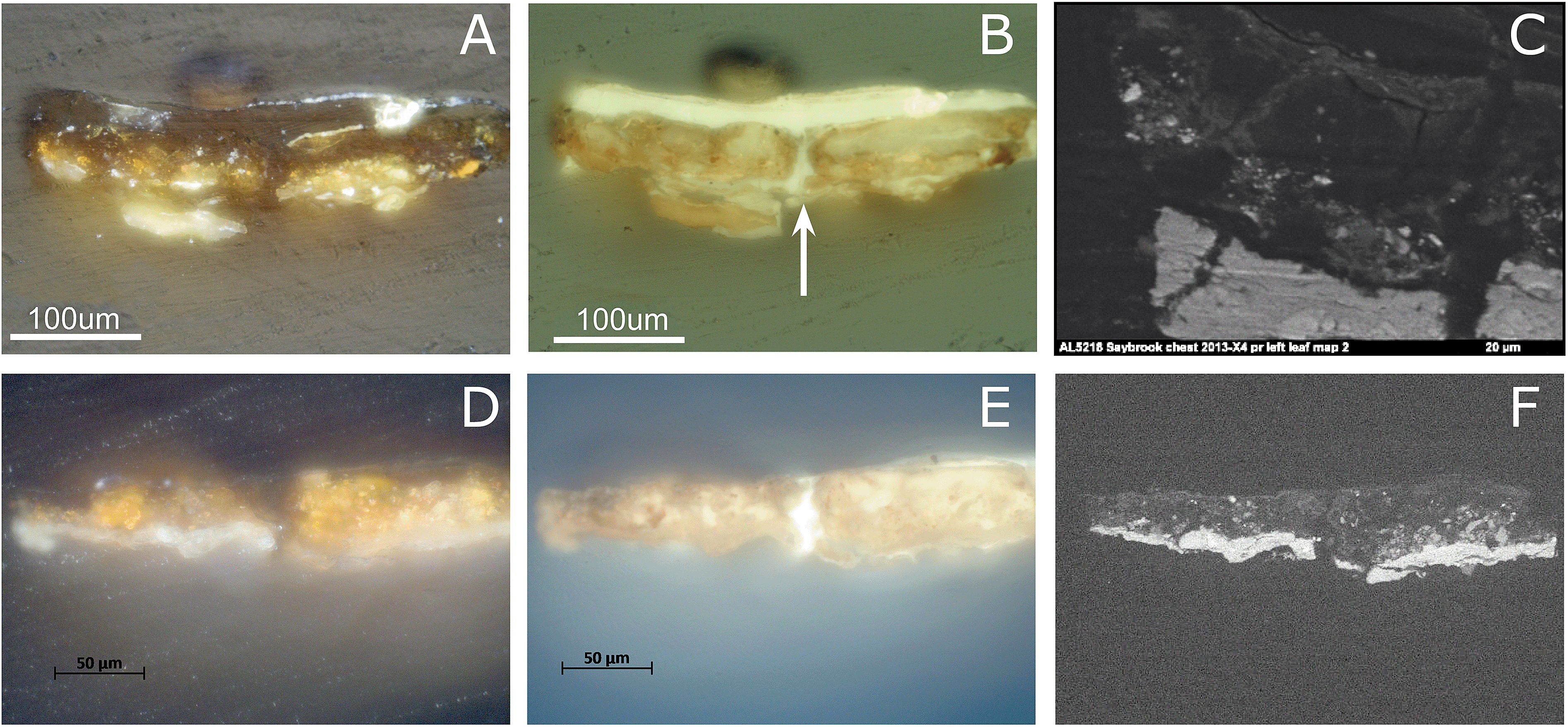 | ||
| Fig. 7 Light microscopic image of the paint cross-section X4 and X8 of the Saybrook chest under normal (A and D) and UV light conditions (B and E), backscattered electron image (C and F). Microscopy images of sample X4 are shown in A–C and sample X8 in D-F. (C) illustrates the details of the left side of the sample X4 shown in (A) and (B). | ||
Saybrook X8 is far less photo-degraded than X4, and shows a more intact lead white ground, followed by a 30–40 μm thick yellow paint layer (Fig. 7D–F). A much larger percentage of the pigment particles in this sample retain their original yellow hue in this layer, although some white and colorless particles can be observed in the right half of the section. Under UV illumination, a crack is also revealed through the center of this section, and at least two plant resin-containing varnish layers are observed. Again, the overall bright fluorescence of this layer is suggestive of a proteinaceous binder for the AsxSy pigmented layer, which μ-FTIR and GC-MS analyses confirm. Cracks in painted surfaces are commonly observed on furniture finishes due to the dimensional changes of the wood with temperature and relative humidity that are not echoed in the dimensional changes of the paint. It should also be noted that it is not common practice to remove finish layers in early American furniture collections, even when they have become photo-oxidized, as this compromises the ability to use finish history to interpret the history of any alterations or restorations that have been carried out on the piece.
Saybrook X4 was studied by μ-FTIR and XRD prior to and following the synchrotron studies, respectively. μ-FTIR revealed an arsenate stretching band corresponding to lead arsenate at 803 cm−1 (Fig. 3), and XRD revealed the presence of arsenolite and schultenite (PbHAsO4).
Sample X8 was removed from the Saybrook chest on stand for study after it became clear from the S K-edge XANES study of sample X4 that essentially all of the arsenic sulfides had been oxidized, and so a sample that was in a more intermediate state of photo-alteration was required.
Saybrook sample – synchrotron-based techniques
The μ-XRF maps of the Saybrook sample X4 reveal a heterogeneous distribution of the main elements arsenic, sulfur and lead, which extends up into the sample's multiple varnish layers (Fig. 8). To ease the visualisation of the different layers, elemental profiles have been extracted, by averaging the signal over a width of 5 μm (Fig. 8E). Smaller and larger areas of arsenic are found in the paint layer where sulfur is absent. This indicates that arsenic is present predominately as a photo-degraded product, such as an arsenic oxide or an arsenate, in what was originally the paint layer. This is consistent with the almost complete lack of colored particles remaining in this layer. Arsenic is homogeneously dispersed throughout the ground layer (in a lower intensity than in the paint layer) and found as a horizontal band in several (at least three) of the varnish layers in lower yield (Fig. 8B–D). Sulfur is located in distinct small areas in the paint layer, especially near the lower part of the layer, at the interface with the ground. Another thin layer of sulfur is present on top of the varnish layer, most likely deposited at the varnish surface from the atmosphere. This layer, likely containing surface dirt, also contains arsenic, while the other thin arsenic layers below it do not contain sulfur. Chlorine is also found in the upper varnish layers. This distribution suggests an external chlorine source – not surprising as Saybrook is a coastal Connecticut town on the Atlantic Ocean. In the ground, particles of chlorine and lead are present, with chlorine being possibly an impurity of lead white. At least one of the varnish layers (an internal one) also appears to contain a lead drier. A homogeneous distribution of lead is observed in the ground. Tiny particles of lead are also found in the paint layer, indicative of a lead pigment or a lead drier. A few particles containing aluminium and silicon are found in the ground and paint layer, but not in sufficient quantities to suggest an intentional aluminosilicate-based filler. Low intensities of phosphorus-containing compounds are found in the ground layer, possibly indicative of the proteinaceous binder. A few spots in the paint layer correlate with sulfur, suggesting the presence of an as-yet unidentified sulfur-containing compound. | ||
| Fig. 8 Details of Saybrook sample X4: a light microscopy image (A) and atomic distribution of As (green)/S (red)/Pb (blue) (B), As (green)/Si (red)/Al (blue) (C) and P (green)/S (red)/Cl (blue) (D) obtained with μ-XRF (data collection at ESRF ID21; μ-XRF maps excited at 3 keV, beam size 0.4 v × 0.7 h μm2; step size: 0.5 v × 0.7 h. Map size: 130 μm v × 40 μm h. The points represent the location of μ-XANES presented in Fig. 6. The vertical bar in the false color map of the As distribution (E) indicates the area of the line scan (F). The elemental profiles of Cl, Pb, As, S, from bottom to top of the paint sample, are averaged over a width of 5 μm. The profiles have been normalized to 1, for readability. | ||
63 As K-edge μ-XANES spectra were taken from Saybrook sample X8. Two spectra, taken from the ground and paint layers, are shown in Fig. 4e and f. The arsenic in the lead-containing ground is present in the most oxidized form, as an arsenate (As5+). The white-line at 11876.1 eV lies at a slightly higher energy compared to the reference compound As2O5 at 11876.0 eV and the peak corresponding to arsenate taken from the ground layer in the Coorte sample at 11875.2 eV. μXANES spectra taken from the paint layer reveal, besides the presence of an arsenate contribution, an absorption feature representative of the arsenite species (As3+) with a white-line occurring at 11871.7 eV. A representative XANES spectrum for the intact arsenic sulfide pigment could not be unambiguously identified in any of the 63 As-XANES spectra recorded. This is remarkable as the light microscopy image still reveals numerous orange-coloured particles. It is possible that the small spot size of the X-ray beam (2 μm) missed these particles or that an unambiguous identification was hampered by self-absorption effects in the XANES data, which resulted from high As intensities in the arsenic sulfide regions.
Fig. 9B and C show the result of the image segmentation by k-means clustering in the PC space of the multi-energy XRF maps of sample X8 using seven clusters. Clusters 2, 3 and 6 represent an arsenate species; however, their absorption edge varies slightly. The absorption edge of cluster 2 lies at a higher energy compared to clusters 3 and 6. Cluster 2 and clusters 3 and 6 are very different in terms of their location within the sample. Cluster 2 can be found in the ground layer, while clusters 3 and 6 are present above the paint layer, in the plant resin/varnish layers. A slight shift in the absorption edge of the arsenate species was already observed in the As-XANES (Fig. 4f). Based on both results, it can be concluded that the arsenate is present in at least two different species. In the lead-containing ground, this points to the presence of lead arsenate (also confirmed by FTIR). In the resin layer, the arsenate is bound to a different element or is perhaps present as arsenic pentoxide.
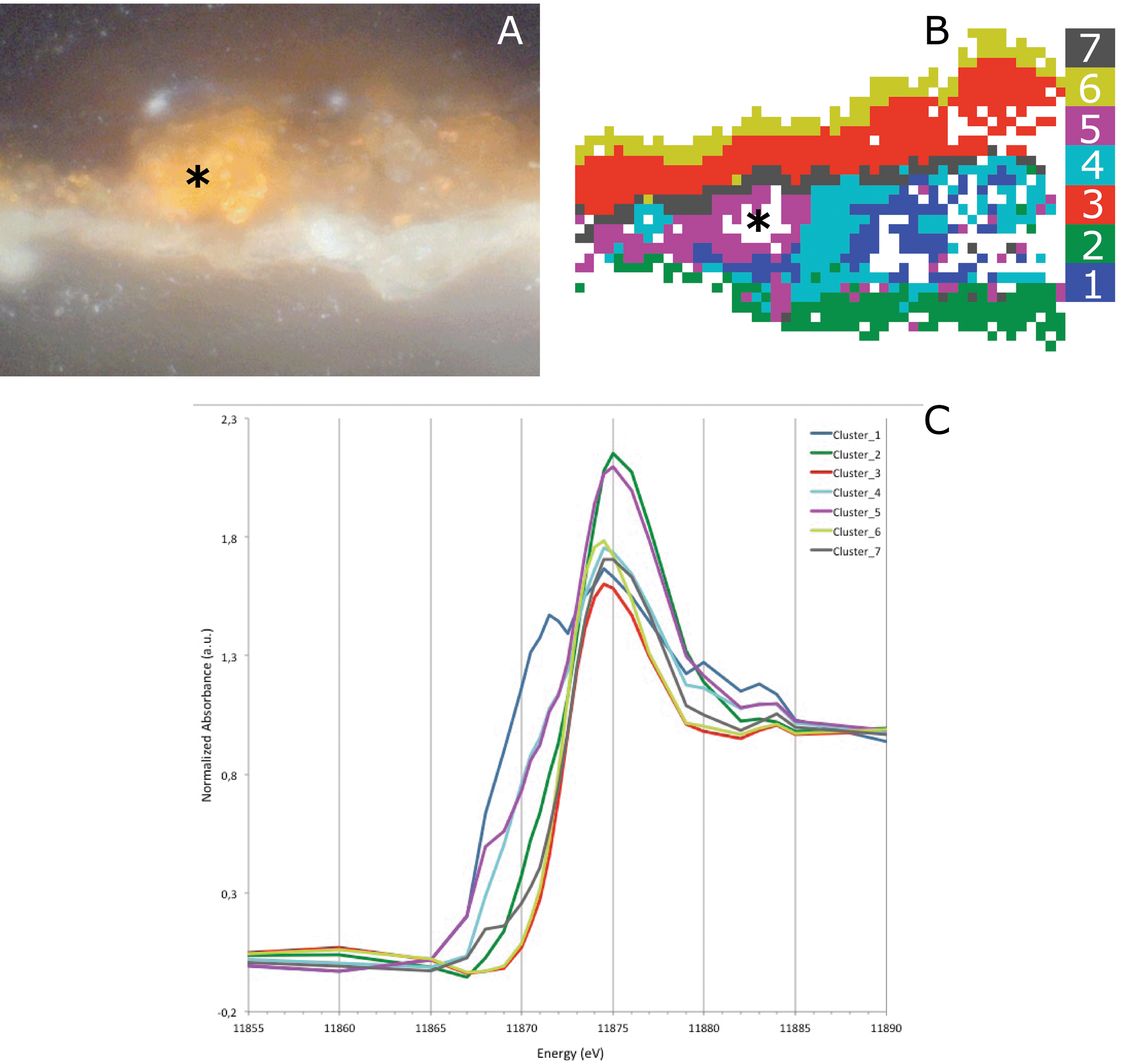 | ||
| Fig. 9 Light microscopic image of Saybrook sample X8 corresponding to the area measured for the multi-energy maps (A). Image segmentation based on PCA and k-means clustering (B) and average XANES of each cluster (C). | ||
Clusters 1 and 4 indicate the presence of an arsenite (As3+) species, while clusters 5 and 7 hint at the existence of the intact arsenic sulfide pigment. Clusters 1 and 4 are distributed over the bulk of the paint layer, while clusters 5 and 7 are located in the left edge of the paint layer and in the interface between the paint and varnish layers. The distribution of the arsenite species here matches the observation in the light microscopic image, where an intact arsenic sulfide pigment is still present. Some pixels are missing due to some self-absorption caused by the high As concentration in the particle. A similar distribution of the As3+ and As5+ is found for the Coorte sample, i.e. the more reduced arsenic is found close to the original AsxSy paint, while the As5+ is found above and below this original paint layer.
The S K-edge XANES spectra taken in an arsenic-rich area in the paint layer of the Saybrook sample X4 show besides sulfate features (at 2482.8 eV, no clear fine structure), a peak at 2473.9 eV, and a small shoulder at 2478.2 eV (Fig. 6g). This is similar to what was observed in the Coorte sample. The spectra taken from the region below the arsenic-rich area, correlating with the concentrated sulfur zones, show primarily sulfate characteristics (Fig. 6h). It has to be mentioned that elemental analyses showed some calcium-rich areas around the arsenic, so the presence of calcium sulfate cannot be excluded.
Both the spectra taken from the interface in the Coorte sample and in the arsenic-rich areas in the Saybrook sample show spectral features that are characteristic for neither the original arsenic sulfide pigments nor for pure sulfates. Decomposition of spectra as LCLS was carried out using the ID21 sulfur XANES database, but the results were not convincing, mainly because of the wide band in the reduced sulfur energy range. Alternatively, and in a more qualitative way, without making any assumption of the species in presence, we decomposed the Saybrook spectrum from Fig. 6f and g as the sum of Gaussian peaks and an arctangent, using the ATHENA software (Fig. 10A and B). Five peaks were considered for the Gaussian peaks, of which energy position and attribution (S form and oxidation state) were made following ref. 27 (namely 2471.6 eV for disulfide (−2), 2473.5 eV for less reduced sulfide, possibly an organic disulfide (+0.2), monodisulfide or thiol (+0.5), 2476.5 eV for sulfoxide (+2), 2478.3 eV for sulfite (+4), 2481.7 keV for sulfonate (+5) and 2482.9 keV for sulfate (+6); absorption edge at: 2478.9 eV). The graphs show the presence of different oxidation states, mainly reduced sulfur (but more similar to organic di/monosulfide than to arsenic disulfides). Complementary analyses will be needed to better determine the present species.26,27 The very low contribution of the features characteristic of arsenic sulfides and the presence of the more oxidized sulfur-species indicate that the sulfur undergoes transformation into various stable sulfur-species before being converted into the sulfate group.
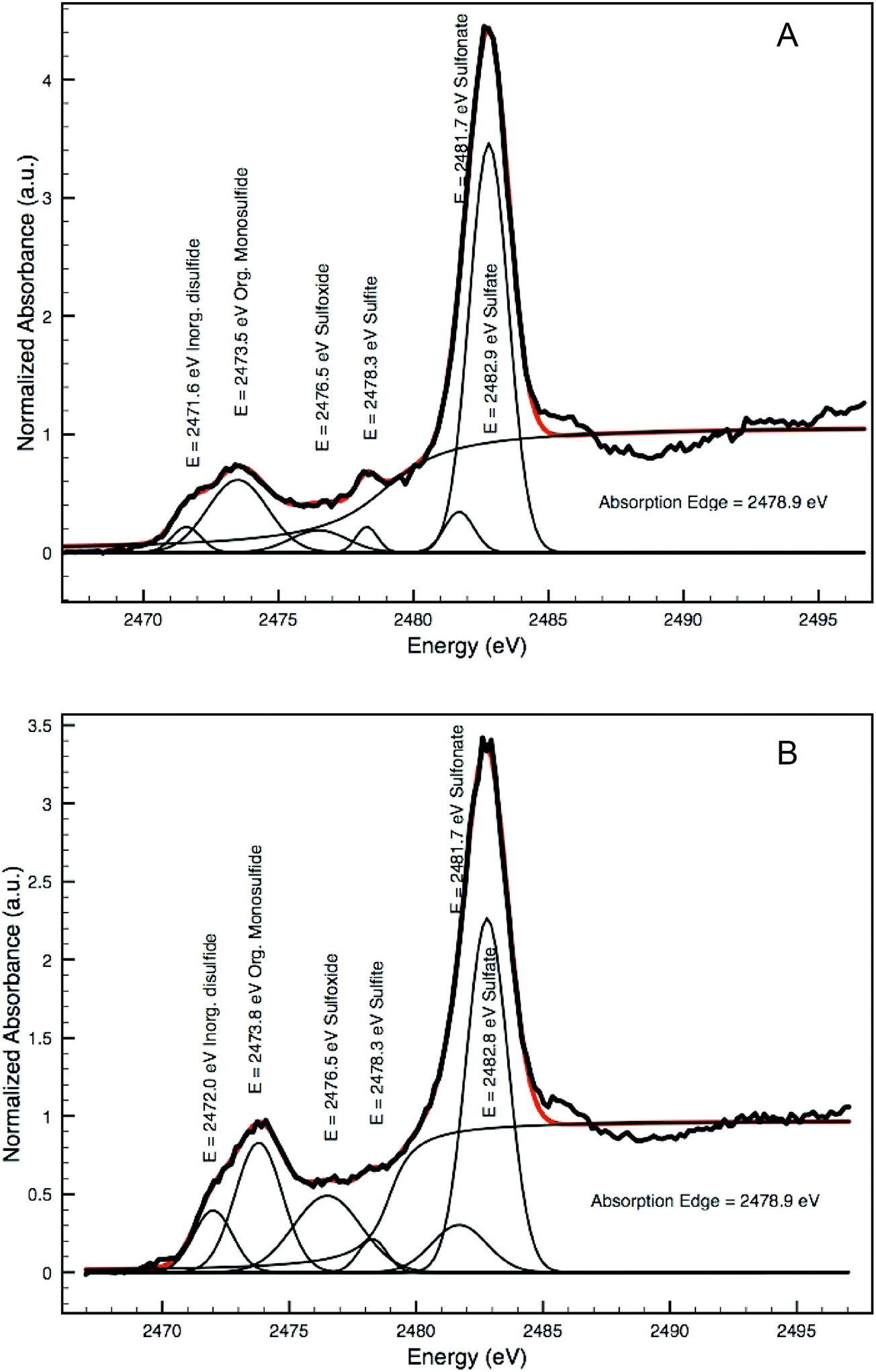 | ||
| Fig. 10 Decomposition of two XANES spectra shown in Fig. 6f (A) and g (B) as a sum of Gaussian peaks and an arctan function illustrating the presence of various sulfur species. | ||
Discussion
The XRF arsenic maps show that the arsenic photo-alteration products have mobilized and moved throughout the depth of the paint. In the case of the Saybrook chest, these photo-alteration products have even accumulated in the multiple varnish layers. The migration of arsenic occurs independently of the period in which the work was created, the materials used, and the creator's working method, as these changes are observed in an early 18th century oil painting as well as in samples from a polychromed chest on stand with a proteinaceous paint binder. What these two pieces may have in common is exposure to a high relative humidity environment, facilitating the formation of hydrated and mobile forms of the arsenic oxides.The XANES analyses indicate the presence of arsenic sulfide, arsenite- and arsenate-containing species. The data processing of the multi-energy μ-XRF maps and high resolution μ-XANES made it possible to visualize the spatial distribution of these various arsenic-containing species. In general, the arsenite species are mainly found close to the location of the originally intact arsenic sulfide pigment. However, in the Coorte sample, a thin layer of arsenite is also deposited at the interface between the paint and ground layer. The arsenate-containing species are located at larger distances from their original source. They are particularly found in the ground and the uppermost layers (glaze and varnish), but are also present within the paint layer itself.
Arsenic is well known to be transported in the environment as an oxide via water.28 Depending on the relative humidity, water is spatially not fixed within a painted surface, but is in equilibrium with the environment and thus in motion. It is hypothesized that arsenic is dispersed via a water transport mechanism in the paint system.18 The way in which this water transport occurs in paintings, via nano- or microporous channels as monolayers of water, or as liquid water or vapor, is not precisely known. Arsenic trioxide is the final photo-oxidation product of both realgar and orpiment. In the second stage, arsenic trioxide itself is oxidized into arsenates. It is difficult to determine in the paint samples whether or not the oxidation of arsenite takes place in the solid phase or in aqueous solutions. In aqueous systems, arsenic trioxide is slowly oxidized by oxygen.29,30 Oxides of iron or manganese increase the rate of As3+ oxidation. In the solid phase, arsenic trioxide requires strong oxidizing agents such as ozone, hydrogen peroxide, and nitric acid to convert it to pentoxide.31 These strongly oxidizing compounds are not expected to be present in a mature oil or in a proteinaceous paint system. It is therefore very likely that arsenic trioxide needs to be dissolved in water first before being oxidized to arsenate.
The As3+/As5+ ratio present is also influenced by the redox conditions and pH of water.32 In an As–O–H system at 25 °C and 1 bar, the arsenite species are stable under mildly oxidizing and reducing conditions, while the arsenate species dominate under oxidizing conditions.33 The value of the As3+/As5+ ratio as an indicator of the local pH condition in the paint systems remains to be seen, as paints are highly heterogeneous and non-aqueous systems. Trace elements and minerals present can also influence the chemical speciation of arsenic. Arsenates are strongly adsorbed onto the surface of iron, manganese and aluminium hydroxides found in clay minerals, while arsenites have a lower affinity to minerals and adsorb less strongly.34–37 The low affinity should make arsenite more mobile. However, the As3+ and As5+ distributions in the Coorte and Saybrook samples show the opposite, as arsenates are found throughout the paint sample, while the arsenites remain localized close to the original area of the arsenic sulfide pigment. A plausible explanation would be that the detected arsenic trioxide has not yet been dissolved by the water and thus has not moved. This would imply that all the dissolved arsenites have been oxidized to arsenates, which then spread out. However, the arsenite-rich deposit layer at the paint–ground interface in the Coorte sample indicates that arsenite dissolved and migrated to that interface. In this case, the oxidizing and pH conditions of the water in the paint layer are favorable enough to preserve the arsenite. The presence of an arsenite-containing deposit at the paint–ground interface would suggest that the local conditions of water change when entering the ground layer composed, which is composed of iron oxides and clay minerals. Presumably, both explanations are valid here.
The data analyses of the Saybrook multi-energy maps made it possible to reveal the presence of arsenates with different counter-ions. In the lead white ground layer, the arsenates have formed schultenite (PbHAsO4, identified by μ-XRD and μ-FTIR as well as μ-XANES), while in the upper part the arsenates are bound to another element or are present in an oxide form. In the paint layer of the Coorte sample, the calcium from gypsum is the main element present. The arsenate ion is known to substitute for the sulfate group in the gypsum crystal.38,39 This process is favorable under highly alkaline conditions. The sulfate group is replaced by not only As5+ as [HAsO42−] or [H2AsO4−], but also by the As3+ ion as (AsO3)− or (AsO4)3−, in lower amounts.38 Calcium arsenates are highly insoluble and thus form a stable end product.40 The arsenates in the ground layer are probably a mixture of iron, aluminium and calcium arsenates. The subtle differences in As-XANES of arsenate with different counter-ions are not observed in the PCA and clustering in the ground layer. The characterization and localization of arsenates with different counter-ions in the paint system is beyond the scope of this paper.
Experimental
Sample preparation
The Coorte paint sample was embedded in polyester resin Polypol PS230 using the Easysection™ system and dry polished with Micro-mesh® polishing cloths (final step 12000 mesh) (Scientific Instruments Services Inc., Minnesota).
The Saybrook samples were cast separately in mini-cubes, of approximately half-inch widths, with polyester resin (Extec polyester clear resin with methyl ethyl ketone peroxide catalyst, Extec Corporation®, Enfield, CT). The resin was allowed to cure for 24 h at room temperature and under ambient light. The cubes were then dried and hand-polished successively with 400- and 600-grit sandpaper and 1500- to 12000-grit micro-mesh polishing cloths to expose the cross-section.
Instrumentation
The μ-XRF mapping and μ-XANES (S K-edge) were carried out at ID21 at ESRF and μ-XRF mapping and μ-XANES (As K-edge) at the X-ray microprobe at BL 2–3 at SSRL. This allowed the distribution and speciation of the As- and S-containing compounds in the paint cross-sections from the painting and chest on stand to be imaged at the submicron resolution.The ID21 Scanning X-ray Microscope (SXM) operates in an energy range from 2 to 9.2 keV for μ-XRF and μ-XANES measurements. X-rays are generated by different undulators (U42 and U32). The microscope consists of a fixed-exit double crystal multilayer monochromator (here Si (111)) used for selection and scanning of the X-ray beam energy. μ-XRF and μ-XANES experiments were performed in a vacuum to minimize air absorption of the important low energy XRF lines such as S K, and Pb M. The monochromatic incident beam was focused with a Kirkpatrick Baez mirror system to a size of 0.36 μm (ver.) × 0.70 (hor.) μm2 at the S K-edge (2.5 keV). The samples were raster-scanned in the micro-beam to collect 2D XRF images using a Bruker SDD XFLASH 5100 detector. The incident X-ray flux at the samples was 1.1 × 1010 photons per s per Si (111) bandwidth in the focused mode. μ-XANES spectra were acquired by scanning the primary energy around the S K-edge (2.46–2.53 keV with a step of 0.2 eV). The monochromator was calibrated using a gypsum reference (maximum absorption at 2482.6 eV). The PyMca software package was used to fit XRF spectra and to map the different elemental positions for μ-XRF.33 Decomposition of spectra as the sum of Gaussian peaks was performed using the ATHENA software package.
Prior to the analysis of the paint cross-sections, powdered reference samples (described below) were prepared for bulk XANES analysis by dusting the finely ground powder onto sulfur-free tape and using a 200 μm diameter X-ray beam in transmission and XRF modes. Samples were maintained in a vertical plane, oriented at 30° with respect to the incident beam.
BL 2–3 at SSRL is a bending magnet side station dedicated to X-ray imaging and micro-X-ray absorption spectroscopy operating at an energy range from 5 to 24 keV. A Kirkpatrick-Baez mirror system is used to achieve micro-focus with a beam size of ∼2 × 2 μm2. The microscope consists of a double crystal monochromator (Si (111)) used for energy selection and scanning of the X-ray energy. Beamline 2–3 is equipped with a Vortex silicon drift detector and ionization chambers. Sample positions are controlled with submicron accuracy using a Newport Micro sample positioner. The samples were placed with double side tape (Scotch® tape) in the sample holder and measured under ambient conditions and at room temperature. Prior to the analysis of the paint cross-sections, powdered reference samples (described below) were prepared for bulk XANES analysis by dusting the finely ground powder onto sulfur-free Kapton tape. Cross-section samples were maintained in a vertical plane, oriented at 90° with respect to the incident beam.
The Coorte paint cross-section was examined under a Zeiss Axioplan 2 microscope, both with incident polarized light and incident UV light (from a xenon-lamp and a mercury short arc photo optic lamp HBO, respectively). The filter set ‘UV H365’ used for examination in UV light consists of the following filters: excitation BP 365/12, beam splitter FT 395 and emission LP 397.
The Saybrook samples were examined under reflected visible and UV light using a Nikon Eclipse 80i microscope at 200× magnification with the X-cite® 120 Fluorescence Illumination System with a BV-2A cube. Images were taken with a Nikon digital camera DXM 1200F using ACT-1 software. 10× and 20× objectives were employed with a 10× ocular.
Scanning electron microscopy studies in combination with energy dispersive X-ray analysis (SEM-EDX) of the Coorte sample were performed on a XL30 SFEG high vacuum electron microscope (FEI, Eindhoven, The Netherlands) with an EDX system with spot analysis and elemental mapping facilities (EDAX, Tilburg, The Netherlands). Backscattered electron images of the cross-sections were taken at a 20 kV accelerating voltage, a 5 mm eucentric working distance and a spot size of 3, which corresponds to a beam diameter of 2.2 nm with a current density of approximately 130 pA. Prior to SEM-EDX analysis, samples were gold coated (3 nm thickness) in a SC7640 gold sputter coater (Quorum Technologies, Newhaven, East Sussex, UK) to improve the surface conductivity.
Electron microscopy of the Saybrook samples was carried out using a Topcon ABT-60 SEM with a Bruker Quantax EDS detector, a 20 kV accelerating voltage, a 22 mm working distance for backscattered electron imaging, and a beam current of approximately 100 pA. A 20° stage tilt was used for EDS analysis and elemental mapping. All samples were mounted on carbon stubs prior to analysis and their perimeters painted with SPI Supplies carbon paint, but analyses were performed in variable pressure mode to avoid carbon coating.
The FTIR spectral data from the Coorte sample were collected on a Perkin Elmer Spectrum 100 FTIR spectrometer combined with a Spectrum Spotlight 400 FTIR microscope equipped with a 16 × 1 pixel linear mercury cadmium telluride (MCT) array detector. A PerkinElmer ATR imaging accessory consisting of a germanium crystal was used for ATR imaging.
FTIR spectral data on the Saybrook samples were collected using a Thermo Nicolet 6700 FTIR bench and a Continuum microscope with a 15× IR objective with the sample mounted on a diamond half cell. 128 spectra were collected over 4000 to 650 cm−1 and the spectral resolution was 4 cm−1. A mercury cadmium telluride (MCT) detector was used.
XRD patterns on the Saybrook samples were collected using a Rigaku Rapid II with a Cu anode tube operating at 40 kV and 30 mA. An image plate detector was used to collect data on a cross-section sample mounted on an aluminium stub with adhesive tape, oscillating 15° in ω over the course of 20 minutes. The ICDD powder diffraction file was used for data interpretation.
Raman spectroscopy on the Saybrook samples and the Coorte sample was carried out with a Renishaw InVia Raman spectrometer using a 785 nm laser, a frequency range of 2200 to 100 cm−1, and a spectral resolution of 3 cm−1. The laser spot size used was 1 × 20 μm2 and the laser power used was 3 mW, with a 1200 l mm−1 grating and a 50× objective.
Reference samples
Reference standards employed for the XANES and μ-XANES mapping experiments at SSRL and ESRF included powders of realgar (Sigma-Aldrich 95%), orpiment (Sigma-Aldrich >99.99%), As2O3 (Sigma-Aldrich 99.995%), As2O5 (Sigma-Aldrich 99%), and para-realgar (light exposed realgar).Principal component analysis (PCA) and k-means clustering
PCA was carried out using the mean-centered data matrix (consisting of p pixels and E energies) and singular value decomposition (SVD) reducing the dimensionality of the dataset from E to N dimensions. This reduction is achieved by using only the first N principal components (PCs), which explain most of the data's variance, i.e., without losing significant information because higher PCs describe mainly the noise in the dataset. Therefore, and because the PCs are oriented to best describe the spread in the data, projecting the data to this N-dimensional principal component space highlights the pattern explained by the captured variance and effectively reduces noise. In this reduced space the distance between data points is a direct measure of the similarity of the XANES, i.e. the chemical phase, and can be used to cluster pixels according to their (Euclidean) distances from cluster centers (centroid linkage method, k-means clustering). This results in an effective grouping of pixels with similar XANES into k groups resulting in image segmentations like the ones displayed in Fig. 5G and 9C. The number of clusters k has to be at least equal to N (in this study 3) in order to exploit all the information provided by the reduced PC space, but the data were intentionally over-clustered using k = 6 or 7 to achieve a finer image segmentation in order to investigate different stages of oxidation state transitions. A more detailed description of the applied approach to process XANES image data can be found in ref. 23 and 24.Conclusion
The arsenic sulfide paints in the Still Life with Five Apricots and the Saybrook chest on stand are photo-degraded, which has resulted in discolored and vulnerable paints. PCA combined with k-means clustering of As K-edge XANES data and multi-energy μ-XRF maps showed the presence of arsenates throughout the entire multi-layered paint system. Arsenite is located close to the intact arsenic sulfide pigment particles and at the interface between the paint and ground layer of the Coorte sample. The final photo-degradation product of arsenic sulfide pigments is arsenic trioxide, which implies that arsenites are photo-oxidized to arsenates. The oxidation of arsenic trioxide is not expected to take place in the solid-state as strong oxidation agents are required. An oxidation of arsenic trioxide in an aqueous form (As(OH)3) is more likely. The arsenate and arsenite products are both water-soluble and therefore mobile. Arsenite and arsenate are transported via water in painted works of art and are adsorbed by chalk, gypsum, lead white, iron oxide and earth pigments in the paint. These findings expand upon our current knowledge of the degradation of AsxSy paints, identifying how water can act as a carrier for the movement of inorganic species through paintings and painted finishes. This is a starting point for suggesting a means of preservation and/or preventive conservation for these pieces.Acknowledgements
This work is part of the PAinT project, supported by the Science4Arts program of the Dutch Organization for Scientific Research (NWO). Some of the measurements reported here were performed on beamline 2–3 at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operating for the U.S. Department of Energy Office of Science by Stanford University. Other measurements were performed on beamline ID21 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. The authors would like to thank Sam Webb (SSRL, USA) and Suzan de Groot (Cultural Heritage Agency of the Netherlands) for their technical support and Catherine Matsen (Scientific Research and Analysis Laboratory, Winterthur Museum), Marc Vermeulen (Royal Institute for Cultural Heritage, Brussels), W. Christian Petersen (Scientific Research and Analysis Laboratory, Winterthur Museum), and Jessica Arista (Conservation Department, Museum of Fine Arts, Boston), who played important roles in determining the palette and binding medium for the Saybrook chest.References
- L. Bindi, V. Popova and P. Bonazzi, Can. Mineral., 2003, 41, 1463–1468 CrossRef CAS.
- P. Bonazzi, S. Menchetti, G. Pratesi, M. Muniz-Miranda and G. Sbrana, Am. Mineral., 1996, 81, 874–880 CAS.
- P. Bonazzi, S. Menchetti and G. Pratesi, Am. Mineral., 1995, 80, 400–403 CAS.
- A. H. Clark, Am. Mineral., 1970, 55, 1338–1344 CAS.
- A. Kyono, M. Kimata and T. Hatta, Am. Mineral., 2005, 90, 1563–1570 CrossRef CAS.
- A. C. Roberts, H. G. Ansell and M. Bonardi, Can. Mineral., 1980, 18, 525–527 CAS.
- D. L. Douglass, C. Shing and G. Wang, Am. Mineral., 1992, 77, 1266–1274 CAS.
- P. Ballirano and A. Maras, Plinius, 2002, 28, 35–36 Search PubMed.
- K. Trentelman, L. Stodulski and M. Pavlosky, Anal. Chem., 1996, 68, 1755–1761 CrossRef CAS.
- C. Rötter, G. Grundmann, M. Richter, A. van Loon, K. Keune, A. Boersma and K. Rapp, in Auripigment/Orpiment: Studien zu dem Mineral und den küstlichten Produkten, ed. M. Schuller, E. Emmerling and W. Nerdinger, Verlag Anton Siegl, Fachbuchhandlung GmbH, München, 2007 Search PubMed.
- A. Van Loon, PhD Thesis, University of Amsterdam, MOLART Report 14, Archetype, London, 2008.
- L. Sheldon, S. Woodcock and A. Wallert, in ICOM Committee for Conservation, 14th Triennial Meeting, The Hague, 12–16 September 2005, Preprints, ed. I. Verger, James & James, London, 2005, vol. 1, p. 529 Search PubMed.
- C. Rötter, Restauro, 2003, 109(6), 408–413 Search PubMed.
- J. Dubois, S. Meloni, E. Metz, B. Schoonhoven and A. Wallert, in Deterioration of Artists' Paints: Effects and Analysis – A Joint Meeting of ICOM-CC Working Groups Paintings 1 and 2 and the Painting Section, ed. A. Phenix, UKIC, British Museum, London, 2001, pp. 75–97 Search PubMed.
- E. Hendriks and A. Wallert, in Art et Chimie, la Couleur: Actes du Congrès, ed. J. Goupy and J. P. Mohen, CNRS Editions, Paris, 1998, pp. 111–112 Search PubMed.
- E. W. FitzHugh, in Artists' Pigments: A Handbook of Their History and Characteristics, ed. E.W. FitzHugh, National Gallery of Art, 1997, vol. 3, pp. 47–79 Search PubMed.
- A. Wallert, Maltechnik-Restauro, 1984, 90(4), 45–57 Search PubMed.
- K. Keune and J. J. Boon, in Preprints ICOM Committee for Conservation, 16th Triennial Meeting, Lisbon, September 19–23, ed. J. Bridgland, 2011 Search PubMed.
- K. Keune and J. J. Boon, Stud. Conserv., 2013, 58(3), 199–210 CrossRef CAS PubMed.
- C. Pottasch and K. Mensch, in Historical Technology, Materials and Conservation: SEM and Microanalysis, ed. N. Meeks, C. Cartwright, A. Meek and A. Mongiatti, Archetype Publications, London, 2012, pp. 100–106 Search PubMed.
- S. Goldberg and C. T. Johnston, J. Colloid Interface Sci., 2001, 234, 204–216 CrossRef CAS PubMed.
- S. C. B. Myneni, S. J. Traina, G. A. Waychunas and T. J. Logan, Geochim. Cosmochim. Acta, 1998, 62, 3499–3514 CrossRef CAS.
- Y. Liu, F. Meirer, P. Williams, J. Wang, J. C. Andrews and P. Pianetta, J. Synchrotron Radiat., 2012, 19, 281–287 CrossRef CAS PubMed.
- F. Meirer, Y. Liu, E. Pouyet, B. Fayard, M. Cotte, C. Sanchez, J. C. Andrews, A. Mehta and P. Sciau, J. Anal. At. Spectrom., 2013, 28, 1870–1883, 10.1039/c3ja50226k.
- U. Boesenberg, F. Meirer, Y. Liu, A. Khushalchand Shukla, R. Dell'Anna, T. Tyliszczak, G. Chen, J. C. Andrews, T. J. Richardson, R. M. Kostecki and J. Cabana, Chem. Mater., 2013, 25, 1664–1672 CrossRef CAS PubMed.
- A. Vairavamurthy, Spectrochim. Acta, Part A, 1998, 54, 2009–2017 CrossRef.
- J. Prietzel, J. Thieme, U. Neuhausler, J. Susini and I. Kogel-Knabner, Eur. J. Soil Sci., 2003, 54, 423–443 CrossRef CAS.
- Arsenic in ground water; geochemistry and occurrence, ed. A. H. Welch and K. G. Stollenwerk, Kluwer Academic Publishers, Boston, 2003 Search PubMed.
- J. A. Cherry, A. U. Shaikh, D. E. Tallman and R. V. Nicholson, J. Hydrol., 1979, 43, 373–392 CrossRef CAS.
- L. E. Eary and J. A. Schramke, in Chemical Modeling of Aqueous Systems II, ed. D. C. Melchior and R. L. Bassett, American Chemical Society, 1990, ch. 30 Search PubMed.
- J. H. T. Luong, E. Lam and K. B. Male, Anal. Methods, 2014, 6, 6157–6169 RSC.
- Arsenic in Ground Water; Geochemistry and Occurrence, ed. A. H. Welch and K. G. Stollenwerk, Kluwer Academic Publishers, Boston, 2003, pp. 67–72 Search PubMed.
- P. Lu and C. Zhu, Environ. Earth Sci., 2011, 62, 1673–1683, DOI:10.1007/s12665-010-0652-x.
- Arsenic in Ground Water; Geochemistry and Occurrence, ed. A. H. Welch and K. G. Stollenwerk, Kluwer Academic Publishers, Boston, 2003, ch. 2 Search PubMed.
- P. A. O'Day, Elements, 2006, 2, 77–83 CrossRef.
- P. L. Smedley and D. G. Kinniburgh, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- R. S. Oremland and J. F. Stolz, Science, 2003, 300(5621), 939–944 CrossRef CAS PubMed.
- J. Lin, N. Chen, M. J. Nilges and Y. Pan, Geochim. Cosmochim. Acta, 2013, 106, 524–540 CrossRef CAS PubMed.
- F. Bardelli, M. Benvenuti, P. Costagliola, F. Di Benedetto, P. Lattanzi, C. Meneghini, M. Romanelli and L. Valenzano, Geochim. Cosmochim. Acta, 2011, 75, 3011–3023 CrossRef CAS PubMed.
- Y. N. Zhu, X. H. Zhang, Q. L. Xie, D. Q. Wang and G. W. Cheng, Water, Air, Soil Pollut., 2006, 169, 221–238 CrossRef CAS; V. A. Solé, E. Papillon, M. Cotte, Ph. Walter and J. Susini, Spectrochim. Acta, Part B, 2007, 62(1), 63–68 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ja00424h |
| This journal is © The Royal Society of Chemistry 2015 |