
An internal standardisation strategy for quantitative immunoassay tissue imaging using laser ablation inductively coupled plasma mass spectrometry†
Daniel A.
Frick
a,
Charlotte
Giesen
b,
Teresa
Hemmerle
c,
Bernd
Bodenmiller
b and
Detlef
Günther
*a
aDepartment of Chemistry and Applied Biosciences, Laboratory of Inorganic Chemistry, ETH Zürich, Zürich, Switzerland. E-mail: guenther@inorg.chem.ethz.ch
bInstitute of Molecular Life Sciences, University of Zürich, Zürich, Switzerland
cDepartment of Chemistry and Applied Biosciences, Institute of Pharmaceutical Sciences, ETH Zürich, Zürich, Switzerland
First published on 29th September 2014
Abstract
Laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) for imaging of biological materials, such as tissue sections, has gained growing attention in the last few years, and even more since the coupling with isotopically enriched and labelled antibodies for quantitative immunoassays became available. In this work, an internal standardisation strategy for the quantitative imaging analysis of biological samples by LA-ICP-MS using labelling with an iridium compound is proposed. The utilisation of an iridium intercalator offers the advantage that the internal standard does not only correct for drift and matrix effects but also correct for differences in the ablated mass at the same time. For this, the applicability of the internal standard was investigated for formalin fixed paraffin embedded tissues (FFPEs) and for cryosections. Background contamination and thickness correlation were studied. As a proof of concept, the quantification of an isotope labelled mouse anti-E-Cadherin antibody was performed on a FFPE kidney section of a mouse at different spatial resolutions.
Introduction
Tissue imaging
Imaging of soft tissues by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) has emerged over the last few years since the first LA-ICP-MS analyses of soft tissues were presented in the late 1990s.1 At that time, the focus in quantitative tissue imaging lay on the determination of spatially resolved concentrations of Cu, Zn, Mo, Cd and Pb in various samples such as kidney and liver from sheep and pigs.2 After that two different directions in research have been pursued. The first has dealt with the determination of trace element concentrations2,3 or isotope ratios4,5 in soft biological tissues. A recent review by Konz et al. has provided an extensive overview of the use of LA-ICP-MS for biomedical applications.6 Recently, the second direction, called quantitative immunoassays,7 has emerged, which makes use of isotope labelled antibodies to determine the corresponding antigen concentrations. This strategy is based on a concept developed by Baranov et al.,8 who presented their first results at the 2001 European Winter Conference on Plasma Spectrochemistry, Quinn et al.9 and Zhang et al.10 The concept was lately further extended by Tanner, Ornatsky and co-workers.11,12 Most recently Giesen et al. measured human breast cancer formalin-fixed paraffin-embedded (FFPE) samples and mapped 32 proteins and protein modifications for each cell in a multiplexing immunoassay.13 Common to both techniques, immunoassay and elemental imaging, is the need for suitable external calibration standards and internal standardisation strategies to obtain quantitative results.Considerations for internal standards
The basic considerations for a suitable internal standard for LA-ICP-MS can be found in the publication by Longerich et al.14 It is stated that an internal standard should correct for the drift in sensitivity, matrix effects, and the ablated mass. Currently, there are four different internal standardisation procedures described in elemental imaging of biological materials.The first one relies on the addition of a standard solution, either desolvated or directly aspirated, into the ablation cell or directly in front of the torch, containing the internal standard element at a known concentration.15 This approach can correct for sensitivity drift of the instrument and eventually changes in analyte sensitivity due to, e.g., mass load in the plasma,16 but cannot correct for changes in the ablated mass and transport efficiency since the internal standard is introduced separately from the tissue sample.
The second approach uses an additional layer of a suitable standard, located either between the support and the tissue17 or on top of the tissue.18 This allows correcting for transport efficiency and sensitivity drift, but thickness variations within a tissue sample can only be corrected for highly specific conditions. These approaches propose to remove the entire layers (internal standard and tissue), therefore the internal standard layer on top or between the sample and support material cannot correct for the thickness variation of the tissue.
The third correction procedure utilises an element, which is homogenously distributed in the samples. Here, carbon has been most frequently selected as the internal standard.2,3 Unfortunately, carbon is not suited as an internal standard as it was shown that even small variations in the oxygen/carbon content of a sample to the standard changes the degree to which carbon containing gaseous species (CCGS) are formed. Therefore, the internal standard as well as analytes enter the plasma partly as different species, giving rise to different diffusion processes.19 If the external standard has the same composition and macroscopic consistency, the third approach is adequate for internal standardisation since it can correct for ablated mass, transport efficiency and drifts in the plasma. However, further drawbacks are that carbon has a high ionization potential which leads to low sensitivity and that the background intensity of carbon is very high due to the atmospheric plasma (entrainment of CO2 into the plasma from the atmosphere). These are severe limitations for high spatially resolved analyses.
The fourth approach has been reported by Becker et al.15 using no internal standard but total consumption of the sample and standard. A standard has been prepared by taking a sample from the same tissue. The sample is homogenized and various spiking concentrations of the elements of interest can be added. Afterwards, the material is frozen and cryo-sectioned using the same preparation as the sample of interest and at the same thickness for the sample and standard. The only assumption in using such a procedure is similar transport efficiency for the sample and standard. This can be achieved when the entire thin section is ablated for analysis and an appropriate ablation cell is used. For elements naturally occurring in the tissue samples, this method lacks a true blank.
Due to the texture of biological tissues, homogenous distribution of elements can usually not be assumed. The concentrations of ubiquitous elements such as sodium and potassium can be altered during the preparation of the tissue and are notoriously difficult to determine by ICP-MS because of high instrumental background levels as a result of contamination or spectral interference. Based on the 3rd quantification approach, iodine was successfully applied as an elemental marker to correct for drifts of the ICP and tissue thickness inhomogeneities. Iodine binds covalently to two amino acids (tyrosine and histidine) and has been applied using reduction of iodinate directly in proteins, proteomes and antibody samples.20,21 Even though iodine is applicable it has some disadvantages with respect to wash-out behaviour and cross-contamination (see ESI†). Further efforts were made to mark single cells with a metal intercalator for immunoassay using single cell ICP-MS.22 A possible disadvantage of using metal intercalation could be that the intercalator binds to DNA, which is confined in the cell nucleus and might not necessarily be present to the same extent in all cell types. Since in the experiments described below the targeted spatial resolution was chosen greater than the size of a single cell, this problem will not pose a significant limitation. Therefore, an iridium intercalator ((η5-pentamethylcyclopentadienyl)-iridium(III)-dipyridophenazine) was investigated on fixed tissue thin sections for its suitability as the internal standard for quantitative immunoassay imaging.
Results and discussion
Background contamination
Since the iridium intercalator ((η5-pentamethylcyclopentadienyl)-iridium(III)-dipyridophenazine, the structure is shown by Ornatsky et al.22) is applied in a later step onto the tissue section, a contamination of the embedding material with the iridium intercalator is theoretically possible. For both types of thin sections, the support material next to the tissue was ablated with the same settings as the tissues and the contamination of the support material was evaluated. ESI, Fig. 1a† shows background corrected signal intensities of 193Ir for the FFPE tissue and the support. The 193Ir signal intensities are at background level, indicating no significant contamination. ESI, Fig. 1b† shows the background corrected signal intensities of 193Ir for the cryo tissue and the support, where the 193Ir signal intensities are increased to an average of 200 cps compared to a gas background of 30 cps. During the sample preparation steps, the cryo embedding medium was not entirely removed and the iridium intercalator binds to this residue and leads therefore to a higher background signal. Since the signal/background ratio is above a factor of 50, this could affect the internal standardisation. The signal/background ratio for the iodine stained tissue was between 20 and 40 depending if measured on the support material which was in direct contact with iodine or just carried over during the washing steps.Thickness correlation
To correct for the ablated mass, the iridium signal intensities have to correlate with the sample thickness. For this, five liver cryosections were cut at different thicknesses, ranging from 2 μm to 14 μm. The samples were fixed with acetone and treated with the iridium intercalator. The tissue sections collapse after drying, therefore the thickness was determined by white-light interferometry and it was shown that the thickness after drying correlates with the cut size of the cryosections (Fig. 1a). Successively two paths were ablated across the tissue and the average background corrected signal intensities were plotted against the cut tissue thickness. The correlation is shown in Fig. 1b, both isotopes of iridium show a good correlation between average background corrected signal intensities and tissue thickness.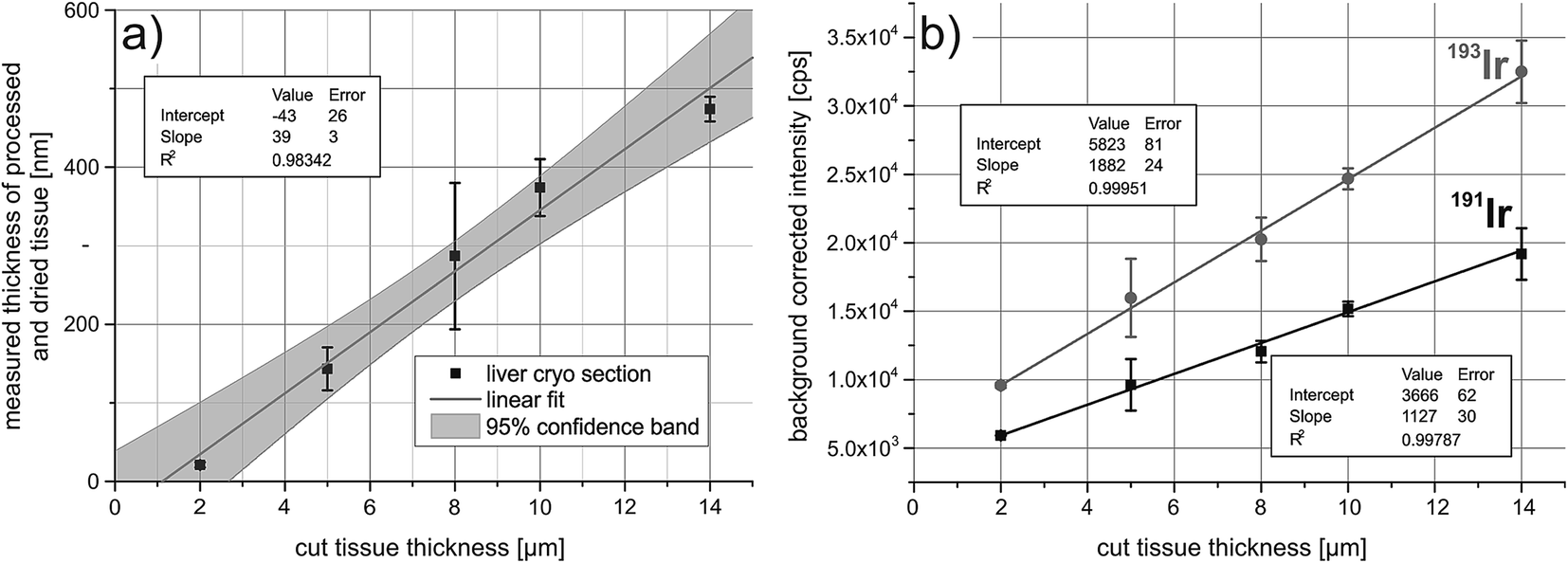 | ||
