
C18-coated stir bar sorptive extraction combined with HPLC-ICP-MS for the speciation of butyltins in environmental samples
Xiangju
Mao
,
Wenying
Fan
,
Man
He
,
Beibei
Chen
and
Bin
Hu
*
Key Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), Department of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: binhu@whu.edu.cn; Fax: +86 27 68754067; Tel: +86 27 68752162
First published on 22nd October 2014
Abstract
In this paper, a new approach of C18-coated stir bar sorptive extraction (SBSE) coupled with high performance liquid chromatography-inductively coupled plasma mass spectrometry (HPLC-ICP-MS) was developed for the speciation of butyltins in environmental samples. The butyltin compounds, including monobutyltin trichloride (MBT), dibutyltin dichloride (DBT) and tributyltin chloride (TBT), were first extracted with a C18-coated stir bar, and then desorbed with 40% (v/v) methanol for subsequent HPLC-ICP-MS analysis. The factors affecting C18-SBSE were systematically studied. To reduce the percentage of organic solvents in the mobile phase loaded on the plasma and to improve the separation resolution, a CN column with carboxylic acids as the mobile phase additives was used for butyltin separation. Quick separation (<8 min) was accomplished using a methanol–formic acid–water (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:76, v/v/v) mobile phase containing 5 mmol L−1 mercaptoacetic acid, and the mobile phase was compatible with the conventional quadrupole ICP-MS detector without addition of oxygen gas or desolvation and post-column dilution systems. Under the optimal conditions, the limits of detection for three target butyltins ranged from 15.6 to 29.4 ng L−1, with a linear range of 0.05–50 μg L−1. The enrichment factors (EFs) were 85- to 127-fold (the theoretical EF is 200-fold). The developed method is simple and rapid, with no derivatization involved, and is successfully applied to the speciation of butyltins in the Certified Reference Material of PACS-2 sediment and real water as well as sediment samples from the East Lake, Yangtze River and Bohai Sea in China.
:8:76, v/v/v) mobile phase containing 5 mmol L−1 mercaptoacetic acid, and the mobile phase was compatible with the conventional quadrupole ICP-MS detector without addition of oxygen gas or desolvation and post-column dilution systems. Under the optimal conditions, the limits of detection for three target butyltins ranged from 15.6 to 29.4 ng L−1, with a linear range of 0.05–50 μg L−1. The enrichment factors (EFs) were 85- to 127-fold (the theoretical EF is 200-fold). The developed method is simple and rapid, with no derivatization involved, and is successfully applied to the speciation of butyltins in the Certified Reference Material of PACS-2 sediment and real water as well as sediment samples from the East Lake, Yangtze River and Bohai Sea in China.
Introduction
The main organotin compounds (OTs) in the environment originate from the high amount of tributyltin chloride (TBT) in anti-fouling paints and triphenyltin chloride (TPhT) in pesticides. Once OTs enter the environment, they can be degraded into di-derivatives and mono-derivatives by UV light and microorganisms and ultimately converted to inorganic tin. OTs in aquatic environments tend to accumulate in sediments and organisms.1 The toxicity of different OTs strongly depends on their species. Although inorganic tin is considered harmless, TBT and TPhT are very toxic and can promote harmful effects on non-target aquatic organisms, even at a low ng L−1 level.2 Organotin pollutions have attracted the attention of many governments and environmental protection organizations, and OTs are the priority in the “blacklist” of pollutants in many countries. Therefore, developing simple, fast, and sensitive methods for speciation analysis of OTs is very important.Highly efficient separation techniques coupled with highly sensitive and selective detection, such as inductively coupled plasma mass spectrometry (ICP-MS), meet the requirements for organotin (OT) speciation analysis. Gas chromatography (GC) combined with atomic/mass spectrometry has become one of the most valuable approaches in OT speciation analysis.3–5 Unfortunately, most OTs are non-volatile compounds; thus, an extra derivatization step is necessary prior to GC analysis. For this reason, high performance liquid chromatography (HPLC) separations have gained growing interest. The HPLC separation of OTs has several advantages over GC, such as the direct separation of non-volatile OTs without tedious derivatization steps, the variety of stationary and mobile phases available, and the possibility of separation at ambient temperature. These advantages reduce possible losses of target species during separation.6
Several HPLC separation modes have been proposed for OT speciation, including ion exchange (IE), reversed phase (RP), reversed phase ion pair (RP-IP) and micellar modes.7–11 However, HPLC is not as widely applied as GC in OT speciation analysis, mainly because of the incompatibility between the HPLC mobile phase and ICP-MS detection.1 Complex mobile phases, such as those used in a pH-gradient elution or those with high percentages of methanol, must be used because OTs are likely to be strongly adsorbed onto the HPLC stationary phase. Typical mobile phase components include citrate, acetate, or oxalate buffers at pH 4 to 5 and a methanol percentage exceeding 70%.6,12 For example, Vela and Caruso13 used up to 95% methanol to elute trimethyltin chloride (TMT), TBT, and TPhT, and the effluent was introduced in ICP-MS with oxygen as a secondary nebulizer gas. When HPLC is on-line hyphenated with ICP-MS, the high percentages of organic solvents in the mobile phase will result in serious problems, such as plasma instability or even extinguishment and carbon deposits on the sampler and skimmer cones.14,15 Thus, when a mobile phase contains high percentages of organic solvents, several special adaptations are needed for ICP-MS: (1) the addition of oxygen gas to the nebulizer argon gas flow to allow the combustion of the organic solvent in the plasma, which contributes to plasma stability; (2) the application of a high forward power to the plasma; (3) the use of a refrigerated spray chamber (−5 to −20 °C), which reduces the solvent loading on the plasma; and (4) the use of desolvating systems to remove the organic solvents before they reach the plasma.7,8,16,17 However, these requirements increase the analytical cost of the experiment. To reduce the percentages of organic solvent in the HPLC mobile phase, micellar HPLC mode has been studied for OT separation.10,11 Using an ODS short column (4.6 mm × 50 mm, 5 μm) as the stationary phase and sodium dodecyl sulfate (0.1 mol L−1)–isopropanol (3%)–acetic acid (3%) as the mobile phase, TMT, triethyltin bromide (TET), and tripropyltin chloride (TPT) were successfully separated. However, butyltins cannot be eluted under these conditions.10 Dimethyltin dichloride (DMT), TMT, dibutyltin dichloride (DBT), TBT, diphenyltin dichloride (DPhT), and TPhT were completely separated within 20 min via micellar liquid chromatography with tris(hydroxymethyl)aminomethane dodecylsulfate (0.05 mol L−1)–acetic acid (3%)–ethanol (15%) as the mobile phase and butyl-group-bonded silica gel as the stationary phase (4.6 mm × 50 mm, 5 μm).11 However, the ICP torch and the sampling orifice may become blocked after a few hours under the continuous introduction of the micellar phase. To the best of our knowledge, with the exception of methods based on micellar HPLC, no study on the reduction of the percentage of organic solvent in the HPLC mobile phase for the separation of OTs has been reported to date.
In addition, for the speciation of organotins in environmental samples, a sample pretreatment procedure is necessary to concentrate the target species, remove the interfering compounds, and make them suitable for the subsequent determinations. Different sample pretreatment techniques have been used for OT speciation analysis, including classical liquid extraction (LLE), solid-phase extraction (SPE) and the newly emerging supercritical fluid extraction (SFE), microwave/ultrasonication-assisted extraction, liquid-phase microextraction (LPME), solid-phase microextraction (SPME), and stir bar sorptive extraction (SBSE).18–20 Among these sample pretreatment techniques, SBSE has the advantages of high extraction efficiency, good reproducibility, high sensitivity, and less organic solvent consumption and has been successfully applied to the extraction of trace organic compounds, inorganic elements, and their species in environmental, food, and biological samples.21–26 The methods that involve the use of SBSE for OT analysis are usually combined with derivatization-GC-ICP-MS26 or GC-MS.25 In these methods, the OTs adsorbed on the stir bar are thermally desorbed and introduced to the GC system. To date, the direct coupling of SBSE with HPLC-ICP-MS for OT analysis without derivatization has not been reported.
The purpose of this work is to establish a non-derivatization SBSE-HPLC-ICP-MS method for butyltin speciation analysis. A C18-silica-particle-coated stir bar was prepared by adhesion. The factors affecting the extraction of monobutyltin trichloride (MBT), DBT, and TBT by SBSE were studied in detail. In addition, the HPLC separation of butyltins was also studied, and rapid separation was successfully achieved with a relatively low percentage of the organic solvent applied in the mobile phase. Certified Reference Material (CRM) of PACS-2 sediment was analyzed for validating the accuracy of the proposed method. The proposed SBSE-HPLC-ICP-MS method was applied for the speciation of butyltins in seawater, river water, lake water and corresponding sediment samples successfully.
Experimental
Standard solutions and reagents
Monobutyltin trichloride (MBT, 95%), dibutyltin dichloride (DBT, 95%), and tributyltin chloride (TBT, 96%) were purchased from Alfa Aesar (Ward Hill, MA, USA). The individual standard stock solutions (1 g L−1 as tin) of these butyltins were prepared by dissolving a specific amount of their corresponding salts in methanol and were then stored at 4 °C in the dark. The working solutions were diluted daily from the standard stock solution using high-purity water. C18 silica (100, 10, and 5 μm) was purchased from Qingdao Haiyang Chemical Co., Ltd (Qingdao, Shandong, China). Other solvents, acids, and common laboratory reagents were of analytical grade. High-purity water obtained from a Milli-Q water purification system (18.25 MΩ cm, Millipore, Molsheim, France) was used throughout all experiments.Glassware was rinsed with high-purity deionized water, decontaminated overnight in 1:1 nitric acid solution, and then rinsed thrice with high-purity deionized water.
Instrumentals
Quadrupole ICP-MS (HP 7500a, Tokyo, Japan) was combined with HPLC for the online determination of butyltins. An HPLC system consisting of an LC-10AD high-pressure pump, CTO-10A column oven (Shimadzu, Japan), and Hypersil CN column (5 μm, 150 mm × 4.6 mm i.d., Dalian Elite Analytical Instruments Co., Ltd, China) was used for the separation of butyltins. The typical operating conditions of the HPLC and ICP-MS are summarized in Table 1. An X-650 scanning electron microscope (SEM) (HITACHI, Japan) at an acceleration voltage of 30 kV was used for the characterization of the morphology of the C18-coated stir bar.| HPLC | |
| Column | CN column (Hypersil, 4.6 × 150 mm, 5 μm particle size) |
| Mobile phase | Methanol–formic acid–water (16:8:76, v/v) containing 5 mmol L−1 of mercaptoacetic acid, pH 2.5 |
| Temperature | 55 °C |
| Flow rate | 1.5 mL min−1 |
|
|
| ICP-MS plasma | |
| Reflection power | 1200 W |
| Nebulizer gas flow | 0.9 mL min−1 |
| Sample introduction | Babington nebulizer |
| Sample and skimmer cones | Nickel |
| Sample depth | 6.0 mm |
| Refrigerated spray chamber | −3 °C |
| Isotope monitored | 116Sn, 117Sn, 118Sn (for quantification), 120Sn |
| Acquisition mode | Time-resolved analysis |
Preparation of C18-coated stir bar
A “dumbbell-shaped” bare stir bar was prepared and activated based on Yu's work.27 The polydimethylsiloxane (PDMS) sol used for coating the stir bar consisted of component A (mixture of oligomers and catalyst) and component B (curing crosslinker) (GE RTV 615, Momentive, USA), and their mixing ratio was 10:1 (A:B). The C18 silica particles were adhered onto the bare glass stir bar by the PDMS sol using the procedure detailed as follows. The bare glass bar was dipped into the PDMS sol. and then removed and placed vertically in a plastic tube, where it was allowed to stand for 5 min. The glass bar coated with the PDMS sol was subsequently placed on a culture dish covered with C18 silica particles. The culture dish was then shaken up and down at an amplitude of approximately 45° to roll the glass bar back and forth, ensuring the uniform and complete adhesion of C18 silica particles on the glass bar surface. Lastly, the stir bar was incubated in a constant-temperature drier at 60 °C for 24 h. Prior to use, the stir bar was cleaned in methanol by ultrasonication for 20 min to remove any organic contaminants on the coating.
Sample collection and extraction
Certified Reference Material of PACS-2 sediment containing butyltin species was obtained from the National Research Council of Canada (Ottawa, Canada). Real-world sediment and water samples were collected from the coastal area (Bohai, Dalian, China), East Lake and Yangtze River (Wuhan, China).The water samples were filtered through a 0.45 μm membrane and stored in a refrigerator (4 °C) prior to analysis.
The sediment samples were dried at room temperature, and then homogenized and sieved through a 0.2 mm sieve. Glacial acetic acid was used to extract butyltins in PACS-2 and the sediment samples according to the literatures.28,29 Briefly, PACS-2 (0.5 g) and the dried sediment samples (0.5 g) were weighed in separate polyethylene extraction tubes. The samples were supplemented with 5 mL of glacial acetic acid. The tubes were subjected to ultrasound-assisted extraction for 30 min and centrifuged for 10 min at 4000 rpm. The supernatants were transferred to 50 mL volumetric flasks, and the residues were re-extracted as described above. The twice-extracted supernatants were then combined and diluted with high-purity water and adjusted to the desired pH. The extracts of the sediment were subjected to SBSE-HPLC-ICP-MS.
Results and discussion
SEM of C18-coated stir bar
In the preparation of the C18-coated stir bar, the glass stir bar surface was first silylated with PDMS, which functions as a coupling agent for the C18-coating layer and the glass bar substrate. PDMS can react with exposed silanol groups on the glass stir bar surface and on the incomplete end-capped silanol sites of C18 silica particles through condensation. The prepared C18-coated stir bar was characterized by SEM. As can be seen from Fig. 1, the C18 silica particles were uniformly adhered, and they formed a single layer on the surface of the glass bar. Granular coatings have a high specific surface area and a large number of active sites, which favor sorptive extraction. C18 silica of different particle sizes (100, 10, and 5 μm) were evaluated, and 100 μm C18 silica was found to have the highest extraction efficiency. Since the particle size of the 100 μm C18 silica was larger than the thickness of the PDMS coating (ca. 30 μm), these C18 silica particles were not covered by the PDMS sol. In contrast, the 10 μm and 5 μm C18 silica particles were covered by the PDMS sol, negatively affecting the extraction. Therefore, the 100 μm C18 silica was selected. The preparation process was simple, and the resulting C18-coated stir bar had a good mechanical strength and could be reused for more than 25 times. These characteristics indicate that the C18 silica particles successfully bonded with the glass surface and that the PDMS functioned as a coupling agent. | ||
| Fig. 1 Scanning electron micrographs of 100 μm (A and D), 10 μm (B and E), and 5 μm (C and F) C18-coated stir bar with magnifications of ×40 and ×150, respectively. | ||
Optimization of HPLC separation conditions for butyltins
The organotin species are considered to be readily adsorbed on the HPLC stationary phase.30 High percentages of organic solvent are usually employed in the mobile phase for fast elution.7–11,16,17 In this study, the relatively highly polar C8 and CN silica-based columns were tested for the separation of butyltins. The preliminary results indicate that the retention of butyltins on the CN column was weaker than that on the C8 column. Therefore, the CN column was used as the stationary phase in the following experiments. The effects of different types of mobile phase additives (formic acid, mercaptoacetic acid, oxalic acid, succinic acid, and L-cysteine (L-Cys)) and the percentage of methanol on the peak shape and retention of MBT, DBT, and TBT were studied.| Additives | t R (s) | ||
|---|---|---|---|
| MBT | DBT | TBT | |
| 50 mmol L−1 oxalic acid | 130 | 192 | 4300 |
| 50 mmol L−1 succinic acid | 170 | 1500 | — |
| 50 mmol L−1L-Cys | 310 | 1050 | — |
| 60 mmol L−1 mercaptoacetic acid | 195 | 460 | — |
| 8% (v/v) formic acid | — | 317 | 485 |
According to the hard and soft acid–base rule, hard acid OTs can readily react with hard/medial base ligands to form coordinative compounds. The preparation of organotin carboxylates can be carried out by the reaction of organotin halides with metal/ammonium carboxylates (eqn (1)) or by adding a tertiary amine to a solution of carboxylic acid and tin chloride (eqn (2)).32 For TBT, which has the highest steric hindrance effect, only small molecular compounds, such as formic acid, can react with TBT. The formed organotin carboxylates exhibited good water solubility;33 thus, the adsorption of organotin species on the stationary phase decreased. Furthermore, the quick elution of OTs can be achieved using a mobile phase containing relatively low percentages of organic solvents.
| RnSnCl4−n + 4 − n R′COONH4 → RnSn(OOCR′)4−n + 4 − n NH4Cl | (1) |
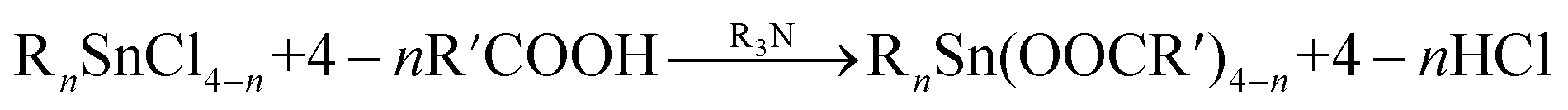 | (2) |
Based on the above mentioned results, the mercaptoacetic acid–formic acid system was selected to further optimize the conditions of the mobile phase for the separation of OTs.
The effect of mercaptoacetic acid concentration (0, 2.5, 5, 10 and 20 mmol L−1) in the mobile phase on the retention behavior of the target OTs was also studied by fixing the methanol percentage in the mobile phase at 16% (v/v), formic acid concentration at 8% (v/v) (pH 2.5), mobile phase flow rate at 1.5 mL min−1 and column temperature at 55 °C. It was found that mercaptoacetic acid can promote the elution of MBT and DBT, and the retention time of MBT and DBT decreased from larger than 700 s to around 150 s and from 320 s to around 190 s with the addition of 2.5 mmol L−1 mercaptoacetic acid in the mobile phase, respectively. However, the retention of TBT was prolonged from 405 s to 680 s with an increasing mercaptoacetic acid concentration from 0 to 20 mmol L−1. A good resolution of 1.88 was obtained for MBT and DBT at 5 mmol L−1 mercaptoacetic acid in the mobile phase. Therefore, 5 mmol L−1 mercaptoacetic acid was selected in the mobile phase for the separation of OTs. By keeping other chromatographic conditions unchanged, the effect of formic acid concentration (0%, 2.5%, 5%, 8% and 10%) in the mobile phase on the retention behavior of the target OTs was studied. It was found that the addition of formic acid had a remarkable effect on the retention times of MBT and TBT. When formic acid was not added, TBT could not be eluted within 3600 s, while the retention times of TBT was significantly shortened with the addition of formic acid. With the increase in the concentration of formic acid from 2.5% to 10% (v/v), the retention times of MBT, DBT, and TBT were decreased, and the highest resolution of MBT and DBT (1.92) was obtained at 8% (v/v) formic acid in the mobile phase. Finally, 8% (v/v) formic acid in the mobile phase was selected for the separation of OTs.
With methanol–formic acid–water (16:8:76, v/v/v) containing 5 mmol L−1 mercaptoacetic acid (pH 2.5) as the mobile phase, the effect of column temperature in the range of 25–55 °C on the retention time of the target OTs and the resolution of MBT/DBT was studied. It was found that when the column temperature increased from 25 to 55 °C, the retention times of MBT, DBT, and TBT decreased, while the resolution of MBT/DBT remained almost constant. To achieve a quick elution, 55 °C was selected as the optimum column temperature.
In summary, the optimized separation conditions are as follows: the mobile phase was methanol–formic acid–water (16:8:76, v/v/v) containing 5 mmol L−1 mercaptoacetic acid (pH 2.5, adjusted by ammonia), and the column temperature was 55 °C. Fig. 2 is the typical HPLC-ICP-MS chromatogram for butyltins separation. As can be seen, a quick separation (<8 min) was successfully achieved using a relatively low amount of organic solvent.
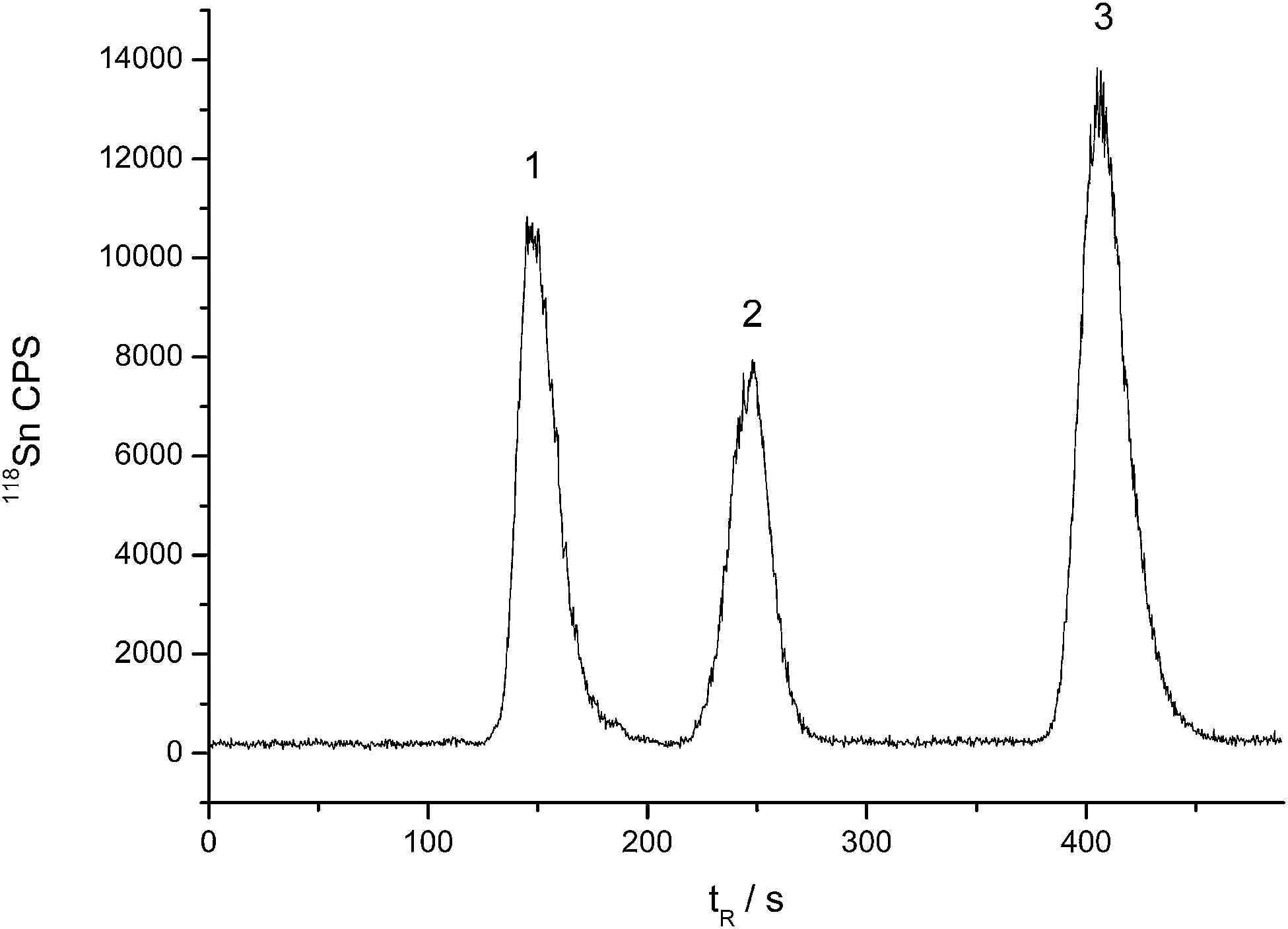 | ||
| Fig. 2 Chromatogram of butyltins by HPLC-ICP-MS. Concentrations: 200 μg L−1; (1) MBT; (2) DBT; (3) TBT. | ||
Optimization of SBSE conditions
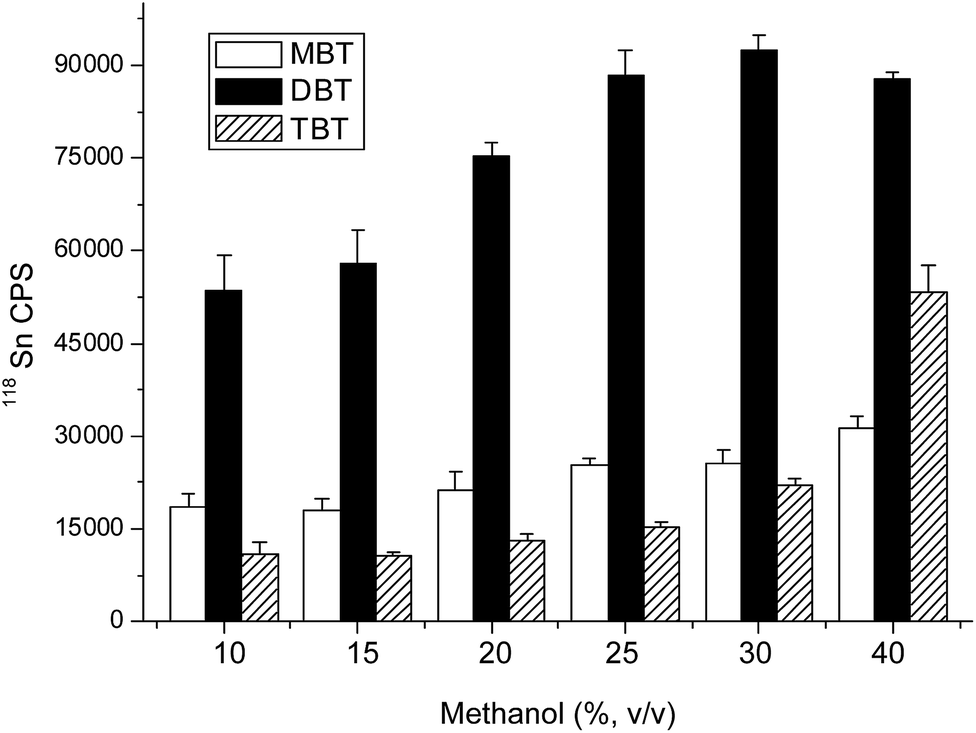 | ||
| Fig. 3 Effect of desorption solution on SBSE extraction efficiency of MBT, DBT and TBT. Conditions: MBT, DBT, TBT 2 μg L−1, 10 mL; extraction 20 min; desorption 15 min; 600 rpm stirring rate; desorption volume 150 μL. | ||
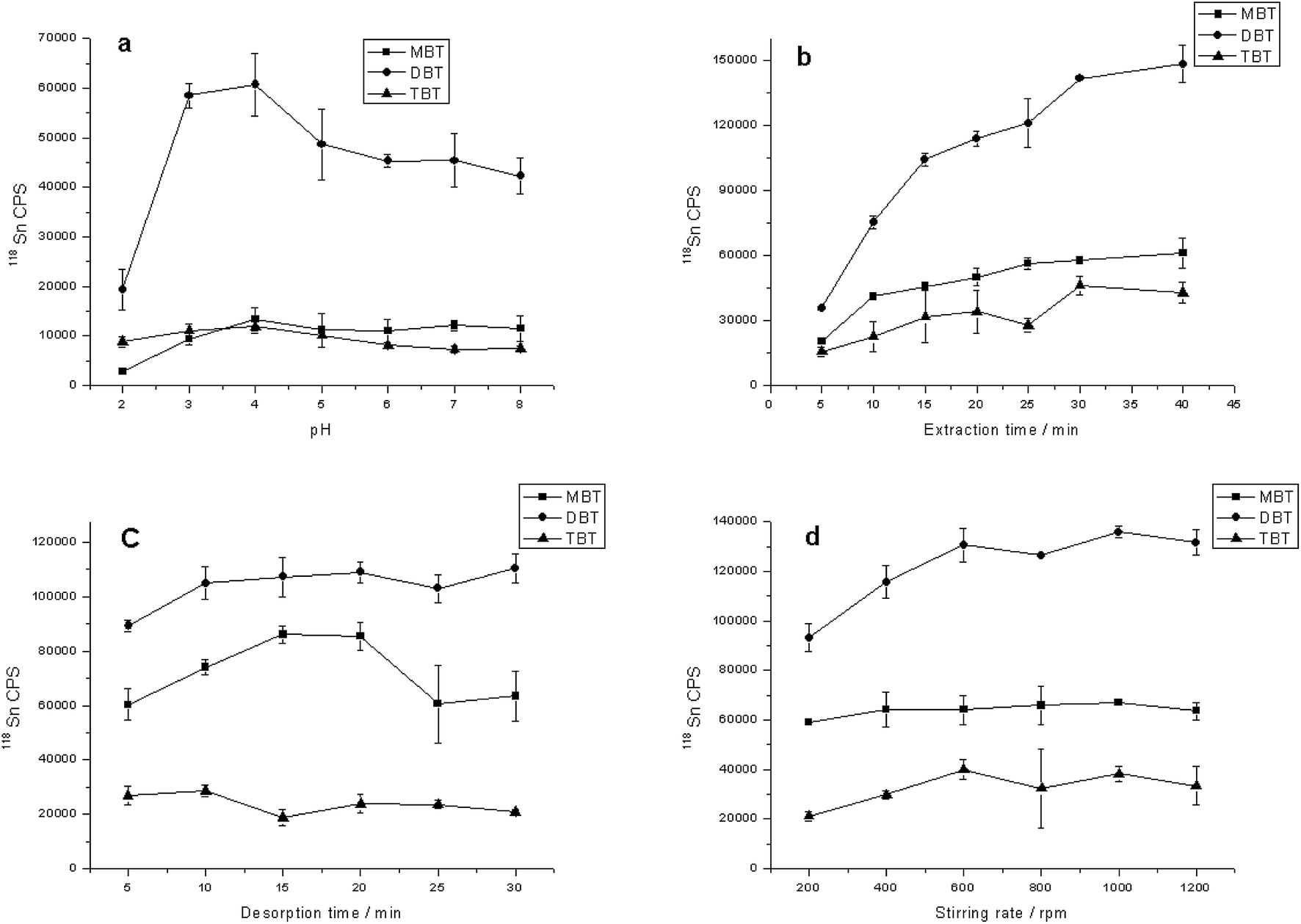 | ||
| Fig. 4 Effect of (a) sample pH; (b) extraction time; (c) desorption time and (d) stirring speed on SBSE extraction efficiency of MBT, DBT and TBT. | ||
Analytical performance
Under the optimized conditions, the analytical performance of the developed C18-SBSE-HPLC-ICP-MS method was evaluated. The results are listed in Table 3. The linearity of the proposed method was tested using standard solutions with increasing concentrations of MBT, DBT, and TBT from the limit of quantification to 50 μg L−1. The calibration curves (peak area vs. concentration) showed a linear response over the complete range, with correlation coefficients (R) ranging from 0.9996 to 0.9999. The limits of detection (LODs) calculated according to the IUPAC guidelines (three times the standard deviation of the background for 11 runs divided by the slope of the calibration curve) ranged from 15.6 to 29.4 ng L−1. The calculated enrichment factor (EF), which is defined as the slope ratio of the calibration curve obtained with and without SBSE, ranged from 85- to 127-fold (the theoretical EF is 200-fold).| Analytes | Linear range (μg L−1) | Linear equation | R | Enrichment factora | LODsb (ng L−1) | Bar to bar RSDsc % | Batch to batch RSDsc % | Reuse times |
|---|---|---|---|---|---|---|---|---|
| a Theoretical enrichment factor was 200 (20 mL sample solution, 100 μL desorption solution). b 118Sn for quantification. c Conditions: MBT, DBT 2 μg L−1, TBT 10 μg L−1, n = 7. | ||||||||
| MBT | 0.10–50 |
y = 43976x − 9120.5 |
0.9999 | 109 | 29.4 | 11.8 | 12.8 | >25 |
| DBT | 0.05–50 |
y = 33132x − 4316.0 |
0.9999 | 85 | 15.7 | 5.6 | 15.2 | |
| TBT | 0.05–50 |
y = 56949x − 1181.4 |
0.9996 | 127 | 15.6 | 9.2 | 15.4 | |
The LODs for the butyltins obtained by C18-SBSE-HPLC-ICP-MS were compared with those obtained using other approaches reported in the literature. As can be seen in Table 4, the LODs of the proposed method were lower than those of direct injection HPLC-ICP-MS7,34,35 and SPME-HPLC-ICP-MS,36 but higher than those of HS-SDME-GC-ICP-MS,37 SPME-GC-ICP-TOF-MS,38 SBSE-GC-ICP-MS,26 and SBSE-GC-MS.25 Compared with nearly 100% sample introduction of GC-ICP-MS, the extremely low sample introduction efficiency of the conventional pneumatic nebulizer (<5%) used in HPLC-ICP-MS resulted in higher LODs in the present study than in the ref. 25, 26, 37 and 38. However, the proposed method is a simple, rapid, and solvent-less technique that does not require any preliminary derivatization step. In addition, the hyphenation of HPLC with ICP-MS is considerably simpler than that of GC-ICP-MS. The HPLC separation conditions for the butyltins obtained in this work were also compared with those in other works. As observed in Table 5, the percentage of organic solvent employed in this work was less than that employed in RP, RP-IP, and IE-HPLC. Although the organic solvent percentage in the micellar phase was relatively low, the ICP torch and the sampling orifice could become blocked after several hours under continuous introduction of the micellar phase. Quick HPLC separation (<8 min) was achieved in this work, and the mobile phase was compatible with the conventional ICP-MS detector without special adaptations.
| Analyte | Pretreatment techniques/coating | Derivatizing reagents | Detection | LOD (ng L−1) | Ref. |
|---|---|---|---|---|---|
| a pg g−1. b ng g−1. | |||||
| MBT, DBT, TBT | HS-SDME | NaBEt4 | GC-ICP-MS | 1.4, 1.8, 0.8 | 37 |
| MBT, DBT, TBT | SPME/PDMS-DVB | NaBEt4 | GC-ICP-TOF-MS | 0.77, 0.99, 0.62a | 38 |
| TBT | SBSE/PDMS | NaBEt4 | GC-ICP-MS | <0.2 | 26 |
| MBT, DBT, TBT | HS-SBSE/PDMS | NaBEt4 | TD-GC-MS | 2, 0.4, 0.8 | 25 |
| TBT | SPME/PDMS-DVB | — | HPLC-ICP-MS | 185 | 36 |
| MBT, DBT, TBT | — | — | HPLC-ICP-MS | 80, 50, 30b | 7 |
| MBT, DBT, TBT | — | — | HPLC-ICP-MS | 360, 500, 370 | 34 |
| DBT, TBT | — | — | HPLC-ICP-MS | 270, 280 | 35 |
| MBT, DBT, TBT | SBSE/C18 | — | HPLC-ICP-MS | 29.4, 15.7, 15.6 | This work |
| Analyte | HPLC modes/column type | Mobile phase | t R (min) | Nebulizer type | Rf power (w) | Spray chamber temperature (°C) | O2 gas | Desolvation | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| TPhT, TBT | RP-IP/C18 | MeOH–H2O–CH3COOH (70:29:1) (v/v), and 4 mmol L−1 ion pair reagent |
∼4.2, 7.0 | Glass concentric, Micro Mist, MCN | 1500 | 0 | ✓ | × | 8 |
| MBT, DBT, TBT | RP-IP/ODS | MeOH–CH3COOH–H2O (80:2:18), 4 mmol L−1 sodium 1-pentanesulfonate and 0.1% tropolone as the ion pairing reagent |
<4 | DIN | 1200 | — | ✓ | × | 34 |
| TMT, TET, TPhT, TBT, TPhT | RP-IP/C8 | MeOH–CH3COOH–H2O (50:5:45) with 5 mmol L−1 sodium 1-pentanesulfonate |
<6 | USN | 1150 | −10 | ✓ | ✓ | 17 |
| MBT, DBT, TBT | IE/SCX | MeOH–water (60:40), 0.16 mol L−1 ammonium citrate buffer |
<7 | Cross-flow nebulizer | 1600 | — | × | × | 7 |
| MBT, DBT, TBT | IE/SCX | MeOH–H2O (60:40) with 0.16 mol L−1 ammonium citrate, pH = 4.8 |
<8 | Cross-flow nebulizer | 1600 | Room temperature | × | × | 39 |
| MBT, DBT, TBT | IE/SCX | MeOH–CH3COOH–H2O (30:5:65) with 0.05 mol L−1 ammonium citrate |
<12 | — | 1800 | — | × | × | 16 |
| DBT, TBT, DPhT, TPhT | RP/C18 | CH3CN–H2O–CH3COOH (65:23:12), 0.05% TEA, pH 3.0 |
<43 | PFA | 1500 | −5 | ✓ | × | 35 |
| MBT, DBT, TBT, MPhT, DPhT, TPhT | RP/TSK gel ODS | MeOH–CH3COOH–H2O (72.5:6:21.5), with 0.075% tropolone and 0.1% TEA |
<20 | — | 1350 | −4 | ✓ | × | 40 |
| DBT, TBT, DPhT, TPhT | RP/C18 | CH3CN–CH3COOH–H2O (65:10:25) with 0.05% TEA |
<13 | — | 1125–1250 | −10 to −15 | ✓ | × | 41 |
| DBT, TBT, TPhT | RP/C18 | (A): CH3CN–H2O–CH3COOH with 0.05% TEA (65:25:10), pH 3.4 ± 0.1 (B) and (C): CH3CN–H2O–CH3COOH with 0.05% TEA (65:23:12), pH 3.1 ± 0.1 |
5.14–13.48 | Micro flow PFA concentric nebulizer | 1350–1550 | −5 | ✓ | × | 9 |
| TET, TBT, TPhT | RP/capillary ODS columns | CH3CN 100%, or MeOH–CH3COOH–pyridine–H2O (85:4:1:10) |
<10 | Laboratory-made MCN | 1000 | — | × | × | 42 |
| TBT, TPhT | RP/C18 | MeOH–CH3COOH–TEA–H2O (82:2.5:0.3:15), with 10 mg L−1 oxalic acid |
<10 | — | 1250 | −15 | ✓ | × | 31 |
| TMT, TEtT, TPrT | Micellar HPLC/Spherisorb ODS | Propanol–CH3COOH–H2O (3:3:94) with 0.1 mol L−1 SDS and 5 mmol L−1 KF−1 |
<8 | Concentric nebulizer | 1300 | 5 | × | × | 10 |
| TMT, DMT, DPhT, TPhT, DBT, TBT | Micellar HPLC/SIL-TMS/5B, SIL-C4A/5B, SIL-PHE-5B, SIL-CNP/5B | Ethanol–CH3COOH–H2O (15:3:82) with 0.05 mol L−1 TDS and 0.05 mol L−1 ammonium nitrate |
<20 | Concentric glass nebulizer | 1300 | 0 | × | × | 11 |
| MBT, DBT, TBT | CN | MeOH–formic acid–water (16:8:76) containing 5 mmol L−1 of mercaptoacetic acid |
<10 | Babington nebulizer | 1200 | −3 | × | × | This work |
Real sample analysis
Certified Reference Material of PACS-2 sediment was analyzed to evaluate the accuracy of the proposed method. The analytical results are listed in Table 6. The determined values coincide with the certified ones. Real samples, which include water and sediment from the Yangtze River, East Lake and the coastal areas of Bohai, were analyzed to investigate the applicability of the proposed method. The analytical results, along with the recoveries for the spiked samples, are given in Tables 7 and 8, and Fig. 5 shows the C18-SBSE-HPLC-ICP-MS chromatograms for real-world samples. As can be seen, MBT, DBT, and TBT were not detected in water and sediment samples collected from the East Lake and Yangtze River, but 0.231 ± 0.010 μg L−1 and 46.6 ± 3.1 ng g−1 of TBT were found in the seawater and coastal sediment collected from Bohai, Dalian. The recoveries are mostly around 90% (Tables 7 and 8), suggesting that there might be ∼10% loss of the target butyltin species in the sample extraction/preparation process.| Analytes | East Lake | Yangtze River | Seawater | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Added (μg L−1) | Found (μg L−1) | Recovery (%) | Added (μg L−1) | Found (μg L−1) | Recovery (%) | Added (μg L−1) | Found (μg L−1) | Recovery (%) | |
| MBT | 0 | — | — | 0 | — | — | 0 | — | — |
| 0.2 | 0.177 ± 0.008 | 88.5 | 0.2 | 0.213 ± 0.008 | 106.5 | 0.2 | 0.163 ± 0.009 | 81.5 | |
| 1 | 1.003 ± 0.071 | 100.3 | 1 | 0.910 ± 0.049 | 91.0 | 1 | 0.874 ± 0.039 | 87.4 | |
| 5 | 4.485 ± 0.278 | 89.7 | 5 | 4.548 ± 0.337 | 91.0 | 5 | 5.220 ± 0.215 | 104.4 | |
| DBT | 0 | — | — | 0 | — | — | 0 | — | — |
| 0.2 | 0.181 ± 0.006 | 90.5 | 0.2 | 0.212 ± 0.009 | 106.0 | 0.2 | 0.208 ± 0.013 | 104.0 | |
| 1 | 0.887 ± 0.016 | 88.7 | 1 | 0.959 ± 0.078 | 95.9 | 1 | 0.939 ± 0.031 | 93.9 | |
| 5 | 4.614 ± 0.178 | 92.3 | 5 | 4.532 ± 0.318 | 90.6 | 5 | 4.731 ± 0.161 | 94.6 | |
| TBT | 0 | — | — | 0 | — | — | 0 | 0.231 ± 0.010 | — |
| 0.2 | 0.167 ± 0.011 | 83.5 | 0.2 | 0.187 ± 0.007 | 93.5 | 0.2 | 0.414 ± 0.014 | 91.5 | |
| 1 | 0.941 ± 0.066 | 94.1 | 1 | 0.883 ± 0.056 | 88.3 | 1 | 1.096 ± 0.020 | 86.5 | |
| 5 | 4.395 ± 0.094 | 87.9 | 5 | 4.151 ± 0.270 | 83.0 | 5 | 5.033 ± 0.279 | 96.0 | |
| Analytes | East Lake sediment | Yangtze River sediment | Seawater sediment | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Added (ng g−1) | Found (ng g−1) | Recovery (%) | Added (ng g−1) | Found (ng g−1) | Recovery (%) | Added (ng g−1) | Found (ng g−1) | Recovery (%) | |
| MBT | 0 | — | — | 0 | — | — | 0 | — | — |
| 20 | 16.3 ± 1.2 | 81.5 | 20 | 17.3 ± 0.3 | 86.5 | 20 | 17.1 ± 0.5 | 85.5 | |
| 100 | 82.7 ± 3.7 | 82.7 | 100 | 78.8 ± 6.1 | 78.8 | 100 | 83.4 ± 2.6 | 83.4 | |
| 500 | 406.8 ± 31.0 | 81.4 | 500 | 379.6 ± 32.6 | 75.9 | 500 | 377.3 ± 16.8 | 75.4 | |
| DBT | 0 | — | — | 0 | — | — | 0 | — | — |
| 20 | 15.9 ± 0.7 | 79.6 | 20 | 15.4 ± 0.9 | 77.0 | 20 | 17.5 ± 0.8 | 87.6 | |
| 100 | 80.5 ± 7.9 | 80.5 | 100 | 89.6 ± 5.5 | 89.6 | 100 | 76.5 ± 7.1 | 76.5 | |
| 500 | 393.4 ± 30.7 | 78.7 | 500 | 411.5 ± 32.2 | 82.3 | 500 | 441.6 ± 26.4 | 88.3 | |
| TBT | 0 | — | — | 0 | — | — | 0 | 46.6 ± 3.1 | — |
| 20 | 17.0 ± 0.9 | 85.1 | 20 | 18.9 ± 1.4 | 94.5 | 20 | 62.3 ± 3.5 | 78.7 | |
| 100 | 102.7 ± 9.4 | 102.7 | 100 | 92.6 ± 6.2 | 92.6 | 100 | 130.1 ± 10.8 | 83.5 | |
| 500 | 431.9 ± 29.7 | 86.4 | 500 | 373.3 ± 9.6 | 74.7 | 500 | 478.5 ± 17.1 | 86.4 | |
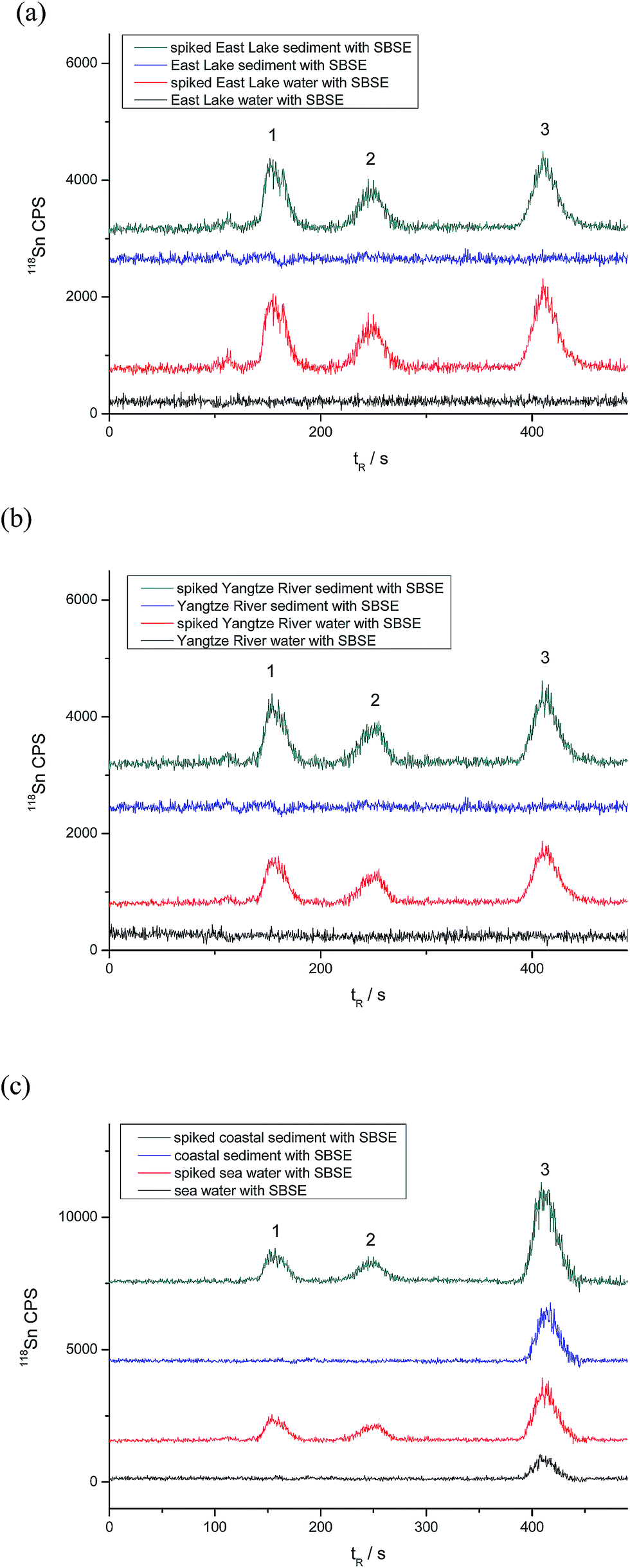 | ||
| Fig. 5 C18-SBSE-HPLC-ICP-MS chromatograms for real-world water and sediment samples from (a) East Lake, (b) Yangtze River and (c) Bohai, Dalian. Conditions: sample volume: 10 mL; pH: 4.0; extraction time: 30 min; desorption time: 20 min; stirring rate: 600 rpm; desorption volume: 150 μL; no salt addition. Spiked concentration: 0.2 μg L−1 of MBT, DBT and TBT for waters and 20 ng g−1 for sediments. | ||
Conclusions
The development of C18-SBSE-HPLC-ICP-MS has proven that the coupling of C18-SBSE to HPLC-ICP-MS is feasible and can be successfully applied for butyltin analysis. The optimized method based on this coupling offers an attractive new approach for the quantification of MBT, DBT, and TBT in both water and sediment samples. The developed method is a simple, rapid, and solvent-less technique that does not require any preliminary derivatization step. Quick HPLC separation (<8 min) was successfully achieved with a relatively low percentage of organic solvent in the mobile phase, and the mobile phase is compatible with conventional ICP-MS without special adaptations.Acknowledgements
Financial supports from the National Nature Science Foundation of China (no. 21175102), the Science Fund for Creative Research Groups of NSFC (no. 20621502, 20921062), the Fundamental Research Funds for the Central Universities (no. 114009, MOE China) and Large-scale Instrument and Equipment Sharing Foundation of Wuhan University are gratefully acknowledged.References
- E. González-Toledo, R. Compañó, M. Granados and M. D. Prat, TrAC, Trends Anal. Chem., 2003, 22, 26 CrossRef.
- K. Fent, Crit. Rev. Toxicol., 1996, 26, 3 CrossRef.
- M. Bravo, A. Valenzuela, W. Quiroz, M. Pinto, M. Flores and H. Pinochet, Talanta, 2011, 81, 1034 CrossRef PubMed.
- P. Canosa, R. Montes, J. P. Lamas, M. Garcia-Lopez, I. Orriols and I. Rodriguez, Talanta, 2009, 79, 598 CrossRef CAS PubMed.
- M. Vahcic, R. Milacic and J. Scancar, Anal. Chim. Acta, 2010, 694, 21 CrossRef PubMed.
- E. Pobozy, B. Glod, J. Kaniewska and M. Trojanowicz, J. Chromatogr. A, 1995, 718, 329 CrossRef CAS.
- L. Yang and J. W. H. Lam, J. Anal. At. Spectrom., 2001, 16, 724 RSC.
- K. L. Ackley, K. L. Sutton and J. A. Caruso, J. Anal. At. Spectrom., 2000, 15, 1069 RSC.
- R. Wahlen and T. Catterick, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 783, 221 CrossRef CAS.
- H. Suyani, D. Heitkemper, J. Creed and J. Caruso, Appl. Spectrosc., 1989, 43, 962 CrossRef CAS.
- Y. Inoue, K. Kawabata and Y. Suzuki, J. Anal. At. Spectrom., 1995, 10, 363 RSC.
- J. Szpunar-Lobińska, C. Witte, R. Łobinski and F. C. Adams, Fresenius' J. Anal. Chem., 1994, 351, 351 CrossRef.
- N. P. Vela and J. A. Caruso, J. Anal. At. Spectrom., 1996, 11, 1129 RSC.
- R. F. Browner, A. Canals and V. Hernandis, Spectrochim. Acta, Part B, 1992, 47, 659 CrossRef.
- A. W. Boorn and R. F. Browner, Anal. Chem., 1982, 54, 1402 CrossRef CAS.
- J. I. Garcia-Alonso, A. Sanz-Medel and L. Ebdon, Anal. Chim. Acta, 1993, 283, 261 CrossRef CAS.
- H. J. Yang, S. J. Jiang, Y. J. Yang and C. J. Hwang, Anal. Chim. Acta, 1995, 312, 141 CrossRef CAS.
- J. Heroult, T. Zuliani, M. Bueno, L. Denaix and G. Lespes, Talanta, 2008, 75, 486 CrossRef CAS PubMed.
- C. Dietz, J. Sanz, E. Sanz, R. Munoz-Olivas and C. Camara, J. Chromatogr. A, 2007, 1153, 114 CrossRef CAS PubMed.
- T. Zuliani, G. Lespes, R. Milacic and J. Scancar, Talanta, 2010, 80, 1945 CrossRef CAS PubMed.
- J. Y. Barletta, P. Gomes, A. J. dos Santos-Neto and F. M. Lancas, J. Sep. Sci., 2011, 34, 1317 CrossRef CAS PubMed.
- S. Bazhdanzadeh, Z. Talebpour, N. Adib and H. Y. Aboul-Enein, J. Sep. Sci., 2011, 34, 90 CrossRef CAS PubMed.
- Y. L. Hu, J. W. Li and G. K. Li, J. Sep. Sci., 2011, 34, 1190 CrossRef CAS PubMed.
- X. J. Mao, B. B. Chen, C. Z. Huang, M. He and B. Hu, J. Chromatogr. A, 2011, 1218, 1 CrossRef CAS PubMed.
- A. Prieto, O. Zuloaga, A. Usobiaga, N. Etxebarria, L. A. Fernandez, C. Marcic and A. de Diego, J. Chromatogr. A, 2008, 1185, 130 CrossRef CAS PubMed.
- J. Vercauteren, C. Peres, C. Devos, P. Sandra, F. Vanhaecke and L. Moens, Anal. Chem., 2001, 73, 1509 CrossRef CAS.
- C. H. Yu, Z. M. Yao and B. Hu, Anal. Chim. Acta, 2009, 641, 75 CrossRef CAS PubMed.
- S. Aguerre, G. Lespes, V. Desauziers and M. Potin-Gautier, J. Anal. At. Spectrom., 2001, 16, 263 RSC.
- J. Carpinteiro, I. Rodríguez and R. Cela, Fresenius' J. Anal. Chem., 2001, 370, 872 CrossRef CAS.
- B. Michalke, TrAC, Trends Anal. Chem., 2002, 21, 154 CrossRef CAS.
- B. Fairman, T. Catterick, B. Wheals and E. Polinina, J. Chromatogr. A, 1997, 758, 85 CrossRef CAS.
- D. L. Alleston and A. G. Davies, J. Chem. Soc., 1962, 2050 RSC.
- A. G. Davies, Organotin Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2nd edn, 2004, p. 207 Search PubMed.
- W. S. Chao and S. J. Jiang, J. Anal. At. Spectrom., 1998, 13, 1337 RSC.
- Z. H. Yu, M. Jing, G. Wang, X. Cao, H. M. Qiu, Y. L. Huang and X. R. Wang, Chin. J. Anal. Chem., 2008, 36, 1035 CAS.
- A. Ugarte, N. Unceta, M. C. Sampedro, M. A. Goicolea, A. Gomez-Caballero and R. J. Barrio, J. Anal. At. Spectrom., 2009, 24, 347 RSC.
- Q. Xiao, B. Hu and M. He, J. Chromatogr. A, 2008, 1211, 135 CrossRef CAS PubMed.
- P. Jitaru, H. G. Infante and F. C. Adams, J. Anal. At. Spectrom., 2004, 19, 867 RSC.
- L. Yang, Z. Mester and R. E. Sturgeon, Anal. Chem., 2002, 74, 2968 CrossRef CAS.
- S. Chiron, S. Roy, R. Cottier and R. Jeannot, J. Chromatogr. A, 2000, 879, 137 CrossRef CAS.
- S. White, T. Catterick, B. Fairman and K. Webb, J. Chromatogr. A, 1998, 794, 211 CrossRef CAS.
- R. Trones, A. Tangen, W. Lund and T. Greibrokk, J. Chromatogr. A, 1999, 835, 105 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |