
Coupled techniques for arsenic speciation in food and drinking water: a review
Bashdar
Sadee
,
M. E.
Foulkes
and
S. J.
Hill
*
School of Geography, Earth and Environmental Sciences, University of Plymouth, Plymouth PL4 8AA, UK. E-mail: sjhill@plymouth.ac.uk
First published on 1st December 2014
Abstract
Arsenic is ubiquitous in nature appearing in various chemical forms. The toxicity, environmental mobility and accumulation of As in living organisms depends on the form in which the element exists, thus requiring techniques which can identify specific forms whilst retaining their integrity during extraction and pre-treatment prior to measurement. Both organic and inorganic arsenic species may be present in food staples of both terrestrial and marine origin as well as natural waters, at sub ng l−1 to high mg l−1 levels. In this review, the speciation steps (sample preparation, species speciation and detection) most commonly used for the determination of As in food are described. High performance liquid chromatography separation with plasma source mass spectrometry is often the technique of choice due to its versatility, robustness and good detection limits. However, detection systems such as atomic absorption spectroscopy, atomic fluorescence spectrometry, and atomic emission spectrometry are also widely used and covered in this review together with some less utilised techniques.
1. Introduction
Elemental speciation is well established as an important discipline in analytical chemistry. Arsenic is a ubiquitous element in the environment having been introduced via both natural and anthropogenic routes.1 It can be found in the atmosphere, the pedosphere, the hydrosphere and the biosphere. In addition to the biological mechanisms, including microbiological processes, physico-chemical processes such as oxido-reduction, precipitation/solubilisation, and adsorption/desorption determine the biogeochemical behaviour of As.2 Routine determination of the As content of a sample can be achieved by measurement of the total As using a quantitative procedure.3 Although arsenic has the reputation of being a toxic element, is also well established that its toxicity critically depends on the chemical form in which it exists and that inorganic species, arsenite (AsIII) and arsenate (AsV), are classified as more toxic than organo arsenic compounds.4 The oxidation state of organic forms also changes the toxicity, so that trivalent methylated forms are likely to be more toxic than previously thought.5 Arsenobetaine (AsB) is the major As species in fish and other seafood, and arsenocholine (AsC) is considered as a precursor of AsB, which is the end product of marine arsenic metabolism.6 These are not considered toxic compounds.7 Other arsenicals such as monomethylarsonic acid (MMA), dimethylarsinic acid (DMA), are less toxic than inorganic arsenic,4 and together with trimethylarsine oxide are often found in marine organisms, together with many arsenosugars and arsenic containing lipids in the case of marine algae and seaweed.8,9The accumulation of arsenic by plants and fauna of marine origin is relatively high compared to other food sources,10,11 therefore, many arsenic speciation studies have focused on these types of food. Even though the majority of ingested arsenic (75%) is contributed by fish and shellfish, it generally represents only a small percentage (2%) of the daily dietary intake.12 Seaweeds used in human foods have a total arsenic content of between 0.031–149 mg kg−1 and inorganic arsenic between <0.014 to 117 mg kg−1.13 In fish, the As contents varies according to the species of fish concerned; average concentrations vary between 5 and 100 mg kg−1,11 although conger and dogfish may contain elevated values of 100 to 250 mg As per kg. In flat fish the values vary between 10 to 60 mg kg−1.14 Nevertheless it has been confirmed that these elevated concentrations in seafood cause little risk to health, since almost 80–90 % of arsenic is in the organic form (AsB, AsC, arsenosugars, and arsenolipids).7 Rattanachongkiat et al.15 in their study of arsenic speciation in sardines, demonstrated that among 95% of As extracted (5.8 mg per kg dry weight), 77% was AsB, 17% DMA and 6% inorganic arsenic.
Because of its widespread nature, arsenic exists in all natural waters and concentrations of arsenic between <0.5 μg l−1 and more than 5000 μg l−1 have been reported. The WHO recommended threshold value for As in drinking water is 10 μg l−1.16 However, freshwater usually contains less than 10 μg l−1 and frequently less than 1.0 μg l−1 of arsenic. In some cases, much higher concentrations in groundwater have been monitored. In such areas, often more than 10% of wells are affected (sometimes up to 90%), with arsenic levels exceeding 50 μg l−1. It has been reported that some countries such as Argentina, Chile, Mexico, China, and Hungary and more recently in West Bengal (India), Bangladesh and Vietnam have high levels of As in ground water.17 The inorganic As species, AsIII and AsV, are the predominant species found in water,18–20 although the concentration of each species varies. A study of thermal waters in New Zealand for example,21 found concentrations up to 8.5 mg l−1 As with the trivalent As form being the dominant species and contributing up to 90% of total As. The concentration of arsenic in seawater is less than 2.0 μg l−1. Baseline concentrations of arsenic in unpolluted surface water and groundwater typically range between 1–10 μg l−1.21 The weathering and dissolution of arsenic-bearing rocks, minerals and ores also lead to occurrence of arsenic in water,22 and the arsenic cycle through the groundwater compartment has an important impact on human toxicology.23 It has been concluded by the International Agency for Research on Cancer that there is sufficient evidence in humans to suggest that arsenic in drinking-water causes cancers of the urinary bladder, lung and skin.24 According to a study that has been conducted in West Bengal, 94% of those people exposed to high levels of arsenic in drinking water had leukomelanosis and hyperkeratosis which can lead to skin cancer.22
1.1 Chemistry of arsenic
Arsenic is a metalloid which ranks 20th in natural abundance and 12th in the human body.25 It has been used as a medicine, and it has also been utilized in various field such as electronics, agriculture, livestock, metallurgy, industry,21 pesticides,26 and fertilizers.27 More than 245 minerals contain arsenic, the most important arsenic bearing minerals being orpiment (As2S3), realgar (AsS), mispickel (FeAsS), loelling-ite (FeAs2), niccolite (NiAs), cobaltite (CoAsS), tennantite (Cu12As4S13), and enargite (Cu3AsS4).28 The origins of high arsenic concentrations in the environment are through volcanic eruption and other natural processes, and human activities such as the disposal of industrial waste chemicals, the smelting of arsenic bearing minerals, the burning of fossil fuels, and the application of arsenic compounds in many products over the past hundred years.29 Mining operation contribute high level of As and other heavy metals which are mobilized in the soil and then accumulated in the food chain via plants.30–32 Arsenic exists in four oxidation states, +V (arsenate), +III (arsenite), 0 (arsenic), and −III (arsine and arsenide). The most common species in nature are the two highest oxidation states, while the two lowest are rare.33 Apart from arsenite, arsenate and their methylated derivatives, there are also other compounds such as “fish arsenic” (arsenobetaine and arsenocholine), and arsenosugars; all of which are compounds of environmental interest. Fig. 1 shows examples of some common arsenic compounds.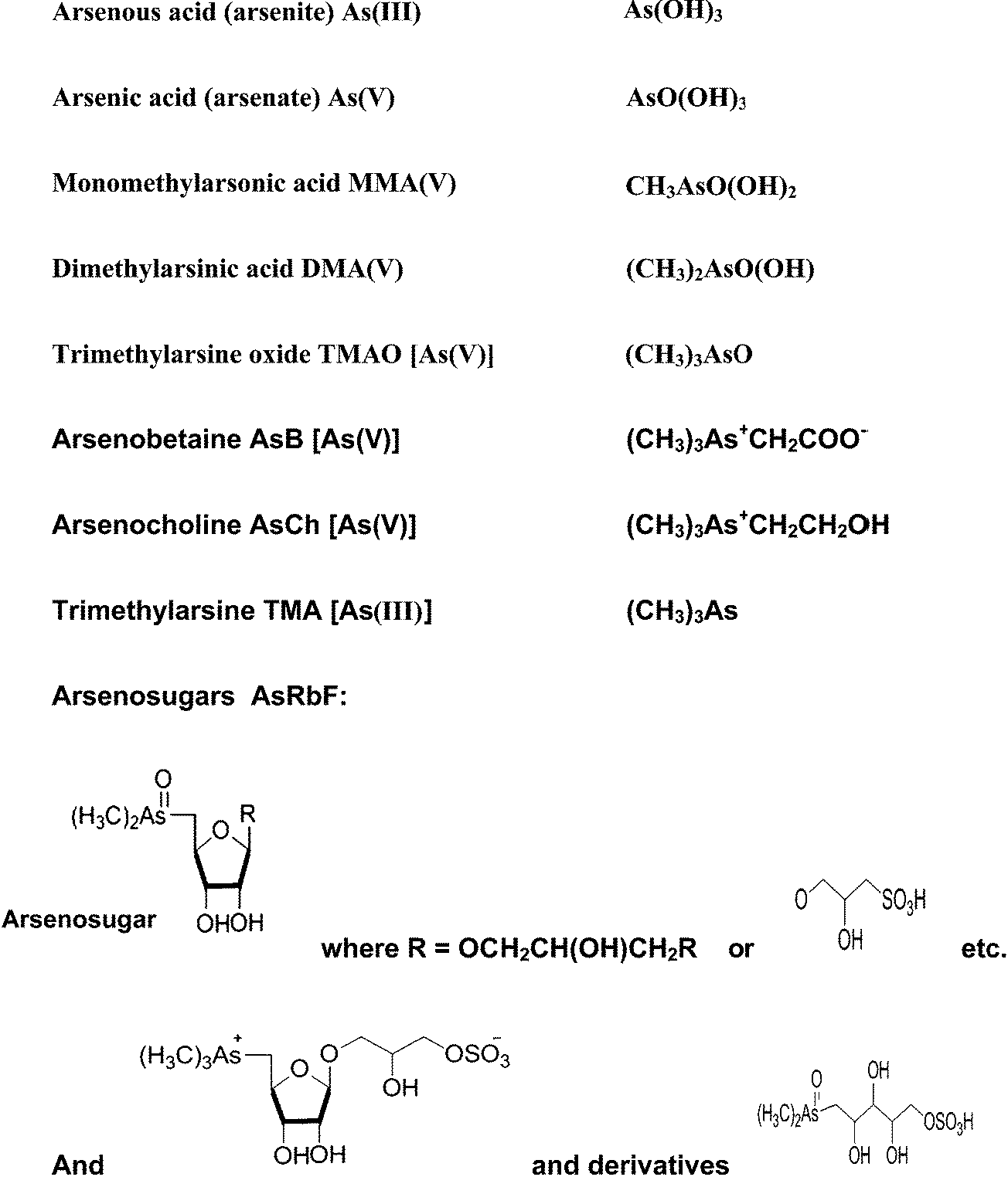 | ||
| Fig. 1 Examples of some common arsenic species. | ||
1.2 Toxicity
Toxicity of arsenic in humans depends on chemical speciation and the oxidation state of the As.34,35 It is considered that the toxicity of As increases in the order of arsenobetaine; arsenosugar, dimethylarsinic acid; monomethylarsonic acid, arsenate and arsenite.36 To humans, trivalent arsenic is about 60 times more toxic than the oxidized pentavalent state, because the arsenite can react with sulfydryl groups, whereas the arsenate does not.37 Inorganic As compounds are about 100 times more toxic than organic As compounds (DMA and MMA).38 The 50% lethal dose (LD50) values in rat for some arsenical species are illustrated in Table 1. It can be seen from the table that AsIII is more toxic by a factor of between 200 and 300 times than arsenocholine and trimethylarsine oxide, respectively while trimethylated compounds are virtually non-toxic.34,39| Arsenic species | Dose (mg kg−1) |
|---|---|
| Arsine | 3.0 |
| AsIII | 14.0 |
| AsV | 20.0 |
| TMA+ | 890 |
| MMA | 700–1800 |
| DMA | 700–2600 |
| AsB | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| AsC | 6500 |
1.3 Toxicity in food
The most toxic As species in food are inorganic As, AsIII and AsV, followed by organic arsenic such as MMAV, DMAV and TMA+ which are considered less toxic. However, some organic As species found in food as major or constituent like AsB, AC, TMAO, and arsenosugers are considered harmless. Trivalent methylated species such as MMAIII and DMAV have been detected in the human urine.41 These methylated arsenicals are more toxic than inorganic forms42,43 but they have not been found in any foodstuffs possibly because of lack of a suitable extraction method.1.4 Arsenic in the diet
Today, inorganic As is not intentionally used as a preservative added to food as it was in the late 1800s and early 1900s.44 It is, however, well known that the diet contains mainly inorganic and organic As compounds.44,45 The WHO has established a provisional maximum tolerable daily intake (PMTDI) of 2.1 μg inorganic As per kg per day body weight to cover risks from both water and food, although these guidelines are not for a specific food.46 Estimates of inorganic As in diet are varied. In the UK, according to the survey by Rose et al.47 it has been found that the amount of inorganic As consumed by an adult is 0.03–0.09 pg per kg body-weight per day. In the United State, it is estimated that the average adult intake is 3.2 μg per day, with a range of 1–20 μg per day.48 Similar estimates have been observed in children diet.49 Recently, a higher intake level has been estimated by the European Food Safety Authority (EFSA). However, simplifying assumptions which are related to the ratio of inorganic As to total As in food are used to determine these estimates.45 It has been reported by EFSA that the national As exposure from food and water across 19 European countries utilizing lower bound and upper bound concentrations have been measured to be in the range 0.13–0.56 μg per kg body weight.50 It has also been shown that some of our foodstuffs are contaminated with As. Most foodstuffs contain organic arsenic compounds at a total concentration of less than 1 mg kg−1.51 Rice can contain a relatively high amount of As.52,53 Rice provides 70% of energy of daily food intake of over half of the world's population especially in Asian developing countries53 and can accumulate typically between 100–400 μg kg−1 As.11,54,55 The arsenic species determined in rice include AsIII, MMA, DMA and AsV.56,57 Raber et al.56 have demonstrated that inorganic As and total As of 10 rice sample was 25–171 μg kg−1 and 36–218 μg kg−1, respectively. When the diet is not rice-based wheat will be the major contributor to the consumption of inorganic As. It has been found the total As concentration in wheat samples ranged between 8.6–166 μg per kg dry weight and about 91–95% of the As was found to be in inorganic form, while the rest was mainly DMA.56,58 However, seafood is the main source of As in diet,3,59,60 with AsB being the major species in fish and seafood.61 Other arsenic species such as AsIII, AsV, AsC, MMA, DMA, TMAO and arsenic containing lipids are also present in aquatic organisms, as well as arsenosugars in marine algae and seaweed.34,62–64Table 2 shows the total As and inorganic As concentrations reported in 20 different food stuffs in the UK from a study by Rose et al.47| Food group | Inorganic arsenic mg kg−1 | Total arsenic mg kg−1 |