| Fig. 1 (a) Correlation between the cut tissue height (frozen, set value from the microtome) and the measured thickness of the processed (staining) and dried tissue using white light interferometry. (b) Iridium signal intensity correlation with the cut thickness of the tissues. | ||
Quantitative immunoassay imaging
For the proof of concept, a FFPE section of a mouse kidney was chosen which contains a folding. Fig. 2a shows the topology of the measured kidney obtained by white light interferometry. A section containing this folding (Fig. 2b is rectangle I from Fig. 2a) was then analysed using LA-ICP-MS and the correlation of the internal standard with the thickness was tested. Fig. 2c shows the 193Ir signal intensities for the same inset (rectangle I) in Fig. 2a and the three different height levels are visible. The concentration of the internal standard was determined by standard addition of desolvated aerosol.15,23 ESI, Fig. 2† shows the result from the standard addition. Notably in Fig. 2c a region with apparent lower iridium intensities is highlighted with a ↓. During the ablation of this laser ablation track the positioning was misaligned by 6 μm, meaning that this track ablated less material than the other ablation tracks.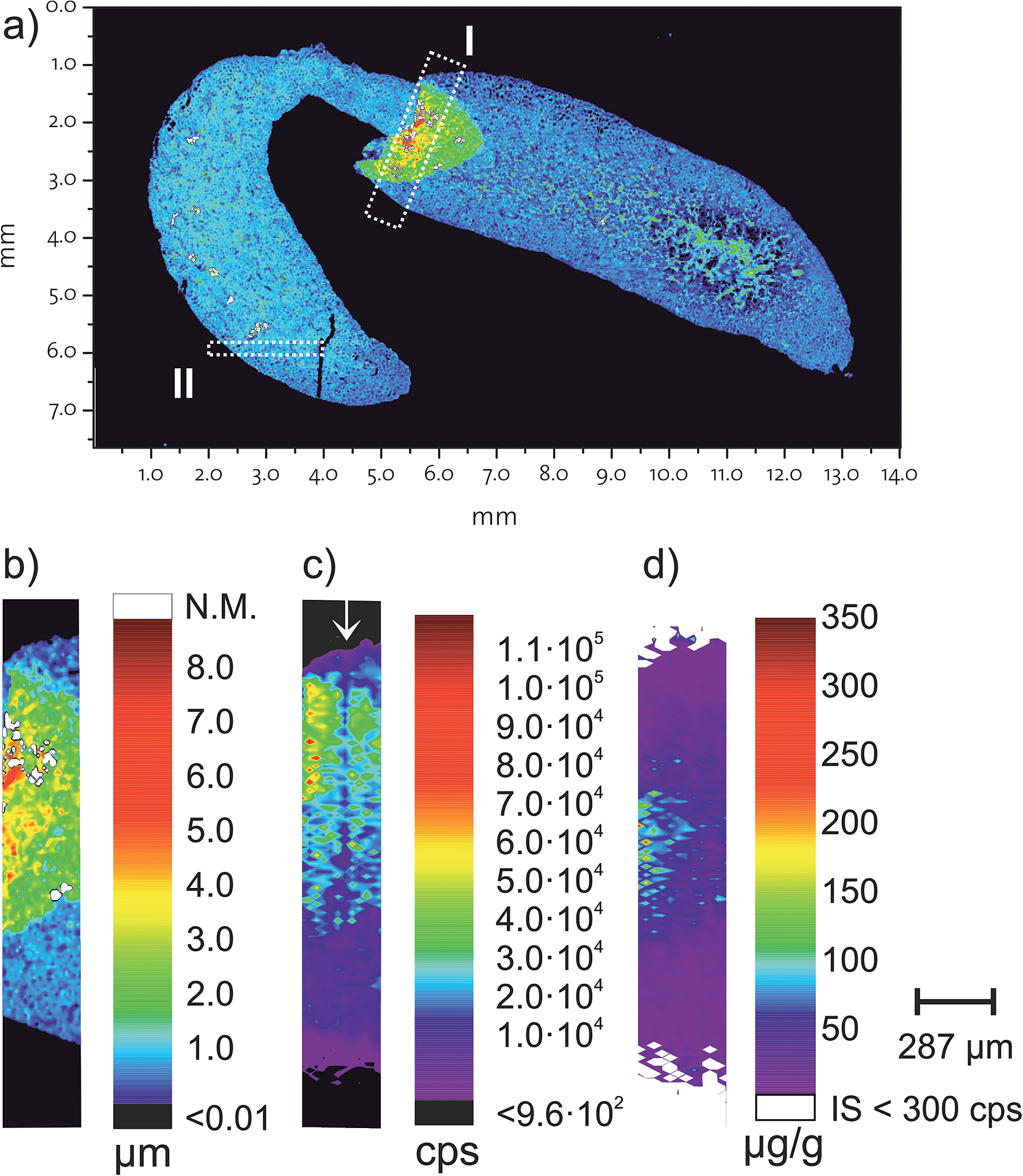 | ||
| Fig. 2 (a) Overview of the mouse kidney FFPE section measured by white light interferometry (the scale bar is the same as under (b) with the inset of the investigated areas. Section I was measured with a 24 μm crater size and section II with 5 μm resolution. (b–d) show the results from section I. (b) is the thickness measured by white light interferometry, (c) is the measured 193Ir signal intensity and (d) is the concentration of the 167Er (polymer tag with E-Cadherin antibodies). In (c) marked with a ↓ is the region where the laser stage was misaligned by 6 μm, see text for more details. | ||
The tissue sample was tagged with two different antibodies, anti-E-Cadherin (transmembrane proteins which are distributed on the cell surface) and histone H3 (a DNA packaging protein). Each antibody was labelled with a specific, isotopically enriched elemental polymer tag. The obtained signal intensities were quantified using the cellulose based external standards and iridium as the internal standard. Fig. 2d shows the corresponding concentration of the polymer tag on E-Cadherin antibodies. The region in Fig. 2c (highlighted with a ↓), with the apparent lower iridium concentration due to a misaligned laser ablation track is not visible in Fig. 2d. Presumably also less antibodies are ablated in this track, however iridium as the internal standard does correct for the lower ablated mass and a similar concentration as the surrounding area is visible.
With this method, it is possible to assess the concentration of tissue samples using non-matrix matched standards. The internal standard can be added as an intercalator in an independent, secondary step into the tissues which corrects for differences in ablated mass. Under the experimental conditions, iridium forms no gas phase which is exchanged in the gas exchange device (GED) or penetrates the membrane filters and thus undergoes the same excitation processes as the analytes in the plasma. In contrast, iodine formed a gas phase which was detected after the membrane filters (see ESI, Fig. 3†).
Homogeneity of the intercalator
To estimate the homogeneity of the iridium intercalator at a subcellular level, a section from the kidney FFPE section was analysed with a higher spatial resolution. Section II shown in Fig. 2a was analysed with 5 μm resolution. The intensity map for 193Ir is shown in Fig. 3. Close inspection reveals that at the 5 μm resolution level already certain structures in the intensity profiles are recognized. However, the homogeneity of the intercalator at a sub-cellular level was not assumed, thus the suitability of using iridium as the internal standard at a lower spatial resolution is given. | ||
| Fig. 3 Homogeneity of the iridium intercalator was assessed with a high spatially resolving laser system on a mouse kidney. The intensity map shown corresponds to section II from Fig. 2a and shows the iridium signal intensities of 193Ir acquired on Element 2 with 5 μm resolution. | ||
Instrumentation
All experiments with cells were conducted in a biosafety level 1 (BSL-1) laboratory under sterile conditions. Animals were treated according to animal welfare of the canton of Zürich, Switzerland. Sample preparation (fixation, staining and digestions) and measurements were made under clean room conditions.Tissue samples and cell lines
Female ICR (imprinting control region) mice were obtained from Charles River (Sulzfeld, Germany) and Balb/c mice from Elevage Janvier (Le Genest Saint-Isle, France). Animal experiments were performed under a project license granted by the Verterinäramt des Kantons Zürich, Switzerland (42/2012).For formalin fixed paraffin embedded tissue (FFPE) sections, organs (liver and kidney) were cross-linked in 4% formaldehyde in phosphate buffered saline (PBS) solution, embedded into paraffin, cut on a microtome (Leica RM2245, Leica Microsystems (Schweiz) AG, Heerbrugg, Switzerland) and placed on SuperFrost slides (Thermo Scientific, Braunschweig, Germany).
For rehydration, the FFPE sections were placed in a p.a. xylene bath overnight, and subsequently the paraffin was leached out using a sequence of ethanol–water mixtures (100%, 90%, 80%, 75% and 50% EtOH p.a./deionized water (DIW), 100% DIW) for 20 min each. The FFPE tissues were then reactivated at 85 °C in a tris(hydroxymethyl)aminomethane (TRIS)/ethylenediaminetetraacetic acid (EDTA) mixture buffered at pH 9.0. After the activation, the tissues were cooled to room temperature (RT) by DIW and afterwards the tissues were stained.
For fresh frozen tissue sections, organs were embedded in Richard-Allan Scientific Neg-50 Frozen Section Medium (Thermo Scientific) and deep-frozen in liquid nitrogen. Tissues were cut on a microtome (HM 505 N, Carl Zeiss AG, Feldbach, Switzerland), placed on SuperFrost slides and stored at −80 °C. For staining procedures, sections were warmed up to RT and fixed in 100% acetone p.a. for 10 minutes. Subsequently the cryo sections were stained.
Antibodies
For the proof of concept, the tissues were incubated with two antibodies. Mouse anti-E-Cadherin (36/E-Cadherin, Becton Dickinson (BD) Transduction Laboratories, Allschwil, Switzerland) and histone H3 (D1H2, Cell Signaling Technology (CST), Danvers, MA, USA) were coupled with metal-chelating polymers as described by Bendall et al.24 using the MaxPAR antibody conjugation kit (Fluidigm, formerly DVS Sciences, Sunnyvale, CA, USA) and protocol. Mouse anti-E-Cadherin was conjugated with enriched 167Er and histone H3 with enriched 176Yb. Enriched isotopes were obtained from Fluidigm, formerly DVS Sciences.Iridium intercalator
The iridium intercalator ((η5-pentamethylcyclopentadienyl)-iridium(III)-dipyridophenazine), was obtained from DVS Sciences and was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 (v/v) in PBS prior to staining.
:1000 (v/v) in PBS prior to staining.
Tissue staining procedure
The tissues were blocked with 1% bovine serum albumin (BSA) in PBS for 30 min and washed three times with DIW. Antibody incubation was conducted overnight at 4 °C and the samples were washed three times with PBS. The tissue sections were immersed in the iridium intercalator solution at RT for 20 min. Finally, the tissues were washed three times with DIW and were dried at RT.Standards
Cellulose acetate standard doped with REE and Rh, Ir and Pt were prepared according to Rege et al.25 using single element ICP-MS standards (Inorganic Ventures, Christiansburg, VA, USA). All solutions were prepared from doubly sub-boiled HNO3 diluted in ultra-pure water (18 MΩ cm, MilliQ water, Millipore, Billerica, MA, USA). Cellulose acetate was chosen because of the similar carbon containing gas phase/carbon particle phase (CCGS/CPP) ratio which was observed during LA.19As a control and to determine the concentration of the internal standard, matrix matched standards were produced. To this purpose, a liver from a mouse was homogenized and the cells were incubated in a solution containing single element standards. The cell pellet was frozen in liquid nitrogen, embedded in cryo medium and cut into 10 μm sections. See ESI† for a detailed procedure.
LA-ICP-MS
An ArF Excimer laser (GeoLasC, MicroLas Lasersystem GmbH, Göttingen, Germany) and a sector field instrument (Element 2, Thermo Scientific, Bremen, Germany) were used and details about the laser ablation experiments and the optimization thereof can be found in the ESI.† A part of the tissue was also analysed using a high spatial resolution LA-ICP-MS system which can provide a crater as small as 1 μm in diameter.26 To assess the homogeneity of the internal standard, a tissue section was evaluated with a spatial resolution of 5 μm.For the quantification with non-matrix matched standards, the concentration of the internal standard in the tissue section has to be known. There are three different methods to assess this concentration. (A) Measuring the thickness of the tissue and correlating this with signal intensities, (B) remobilization of a tissue section and digestion, followed by SN-ICP-MS to assess the concentration and (C) using a standard addition approach with desolvated aerosols as proposed by Günther et al. for soils23 and successfully applied by Becker et al. on human brain samples.15
For the assessment of the iridium concentration, option (C) was chosen. For this, several solutions were continuously aspired using a micro flow nebulizer (PFA-ST, Elemental Scientific, Omaha, NE, USA) connected to an Aridus I desolvation unit (CETAC Technologies, Omaha, NE, USA). The laser aerosol is mixed with the desolvated aerosol using a laminar flow adapter. The laser ablation and nebulization were tuned in conjunction, for stability and high signal intensities whereas maintaining low doubly charged ion formation and oxide rates. 5 different solutions with increasing iridium concentrations were aspired. All solutions contained 1 μg g−1 indium as the internal standard, while several scans across the tissue were performed.
Similar to a previous study19 where the carbon containing gas phase (CCGS) and carbon particulate phase (CPP) were investigated, the formation of gaseous species of iodine and iridium during the analysis of tissue samples was examined. For this, two samples were ablated, a 10 μm cryo kidney tissue and a tonsil tissue sample (provided by C. Giesen, BAM)27 were stained with iodine according to the procedure described by Giesen et al.27 The aerosol was analysed passing through the gas exchange device (GED, based on a design described by Nishiguchi et al.28 and Kovacs et al.29), to remove gaseous species from the laser aerosol, through a membrane filter, to retain the particulate phase and directly, as a comparison.
White-light-interferometry
The thickness of the tissue samples (cryo and FFPE) was measured after the staining procedure by white-light interferometry. For this, the samples were analysed using a 3D profilometer (PLu neox, Sensofar-Tech, Terrassa, Spain) and 3D contour maps were acquired.Conclusion
For the quantitative imaging of the element distribution and the quantitative imaging of the antigen distribution in tissue samples, a correction for the transport to and a drift of sensitivity in the plasma is not sufficient. An internal standard has also to correct for the ablated mass which is introduced into the ICP-MS. To correct for the ablated mass, an element, which is either present initially or added to the tissue sample, has to be homogenously distributed within the tissue and detectable by LA-ICP-MS. Labelling with (η5-pentamethylcyclopentadienyl)-iridium(III)-dipyridophenazine fulfils these requirements when analysed at supracellular levels. It is fast and readily applicable to both, formalin fixed paraffin embedded (FFPE) and cryo tissue sections within the same routine used for antibody labelling. For the investigated range of tissue thicknesses (2–14 μm), the internal standard signal intensities are correlated with the measured thickness of the samples. Application of polymer tagged antibodies prior to the iridium intercalation for quantitative immunoassay is possible. In contrast to other approaches such as using iodine,20,21,27 the iridium intercalator and the polymer tags are transported which enter the plasma in the same particulate phase as the analytes. Furthermore, the rare earth elements and iridium show similar wash-out behaviour. The intercalator binds strongly to the DNA30 and unspecific binding with the coating of the glass slides is reduced. Iodine staining and the iridium intercalation are complementary techniques, which can be applied for internal standardisation of the tissue section. However based on the results shown here, gas phase formation and memory effects are lower for iridium intercalation. A major limitation of quantitative bioimaging is the availability of certified reference materials. Most studies use in-house produced standards for the quantification of biological materials. The intercalation with iridium does not solve the problem to find a suitable external standard but it allows the use of the same internal standard species for the sample and external standard. First results in this work show that it is possible to correct for differences in thickness of tissue samples and to use the internal standard to determine antibodies labelled with polymer tags in tissue samples.Acknowledgements
This study is financially supported by ETH Zürich. The Institute of Molecular Life Sciences, University Zürich and the Institute of Pharmaceutical Sciences, ETH Zürich, especially Prof. Dr Neri, are acknowledged for providing samples and chemicals. J. Scholl is thanked for the embedding and cutting of the paraffin thin sections. The department of Materials at ETH Zürich, Prof. Dr Spencer and C. Cremmel are thanked for the help with white-light interferometry.References
- P. Ek, presented in part at the 1998 Winter Conference on Plasma Spectrochemistry, Scottsdale, AZ, USA, 05–10 January, 1998 Search PubMed.
- J. Feldmann, A. Kindness and P. Ek, J. Anal. At. Spectrom., 2002, 17, 813–818, 10.1039/b201960d.
- A. Kindness, C. N. Sekaran and J. Feldmann, Clin. Chem., 2003, 49, 1916–1923, DOI:10.1373/clinchem.2003.022046.
- D. Hare, S. Tolmachev, A. James, D. Bishop, C. Austin, F. Fryer and P. Doble, Anal. Chem., 2010, 82, 3176–3182, DOI:10.1021/ac902650w.
- B. Wu, Y. Chen and J. S. Becker, Anal. Chim. Acta, 2009, 633, 165–172, DOI:10.1016/j.aca.2008.11.052.
- I. Konz, B. Fernandez, M. L. Fernandez, R. Pereiro and A. Sanz-Medel, Anal. Bioanal. Chem., 2012, 403, 2113–2125, DOI:10.1007/s00216-012-6023-6.
- C. Giesen, L. Waentig, U. Panne and N. Jakubowski, Spectrochim. Acta, Part B, 2012, 76, 27–39, DOI:10.1016/j.sab.2012.06.009.
- V. I. Baranov, Z. Quinn, D. R. Bandura and S. D. Tanner, Anal. Chem., 2002, 74, 1629–1636, DOI:10.1021/ac0110350.
- Z. A. Quinn, V. I. Baranov, S. D. Tanner and J. L. Wrana, J. Anal. At. Spectrom., 2002, 17, 892–896, 10.1039/B202306G.
- C. Zhang, Z. Zhang, B. Yu, J. Shi and X. Zhang, Anal. Chem., 2002, 74, 96–99, DOI:10.1021/ac0103468.
- O. Ornatsky, V. I. Baranov, D. R. Bandura, S. D. Tanner and J. Dick, J. Immunol. Methods, 2006, 308, 68–76, DOI:10.1016/j.jim.2005.09.020.
- S. D. Tanner, O. Ornatsky, D. R. Bandura and V. I. Baranov, Spectrochim. Acta, Part B, 2007, 62, 188–195, DOI:10.1016/j.sab.2007.01.008.
- C. Giesen, H. A. Wang, D. Schapiro, N. Zivanovic, A. Jacobs, B. Hattendorf, P. J. Schuffler, D. Grolimund, J. M. Buhmann, S. Brandt, Z. Varga, P. J. Wild, D. Günther and B. Bodenmiller, Nat. Methods, 2014, 11, 417–422, DOI:10.1038/nmeth.2869.
- H. P. Longerich, S. E. Jackson and D. Günther, J. Anal. At. Spectrom., 1996, 11, 899–904, 10.1039/Ja9961100899.
- J. S. Becker, M. V. Zoriy, C. Pickhardt, N. Palomero-Gallagher and K. Zilles, Anal. Chem., 2005, 77, 3208–3216, DOI:10.1021/ac040184q.
- I. Kroslakova and D. Günther, J. Anal. At. Spectrom., 2007, 22, 51–62, 10.1039/B606522h.
- C. Austin, D. Hare, T. Rawling, A. M. McDonagh and P. Doble, J. Anal. At. Spectrom., 2010, 25, 722–725, 10.1039/b911316a.
- I. Konz, B. Fernandez, M. L. Fernandez, R. Pereiro, H. Gonzalez, L. Alvarez, M. Coca-Prados and A. Sanz-Medel, Anal. Bioanal. Chem., 2013, 405, 3091–3096, DOI:10.1007/s00216-013-6778-4.
- D. A. Frick and D. Günther, J. Anal. At. Spectrom., 2012, 27, 1294–1303, 10.1039/C2ja30072a.
- N. Jakubowski, J. Messerschmidt, M. G. Anorbe, L. Waentig, H. Hayen and P. H. Roos, J. Anal. At. Spectrom., 2008, 23, 1487–1496, 10.1039/b718074h.
- L. Waentig, N. Jakubowski, H. Hayen and P. H. Roos, J. Anal. At. Spectrom., 2011, 26, 1610–1618, 10.1039/C1ja10090d.
- O. I. Ornatsky, X. Lou, M. Nitz, S. Schafer, W. S. Sheldrick, V. I. Baranov, D. R. Bandura and S. D. Tanner, Anal. Chem., 2008, 80, 2539–2547, DOI:10.1021/ac702128m.
- D. Günther, H. Cousin, B. Magyar and I. Leopold, J. Anal. At. Spectrom., 1997, 12, 165–170, 10.1039/a604531f.
- S. C. Bendall, E. F. Simonds, P. Qiu, E.-a. D. Amir, P. O. Krutzik, R. Finck, R. V. Bruggner, R. Melamed, A. Trejo, O. I. Ornatsky, R. S. Balderas, S. K. Plevritis, K. Sachs, D. Pe'er, S. D. Tanner and G. P. Nolan, Science, 2011, 332, 687–696, DOI:10.1126/science.1198704.
- S. Rege, W. L. Griffin, G. Kurat, S. E. Jackson, N. Pearson and S. Y. O'Reilly, Lithos, 2008, 106, 39–54, DOI:10.1016/j.lithos.2008.06.002.
- H. A. O. Wang, D. Grolimund, C. Giesen, C. N. Borca, J. R. H. Shaw-Stewart, B. Bodenmiller and D. Günther, Anal. Chem., 2013, 85, 10107–10116, DOI:10.1021/Ac400996x.
- C. Giesen, L. Waentig, T. Mairinger, D. Drescher, J. Kneipp, P. H. Roos, U. Panne and N. Jakubowski, J. Anal. At. Spectrom., 2011, 26, 2160–2165, 10.1039/C1ja10227c.
- K. Nishiguchi, K. Utani and E. Fujimori, J. Anal. At. Spectrom., 2008, 23, 1125–1129, 10.1039/b802302f.
- R. Kovacs, K. Nishiguchi, K. Utani and D. Günther, J. Anal. At. Spectrom., 2010, 25, 142–147, 10.1039/b924425e.
- S. Schäfer and W. S. Sheldrick, J. Organomet. Chem., 2007, 692, 1300–1309, DOI:10.1016/j.jorganchem.2006.10.033.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ja00293h |
| This journal is © The Royal Society of Chemistry 2015 |