|---|---|---|
| Bread | <0.01 | <0.005 |
| Miscellaneous cereal | 0.012 | 0.018 |
| Carcase meat | <0.01 | 0.006 |
| Offal | <0.01 | 0.008 |
| Meat products | <0.01 | 0.005 |
| Poultry | <0.01 | 0.022 |
| Fish | 0.015 | 3.99 |
| Oils and fats | <0.01 | <0.005 |
| Eggs | <0.01 | <0.003 |
| Sugars and preserves | <0.01 | 0.005 |
| Green vegetable | <0.01 | 0.004 |
| Potatoes | <0.01 | 0.005 |
| Other vegetables | <0.01 | 0.005 |
| Canned vegetables | <0.01 | 0.005 |
| Fresh fruit | <0.01 | 0.001 |
| Fruit products | <0.01 | 0.001 |
| Beverages | <0.01 | 0.003 |
| Milk | <0.01 | <0.001 |
| Dairy produce | <0.01 | <0.003 |
| Nuts | <0.01 | 0.007 |
1.5 Arsenic in natural waters
Human exposure to elevated As is often associated with drinking water. Drinking water contaminated with As is a major global concern, with over 100 million people affected, including up to 57 million in Bangladesh alone.65 As is present predominately as AsIII and AsV in water.18 A clear link between elevated As exposure via drinking water and the prevalence of skin, lung, and bladder cancer has been reported based on epidemiological studies of populations exposed to high levels of As.66The levels of As in uncontaminated groundwater usually range from 1–2 μg l−1.21 The predominant arsenic species in ground water is AsV while AsIII is a minor As species.67,68 In some contaminated areas the concentrations of As in ground water can reach as high as hundreds of μg l−1 as summarized in Table 3. Contamination of ground water by As has already been demonstrated in 20 countries around the world.69 Millions of people in As-contaminated ground water areas drink water with As concentration ≥50 μg l−1,17,69i.e. significantly higher than the World Health Organization (WHO) maximum permissible limit in drinking water which is 50 μg l−1 and the recommended value is 10 μg l−1.70 Various analytical techniques have been used to measure As in drinking water, some of which are included in Table 5.
| Location | Sampling period | Arsenic source | Concentration μg l−1 | Reference |
|---|---|---|---|---|
| Laos PDR | 2008 | Tube-well water | <0.05–278 | 71 |
| Kandal, Cambodia | Not mentioned | Aquifer, wells | 15–1300 | 72 |
| Shallow wells | 0–1000 | |||
| South Vietnam | 2007 | <1.0–850 | 73 | |
| West Bengal, India | 2000 | Hand tube well | 21–176 | 74 |
| Shallow tube well on agriculture land | 40–182 | |||
| Michigan, USA | 1997 | Shallow groundwater | 0.5–278 | 75 |
| Baseline, UK | Not mentioned | Groundwater | <0.5–10 | 17 |
| Southwest, England | Not mentioned | Groundwater (mining area) | <1.0–80 | 76 |
| Southern Thailand | Not mentioned | Shallow groundwater (mining contaminated) | 1.25–5114 | 77 |
2. Methods to speciate arsenic in food
2.1 Sampling and sample pre-treatment for speciation
Maintaining the concentration and chemical structure of the original species during the sample preparation and extraction steps are critical requirements for obtaining information on accurate As speciation.78 During these procedures problems may result from losses during sampling, unrepresentative samples,79 contamination, inter conversion between species, inefficient extraction of the analyte, and the possibility of precipitation and wall effects from the sample container.80–82 The possible risk of a redox interconversion of inorganic As forms to other species can be minimized using microwave-assisted extraction.81 Microorganisms can participate in a range of element transformations including a change in valence (i.e. oxidation/reduction) or chemical form (i.e. solid, liquid and gas).83 It is well-known that many microorganisms (bacteria, fungi and yeast) have ability of biomethylate arsenic and both volatile (e.g., methylarsines) and nonvolatile (e.g., methylarsonic acid and dimethylarsinic acid) compounds are formed.84 Biological sample should be kept at low temperatures as bacteria can degrade the integrity of the sample. Drying is often used for the stabilization of samples particularly freeze-drying or lyophilisation which tend to reduce analyte loss.852.2 Extraction
Sample extraction is one of the crucial steps in the analysis of food samples. It is important to avoid chemical transformation of the species during the extraction process, and to ensure the full extraction of each species. Extraction procedures employ a range of approaches including solid–liquid extraction86 liquid–liquid extraction,82 solid phase extraction (SPE)87 and solid phase microextraction (SPME).88 Solid sample preparation generally includes milling, grinding, freeze drying or sieving following by some forms of extraction. Enhanced techniques such as soxhlet,89 sonication,90 pressurized liquid extraction (PLE),91 microwave-assisted extraction (MWA)92 and supercritical fluid extraction (SFE)93 have also been utilized for the determination of As in food, although as discussed below, some of these approaches may be problematic for some matrices.:150 (v/v). A summary of research papers focusing on extraction methods for arsenic species in food is shown in Table 4.
| Extraction process | ||||||
|---|---|---|---|---|---|---|
| Extraction solution | Shaking/mixing | Sonication | MW-assisted heating | Sub/supercritical fluid | PLE | Soxhlet |
| Water | 10 and 101–109 | 10, 98, 103, 106 and 110 | 10, 103, 106 and 111–113 | 106 and 114 | 40, 105 and 115 | 10 and 106 |
| Methanol | 10 and 116 | 10, 117 and 118 | 10 and 117 | 115 and 40 | 10, 106 and 117 | |
| Methanol–water mixture | 10, 101, 101, 103–106 and 119 | 10, 18, 98, 103, 106, 115, 117 and 119 | 10, 18, 92, 103, 106, 120, 121 and 121–124 | 125 | 40, 105, 115 and 126–129 | 10 and 117 |
| Ionic extractants | 101, 104, 104, 106 and 115 | 18, 57, 62, 98, 103, 106 and 130 | 57, 92, 103, 103, 106, 131 and 132 | 102 | ||
| Enzymes | 15, 98, 115, 133 and 134 | 18, 107 and 135 | 136 | |||
| Others | 10, 119, 137 and 138 | 10, 18, 62 and 139–142 | 10, 92, 143 and 144 | 102, 106 and 145–148 | 128 and 149–151 | 10 |
Ultrasound probe sonication can be used to aid the removal of the analyte from the sample matrix. A standard ultrasonic bath operating at a frequency of 40 kHz may often be used to extract from solids faster than by using classical methods.93,154 Insoluble arsenic fractions such as protein bound arsenic and/or lipid arsenic have traditionally been little researched due to the absence of a suitable analytical methods and difficulties of a total recovery of species.93 These drawbacks have been tackled by combining enzymatic treatment with ultrasonic probe sonication in more recent studies.135
Supercritical fluid extraction (SFE) has some favourable characteristics which make it attractive as an extraction technique, including the low viscosity and diffusion coefficients.121 However, it has not found widespread use for speciation studies due to is low extraction efficiency for highly polar or ionic compounds.122 The addition of complexing agents and/or modifiers may partly address these problems and enhance extraction efficiencies.155
Pressurized liquid extraction (PLE) is another automated approach which can provide fast extractions using low solvent volumes and avoiding filtration.156,157 This method has been reported for As speciation in marine biological materials including mussels and fish samples.124 However, PLE is not without its problems for speciation studies since dispersion of the sample in an inert medium is a fundamental step. When this dispersal is not homogenous a large reduction in extraction efficiency will be observed.128
Microwave digestion is a viable replacement to conventional techniques for many matrices, offering acceptable and reproducible efficiencies, together with a reduction in extraction times, low solvent volumes, and the opportunity of fast and multiple extraction.126,156 This approach has found widespread application in speciation studies for As. Optimisation is straight forward because of the low number of parameters involved, such as choice of solvent, solvent volume, temperature, extraction time, power and matrix characteristic.156
2.3 Methods of separation
Liquid chromatography (LC) is a method often used for arsenic speciation in food. It provides separation of both inorganic and organic forms of As. The coupling of ICP-MS, ICP-AES and HG-AAS with liquid chromatography has also been widely used for arsenic speciation, since LC offers good separation of many arsenic species using a simple interface for real time measurement.158,159 Arsenical species have been separated using several techniques including anion-exchange HPLC with either isocratic or gradient-step elution or cation-exchange HPLC with isocratic elution. Ion-pair HPLC has also been utilized.160 Since there is sometimes a requirement for the separation of anions and cations of As in a single analysis, column-switching systems, which involve a combination of anion-exchange and reversed-phase separation, have been developed.161,162 The coupling of gas chromatography (GC) with ICP-MS has also been used,163 for example the detection of a range of As-containing hydrocarbons in commercial fish oils200 and seafood.149 Speciation analysis of organometallic compounds in complex environmental and industrial samples have been achieved by combination of capillary GC with ICP-MS to utilise the high resolving power of GC and the sensitivity and specificity of ICP-MS.164 Using GC speciation can be an attractive technique because of the lack of condensed mobile phase although there is often the need for derivatisation of the analyte prior to analysis.165In recent years, the number of reports on the use of capillary electrophoresis (CE) has continued to grow. CE is an attractive technique for elemental speciation since it has several unique characteristic in comparison with GC or HPLC methods i.e. high resolving power, rapid, effectual separations, minimal reagent consumption and the probability of separation with only minor disturbances of the existing equilibrium between different species.166 A wide range of inorganic and organic As species can be separated by this technique.167 Several element-selective detector have been coupled with CE including both ICP-AES and ICP-MS.168,169 Yang et al.170 have analysed seafood using capillary electrophoresis-inductively coupled plasma mass spectrometry. AsIII, AsV, MMA and DMA have been separated and determined in dried Mya arenaria I and shrimp within 10 min. CE has also been coupled to ICP-MS to quantify the As species AsB, AsIII, AsV, DMA, MMA in fish.171
Micro-scale separation has become a popular technique due to the improved separation efficiency, reduced analysis time and reduction in sample consumption.12,172 Micro-bore and narrow-bore have been coupled with ICP-MS as a result of their compatibility with MS ionisation sources.12 Narrow-bore-HPLC column coupled with ICP-MS has been used by Wangkarn and Pergantis173 to analyse several wines. Arsenite at trace levels was found to be the only arsenic species in the analysed wines.
Separation with off line detection depends on the chemical or physical separation of the element of interest. Particular arsenic species are separated selectively before determination as arsenic; for instance, formation of AsCl3 (reasonably volatile, non-polar) from arsenite which is ultimately separated from other organoarsenicals by distillation or solvent partitioning. Off line detection methods have been applied to the separation and determination of inorganic As (AsIII and AsV) and organic arsenic (MMA and DMA) in fish (skate, hake, albacore, blue fin tuna and blue whiting),174–176 plant extracts177 and raw vegetable.178
Organoarsenical compounds have also been quantified by HPLC-MS with LODs below (30 ng ml−1) approaching those of HPLC-ICP-MS. HPLC-MS and HPLC-MS-MS are most often used to characterize arsenicals, such as AsB, AsC, arsenosugars in biota like algae,179 oyster180 and calms.181 Different chromatographic conditions have been used for arsenic speciation in various matrices (Table 5).
| Matrix | Species | Technique | Separation conditions | Time of separation minute | Amount of sample μl | Detection limits (ng ml−1) | References |
|---|---|---|---|---|---|---|---|
| Rice | AsIII, AsV, DMA and MMA | HPLC-ICP-MS | PEEK PRP-X100 anion exchange column; mobile phase, 20 mM ammonium phosphate buffer, pH 4.5, 40 °C | — | 40 | Not given | 131 |
| Rice | AsIII, AsV, DMA | HPLC-ICP-MS | Waters IC-Pak anion HR column; mobile phase, 10 mM (NH4)2CO3, pH 10, Dionex AS7 & AG7 column; mobile phase, 12.5 mM HNO3, pH 1.8, Hamilton PRP-X100 column; mobile phase, 10 mM NH4H2PO4, 10 mM NH4NO3, pH 6.3 | — | 25 | AsIII: 0.10, AsV: 0.10, DMA: 0.13 | 115 |
| Rice | AsIII, AsV, DMA and MMA | HPLC-ICP-MS | PRP-X100 anion-exchange column (Hamilton); mobile phase, 20 mM NH4H2PO4, pH 5.6, 40 °C | 10 | 20 | AsIII: 1.3, AsV: 1.3 DMA: 1.3, MMA: 1.3 | 234 |
| Rice | AsIII, AsV, DMA, MMA | HPLC-ICP-MS | Column X-Select (Charged Surface Hybrid; CSH) C18; mobile phase, 7.5 mM tetrabutylammonium hydroxide, 10 mM ammonium phosphate monobasic, 5% methanol, pH 8.25 | 9 | 25 | AsIII: 0.1, AsV: 0.2, DMA: 0.1, MMA: 0.2 | 134 |
| Rice, straw | AsB, AsIII, DMA, MMA, AsV | HPLC-ICP-MS | Hamilton PRP-X100 anion exchange column; mobile phase, 10 mM HPO42−/H2PO4−, 2% (v/v) methanol, pH 8.5 | 11 | 100 | AsB: 0.0136, AsIII: 0.0196, DMA: 0.0127, MMA: 0.0143, AsV: 0.0194 | 235 |
| Rice | AsIII, MMA, DMA AsV | HPLC-HG-AAS | PRP-X100 analytical and guard anion-exchange column (Hamilton, Reno, NV, USA); mobile phase, 10 mM HPO42−/H2PO4−, pH 6.0 | — | — | AsIII: 0.015, MMA0.06, DMA: 0.06, AsV: 0.06 | 135 |
| Rice | AsIII, AsV, MMA, DMA | HPLC-HG-AFS | Hamilton PRP-X 100 anion-exchange column (250 mm × 4.1 mm I.D. 10 μm); mobile phase, 15 mM phosphate buffer, pH 6 | — | — | Not given | 64 |
| Plant | AsIII, AsV DMA, MA and TMAO | HPLC-ICP-MS | Cation exchange: ZORBAX 300-SCX column; mobile phase, 20 mM pyridine, pH 2.6, anion exchange: PRP-X100 column; mobile phase, 20 mM NH4H2PO4, pH 6, anion exchange: PRP-X100 column; mobile phase, 20 mM NH4HHCO3, pH 10.3 | 7–12 | 20 | Not given | 236 |
| Plant | AsIII, AsV, DMA and MMA | HPLC-ICP-MS | Hamilton PRP-X100 anion-exchange column; mobile phase, 30 and 100 mM TRIS acetate buffer, pH 7 | 13 | 200 | Not given | 237 |
| White mustard (Sinapis alba) | AsIII, AsV, DMA and MMA | HPLC-ICP-MS | Anion exchange column PRP-X100; mobile phase, 0.01 M Na2HPO4 (80%), 0.01 M NaH2PO4 (20%), pH 6 | — | 100 | Not given | 238 |
| Carrots | AsIII, AsV, MMA, DMA, AsB | HPLC-ICP-MS | Column, Waters IC-Pak Anion HR; mobile phase, 10 mM ammonium carbonate, pH 10 | 7 | 20 | AsIII: 0.15, AsV: 0.11, MMA: 0.13, DMA: 0.24, AsB: 0.14 | 40 |
| Fruit and vegetable | AsIII, AsV, DMA and MMA | HPLC-ICP-MS | PRP-X100 anion exchange column; mobile phase, ammonia phosphate buffer (6.6 mM ammonium dihydro-phosphate, 6.6 mM ammonium nitrate), pH 6.2 | — | 100 | Not given | 132 |
| Apple | AsIII, DMA, MMA, AsV | HPLC-ICP-MS | Hamilton PRP-X100 anion exchange column with mobile phase A: 12.5 mM (NH4)2CO3; pH 8.5: Mobile phase B: 50 mM (NH4)2CO3 | 30 | 200 | AsIII: 0.089, DMA: 0.034, MMA: 0.063, AsV: 0.19 | 239 |
| Xerocomus badius (Mushroom) | AsIII, AsV, and DMA | HPLC-HG-AAS | A-First analytical system: Column Supelco LC SAX-1; mobile phase, phosphate buffer (50 mM Na2HPO4 and 5 mM KH2PO4·2H2O), B-Second analytical system: Column, Zorbax SAX, mobile phase, phosphate buffer (100 mM Na2HPO4 and 10 mM KH2PO4·2H2O) | — | — | Not given | 240 |
| Plant (bean, rice, hot pepper) | AsIII, AsV, and DMA | HPLC-HG-AFS | Hamilton PRP-X100 anion-exchange column; mobile phase, 5 mM ammonium phosphate buffers, pH 4.7 for 4.1 min; 30 mM at pH 8.0 for 6.0 min; 5 mM at pH 4.7 again for 10 min, in order to equilibrate the column before the following analysis) | 21 | 100 | AsIII: 1.5, DMA: 2.4, MMA: 2.1, AsV: 1.8 | 103 |
| Feed additive | AsIII, AsV, DMA, MMA, Roxarsone (ROX) and p-arsanilic acid (ASA) | HPLC-ICP-MS | PRP-X100 anion exchange chromatographic column (Hamilton, USA); ZORBAX Eclipse XDB-C18 chromatographic column (Agilent, USA); mobile phase, A: H2O; B: 50 mM (NH4)2HPO4, pH 6.0 | 20 | 15–25 | AsIII: 0.04, AsV: 0.15 DMA: 0.24, MMA: 0.36, ROX: 0.5, ASA: 0.092 | 241 |
| Algae and freshwater plant | Glycerol-arsenosugar (gly-sug), AsIII, AsV, DMA and MMA | HPLC-ICP-MS | PRP-X100 (Hamilton, USA) column; mobile phase, 20 mM NH4H2PO4, and Zorbax SCX300 (Agilent, Germany) column; mobile phase, 20 mM pyridine | 10 | 20 | AsIII: 2, AsV: 8, MMA: 5, DMA: 3, gly-sug: 15 | 108 |
| Seaweed | AsB, AsIII, AsV, DMA, ribose-OH, ribose-PO4, ribose-SO3 | HPLC-ICP-MS | Anion-exchange Hamilton PRP-X100 anion-exchange; mobile phase, 20 mM NH4HCO3, pH 9.0, 1% MeOH | 25 | 50 | Not given | 62 |
| Clams and seaweed | AsIII, AsV | HPLC-HG-AAS | Hamilton PRP-X100 anion exchange column; mobile phase, 20 mM ammonium phosphate pH 6 | — | — | Not given | 53 |
| Porphyra | AsIII, AsV, MMA, DMA and AsB | HPLC-(UV)-HG-AFS | Hamilton PRP-X100 anion exchange column; mobile phase, 3 mM (NH4)2HPO4, pH 8.7 | — | — | AsIII: 2.7, AsV: 8.3 MMA: 2.1, DMA: 1.8 AsB: 2.1 | 242 |
| Ground water | AsIII, AsV, DMA and MMA | HPLC-ICP-MS | Strong cation exchange (SCX); strong anion exchange (SAX) cartridge; mobile phase, 1 M HNO3 for DMA, and 5 ml of 80 mM acetic acid, 5 ml of 1 M HNO3. | — | — | AsIII: 0.12, AsV: 0.02, MMA: 0.02, DMA: 0.03 | 243 |
| Water | AsB, AsIII, AsV, MMA and DMA. | HPLC-ICP-MS | Column, Dionex AS7 anion-exchange; mobile phase, A: 2.5 mM NH4H2PO4, pH 10.0, B: 50 mM NH4H2PO4 | 30 | 20 | AsB: 0.024, AsIII: 0.017 AsV: 0.026, MA: 0.026, DMA: 0.023 | 244 |
| Fresh water and seawater | AsB, AsIII, DMA, MMA and AsV | HPLC-HG-AAS | Anion exchange column (Hamilton, Reno, NV, USA); mobile phase, 25 mM phosphate, pH 5.8 | — | — | AsB: 0.3, AsIII: 0.08 DMA: 0.1, MMA: 0.1, AsV: 0.3 | 214 |
| Fresh water | AsIII, MMA, DMA AsV | HPLC-HG-AAS | Anionic column (Hamilton PRP-X100), mobile phase (17 mM H2PO4−/HPO4, pH 6.0) | — | — | AsIII: 0.1, AsV: 0.6, MMA: 0.3, DMA: 0.2 | 245 |
| Ground water | AsIII, AsV | HPLC-HG-AAS | Anion-exchange column Supelco LC-SAX1 and thermostatted by column oven (CTO-10ASvp); mobile phase phosphate buffer (50 mM Na2HPO4, 5 mM, KH2PO4; pH 5.4) | — | — | AsIII: 7.8, AsV: 12.0 | 246 |
| Fresh water | AsIII, MMA, DMA, AsV | HPLC-HG-AFS | Hamilton PRP-X100 anion exchange column; mobile phase A; NH4H2PO4/(NH4)2HPO4) 5 mM, pH 4.8, mobile phase B: NH4H2PO4/(NH4)2HPO4) 30 mM, pH 8.0 | 20 | 100 | AsIII: 0.05, AsV: 0.06, MMA: 0.07, DMA: 0.05 | 247 |
| Algae, fish tissue and shellfish | Inorganic arsenic, DMA, AsB, Arseniosugar PO4, Arseninosugar OH, Arsinosugar SO3 | HPLC-ICP-MS | Cation exchange Dionex Ionpac CS-10 column; mobile phase, 5 mM pyridinium, pH 2, anion exchange Hamilton PRP-X100 column; mobile phase, 20 mM NH4HCO3, pH 10.3 | — | 50 | — | 34 |
| Fish and sediment | AsB, AsC, DMA, MMA, AsIII and AsV | HPLC-ICP-MS | Hamilton PRPX-100 column; mobile phase A, 10 mM NH4H2PO4−(NH4)2HPO4, 2% CH3CN, pH 6.5; mobile phase B, 100 mM (NH4)2HPO4, pH 7.95 | 10 | 20 | AsC: 0.5, AsB: 0.5, AsIII: 0.5, DMA: 1.0, MMA: 1.0 AsV: 1.5 | 248 |
| Fish, mussel | AsB, AsC, DMA, MMA, AsIII and AsV | HPLC-ICP-MS | Column, Hamilton PRP-1; mobile phase, 0.5 mM tetrabutylammoniumphosphate–4 mM phosphate buffer, pH 9. | 9 | 20 | AsC: 9, AsB: 6, AsIII: 6, AsV: 25, MMA: 22, DMA: 10 | 249 |
| Dogfish | AsB, DMA, MMA, AsIII and AsV | HPLC-ICP-MS | Anion-pairing column, 10 μm PRP-1; mobile phase, 0.5 mM tetrabutylammonium hydroxide,5% methanol, pH 7, anion-exchange column, PRPX-100 (Hamilton); mobile phase, 8 mM phosphate buffer, pH 7; cation-pairing column PRP-1 (Hamilton); mobile phase, 5% methanol, 2.5% acetic acid and 50 mM sodium dodecylsulphate, pH 2.5 | 9 | 200 | AsB: 5.0, AsIII: 1.0 | 250 |
| Fish tissues | AsB, AsIII, DMA, MMA and AsV | HPLC-ICP-MS | Metrosep™ Anion Dual 3 column; mobile phase, A: 5 mM NH4NO3: B: 50 mM NH4NO3, 2% (v/v) methanol, pH 8.7 | 12 | 100 | AsB: 22, AsIII: 15 DMA: 16, MMA: 14 AsV: 17 | 251 |
| Dorm 2, fish | AsB, DMA, MMA, AsIII and AsV | HPLC-ICP-MS | Hamilton PRP-X100 column; mobile Phase, A: 15 mM (NH4)2CO3, 2% MeOH, pH 9: B: 50 mM (NH4)2CO3, 2% MeOH, pH 9 | 22 | 200 | AsB: 0.003, AsIII: 0.01, DMA: 0.004, MMA: 0.003 | 252 |
| Fish, molluscs and crustaceans | AsB, AsIII, DMA, MMA and AsV | HPLC-ICP-MS | A Hamilton PRPX-100 column, mobile phase, A: 60 mM ammonium carbonate, pH 9, B: H2O | 15 | 60 | Not given | 111 |
| Fish tissue, DORM-2 | AsB, DMA, MMA, AsIII and AsV | HPLC-ICP-MS | Dionex Ionpac AS4A4 column; mobile Phase, A: 0.4 mM HNO3, pH 3.4: B: 50 mM HNO3, pH 1.3 | — | 100 | AsB: 0.042, AsIII: 0.066, AsV: 0.045, MMA: 0.059, DMA: 0.044 | 253 |
| Fish and oyster | AsB, AsC, AsIII, AsV, DMA, MMA | CE-ICP-MS | 15 mM Tris solution containing 15 mM SDS (pH 9.0) was used as the electrophoretic buffer and the applied voltage was set at 122 kV | 0.2 | 0.02 | 0.3–0.5 | 254 |
| Fish, crustacean | AsB, AsIII, AsV, DMA, MMA | HPLC-ICP-MS | Hamilton PRP-X100 anion exchange column; mobile phase, A: 5.0 mM Na2SO4, pH 10–10.5; B: 50 mM Na2SO4, pH 10–10.5 (fish and crustacean), Hamilton PRP-X100 anion exchange column; mobile phase, A: H3PO4, pH 7.5: B: 50 mM, pH 6 (sediment) | 15 | 100 | Not given | 15 |
| Marine organisms | Arsenosugar glycerol, arsenosugar phosphate, arsenosugar sulfonate and arsenosugar sulfate | HPLC-ICP-MS | ZirChrom-SAX column; mobile phase, 1 mM NH4H2PO4, pH 5.6, Hypercarb (Thermo Electron Corporation, Runcorn UK) column; mobile phase, 13.8 mM nitric acid, 2% (v/v) MeOH, pH 8 | 20 | 20 | 1.5–2.0 | 63 |
| Seafood | AsIII, MMA, DMA, AsV, AsB, AC, TMA+ and TMAO | HPLC-ICP-MS | An IonPac AG4 guard column and an IonPac AS4A analytical column (both from Dionex Corpn, USA); mobile phase, A: 0.4 mM HNO3, pH 3.3; B: 50 mM HNO3, pH 1.3 | 15 | 100 | AsIII: 0.03, MMA: 0.05, DMA: 0.05, AsV: 1.6, AsB: 0.08, AC: 0.14, TMA+: 0.09, TMAO: 0.13 | 255 |
| Seafood | AsB, AsC, AsIII, DMA, MMA and AsV | HPLC-ICP-MS | IonPac AS7 anion exchange column; mobile phase, A: 1.0 mM HNO3, 1% (v/v) methanol, pH 2.9: B: 80 mM HNO3, 1% (v/v), pH 1.3 | 9.5 | 50 | AsB: 8.5, AsC: 6.7 AsIII: 5.4, DMA: 10.7 MMA: 10.8, AsV: 6.2 | 80 |
| Oyster tissue | DMA, MMA, AsV, oxo-arsenosugars: O-PO4, S-Gly and S-PO4 | HPLC-ICP-MS | Hamilton PRP-X100 column; mobile phase, A: 20 mM phosphate buffer, pH 5.6; B: 20 mM phosphate, pH 5.6, MeOH50% (v/v), 40 °C | 25 | 10 | Not given | 239 |
| Shrimp | AsB, DMA, AsIII, AsV, OXO-As-SugPO4, Thio-As-SugPO4 | HPLC-ICP-MS | Hamilton PRP-X100 anion exchange column; mobile phase, 20 mM NH4H2PO4, pH 6, 40 °C, Cation exchange Supelcosil LC-SCX column, mobile phase, 20 mM pyridine at pH 2, 40 °C, reverse phase chromatography using a Shisheido Capcell PAK C18 MGII; mobile phase, 10 mM sodium 1-butansulfonate, 4 mM tetramethylammonium hydroxide, 4 mM malonic acid, 0.5% MeOH, pH 3 | 19 | — | Not given | 256 |
| Bivalve mollusks | AsB, As III, MMA, DMA, As V, p-arsanilic acid (p-ASA) | HPLC-ICP-MS | Hamilton PRP-X100 column; mobile phase, A: 20 mM (NH4)2HPO4, pH 6.0; B: 20 mM (NH4)2CO3, pH 8.5 | 15 | 200 | Not given | 123 |
| Edible periwinkles | TMA+, AsB, MMA, glycerol arsenosugar and inorganic As | HPLC-ICP-MS | Hamilton PRP-X100 anion exchange column; gradient mobile phase, A: 4 mM NH4NO3; B: 60 mM NH4NO3, pH 8.65, Hamilton PRP-X200 cation-exchange column; mobile phase, 20 mM pyridine (C5H5N)/pH 2.7, formic acid (CH2O2) | 8 | — | Not given | 158 |
| Biological tissues (certified material TORT-1 and fresh bivalve tissues) | AsB, As III, MMA, DMA AsV | HPLC-HG-AAS | Column, Hamilton PRP X-100 strong anionic exchange column; mobile phase, phosphate buffers (10 mM and 100 mM at pH 5.8) | — | — | AsB: ND, As III: 1.1, DMA: 2.0, MMA: 1.9, As V: 3.9 | 257 |
| Biota sample | AsB, AsIII, DMA, MMA and AsV | HPLC-HG-AAS | Anion exchange column (Hamilton, Reno, NV, USA); mobile phase, 25 mM phosphate, pH 5.8 | — | — | AsB: 0.3, AsIII: 0.08 DMA: 0.1, MMA: 0.1, AsV: 0.3 | 117 |
| Marine organism | AsIII, AsV, MMA, DMA and AsB | HPLC-(UV)-HG-AFS | Hamilton PRP X-100 (25 cm × 4.1 mm) column; mobile phase, 25 mM phosphate buffer, pH 5.8 | — | — | AsIII:AsV:MMA:DMA:AsB = 0.3 |
257 |
| Canned cod liver tissue | Triethylarsine (Et3As), triphenylarsine (Ph3As) | GC-ICP-MS | Column: HP-5MS (30 mm × 0.25 mm × 0.25 μm), carrier gas: He 2 ml min−1, GC program; A: 40 °C, 10 °C min−1 to 60 °C, 30 °C min−1 to 250 °C, 40 °C min−1 to 280 °C, B: 50 °C, 1 min, 50 °C min−1 to 180 °C, 3 °C min−1 to 220 °C 1 min, 15 °C min−1 to 270 °C 8 min | 20 | — | Et3As: 0.00005, Ph3As: 0.00013 | 258 |
2.4 Certified reference materials
The use of CRM materials has been reviewed extensively with respect of quality control, method validation, interlaboratory testing, control charting and evaluation of analytical results using a matrix-matched CRM.182 Several arsenic-containing CRMs have been developed, but most of them are certified for the total-element concentration. Species specific CRM materials are now crucial as a result of the increasing used for species specific measurement.12 Amongst the CRMs available for As are BCR627 (Tuna fish), BCR 710 (oyster tissue), DORM-2 (dog fish muscle), and SRM 1640 (natural water).183 Species specific materials include TORT-3 (lobster) and several from the National Metrology Institute of Japan (NMIJ), including CRM 7405 (seaweed) and CRM 7503a (rice flour).3. Methods of detection
3.1 X-ray spectroscopic techniques
X-ray spectroscopic methods are being increasingly used for As speciation analysis. They are most often used for geological samples184,185 but can also be used for arsenic-rich biological samples.186,187 The possibility of conducting speciation analysis on solid environmental samples without the need for extraction of the element species has been investigated and a number of X-ray spectroscopic techniques have been used to measure total As and As speciation in different solid environmental and biological samples. However these techniques may have limited application for food analysis, due to the relatively poor detection limits and problems from the high intensity of the X-ray beam modifying the samples.188,189 XANES and EXAFS have been used for arsenic speciation in biological environmental samples,190,191Daphnia pulex,192 plant material,193,194 seaweed195 and rice grain.1963.2 Mass spectrometry
MS is the most frequently applied method for identifying and elucidating unknown compounds in foods following speciation. Ionization of the compounds can be achieved by techniques such as ionspray, electrospray, atmospheric pressure chemical ionization (APCI), electron ionization (EI), and fast atom bombardment. Because most As compounds are not volatile, some form of derivatization is require before GC separation. Many As speciation methods are based on conversion of As into the corresponding methylarsine by sodium borohydride, although thioglycolic acid methylester has been used to derivatise methylarsenic to produce lipophilic species,197 and methyl thioglycolate has been used to derivatize MMA, DMA and inorganic As for extraction into cyclohexane prior to chromatographic separation. Mercaptanes/dimercaptanes or thioglycolic acid methyl esters have also been used to derivatize phenylarsinc compounds before injecting into the GC-MS.198,1993.3 Detection by AAS, AFS and AES
In atomic spectrometry, an excitation source is required to atomise or ionise the analyte of interest. The advantage of these techniques is their inherent sensitive and element specific detection. Graphite furnace atomic absorption spectroscopy (GFAAS) has found preference over flame AAS for As studies since the sensitivity is greater by a factor of 10–100 times.201 Both fraction collection and on-line coupling of HPLC with GFAAS have been reported offering detection limit in the range of a few nanogram.174,202–206Due to its low detection limit and high selectivity, hydride generation atomic absorption spectroscopy (HG-AAS) has been traditionally one of the most widely used methods for As speciation.39,207–210 Hydride generation coupled with AAS is a popular method for determining hydride reducible arsenic compounds such as AsIII, AsV, MMA and DMA. The volatile As species is produced using either by zinc/hydrochloric acid or sodium borohydride/acid mixtures and the volatile As species produced are transported to the detection system with argon gas. By forming arsine gas, the analyte is easily and efficiently separated from its sample matrices and transported to the detection system, sometimes via a cryogenic pre-concentration step to obtain better detection limits. However, a number of organo arsenicals, for instance AsB and AC, cannot be detected by this method since they are not able to produce volatile hydrides. In this case, the separation of these species prior to HG-AAS is required followed by conversion of the individual As species via photolysis or chemical destruction.3 As a result of incorporating these techniques, AsB and AC may be determined using hydride generation, although controllable reaction conditions and the reduction of certain interfering elements may be required.211
Total As in sea food has been determined by HG-AAS after performing a dry-ashing to the sample.212 The results in this study were very close to the data achieved by other authors using a range of different methods. HG-AAS has also been widely utilized for the determination of As in water.213 A summary of publications employing HG-AAS and HPLC coupled with HG-AAS is presented in Table 5.
Coupling atomic fluorescence spectrometry (AFS) with HPLC is now a well-established and useful technique for As speciation. AFS can rival ICP-MS regarding performance criteria such as detection limits, reproducibility, repeatability, and sensitivity for As. AFS also offers low purchase and running cost, shorter warm up times prior to analysis and easy handling.214 HPLC-(UV)-HG-AFS has been applied to As speciation for the both NRCC-TORT1 reference material and several environmental samples with the detection limits ranging from 0.1 to 0.3 μg l−1.214
Finally, atomic emission spectroscopy may be used as an alternative technique for As speciation. Chausseau et al.215 concluded that HPLC-ICP-AES is a reliable technique for As speciation, when very low limit of detections are not required; they reported detection limits better than 10 μg l−1 for AsIII, and DMA and 20 μg l−1 for AsV. The technique can also be used in conjunction with HG, although it should be remembered that not all As species may be determined using this approach.
3.4 Detection by ICP-MS
The merits of ICP-MS are well documented,216,217 and this approach is now the method of choice in most laboratories for As speciation. The main advantages that the ICP-MS has over the other techniques are its low detection limits, 1–10 pg ml−1 range for quadrupole instruments, large linear dynamic range, rapid, multi-element capability for many elements and potential to use isotopic studies (although not As).218 Despite all of these advantages there some limitations using ICP-MS for As speciation. The use of ICP-MS alone does not provide direct molecular information and it is impossible to identify individual As species without some form of prior separation usually by HPLC.Interferences can be a problem in ICP-MS, particularly when there is an isobaric overlap due to polyatomic ions formed by combination of two or more atoms. The most significance polyatomic ions are formed from the most abundant isotopes of argon, atmospheric gases, and the solvents or acids used during sample preparation.219 A major polyatomic interference for As [As is monoisotope m/z 75] is 40As35Cl. Incomplete dissociation, or recombination in cooler plasma regions may lead to the formation of refractory oxides, especially in the boundary layer around the sampler cone.220
These interferences problem can be attenuated in ICP-MS by several methods. Polyatomic interferences can be tackled via mathematical correction221 or by adding another gas such as nitrogen, oxygen, air, helium, and hydrogen to the argon plasma, which can minimise the inherent polyatomic interference. Addition of nitrogen gas to an argon plasma has been found very effective due to an increasing in signal and a decrease in the argon and O-based interferences.222 However, a more recent approach utilising collision cell technology is now available on commercial instruments for interferences reduction. For As, a reduction in the 40Ar35Cl+ interference can be achieved using a collision reaction cell including gases such as H2, O2, NH3, CH4, NO, CO2 and C2H4.223–225
Sector field (SF)-ICP-MS is perhaps the ultimate choice for elemental speciation studies due to its sensitivity and ability to resolve isobaric overlaps.226 Some examples of As speciation studies using this technique include arsenic speciation in xylem sap of cucumber,227 freshwater fish228 and fish sample.228
3.5 Carbon enhancement of the As signal
Signal enhancement is a well-known phenomenon in inductively plasma mass spectrometry. The addition of carbon to the argon plasma of an ICP-MS causes an increase in the proportion of As atoms that are ionised by the charge transfer effect. This increases the observed counts per second for the As signal at m/z 75.229–231 Traditionally this has been achieved through the addition of organic solvents to the sample matrix231 or to the mobile phase232 to improve sensitivity. Signal enhancement can also be obtained by addition of aqueous solutions of volatile carbon compounds (acetone, methanol, and acetic acid) directly into the thermostatic spray chamber.2334. Conclusion
Arsenic species can accumulate in both plant derived and marine food stuffs. Arsenic exists in food as AsIII and AsV, organic arsenic (such as MMA, DMA) and tetramethylarsonium ion, AC, TMAO, and arsenosugers. Fauna sources such as fish and seafood are well known to contain relatively high concentration of AsB which is not toxic, whereas cereals for example rice, and drinking water may contain inorganic arsenic which may present a risk to health. This review of the literature suggests that appropriate analytical techniques now exists to determine the most common As species in food and waters to ensure that current health guidelines are met.References
- S. W. Al Rmalli, P. I. Haris, C. F. Harrington and M. Ayub, Sci. Total Environ., 2005, 337, 23–30 CrossRef CAS PubMed.
- D. Lièvremont, P. N. Bertin and M.-C. Lett, Biochimie, 2009, 91, 1229–1237 CrossRef PubMed.
- K. J. Lamble and S. J. Hill, Anal. Chim. Acta, 1996, 334, 261–270 CrossRef CAS.
- E. Sanz, R. Muñoz-Olivas and C. Cámara, J. Chromatogr. A, 2005, 1097, 1–8 CrossRef CAS PubMed.
- M. F. Hughes, Toxicol. Lett., 2002, 133, 1–16 CrossRef CAS.
- A. Moreda-Piñeiro, E. Peña-Vázquez, P. Hermelo-Herbello, P. Bermejo-Barrera, J. Moreda-Piñeiro, E. Alonso-Rodríguez, S. Muniategui-Lorenzo, P. n. López-Mahía and D. o. Prada-Rodríguez, Anal. Chem., 2008, 80, 9272–9278 CrossRef PubMed.
- W. Goessler, W. Maher, K. Irgolic, D. Kuehnelt, C. Schlagenhaufen and T. Kaise, Fresenius. J. Anal. Chem., 1997, 359, 434–437 CrossRef CAS.
- T. Watanabe and S. Hirano, Arch. Toxicol., 2012, 1–11 CAS.
- J. Moreda–Piñeiro, E. Alonso-Rodríguez, V. Romarís-Hortas, A. Moreda-Piñeiro, P. López-Mahía, S. Muniategui-Lorenzo, D. Prada-Rodríguez and P. Bermejo-Barrera, Food Chem., 2012, 130, 552–560 CrossRef PubMed.
- C.-g. Yuan, G.-b. Jiang and B. He, J. Anal. At. Spectrom., 2005, 20, 103–110 RSC.
- K. A. Francesconi, Pure Appl. Chem., 2010, 82, 373–381 CrossRef CAS.
- S. McSheehy, J. Szpunar, R. Morabito and P. Quevauviller, TrAC, Trends Anal. Chem., 2003, 22, 191–209 CrossRef CAS.
- C. Almela, M. Jesús Clemente, D. Vélez and R. Montoro, Food Chem. Toxicol., 2006, 44, 1901–1908 CrossRef CAS PubMed.
- L. Benramdane, F. Bressolle and J. Vallon, J. Chromatogr. Sci., 1999, 37, 330–344 CAS.
- S. Rattanachongkiat, G. E. Millward and M. E. Foulkes, J. Environ. Monit., 2004, 6, 254–261 RSC.
- K. Morita and E. Kaneko, Anal. Sci., 2006, 22, 1085–1090 CrossRef CAS.
- P. L. Smedley and D. G. Kinniburgh, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- L. S. Milstein, A. Essader, C. Murrell, E. D. Pellizzari, R. A. Fernando, J. H. Raymer and O. Akinbo, J. Agric. Food Chem., 2003, 51, 4180–4184 CrossRef CAS PubMed.
- J. M. Ballantyne and J. N. Moore, Geochim. Cosmochim. Acta, 1988, 52, 475–483 CrossRef CAS.
- T. Yokoyama, Y. Takahashi and T. Tarutani, Chem. Geol., 1993, 103, 103–111 CrossRef CAS.
- V. K. Sharma and M. Sohn, Environ. Int., 2009, 35, 743–759 CrossRef CAS PubMed.
- B. Petrusevski, S. Sharma, J. C. Schippers and K. Shordt, Delft: IRC International Water and Sanitation Centre, 2007, vol. 17.
- J. K. Nag, V. Balaram, R. Rubio, J. Albertí and A. K. Das, J. Trace Elem. Med. Biol., 1996, 10, 20–24 CAS.
- W. Anders, R. Bull, K. Cantor, D. Chakraborti, C. Chen, A. DeAngelo, D. DeMarini, C. Ferreccio, S. Fukushima and T. Gebel, Some drinking-water disinfectants and contaminants, including arsenic, World Health Organization, 2004 Search PubMed.
- B. K. Mandal and K. T. Suzuki, Talanta, 2002, 58, 201–235 CrossRef CAS.
- G. X. Sun, P. N. Williams, A. M. Carey, Y. G. Zhu, C. Deacon, A. Raab, J. Feldmann, R. M. Islam and A. A. Meharg, Environ. Sci. Technol., 2008, 42, 7542–7546 CrossRef CAS.
- P. Thomas, J. K. Finnie and J. G. Williams, J. Anal. At. Spectrom., 1997, 12, 1367–1372 RSC.
- J. Matschullat, Sci. Total Environ., 2000, 249, 297–312 CrossRef CAS.
- M. Bissen and F. H. Frimmel, Acta Hydrochim. Hydrobiol., 2003, 31, 9–18 CrossRef CAS.
- E. Moreno-Jiménez, R. Manzano, E. Esteban and J. Peñalosa, J. Soils Sediments, 2010, 10, 301–312 CrossRef PubMed.
- E. Hozhina, A. Khramov, P. Gerasimov and A. Kumarkov, J. Geochem. Explor., 2001, 74, 153–162 CrossRef CAS.
- A. M. Murciego, A. G. Sánchez, M. González, E. P. Gil, C. T. Gordillo, J. C. Fernández and T. B. Triguero, Environ. Pollut., 2007, 145, 15–21 CrossRef CAS PubMed.
- R. S. Oremland and J. F. Stolz, Trends Microbiol., 2005, 13, 45–49 CrossRef CAS PubMed.
- W. Li, C. Wei, C. Zhang, M. Van Hulle, R. Cornelis and X. Zhang, Food Chem. Toxicol., 2003, 41, 1103–1110 CrossRef CAS.
- B. K. Mandal, K. T. Suzuki, K. Anzai, K. Yamaguchi and Y. Sei, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 874, 64–76 CrossRef CAS PubMed.
- M. Van Hulle, C. Zhang, X. Zhang and R. Cornelis, Analyst, 2002, 127, 634–640 RSC.
- J. F. Ferguson and J. Gavis, Water Res., 1972, 6, 1259–1274 CrossRef CAS.
- H. Guo, D. Stüben, Z. Berner and Q. Yu, Appl. Geochem., 2009, 24, 657–663 CrossRef CAS PubMed.
- O. Muñoz, D. Vélez and R. Montoro, Analyst, 1999, 124, 601–607 RSC.
- N. P. Vela, D. T. Heitkemper and K. R. Stewart, Analyst, 2001, 126, 1011–1017 RSC.
- X. C. Le, X. Lu, M. Ma, W. R. Cullen, H. V. Aposhian and B. Zheng, Anal. Chem., 2000, 72, 5172–5177 CrossRef CAS.
- S. Suzuki, L. L. Arnold, K. L. Pennington, B. Chen, H. Naranmandura, X. C. Le and S. M. Cohen, Toxicol. Appl. Pharmacol., 2010, 244, 99–105 CrossRef CAS PubMed.
- A. McKnight-Whitford, B. Chen, H. Naranmandura, C. Zhu and X. C. Le, Environ. Sci. Technol., 2010, 44, 5875–5880 CrossRef CAS PubMed.
- A. H. Buck, Am. J. Med. Sci., 1904, 123, 162 CrossRef PubMed.
- M. F. Hughes, B. D. Beck, Y. Chen, A. S. Lewis and D. J. Thomas, Toxicol. Sci., 2011, 123, 305–332 CrossRef CAS PubMed.
- A. A. Meharg and A. Raab, Environ. Sci. Technol., 2010, 44, 4395–4399 CrossRef CAS PubMed.
- M. Rose, M. Baxter, N. Brereton and C. Baskaran, Food Addit. Contam., Part A, 2010, 27, 1380–1404 CrossRef CAS PubMed.
- R. A. Schoof, L. J. Yost, J. Eickhoff, E. A. Crecelius, D. W. Cragin, D. M. Meacher and D. B. Menzel, Food Chem. Toxicol., 1999, 37, 839–846 CrossRef CAS.
- L. J. Yost, S. H. Tao, S. K. Egan, L. M. Barraj, K. M. Smith, J. S. Tsuji, Y. W. Lowney, R. A. Schoof and N. J. Rachman, Hum. Ecol. Risk Assess., 2004, 10, 473–483 CrossRef CAS.
- European Food Safety Authority (EFSA), 7, 2009, 7, 1351.
- J. Laizu, Bangladesh J. Pharmacol., 2007, 2, 88–94 Search PubMed.
- A. H. Ackerman, P. A. Creed, A. N. Parks, M. W. Fricke, C. A. Schwegel, J. T. Creed, D. T. Heitkemper and N. P. Vela, Environ. Sci. Technol., 2005, 39, 5241–5246 CrossRef CAS.
- S. Torres-Escribano, M. Leal, D. Vélez and R. Montoro, Environ. Sci. Technol., 2008, 42, 3867–3872 CrossRef CAS.
- Y.-G. Zhu, G.-X. Sun, M. Lei, M. Teng, Y.-X. Liu, N.-C. Chen, L.-H. Wang, A. Carey, C. Deacon and A. Raab, Environ. Sci. Technol., 2008, 42, 5008–5013 CrossRef CAS.
- B. P. Jackson, V. F. Taylor, M. R. Karagas, T. Punshon and K. L. Cottingham, Environ. Health Perspect., 2012, 120, 613–626 CrossRef PubMed.
- G. Raber, N. Stock, P. Hanel, M. Murko, J. Navratilova and K. A. Francesconi, Food Chem., 2012, 134, 524–532 CrossRef CAS PubMed.
- I. N. Pasias, N. S. Thomaidis and E. A. Piperaki, Microchem. J., 2013, 108, 1–6 CrossRef CAS PubMed.
- M. D'Amato, F. Aureli, S. Ciardullo, A. Raggi and F. Cubadda, J. Anal. At. Spectrom., 2011, 26, 207–213 RSC.
- I. B. Karadjova, P. K. Petrov, I. Serafimovski, T. Stafilov and D. L. Tsalev, Spectrochim. Acta, Part B, 2007, 62, 258–268 CrossRef PubMed.
- J. M. Llobet, G. Falcó, C. Casas, A. Teixidó and J. L. Domingo, J. Agric. Food Chem., 2002, 51, 838–842 CrossRef PubMed.
- A. Moreda-Piñeiro, J. Moreda-Piñeiro, P. Herbello-Hermelo, P. Bermejo-Barrera, S. Muniategui-Lorenzo, P. López-Mahía and D. Prada-Rodríguez, J. Chromatogr. A, 2011, 1218, 6970–6980 CrossRef PubMed.
- C. García Sartal, M. d. C. Barciela-Alonso and P. Bermejo-Barrera, Microchem. J., 2012, 105, 65–71 CrossRef PubMed.
- A. Terol, F. Ardini, M. Grotti and J. L. Todolí, J. Chromatogr. A, 2012, 1262, 70–76 CrossRef CAS PubMed.
- S. Musil, Á. H. Pétursdóttir, A. Raab, H. Gunnlaugsdóttir, E. Krupp and J. Feldmann, Anal. Chem., 2014, 86, 993–999 CrossRef CAS PubMed.
- M. Argos, T. Kalra, B. L. Pierce, Y. Chen, F. Parvez, T. Islam, A. Ahmed, R. Hasan, K. Hasan and G. Sarwar, Am. J. Epidemiol., 2011, 174, 185–194 CrossRef PubMed.
- X. C. Le, S. Yalcin and M. Ma, Environ. Sci. Technol., 2000, 34, 2342–2347 CrossRef CAS.
- G. W. Page, Environ. Sci. Technol., 1981, 15, 1475–1481 CrossRef CAS.
- F. Robertson, Environ. Geochem. Health, 1989, 11, 171–185 CrossRef CAS PubMed.
- D. Chakraborti, M. M. Rahman, K. Paul, U. K. Chowdhury, M. K. Sengupta, D. Lodh, C. R. Chanda, K. C. Saha and S. C. Mukherjee, Talanta, 2002, 58, 3–22 CrossRef CAS.
- A. H. Smith and M. M. H. Smith, Toxicology, 2004, 198, 39–44 CrossRef CAS PubMed.
- P. Chanpiwat, S. Sthiannopkao, K. H. Cho, K.-W. Kim, V. San, B. Suvanthong and C. Vongthavady, Environ. Pollut., 2011, 159, 567–576 CrossRef CAS PubMed.
- B. D. Kocar, M. L. Polizzotto, S. G. Benner, S. C. Ying, M. Ung, K. Ouch, S. Samreth, B. Suy, K. Phan, M. Sampson and S. Fendorf, Appl. Geochem., 2008, 23, 3059–3071 CrossRef CAS PubMed.
- J. Buschmann, M. Berg, C. Stengel, L. Winkel, M. L. Sampson, P. T. K. Trang and P. H. Viet, Environ. Int., 2008, 34, 756–764 CrossRef CAS PubMed.
- T. Roychowdhury, H. Tokunaga and M. Ando, Sci. Total Environ., 2003, 308, 15–35 CrossRef CAS.
- M.-J. Kim, J. Nriagu and S. Haack, Environ. Pollut., 2002, 120, 379–390 CrossRef CAS.
- D. K. Nordstrom, Science, 2002, 296, 2143–2145 CrossRef CAS PubMed.
- M. Williams, F. Fordyce, A. Paijitprapapon and P. Charoenchaisri, Geo, 1996, 27, 16–33 CrossRef CAS.
- C. B'Hymer and J. A. Caruso, J. Chromatogr. A, 2004, 1045, 1–13 CrossRef PubMed.
- R. Cornells, J. Caruso, H. Crews and K. Heumann, Handbook of elemental speciation: Techniques and methodology, John Wiley and Sons, 2005, pp. 49 Search PubMed.
- J. Moreda-Piñeiro, E. Alonso-Rodríguez, A. Moreda-Piñeiro, C. Moscoso-Pérez, S. Muniategui-Lorenzo, P. López-Mahía, D. Prada-Rodríguez and P. Bermejo-Barrera, Anal. Chim. Acta, 2010, 679, 63–73 CrossRef PubMed.
- M. Montperrus, Y. Bohari, M. Bueno, A. Astruc and M. Astruc, Appl. Organomet. Chem., 2002, 16, 347–354 CrossRef CAS.
- S. HonorÚáHansen, J. Anal. At. Spectrom., 1993, 8, 1075–1084 RSC.
- J. Stolz, P. Basu and R. Oremland, Int. Microbiol., 2002, 5, 201–207 CrossRef CAS PubMed.
- R. Bentley and T. G. Chasteen, Microbiol. Mol. Biol. Rev., 2002, 66, 250–271 CrossRef CAS.
- Z. Mester and R. Sturgeon, Sample preparation for trace element analysis, Elsevier Amsterdam, 2003 Search PubMed.
- S. Van Herreweghe, R. Swennen, C. Vandecasteele and V. Cappuyns, Environ. Pollut., 2003, 122, 323–342 CrossRef CAS.
- J. A. Baig, T. G. Kazi, A. Q. Shah, M. B. Arain, H. I. Afridi, G. A. Kandhro and S. Khan, Anal. Chim. Acta, 2009, 651, 57–63 CrossRef CAS PubMed.
- J. Wu, Z. Mester and J. Pawliszyn, Anal. Chim. Acta, 2000, 424, 211–222 CrossRef CAS.
- K. A. Mir, A. Rutter, I. Koch, P. Smith, K. J. Reimer and J. S. Poland, Talanta, 2007, 72, 1507–1518 CrossRef CAS PubMed.
- I. Pizarro, M. Gómez, C. Cámara and M. Palacios, Anal. Chim. Acta, 2003, 495, 85–98 CrossRef CAS PubMed.
- R. Carabias-Martínez, E. Rodríguez-Gonzalo, P. Revilla-Ruiz and J. Hernández-Méndez, J. Chromatogr. A, 2005, 1089, 1–17 CrossRef PubMed.
- S. Foster, W. Maher, F. Krikowa and S. Apte, Talanta, 2007, 71, 537–549 CrossRef CAS PubMed.
- C. Dietz, J. Sanz, E. Sanz, R. Munoz-Olivas and C. Cámara, J. Chromatogr. A, 2007, 1153, 114–129 CrossRef CAS PubMed.
- D. T. Heitkemper, N. P. Vela, K. R. Stewart and C. S. Westphal, J. Anal. At. Spectrom., 2001, 16, 299–306 RSC.
- J. A. Caruso, D. T. Heitkemper and C. B'Hymer, Analyst, 2001, 126, 136–140 RSC.
- K. L. Ackley, C. B'Hymer, K. L. Sutton and J. A. Caruso, J. Anal. At. Spectrom., 1999, 14, 845–850 RSC.
- J. Mattusch, M. Cimpean and R. Wennrich, Int. J. Environ. Anal. Chem., 2006, 86, 629–640 CrossRef CAS.
- J. A. Caruso, D. T. Heitkemper and C. B'Hymer, Analyst, 2001, 126, 136–140 RSC.
- S. Branch, L. Ebdon and P. O'Neill, J. Anal. At. Spectrom., 1994, 9, 33–37 RSC.
- J. W. McKiernan, J. T. Creed, C. A. Brockhoff, J. A. Caruso and R. M. Lorenzana, J. Anal. At. Spectrom., 1999, 14, 607–613 RSC.
- B. He, Y. Fang, G. Jiang and Z. Ni, Spectrochim. Acta, Part B, 2002, 57, 1705–1711 CrossRef.
- S. L. Cleland, L. K. Olson, J. A. Caruso and J. M. Carey, J. Anal. At. Spectrom., 1994, 9, 975–979 RSC.
- Y. Bohari, G. Lobos, H. Pinochet, F. Pannier, A. Astruc and M. Potin-Gautier, J. Environ. Monit., 2002, 4, 596–602 RSC.
- D. Kuehnelt, K. J. Irgolic and W. Goessler, Appl. Organomet. Chem., 2001, 15, 445–456 CrossRef CAS.
- A.-C. Schmidt, W. Reisser, J. Mattusch, P. Popp and R. Wennrich, J. Chromatogr. A, 2000, 889, 83–91 CrossRef CAS.
- J. A. Brisbin and J. A. Caruso, Analyst, 2002, 127, 921–929 RSC.
- V. G. Mihucz, E. Tatár, I. Virág, C. Zang, Y. Jao and G. Záray, Food Chem., 2007, 105, 1718–1725 CrossRef CAS PubMed.
- A. Pell, A. Márquez, J. F. López-Sánchez, R. Rubio, M. Barbero, S. Stegen, F. Queirolo and P. Díaz-Palma, Chemosphere, 2013, 90, 556–564 CrossRef CAS PubMed.
- Z. Šlejkovec, E. Kápolna, I. Ipolyi and J. T. van Elteren, Chemosphere, 2006, 63, 1098–1105 CrossRef PubMed.
- J. Meier, N. Kienzl, W. Goessler and K. A. Francesconi, Environ. Chem., 2006, 2, 304–307 CrossRef.
- A. Ruttens, A. Blanpain, L. De Temmerman and N. Waegeneers, J. Geochem. Explor., 2012, 121, 55–61 CrossRef CAS PubMed.
- S. Karthikeyan, S. Hirata and C. S. P. Iyer, Int. J. Environ. Anal. Chem., 2004, 84, 573–582 CrossRef CAS.
- S. García Salgado, M. A. Quijano Nieto and M. M. Bonilla Simón, J. Chromatogr. A, 2006, 1129, 54–60 CrossRef PubMed.
- J. A. Brisbin, C. B'Hymer and J. A. Caruso, Talanta, 2002, 58, 133–145 CrossRef CAS.
- D. T. Heitkemper, N. P. Vela, K. R. Stewart and C. S. Westphal, J. Anal. At. Spectrom., 2001, 16, 299–306 RSC.
- A. D. Madsen, W. Goessler, S. N. Pedersen and K. A. Francesconi, J. Anal. At. Spectrom., 2000, 15, 657–662 RSC.
- J. Gomez-Ariza, D. Sanchez-Rodas, I. Giraldez and E. Morales, Analyst, 2000, 125, 401–407 RSC.
- J. Alberti, R. Rubio and G. Rauret, Fresenius' J. Anal. Chem., 1995, 351, 420–425 CrossRef CAS.
- M. J. Abedin, M. S. Cresser, A. A. Meharg, J. Feldmann and J. Cotter-Howells, Environ. Sci. Technol., 2002, 36, 962–968 CrossRef CAS.
- M. Vilanó and R. Rubio, Appl. Organomet. Chem., 2001, 15, 658–666 CrossRef.
- A. Leufroy, L. Noël, V. Dufailly, D. Beauchemin and T. Guérin, Talanta, 2011, 83, 770–779 CrossRef CAS PubMed.
- R. Tukai, W. A. Maher, I. J. McNaught and M. J. Ellwood, Anal. Chim. Acta, 2002, 457, 173–185 CrossRef CAS.
- C. M. Santos, M. A. Nunes, I. S. Barbosa, G. L. Santos, M. C. Peso-Aguiar, M. G. Korn, E. M. Flores and V. L. Dressler, Spectrochim. Acta, Part B, 2013, 86, 108–114 CrossRef CAS PubMed.
- C. Han, X. Cao, J.-J. Yu, X.-R. Wang and Y. Shen, Chromatographia, 2009, 69, 587–591 CAS.
- A. Chatterjee, Talanta, 2000, 51, 303–314 CrossRef CAS.
- J. McKiernan, J. Creed, C. Brockhoff, J. Caruso and R. Lorenzana, J. Anal. At. Spectrom., 1999, 14, 607–613 RSC.
- P. A. Gallagher, J. A. Shoemaker, X. Wei, C. A. Brockhoff-Schwegel and J. T. Creed, Fresenius' J. Anal. Chem., 2001, 369, 71–80 CrossRef CAS.
- P. A. Gallagher, S. Murray, X. Wei, C. A. Schwegel and J. T. Creed, J. Anal. At. Spectrom., 2002, 17, 581–586 RSC.
- P. Gallagher, J. Shoemaker, C. Brockhoff and J. Creed, J. Anal. At. Spectrom., 1999, 14, 1829–1834 RSC.
- B. Batista, L. Nacano, S. De Souza and F. Barbosa Jr, Food Addit. Contam., Part A, 2012, 29, 780–788 CrossRef CAS PubMed.
- W. Maher, S. Foster, F. Krikowa, E. Donner and E. Lombi, Environ. Sci. Technol., 2013, 47, 5821–5827 CrossRef CAS PubMed.
- G. Norton, C. Deacon, A. Mestrot, J. Feldmann, P. Jenkins, C. Baskaran and A. A. Meharg, Environ. Sci. Technol., 2013, 47, 6164–6172 CAS.
- S. Branch, L. Ebdon and P. O'Neill, J. Anal. At. Spectrom., 1994, 9, 33–37 RSC.
- S. Nookabkaew, N. Rangkadilok, C. Mahidol, G. Promsuk and J. Satayavivad, J. Agric. Food Chem., 2013, 61, 6991–6998 CrossRef CAS PubMed.
- E. Sanz, R. Muñoz-Olivas and C. Cámara, Anal. Chim. Acta, 2005, 535, 227–235 CrossRef CAS PubMed.
- N. Rubio-Rodríguez, S. M. de Diego, S. Beltrán, I. Jaime, M. T. Sanz and J. Rovira, J. Food Eng., 2012, 109, 238–248 CrossRef PubMed.
- S. Foster, D. Thomson and W. Maher, Mar. Chem., 2008, 108, 172–183 CrossRef CAS PubMed.
- J. Kirby, W. Maher, M. Ellwood and F. Krikowa, Aust. J. Chem., 2004, 57, 957–966 CrossRef CAS.
- S. S. Kannamkumarath, K. Wróbel, K. Wróbel and J. A. Caruso, J. Agric. Food Chem., 2004, 52, 1458–1463 CrossRef CAS PubMed.
- S. Foster, D. Thomson and W. Maher, Mar. Chem., 2008, 108, 172–183 CrossRef CAS PubMed.
- D. Thomson, W. Maher and S. Foster, Appl. Organomet. Chem., 2007, 21, 396–411 CrossRef CAS.
- C. García Sartal, M. d. C. Barciela-Alonso and P. Bermejo-Barrera, Microchem. J., 2012, 105, 65–71 CrossRef PubMed.
- P. M. Yehl, H. Gurleyuk, J. F. Tyson and P. C. Uden, Analyst, 2001, 126, 1511–1518 RSC.
- I. B. Karadjova, P. K. Petrov, I. Serafimovski, T. Stafilov and D. L. Tsalev, Spectrochim. Acta, Part B, 2007, 62, 258–268 CrossRef PubMed.
- B. Wenclawiak and M. Krah, Fresenius' J. Anal. Chem., 1995, 351, 134–138 CrossRef CAS.
- J. S. Wang and K. Chiu, J. Hazard. Mater., 2008, 158, 384–391 CrossRef CAS PubMed.
- J. M. Bayona, TrAC, Trends Anal. Chem., 2000, 19, 107–112 CrossRef CAS.
- N. P. Vela and J. A. Caruso, J. Anal. At. Spectrom., 1996, 11, 1129–1135 RSC.
- R. Wahlen and T. Catterick, Rapid Commun. Mass Spectrom., 2004, 18, 211–217 CrossRef CAS PubMed.
- R. Wahlen, S. McSheehy, C. Scriver and Z. Mester, J. Anal. At. Spectrom., 2004, 19, 876–882 RSC.
- R. Wahlen, J. Chromatogr. Sci., 2004, 42, 217–222 CAS.
- M. Quaghebeur, Z. Rengel and M. Smirk, J. Anal. At. Spectrom., 2003, 18, 128–134 RSC.
- C. Dietz, J. Sanz, E. Sanz, R. Muñoz-Olivas and C. Cámara, J. Chromatogr. A, 2007, 1153, 114–129 CrossRef CAS PubMed.
- P. Bermejo, J. Capelo, A. Mota, Y. Madrid and C. Cámara, TrAC, Trends Anal. Chem., 2004, 23, 654–663 CrossRef CAS PubMed.
- S. Babel and D. del Mundo Dacera, Waste Manage., 2006, 26, 988–1004 CrossRef CAS PubMed.
- C. Sparr Eskilsson and E. Björklund, J. Chromatogr. A, 2000, 902, 227–250 CrossRef CAS.
- E. Björklund, T. Nilsson and S. Bøwadt, TrAC, Trends Anal. Chem., 2000, 19, 434–445 CrossRef.
- K. Whaley-Martin, I. Koch and K. Reimer, Sci. Total Environ., 2013, 456, 148–153 CrossRef PubMed.
- K. L. Linge, Geostand. Geoanal. Res., 2006, 30, 157–174 CrossRef CAS PubMed.
- M. Kumaresan and P. Riyazuddin, Curr. Sci., 2001, 80, 837–846 CAS.
- M. Kumaresan and P. Riyazuddin, Curr. Sci., 2001, 80, 837–846 CAS.
- A. Taboada-de la Calzada, M. C. Villa-Lojo, E. Beceiro-González, E. Alonso-Rodríguez and D. Prada-Rodríguez, TrAC, Trends Anal. Chem., 1998, 17, 167–175 CrossRef CAS.
- Z. Mester and J. Pawliszyn, J. Chromatogr. A, 2000, 873, 129–135 CrossRef CAS.
- B. Bouyssiere, J. Szpunar and R. Lobinski, Spectrochim. Acta, Part B, 2002, 57, 805–828 CrossRef.
- R. Łobiński and F. C. Adams, Spectrochim. Acta, Part B, 1997, 52, 1865–1903 CrossRef.
- X.-B. Yin, X.-P. Yan, Y. Jiang and X.-W. He, Anal. Chem., 2002, 74, 3720–3725 CrossRef CAS.
- R. Naidu, J. Smith, R. G. McLaren, D. Stevens, M. Sumner and P. Jackson, Soil Sci. Soc. Am. J., 2000, 64, 122–128 CrossRef CAS.
- B. Deng and W.-T. Chan, J. Chromatogr. A, 2000, 891, 139–148 CrossRef CAS.
- Q. Tu, J. Qvarnström and W. Frech, Analyst, 2000, 125, 705–710 RSC.
- G. Yang, J. Xu, J. Zheng, X. Xu, W. Wang, L. Xu, G. Chen and F. Fu, Talanta, 2009, 78, 471–476 CrossRef CAS PubMed.
- B. Meermann, M. Bartel, A. Scheffer, S. Trümpler and U. Karst, Electrophoresis, 2008, 29, 2731–2737 CrossRef CAS PubMed.
- V. Tanboonchuy, N. Grisdanurak and C.-H. Liao, J. Hazard. Mater., 2012, 205, 40–46 CrossRef PubMed.
- S. Wangkarn and S. A. Pergantis, J. Anal. At. Spectrom., 2000, 15, 627–633 RSC.
- H. Heng-Bin, L. Yan-Bing, M. Shi-Fen and N. Zhe-Ming, J. Anal. At. Spectrom., 1993, 8, 1085–1090 RSC.
- M. Storelli and G. Marcotrigiano, Food Addit. Contam., 2000, 17, 763–768 CrossRef CAS PubMed.
- M. Storelli, R. G. Stuffler and G. Marcotrigiano, Food Addit. Contam., 2002, 19, 715–720 CrossRef CAS PubMed.
- K. Bluemlein, A. Raab and J. Feldmann, Anal. Bioanal. Chem., 2009, 393, 357–366 CrossRef CAS PubMed.
- O. Muñoz, O. P. Diaz, I. Leyton, N. Nuñez, V. Devesa, M. A. Súñer, D. Vélez and R. Montoro, J. Agric. Food Chem., 2002, 50, 642–647 CrossRef PubMed.
- S. McSheehy, P. Pohl, D. Vélez and J. Szpunar, Anal. Bioanal. Chem., 2002, 372, 457–466 CrossRef CAS PubMed.
- S. McSheehy, P. Pohl, R. Łobiński and J. Szpunar, Analyst, 2001, 126, 1055–1062 RSC.
- S. McSheehy, J. Szpunar, R. Lobinski, V. Haldys, J. Tortajada and J. S. Edmonds, Anal. Chem., 2002, 74, 2370–2378 CrossRef CAS.
- B. de Guillebon, F. Pannier, F. Seby, D. Bennink and P. Quevauviller, TrAC, Trends Anal. Chem., 2001, 20, 160–166 CrossRef CAS.
- B. Radke, L. Jewell and J. Namieśnik, Crit. Rev. Anal. Chem., 2012, 42, 162–183 CrossRef CAS.
- N. Chen, D. Jiang, J. Cutler, T. Kotzer, Y. Jia, G. Demopoulos and J. Rowson, Geochim. Cosmochim. Acta, 2009, 73, 3260–3276 CrossRef CAS PubMed.
- D. Paktunc, A. Foster and G. Laflamme, Environ. Sci. Technol., 2003, 37, 2067–2074 CrossRef CAS.
- S. M. Webb, J.-F. Gaillard, L. Q. Ma and C. Tu, Environ. Sci. Technol., 2003, 37, 754–760 CrossRef CAS.
- C. J. Langdon, A. A. Meharg, J. Feldmann, T. Balgar, J. Charnock, M. Farquhar, T. G. Piearce, K. T. Semple and J. Cotter-Howells, J. Environ. Monit., 2002, 4, 603–608 RSC.
- G. Meitzner, J. Gardea-Torresdey, J. Parsons, S. Scott and E. Deguns, Microchem. J., 2005, 81, 61–68 CrossRef CAS PubMed.
- K. G. Scheckel, E. Lombi, S. A. Rock and M. J. McLaughlin, Environ. Sci. Technol., 2004, 38, 5095–5100 CrossRef CAS.
- P. G. Smith, I. Koch, R. A. Gordon, D. F. Mandoli, B. D. Chapman and K. J. Reimer, Environ. Sci. Technol., 2004, 39, 248–254 CrossRef.
- J. Parsons, M. Lopez, H. Castillo-Michel, J. Peralta-Videa and J. Gardea-Torresdey, Appl. Spectrosc., 2009, 63, 961–970 CrossRef CAS PubMed.
- G. Caumette, I. Koch, M. Moriarty and K. J. Reimer, Sci. Total Environ., 2012, 432, 243–250 CrossRef CAS PubMed.
- P. G. Smith, I. Koch and K. J. Reimer, Sci. Total Environ., 2008, 390, 188–197 CrossRef CAS PubMed.
- M. Hatayama, T. Sato, K. Shinoda and C. Inoue, J. Biosci. Bioeng., 2011, 111, 326–332 CrossRef CAS PubMed.
- I. Koch, K. McPherson, P. Smith, L. Easton, K. G. Doe and K. J. Reimer, Mar. Pollut. Bull., 2007, 54, 586–594 CrossRef CAS PubMed.
- E. Lombi, K. G. Scheckel, J. Pallon, A. M. Carey, Y. G. Zhu and A. A. Meharg, New Phytol., 2009, 184, 193–201 CrossRef CAS PubMed.
- B. Beckermann, Anal. Chim. Acta, 1982, 135, 77–84 CrossRef CAS.
- R. Haas, T. C. Schmidt, K. Steinbach and E. von Löw, Fresenius' J. Anal. Chem., 1998, 361, 313–318 CrossRef CAS.
- K. Thurow, A. Koch, N. Stoll, C. Haney, R. McGuire and J. Compton, Environmental aspects of converting CW facilities to peaceful purposes, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2002, pp. 123–138 Search PubMed.
- V. Sele, H. Amlund, M. G. Berntssen, J. Berntsen, K. Skov and J. Sloth, Anal. Bioanal. Chem., 2013, 405, 5179–5190 CrossRef CAS PubMed.
- E. H. Larsen, J. Anal. At. Spectrom., 1991, 6, 375–377 RSC.
- D. Velez, N. Ybanez and R. Montoro, J. Anal. At. Spectrom., 1996, 11, 271–277 RSC.
- H. Heng-bin, L. Yan-bing, M. Shi-fen and N. Zhe-ming, J. Anal. At. Spectrom., 1993, 8, 1085–1090 RSC.
- B. Welz, J. Anal. At. Spectrom., 1998, 13, 413–417 RSC.
- W. Chintakovid, P. Visoottiviseth, S. Khokiattiwong and S. Lauengsuchonkul, Chemosphere, 2008, 70, 1532–1537 CrossRef CAS PubMed.
- H. M. Anawar, Talanta, 2012, 88, 30–42 CrossRef CAS PubMed.
- R. Torralba, M. Bonilla, L. V. Pérez-Arribas, M. A. Palacios and C. Cámara, Microchim. Acta, 1997, 126, 257–262 CrossRef CAS.
- J. C. Lopez, C. Reija, R. Montoro, M. L. Cervera and M. de la Guardia, J. Anal. At. Spectrom., 1994, 9, 651–656 RSC.
- M. L. Cervera, R. Montoro, O. Muñoz and D. Vélez, 1999, 14.
- A. Shraim, B. Chiswell and H. Olszowy, Talanta, 1999, 50, 1109–1127 CrossRef CAS.
- K. Farzana Akter, Z. Chen, L. Smith, D. Davey and R. Naidu, Talanta, 2005, 68, 406–415 CrossRef CAS PubMed.
- M. Suner, V. Devesa, O. Munoz, F. Lopez, R. Montoro, A. Arias and J. Blasco, Sci. Total Environ., 1999, 242, 261–270 CrossRef CAS.
- A. Chatterjee, D. Das, B. K. Mandal, T. R. Chowdhury, G. Samanta and D. Chakraborti, Analyst, 1995, 120, 643–650 RSC.
- J. L. Gómez-Ariza, D. Sánchez-Rodas, I. Giráldez and E. Morales, Talanta, 2000, 51, 257–268 CrossRef.
- M. Chausseau, C. Roussel, N. Gilon and J. Mermet, Fresenius' J. Anal. Chem., 2000, 366, 476–480 CrossRef CAS.
- R. Clough, C. F. Harrington, S. J. Hill, Y. Madrid and J. F. Tyson, J. Anal. At. Spectrom., 2014, 29, 1158–1196 RSC.
- S. J. Hill, Inductively coupled plasma spectrometry and its applications, Blackwell Publishing Ltd, 2nd edn, 2007, ch. 11, pp. 387–413 Search PubMed.
- E. H. Evans and J. J. Giglio, J. Anal. At. Spectrom., 1993, 8, 1–18 RSC.
- A. Author, Anal. Methods, 2010, 2, 1206–1221 RSC.
- M. Vaughan and G. Horlick, Spectrochim. Acta, Part B, 1990, 45, 1289–1299 CrossRef.
- N. M. Raut, L. Huang, S. K. Aggarwal and K. Lin, J. Chin. Chem. Soc., 2005, 52, 589 CAS.
- S. J. Hill, M. J. Ford and L. Ebdon, J. Anal. At. Spectrom., 1992, 7, 719–725 RSC.
- D. R. Bandura, V. I. Baranov and S. D. Tanner, Fresenius' J. Anal. Chem., 2001, 370, 454–470 CrossRef CAS.
- D. R. Bandura, V. I. Baranov, A. Litherland and S. D. Tanner, Int. J. Mass Spectrom., 2006, 255, 312–327 CrossRef PubMed.
- J. Darrouzès, M. Bueno, G. Lespès, M. Holeman and M. Potin-Gautier, Talanta, 2007, 71, 2080–2084 CrossRef PubMed.
- K. L. Linge, Geostand. Geoanal. Res., 2006, 30, 157–174 CrossRef CAS PubMed.
- V. Mihucz, E. Tatár, I. Virág, E. Cseh, F. Fodor and G. Záray, Anal. Bioanal. Chem., 2005, 383, 461–466 CrossRef CAS PubMed.
- J. Zheng and H. Hintelmann, J. Anal. At. Spectrom., 2004, 19, 191–195 RSC.
- S. Pergantis, J. Anal. At. Spectrom., 1999, 14, 657–662 RSC.
- P. Kralj and M. Veber, Acta Chim. Slov., 2003, 50, 633–644 CAS.
- E. H. Larsen and S. Stürup, J. Anal. At. Spectrom., 1994, 9, 1099–1105 RSC.
- U. Kohlmeyer, J. Kuballa and E. Jantzen, Rapid Commun. Mass Spectrom., 2002, 16, 965–974 CrossRef CAS PubMed.
- M. Kovačevič and W. Goessler, Spectrochim. Acta, Part B, 2005, 60, 1357–1362 CrossRef PubMed.
- J. H. Huang, P. Fecher, G. Ilgen, K. N. Hu and J. Yang, Food Chem., 2012, 130, 453–459 CrossRef CAS PubMed.
- E. Sanz, R. Muñoz-Olivas, C. Cámara, M. K. Sengupta and S. Ahamed, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2007, 42, 1695–1705 CrossRef CAS PubMed.
- M. J. Ruiz-Chancho, J. F. López-Sánchez, E. Schmeisser, W. Goessler, K. A. Francesconi and R. Rubio, Chemosphere, 2008, 71, 1522–1530 CrossRef CAS PubMed.
- R. Larios, R. Fernández-Martínez, I. LeHecho and I. Rucandio, Sci. Total Environ., 2012, 414, 600–607 CrossRef CAS PubMed.
- L. Jedynak, J. Kowalska, M. Kossykowska and J. Golimowski, Microchem. J., 2010, 94, 125–129 CrossRef CAS PubMed.
- C. B'Hymer and J. Caruso, J. Liq. Chromatogr. Relat. Technol., 2002, 25, 639–653 CrossRef PubMed.
- P. Niedzielski, M. Mleczek, Z. Magdziak, M. Siwulski and L. Kozak, Food Chem., 2013, 141, 3571–3577 CrossRef CAS PubMed.
- X. Liu, W. Zhang, Y. Hu and H. Cheng, Microchem. J., 2013, 108, 38–45 CrossRef CAS PubMed.
- C. Zhang, Y. Wang and Y. Ge, Anal. Lett., 2013, 46, 1573–1586 CrossRef CAS.
- M. Christodoulidou, C. Charalambous, M. Aletrari, P. Nicolaidou Kanari, A. Petronda and N. Ward, J. Hydrol., 2012, 468, 94–100 CrossRef PubMed.
- S. N. Ronkart, V. Laurent, P. Carbonnelle, N. Mabon, A. Copin and J.-P. Barthélemy, Chemosphere, 2007, 66, 738–745 CrossRef CAS PubMed.
- M. Gomez, C. Cámara, M. Palacios and A. Lopez-Gonzalvez, Fresenius' J. Anal. Chem., 1997, 357, 844–849 CrossRef CAS.
- P. Niedzielski, Anal. Chim. Acta, 2005, 551, 199–206 CrossRef CAS PubMed.
- Y. Bohari, A. Astruc, M. Astruc and J. Cloud, J. Anal. At. Spectrom., 2001, 16, 774–778 RSC.
- C. Demesmay, M. Olle and M. Porthault, Fresenius' J. Anal. Chem., 1994, 348, 205–210 CrossRef CAS.
- P. Thomas and K. Sniatecki, J. Anal. At. Spectrom., 1995, 10, 615–618 RSC.
- D. Beauchemin, K. Siu, J. W. McLaren and S. S. Berman, J. Anal. At. Spectrom., 1989, 4, 285–289 RSC.
- L. H. Reyes, J. L. G. Mar, G. M. M. Rahman, B. Seybert, T. Fahrenholz and H. M. S. Kingston, Talanta, 2009, 78, 983–990 CrossRef CAS PubMed.
- R.-Y. Wang, Y.-L. Hsu, L.-F. Chang and S.-J. Jiang, Anal. Chim. Acta, 2007, 590, 239–244 CrossRef CAS PubMed.
- S. Karthikeyan, S. Hirata and C. S. Iyer, Int. J. Environ. Anal. Chem., 2004, 84, 573–582 CrossRef CAS.
- C. F. Yeh and S. J. Jiang, Electrophoresis, 2005, 26, 1615–1621 CrossRef CAS PubMed.
- S. Karthikeyan and S. Hirata, Appl. Organomet. Chem., 2004, 18, 323–330 CrossRef CAS.
- V. F. Taylor, B. P. Jackson, M. R. Siegfried, J. Navratilova, K. A. Francesconi, J. Kirshtein and M. Voytek, Environ. Chem., 2012, 9, 130–138 CrossRef CAS PubMed.
- J. L. Gómez-Ariza, D. Sánchez-Rodas, R. Beltrán and I. Giráldez, Int. J. Environ. Anal. Chem., 1999, 74, 203–213 CrossRef.
- U. Arroyo-Abad, J. Mattusch, S. Mothes, M. Möder, R. Wennrich, M. P. Elizalde-González and F.-M. Matysik, Talanta, 2010, 82, 38–43 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |